

Abstract
Proline dehydrogenase (PRODH) catalyzes the FAD-dependent oxidation of l-proline to Δ1-pyrroline-5-carboxylate and is a target for inhibitor discovery because of its importance in cancer cell metabolism. Because human PRODH is challenging to purify, the PRODH domains of the bacterial bifunctional enzyme proline utilization A (PutA) have been used for inhibitor development. These systems have limitations due to large polypeptide chain length, conformational flexibility and the presence of domains unrelated to PRODH activity. Herein, we report the engineering of minimal PRODH domains for inhibitor discovery. The best designs contain one-third of the 1233-residue PutA from Sinorhizobium meliloti and include a linker that replaces the PutA α-domain. The minimal PRODHs exhibit near wild-type enzymatic activity and are susceptible to known inhibitors and inactivators. Crystal structures of minimal PRODHs inhibited by S-(−)-tetrahydro-2-furoic acid and 2-(furan-2-yl)acetic acid were determined at 1.23 and 1.72 Å resolution. Minimal PRODHs should be useful in chemical probe discovery.
Keywords: enzyme inhibition, enzyme kinetics, protein engineering, X-ray crystallography
Graphical abstract

Introduction
Proline dehydrogenase (PRODH) is the first enzyme of proline catabolism and catalyzes the FAD-dependent oxidation of l-proline to Δ1-pyrroline-5-carboxylate (P5C) (Fig. 1A; Tanner, 2019). The hydrolysis of P5C generates the semialdehyde substrate for l-glutamate-γ-semialdehyde dehydrogenase (GSALDH), which completes the pathway by catalyzing the NAD+-dependent oxidation of l-glutamate-γ-semialdehyde to l-glutamate. This pathway is highly conserved in eukaryotes and bacteria. Proline catabolism in eukaryotes occurs in the mitochondria, where PRODH is localized to the inner mitochondrial membrane and GSALDH is in the matrix. In some bacteria, PRODH and GSALDH are combined into the bifunctional enzyme known as proline utilization A (PutA) (Liu et al., 2017).
Fig. 1.

Proline catabolism and PRODH inhibitors and inactivators. (A) Reactions of proline catabolism. (B) Inhibitors and inactivators used in this study. The covalent adducts with FAD resulting from 2 and 3 are shown.
Proline catabolism and biosynthesis have been implicated as metabolic pathways selectively altered in cancer cells providing ATP, macromolecules and redox cofactors (Phang et al., 2012, 2015; Phang and Liu, 2012; Tanner et al., 2018; Phang, 2019; D'Aniello et al., 2020). PRODH has context-dependent anti-tumor and protumor roles. The PRODH gene is induced by p53, and its expression is downregulated in many tumors, most likely those carrying inactivated/mutated p53 variants (Polyak et al., 1997; Maxwell and Davis, 2000; Donald et al., 2001; Maxwell and Rivera, 2003). In contrast, PRODH expression is induced under hypoxic conditions in different tumor cell lines and in a mouse xenograft model of human breast tumor and contributes to cancer cell survival by inducing autophagy (Liu and Phang, 2012a, 2012b). PRODH may play an especially important role in breast cancer, where it is upregulated in a 3D spheroidal cell culture model of breast cancer compared to the 2D culture, as well as in metastases compared to primary tumors in breast cancer patients (Elia et al., 2017).
The central role of PRODH in the altered metabolism of certain cancer cells has motivated the discovery of PRODH inhibitors (Tanner et al., 2018; Bergers and Fendt, 2021). Four classes of PRODH inhibitors have been discovered, each with a different mechanism of inhibition. Noncovalent proline analogs bind in the proline/P5C site and inhibit via the classical competitive mechanism. The best characterized example is S-(-)-tetrahydro-2-furoic acid (1 in Fig. 1B), which has proven to be useful in both in cellulo and in vivo studies of cancer. 1 has been shown to reduce the growth of cancer cell spheroids and metastasis in a mouse model of breast cancer (Elia et al., 2017). Also, 1 has been used to study the role of PRODH in ATP production by the F1FO-ATPase (Pallag et al., 2022). N-propargylglycine (2) is a mechanism-based inactivator that covalently links the FAD N5 atom to an active site lysine with the loss of the inhibitor glycine moiety, effectively locking the FAD in a reduced, inactive state (Fig. 1B; White et al., 2008; Srivastava et al., 2010). The anticancer potential of 2 has been studied and was found to be synergistically lethal when used in combination with glutaminase inhibitors or p53-upregulating MDM2 antagonists (Scott et al., 2019, 2021). The third class is represented by 1,3-dithiolane-2-carboxylate (3), which inactivates PRODH via a light-mediated mechanism resulting in covalent modification of the FAD N5 with 1,3-dithiolane (Fig. 1B; Campbell et al., 2021b). The fourth mechanism has been observed with only one compound, thiazolidine-2-carboxylate. Incubation of PRODH with thiazolidine-2-carboxylate for very long times—days to weeks—eventually results in a covalent bond between the C5 of thiazolidine-2-carboxylate and the N5 of the FAD (Campbell et al., 2020). The mechanism of inactivation by thiazolidine-2-carboxylate is uncertain.
X-ray crystallography has been essential for the development of PRODH inhibitors and inactivators. Production of human PRODH—an inner mitochondrial membrane protein—is challenging; therefore, the field has relied on bacterial surrogates for crystallization. Sequence analysis suggests that the PutA PRODH domain is a good model for the active site of human PRODH (Tanner et al., 2018), and most of the key inhibitor complex structures have been determined using PutA PRODH domain constructs or full-length PutAs. Several crystal structures of bacterial PutAs/PRODHs complexed with 1 and other proline analogs revealed conserved aspects of inhibitor recognition and insights into structure-affinity relationships (Zhang et al., 2004; Luo et al., 2012; Luo et al., 2016; Bogner and Tanner, 2022). The crystal structure of PRODH inactivated by 2 was essential for determining the chemical structure of the covalently modified enzyme and hence the mechanism of inactivation (White et al., 2008). The discoveries of 3 and thiazolidine-2-carboxylate as covalent inactivators were made serendipitously with PutA in crystallo and later verified in solution (Campbell et al., 2020, 2021b).
Despite the success of X-ray crystallography in aiding the development of PRODH inhibitors, the enzymes used for crystallization have some drawbacks. Most of the work on the noncovalent inhibitors and compound 2 used C-terminal truncation variants of PutA from Escherichia coli containing the PRODH domain, as well as other domains known as the arm and α-domain (EcPutA86–630) (Zhang et al., 2004; Ostrander et al., 2009; Bogner and Tanner, 2022). The α-domain is largely disordered in the crystal structure, which contributes to inconsistencies in crystallization and crystallographic resolution. More recently, the full-length PutA from Sinorhizobium meliloti (SmPutA) has been used because of its exceptionally consistent crystallizability and routinely high resolution (1.4–1.9 Å) (Campbell et al., 2021a, 2021b). However, the large size of SmPutA (1233 residues) makes structure completion tedious, and the presence of domains not directly involved in PRODH activity and the potential for allosteric communication between the active sites can complicate the interpretation of inhibitor screening assays.
Herein, we report the engineering of minimal SmPutA PRODH domain constructs for inhibitor discovery. The best designs include less than one-third of full-length SmPutA and are catalytically active and highly crystallizable. Using one of them, referred to as SmPutAΔα2, we characterized the kinetics of inhibition by reversible and irreversible inhibitors and determined the crystal structures of PRODH inhibited by 1 and a new inhibitor, 2-(furan-2-yl)acetic acid (4). SmPutAΔα enzymes should facilitate future investigations of PRODH inhibitors.
Materials and Methods
Protein production
The expression plasmids used in this study were generated by GenScript from the parent plasmid (pNIC28-SmPutA), which encodes full-length SmPutA (UniProtKB F7X6I3, 1233 residues) with an N-terminal His6 tag and TEVP cleavage site. Supplementary Fig. S1, available at PEDS online, shows the amino acid sequence of full-length SmPutA.
Six domain deletion variants of SmPutA were generated. Supplementary Fig. S2, available at PEDS online, shows the amino acid sequences of the variants. Supplementary Fig. S3, available at PEDS online, shows a sequence alignment of the variants. Supplementary Table S1, available at PEDS online, summarizes the amino acid compositions of the variants.
The four SmPutAΔα enzymes were expressed and purified as follows. The expression plasmid was transformed into BL21 (DE3) competent cells and plated onto LB agar plates with 50 μg/ml kanamycin. A 20 ml LB culture, shaken overnight at 37°C and 200 rpm, was used to inoculate a 1 l culture, all containing 50 μg/ml kanamycin. The 1 l culture was grown at 37°C and 250 rpm, and once the OD600 reached ~0.6–0.8, the culture was induced with 0.5 mM IPTG and grown with continued shaking at 18°C overnight. Centrifugation at 5000 rpm for 30 min at 4°C was used to collect the cells and the cell pellet was stored at −20°C until ready for purification. The cell pellet was thawed on ice and resuspended in 50 mM Tris, 300 mM NaCl and 2% glycerol at pH 7.5 (buffer A), with 0.1 mM FAD and two Pierce™ EDTA-free protease inhibitor tablets (Thermo Fisher). The cells were lysed with sonication on ice at an amplitude of 50%, 2 min on and 2 min off, for about 10 min and centrifuged at 16 500 rpm for 1 h at 4°C using an SS-34 rotor. The lysate was then purified by gravity flow chromatography on a column containing Ni2+-NTA resin (Qiagen) pre-equilibrated with buffer A. The column was washed with buffer A supplemented with 20, 40 and 100 mM imidazole. The protein was eluted with buffer A supplemented with 250 mM imidazole and then incubated with TEVP for 1.5 h at room temperature to remove the His tag at a ratio of 0.2 mg of protease per 10 mg of target protein. The protein was then dialyzed overnight in buffer A and poured over a Ni2+-NTA column the next day. The flow-through and washes with 10 mM imidazole were collected, pooled based on SDS-PAGE and subjected to size exclusion chromatography (HiLoad 16/600 Superdex 200 pg column) using a running buffer of 50 mM Tris, 200 mM NaCl, 0.5 mM TCEP and 2% glycerol at pH 7.5 (buffer B). The purest fractions as judged by SDS-PAGE were pooled and dialyzed into a pre-crystallization buffer of 50 mM Tris, 100 mM NaCl, 1 mM TCEP, and 1% glycerol at pH 7.5. The protein was then concentrated to ~12 mg/ml, aliquoted and stored at −80°C. Typically, 30–100 mg of purified protein was obtained from a 1 l culture.
Crystallization of SmPutAΔα proteins
Crystal screening using the INDEX and Crystal Screen kits (Hampton Research) was done with all four SmPutAΔα constructs at 20°C using sitting drop vapor diffusion and an Oryx8 robot. The protein stock solution used for screening contained the protein at 10–12 mg/ml. SmPutAΔα1–3 were co-crystallized with 1 at 10 mM. The stock solution of 1 contained 1 M of 1 in 100 mM Tris pH 7.5. Swissci MRC 2 Well sitting drop trays were used with protein:reservoir drop ratios of 1:1 in one well and 2:1 in the other well.
The structure of SmPutAΔα1 complexed with 1 was determined from a crystal form optimized from Index screen condition H11 (0.1 M potassium thiocyanate and 25% (w/v) PEG MME 2000). The protein (6 mg/ml) was co-crystallized with 10 mM 1 in a Hampton 48 well MRC Maxi tray using 0.05–0.08 M potassium thiocyanate and 19–24% (w/v) PEG MME 2000. Crystals were cryoprotected in the reservoir solution supplemented with 20% PEG 200 and flash-cooled in liquid nitrogen.
The structure of SmPutAΔα2 complexed with 1 was determined from a crystal harvested directly from Index screen condition H6 (0.2 M sodium formate and 20% (w/v) PEG 3350). The crystal was soaked in a cryobuffer made from the reservoir containing 20% PEG 200 and flash cooled in liquid nitrogen. Additional 1 was not included during soaking.
Index H6 condition was used as the starting point for optimization of SmPutAΔα2 using the sitting-drop vapor diffusion method with drops containing 2 μl each of the protein and reservoir. Optimization consisted of systematic variation of sodium formate in the range of 0.05–0.2 M and PEG 3350 in the range of 19–24% (w/v). Crystals from optimization trials were crushed to make a microseed stock, which was used in subsequent trials. The seeds were introduced via seed-streaking with horsehair. The crystals typically grew to diffraction quality size overnight.
The PEG 3350/formate recipe was used to determine a structure of SmPutAΔα2 complexed with 4 via co-crystallization with a protein solution containing 10 mg/ml SmPutAΔα2 and 10 mM of 4. A crystal was cryoprotected in the reservoir supplemented with 20 mM 4 and 20% PEG 200, and after a few minutes of soaking, it was flash cooled in liquid nitrogen.
The in crystallo inactivation of SmPutAΔα2 by 3 was performed as follows. Crystals of the noncovalent complex were grown by co-crystallization (8 mg/ml protein, 10 mM 3) using a reservoir of 0.05–0.2 M sodium formate and 17–22% (w/v) PEG 3350. Yellow crystals were obtained, and exposure to the microscope light for a few minutes bleached the yellow color, indicating that inactivation had occurred. The bleached crystals were cryoprotected in a solution containing 20% PEG 200, 10 mM 3, and reservoir solution and flash cooled in liquid nitrogen after a few minutes.
X-ray diffraction data collection, phasing and refinement
Diffraction experiments were performed at beamlines 24-ID-C and 24-ID-E of the Advanced Photon Source using Eiger detectors. The data were processed by the NECAT automated processing pipeline, which uses XDS (Kabsch, 2010) for autoindexing, integration and scaling, and the CCP4 modules POINTLESS for space group determination, AIMLESS for merging and CTRUNCATE for conversion of intensities to amplitudes and the generation of a test set for Rfree calculations (Evans, 2011). Data processing revealed two P1 crystal forms, distinguished mainly by a difference in γ (108° vs 114°) (Supplementary Table SII available at PEDS online). The asymmetric unit contains two proteins with solvent content of 37.4% and VM of 1.97 Å3/Da (Matthews, 1968). Data processing statistics are listed in Supplementary Table SII, available at PEDS online.
Initial phases for the structure of SmPutAΔα1–1 were calculated using molecular replacement as implemented in the program Phaser (McCoy et al., 2007). The search model was derived from a structure of full-length SmPutA (PDB 6X9A) and included residues 26–78 and 196–538. The model from molecular replacement was used as the starting point for rounds of iterative model building in Coot and refinement in PHENIX (Emsley et al., 2010; Afonine et al., 2012). Crystallographic refinement calculations of SmPutAΔα2 structures were started from refined models of SmPutAΔα1. Restraint files for noncovalent ligands were obtained with eLBOW from SMILES strings (Moriarty et al., 2009). A consistent definition of the asymmetric unit was used for all structures and corresponds to the two-body assembly having the highest interfacial surface area (~800 Å2) and C2 symmetry. Analysis of protein-protein interfaces in the crystal lattice using PDBePISA (Krissinel and Henrick, 2007) suggested that this assembly may be stable in solution. Non-crystallographic symmetry restraints were used in the refinement of SmPutAΔα2 inactivated by 3 because of its modest resolution (2.6 Å). The structures were validated using MolProbity, the wwPDB validation service and polder omit maps (Chen et al., 2010; Gore et al., 2017; Liebschner et al., 2017). Refinement statistics are listed in Supplementary Table SII, available at PEDS online.
The geometrical restraints for the covalent FAD adduct in the refinement of SmPutAΔα2 inactivated by 3 were set up as follows. Restraints for 1,3-dithiolane and reduced FAD (chemical ID FDA) were generated using eLBOW. A model of 1,3-dithiolane was positioned into density in Coot to form the correct covalent bond. Explicit bond and angle terms involving the atoms connecting the FDA to 1,3-dithiolane were added using the phenix.refine option ‘geometry_restraints.edits’. The ideal bond length between the FDA N5 and 1,3-dithiolane C2 was set to 1.47 Å (σ = 0.02 Å), based on knowledge of C-N bond distances from small-molecule X-ray crystallography (Allen et al., 1987). The four bond angles involving these two atoms were also defined and assumed to be 109.5° (σ = 10°). The occupancy of the 1,3-dithiolane adduct refined to 0.75.
The coordinates and structure factor amplitudes for the noncovalent inhibitor complexes have been deposited in the Protein Data Bank under the accession codes 8DKO for SmPutAΔα1–1, 8DKP for SmPutAΔα2–1 and 8DKQ for SmPutAΔα2–4. The structure of SmPutAΔα2 inactivated by 3 was not deposited because of the poor refinement statistics and the fact that a much higher resolution example of the complex using full-length SmPutA is available from the PDB (PDB ID 7MYA, 1.56 Å resolution) (Campbell et al., 2021a, 2021b).
PRODH activity assays and analysis of noncovalent inhibition
Kinetic measurements were performed using the ortho-aminobenzaldehyde (o-AB) assay in a 96-well plate in a BioTek Epoch 2 microplate spectrophotometer at 23°C. The assay monitors the production of P5C as an adduct formed with o-AB, which is detected by absorbance at 443 nm (ε443 = 2.59 mM−1 cm−1). The electron acceptor menadione was used to reoxidize the reduced FAD of PRODH, enabling catalytic cycling. The assay buffer contained 20 mM MOPS pH 7.5 and 10 mM MgCl2. Kinetic measurements were performed with l-proline as the variable substrate (0–350 mM), 4 mM o-AB, 0.15 mM menadione, and 63 nM of enzyme. l-proline was spotted on the plate and a master mix including enzyme, menadione, o-AB and buffer was added to the plate by multichannel pipette to initiate the reaction. The initial rate was determined from linear regression of the first 5 min of the progress curve using Origin v9.7.0.188 software. The kinetic constants Km and kcat are listed in Supplementary Table SIII, available at PEDS online. Inhibition kinetics was performed similarly with the addition of inhibitors also spotted on the plate with l-proline. Final concentrations of the inhibitor were 0–2 mM 1 or 4. The initial rate data as functions of both substrate and inhibitor concentrations were fit globally to a competitive or mixed inhibition model using Origin.
Kinetic analysis of covalent inactivation by 2
A progress curve approach (Salminen et al., 2011) was used to measure the time-dependent inactivation of SmPutAΔα2 by 2. A 96-well plate was spotted with l-proline and various concentrations of 2. The reaction was initiated by the addition of a master mix containing o-AB, menadione, enzyme and o-AB assay buffer. Final concentrations of reagents were 350 mM l-proline (~10× Km value), 0–40 mM 2, 4 mM o-AB, 0.15 mM menadione and 525 nM SmPutAΔα2. The reaction was measured for 35 min at 23°C. The resulting progress curves were fit to equation (1) using Origin software to calculate kobs as a function of inhibitor concentration.
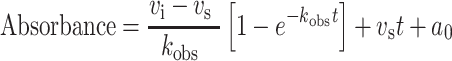 |
(1) |
In equation (1), vi is the initial velocity, vs is the steady state velocity, t is time, a0 is the initial absorbance and kobs is the observed rate of inactivation. A replot of the data using equation (2) was used to obtain kinetic parameters.
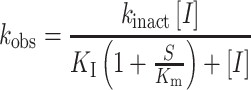 |
(2) |
In equation (2), [I] is the inactivator concentration, S is the substrate concentration (l-proline), Km is the Michaelis constant for the substrate, and kinact and KI are the kinetic parameters of inactivation. The plot did not exhibit saturation, which prevented the determination of kinact and KI separately. Therefore, linear regression was used to estimate the apparent second order rate constant for enzyme inactivation, kinact/KI.
Kinetic analysis of photoinduced covalent inactivation by 3
The method of Kitz and Wilson was used to measure the inactivation of SmPutAΔα2 by 3 (Kitz and Wilson, 1962). SmPutAΔα2 and 3 were incubated together in a 96-well plate on ice and exposed to white light with a 700 lumen LED bulb from a distance of 20 cm for 0–12 min. The total volume of the inactivation reactions was 20 μl and contained 1.25 μM of SmPutAΔα2 and 0–10 μM of 3. At 12 min, 180 μl of an enzyme activity assay master mix containing l-proline, o-AB, menadione and o-AB assay buffer was added to the plate to initiate the PRODH activity assay. The final concentrations of reagents in the activity assay (total volume of 200 μl) were 50 mM l-proline, 4 mM o-AB, 0.15 mM menadione, 125 nM SmPutAΔα2 and 0–1 μM 3. Initial rates were measured at 23°C, expressed as percent activity (normalized by the rate of SmPutAΔα2 that had not been treated with 3) vs time, and fit to an exponential decay function using Origin software. The time constants from the exponential decay fitting were plotted against the reciprocal of the concentration of 3 on a Kitz and Wilson replot to estimate the kinetic parameters of inactivation, kinact and KI.
Small-angle X-ray scattering
Purified SmPutAΔα2 was dialyzed vs a buffer containing 25 mM HEPES pH 7.6, 150 mM NaCl, and 1 mM TCEP, and then pipetted into a 96-well plate at six concentrations in the range of 1.1–6.8 mg/ml. The dialysate was reserved for measurement of the background small-angle X-ray scattering (SAXS) curve. Shutterless SAXS data collection was performed at 20°C using a Pilatus detector at beamline 12.3.1 of the Advanced Light Source through the SIBYLS Mail-in High Throughput SAXS program (Dyer et al., 2014). The total exposure time was 10 s per sample, framed every 0.33 s (30 frames total). The wavelength was 1.234 Å. Buffer-subtracted frames were averaged using SAXS FrameSlice (Shin and Hura, 2022), which enables the averaging of different numbers of frames in three different q regions corresponding to the Guinier region, Porod region and wide angle region. The number of frames used in averaging is listed in Supplementary Table SIV, available at PEDS online. Radiation-induced artifacts at were evident at q < 0.1 Å−1 for the three highest concentrations, so only the lowest three concentration samples were used for further analysis. PRIMUS (Konarev et al., 2003) was used to perform Guinier analysis, calculate the distance distribution function and estimate the molecular weight. The molecular weight was also estimated with SAXS MoW (Piiadov et al., 2019). Theoretical SAXS curves were calculated using FoXS (Schneidman-Duhovny et al., 2010). Shape reconstructions with the constraint of C2 symmetry were performed with DENSS (Grant, 2018) and DAMMIF (20 models) (Franke and Svergun, 2009). The SAXS data, P(r) curves and best fit models have been deposited in the SASBDB (Kikhney et al., 2020) under the accession codes SASDQ48 for 1.1 mg/ml, SASDQ58 for 2.3 mg/ml and SASDQ68 for 3.4 mg/ml.
Results
Structural layout of SmPutA domains
The design plan for engineering minimal PRODHs was based on crystal structures of SmPutA (e.g. PDB ID 7MY9, 5KF6). A PutA was chosen rather than a bacterial monofunctional PRODH because human PRODH is predicted to be more similar to PutA in terms of the fold of the PRODH domain and flavin cofactor preference (Tanner et al., 2018). Among PutAs, SmPutA was chosen as the template because of its robust expression, purification and crystallization properties. Also, SmPutA is primarily monomeric at concentrations used in crystallization, and thus, the design was not complicated by quaternary structure considerations.
For the purpose of protein engineering, we considered SmPutA to consist of several structural elements and domains (Fig. 2). The structure begins with an N-terminal extension of 28 residues, the first 13 of which are disordered in the crystal structure. The N-terminal extension connects to a 3-helix arm-like domain (residues 29–78), followed by a domain consisting of six α-helices (α-domain, residues 79–197). The PRODH active site is located in a (βα)8 barrel domain consisting of residues 198–497. Residues 498–554 connect the PRODH barrel to the second half of the protein, which contains the Rossmann NAD+-binding domain, GSALDH catalytic domain and a structural Rossmann fold domain denoted ALDHSF for its similarity to aldehyde dehydrogenase superfamily domains.
Fig. 2.

Structure of SmPutA and domain diagrams. (A) Tertiary structure of SmPutA (PDB ID 7MY9). Structural elements and domains are color coded according to the legend in panel B. FAD and NAD+ are shown in yellow and pink sticks, respectively. (B) Domain diagrams of SmPutA and the variants studied.
The relationship of the arm to the PRODH barrel is notable. The arm domain consists of three α-helices that wrap tightly around the outside of the PRODH barrel (Fig. 2A). The helices of the arm are amphipathic, and their nonpolar faces pack against the nonpolar surface of the barrel, while the polar faces contact solvent. This intimate relationship suggests that the barrel may not fold properly without the arm or could have low solubility due to the exposure of hydrophobic surface area. Thus, for design purposes, we considered the arm and barrel to be inseparable.
N- and C-terminal truncations
The first designs tested the tolerance of SmPutA to removing large sections at the N- and C-termini. SmPutA1–571 and SmPutA1–538 lack the GSALDH module, whereas SmPutA26–538 also lacks the N-terminal extension (Fig. 2B). The rationale for 26 and 571 as truncation points is that they correspond to the termini of an E. coli PutA PRODH domain construct used previously (EcPutA86–630) (Ostrander et al., 2009; Srivastava et al., 2010; Zhu et al., 2013). The rationale for 538 as an endpoint is that it aligns to the last residue visible in electron density maps of EcPutA86–630 structures.
All three truncated enzymes were expressed as soluble proteins in E. coli. SmPutA1–571 did not bind to the Ni-NTA column and was purified using ammonium sulfate precipitation in place of affinity chromatography. The purified enzymes were yellow, indicating incorporation of the FAD cofactor, and exhibited PRODH activity. Crystal screens produced no obvious hits. Based on these results, we engineered variants of SmPutA26–538 lacking the α-domain.
First-generation SmPutAΔα design: SmPutAΔα1
Elimination of the α-domain was hypothesized to enhance crystallizability by reducing conformational flexibility. In full-length SmPutA, the α-domain forms tertiary structural contacts with the GSALDH catalytic domain and C-terminus of the protein (Fig. 2A). These contacts presumably contribute to the stability and foldedness of the α-domain, and their absence in the truncation variants may have perturbed the structure of the α-domain. Indeed, the α-domain is mostly disordered in structures of EcPutA86–630, which lack the GSALDH half of the protein. Also, sections of the α-domain tend to be disordered in full-length PutA structures. For these reasons, we designed variants of SmPutA26–538 that lack the α-domain (denoted SmPutAΔα).
The first-generation SmPutAΔα construct (SmPutAΔα1) has a flexible linker inserted between Gly83 and Met190, thus eliminating the α-domain (Fig. 2B). Gly83 is located at the end of the loop that connects the arm and α-domain (Supplementary Fig. S4, available at PEDS online). Electron density for this loop is weak in SmPutA structures, indicating high flexibility and suitability as a linker to the PRODH domain. Met190 is located near the C-terminus of the last helix of the α-domain (Supplementary Fig. S4, available at PEDS online). The rationale for Met190 as an anchor residue was to preserve a few turns of α-helix to allow for a native-like structural connection to the PRODH barrel. The linear distance between anchor residues Gly83 and Met190 is 9.1 Å. Homology modeling with SWISS-MODEL (Arnold et al., 2006) aided the design of linkers to span this distance without significant perturbation to the three-dimensional structure. Models were predicted for several linkers of length 1–7 residues and having Gly and Ser in the sequence. Linkers that did not perturb the α-helical structures around the anchor residues were deemed acceptable. These calculations suggested SSGS as a plausible linker to span the 9.1 Å gap between Gly83 and Met190.
SmPutAΔα1 was expressed in E. coli as a soluble protein and purified. The purified enzyme exhibited PRODH activity with hyperbolic dependence of the rate on proline concentration (Fig. 3A). The kinetic constants of SmPutAΔα1 are kcat of 0.6 s−1 and Km of 24 mM (Supplementary Table SIII, available at PEDS online). For reference, the kinetic constants of full-length SmPutA are kcat of 1.3 s−1 and Km of 46 mM. Thus, the catalytic efficiency of SmPutAΔα1 is within 20% of full-length SmPutA, indicating that removal of two-thirds the protein did not significantly perturb the structure of the PRODH active site.
Fig. 3.

PRODH activity of SmPutAΔα variants and the kinetics of reversible inhibition of SmPutAΔα2 by 1 and 4. (A) PRODH activities of full-length SmPutA and the four SmPutAΔα domain deletion variants. The kinetic constants are listed in Supplementary Table SIII, available at PEDS online. (B) Inhibition of SmPutAΔα2 by 1. The data were globally fit to the competitive inhibition model resulting in a Ki value of 16.2 ± 2.0 μM. (C) Inhibition of SmPutAΔα2 by 4. The data were globally fit to a mixed inhibition model. The inhibition constants for the competitive and uncompetitive components are 198 ± 40 and 970 ± 168 μM, respectively. Analysis of the data using other inhibition models is shown in Supplementary Fig. S7, available at PEDS online.
Crystal screens of SmPutAΔα1 complexed with 1 were very successful. For example, 14 different conditions in the Hampton Research Index screen produced yellow crystals (Supplementary Fig. S5, available at PEDS online). The best crystals were obtained in a condition containing potassium thiocyanate as the salt and PEG MME 2000 as the precipitating agent. Optimization of this condition led to a 1.80 Å resolution structure of SmPutAΔα1 complexed with 1 in space group P1 with two copies of the protein in the asymmetric unit (Supplementary Table SII, available at PEDS online).
SmPutAΔα1 exhibits the expected fold, consisting of the α-helical arm, the (βα)8 barrel containing the PRODH active site and a portion of the polypeptide that normally connects to the NAD+-binding domain in the full-length protein (Fig. 4A). Electron density for the engineered linker between the arm and barrel was weak; eight residues of the linker were omitted from chain A (SGSGSSGS) and seven were omitted from chain B (HSGSGSS). At the C-terminus of SmPutAΔα1, the electron density supported the modeling of the polypeptide chain through Met524 in one chain and Val526 in the other. Thus, 10–14 residues of the C-terminus are conformationally disordered. The N-terminus exhibits less disorder; the complete N-terminus could be modeled in one chain and only the first three residues are disordered in the other. In total, the electron density supported modeling 94% of the expected residues. The RMSD between SmPutAΔα1 and full-length SmPutA (PDB 5KF6) is 0.55 Å for 372 aligned residues. This level of structural similarity is very high, considering that the two SmPutAΔα1 molecules in the asymmetric unit align with an RMSD of 0.39 Å for 378 residues.
Fig. 4.

Structure of SmPutAΔα1 complexed with 1. (A) Cartoon representation of the protein fold of SmPutAΔα1. The protomer is colored in a rainbow scheme with blue at the N-terminus and red at the C-terminus. FAD and 1 are shown in yellow and pink sticks, respectively. The missing residues of the engineered linker are indicated by a dashed curve. The inset shows electron density for the FAD (polder omit map, 4σ). (B) The two-body assembly in the asymmetric unit viewed down the 2-fold axis. FAD and compound 1 are shown in yellow and pink sticks, respectively. The α8 helices in the dimer interface are noted. (C) Electron density (polder omit map, 4σ) and interactions for 1 bound to SmPutAΔα1. (D) Comparison of the active sites of SmPutAΔα1–1 (pink) and full-length SmPutA-1 (blue, PDB ID 5KF6).
The asymmetric unit contains a presumed dimer of SmPutAΔα1 (Fig. 4B). This assembly was chosen because it has the largest protein-protein interface in the crystal lattice (~800 Å2) and is predicted to be stable in solution based on analysis with PDBePISA. The presumed dimer has C2 symmetry with the C-terminal ends of the strands of the barrels facing each other. In this arrangement, the α8 helices contact each other in the dimer interface. This is notable because α8 contains several conserved active site residues that contact proline analog inhibitors. We note that the monofunctional PRODH from Deinococcus radiodurans also exhibits this packing arrangement in the crystal lattice (PDB ID 4H6Q) (Luo et al., 2012).
Electron density for the FAD and 1 is strong and unambiguous (Fig. 4), and the conformations of these ligands and their interactions with the enzyme are nearly identical to those of full-length SmPutA. In particular, as in other PRODH-1 structures, the carboxylate of 1 ion pairs with two arginine residues and one lysine, while the O heteroatom hydrogen bonds with a conserved water molecule (Fig. 4C). Other conserved residues complete the binding pocket by making nonpolar contacts with the inhibitor ring, including two tyrosine residues and a leucine. The inhibitor’s pose and the conformations of the surrounding side chains are nearly identical to those of full-length SmPutA (Fig. 4D). Overall, these results show that removal of two-thirds of the amino acids from the polypeptide chain did not disrupt the structure and catalytic function of the PRODH domain.
Second-generation SmPutAΔα designs
The crystal structure of SmPutAΔα1 was used to guide the design of second-generation SmPutAΔα constructs. The disorder at the C-terminus of SmPutAΔα1 in the crystal structure suggested that further truncation of the polypeptide chain may be advantageous. Arg522 was chosen as the truncation point because it is the last residue of the final secondary structure element observed in the crystal structure (a short α-helix, Fig. 4A). Also, the disorder in the engineered linker between the arm and barrel motivated the design of shorter linkers. These considerations led to three new designs: one having a shorter C-terminus (SmPutAΔα2) and two having both a shorter C-terminus and different linkers (SmPutAΔα3 and SmPutAΔα4) (Fig. 2B).
The second-generation SmPutAΔα constructs were tested for expression, enzymatic activity and crystallizability. All three were amenable to expression in E. coli and purification by metal ion affinity chromatography, and all three exhibited PRODH catalytic activity (Fig. 3A). SmPutAΔα2 has a catalytic efficiency within 16% of SmPutAΔα1 and 3–5 times higher than SmPutAΔα3 and SmPutAΔα4 (Supplementary Table SIII, available at PEDS online). All three second-generation SmPutAΔα constructs were crystallizable based on initial crystal screens (Supplementary Fig. S5, available at PEDS online). SmPutAΔα2 yielded the best looking and quickest forming crystals in the screens. SmPutAΔα2 was deemed to have the best combination of catalytic activity and crystallizability and was therefore used in additional inhibition and structural studies.
Reversible inhibition and structures of SmPutAΔα2
The susceptibility of SmPutAΔα2 to reversible PRODH inhibitors was tested using enzyme kinetics assays. The known proline analog inhibitor 1 was found to be a competitive inhibitor (with l-proline) with a Ki of 16.2 ± 2.0 μM (Fig. 3B). This value is lower than that of full-length SmPutA (Ki = 900 ± 100 μM, Supplementary Fig. S6, available at PEDS online) and other PRODHs (Ki = 0.2–1 mM) (Zhang et al., 2004; White et al., 2007; Bogner and Tanner, 2022).
The structure of SmPutAΔα2 complexed with 1 was determined at 1.23 Å resolution from a crystal harvested directly from a crystal screen (Supplementary Table SII, available at PEDS online). This is the highest resolution structure of a PRODH. Electron density for the FAD and 1 is outstanding, and the conformations of these ligands and their interactions with the enzyme are essentially identical to those of SmPutAΔα1 and full-length SmPutA (Fig. 5A).
Fig. 5.

Active sites of SmPutAΔα2 inhibited by 1, 4 and 3. (A) Electron density for the FAD (left) and 1 (right) bound to SmPutAΔα2. The cages represent polder omit maps (4σ). (B) Electron density (polder omit, 4σ) and interactions for 4 bound to SmPutAΔα2. (C) Superposition of the complexes of SmPutAΔα2 with 1 (cyan) and 4 (salmon). (D) Two views of electron density (polder omit, 3σ) for the reduced FAD (dark gray) covalently modified by 1,3-dithiolane (magenta).
SmPutAΔα2 was also used to characterize a new inhibitor (4). Compound 4 exhibited concentration-dependent inhibition of PRODH activity (Fig. 3C). Fitting of the kinetic data to the competitive model yielded a Ki of ~80 μM, but the quality of the fit was suboptimal (Supplementary Fig. S7A, available at PEDS online). An improved fit was obtained with a mixed model including both competitive and uncompetitive inhibition (Fig. 3C). The inhibition constants for the competitive and uncompetitive components are 0.2 mM and 1 mM, respectively, indicating that the mechanism is mainly competitive with l-proline, and the affinity is lower than 1.
The structure of SmPutAΔα2 complexed with 4 was determined at 1.72 Å resolution (Supplementary Table SII, available at PEDS online). 4 binds in the proline site, with its carboxylate engaging the two conserved Arg residues of the α8 helix (Arg488, Arg489), and its ring packed against the isoalloxazine of the FAD (Fig. 5B). Compared to the complex with 1, the ring of 4 is shifted by 0.7 Å (Fig. 5C). Consequently, the O heteroatom of 4 does not hydrogen bond to the conserved water molecule, and its carboxylate does not ion pair with Lys265 (Fig. 5B). Apparently, these interactions are sacrificed to accommodate the extra methylene group of 4. The loss of these interactions may account for the lower affinity of 4 compared to 1.
Covalent inactivation of SmPutAΔα2
The susceptibility of SmPutAΔα2 to known PRODH covalent inactivators was tested. The mechanism-based inactivator 2 was evaluated using a progress curve approach in which enzyme activity is measured in the presence of the inactivator. The progress curves show time-dependent inhibition and eventual flattening, as expected for an irreversible covalent inactivator (Fig. 6A, left). The apparent inactivation rate constants (kobs) obtained from fitting the progress curves to equation (1) were plotted as a function of inactivator concentration (Fig. 6A, right). The replot did not exhibit saturation, which prevented the determination of kinact and KI separately. Therefore, linear regression was used to estimate an apparent second-order rate constant for enzyme inactivation (kinact/KI) of 0.19 ± 0.02 M−1 s−1. For reference, kinact/KI for 2 with other PRODHs ranges from 1 to 9 M−1 s−1. Crystals of SmPutAΔα2 inactivated by 2 were obtained using the PEG 3350/formate condition, but the diffraction was very weak (~8 Å) and a structure was not determined.
Fig. 6.

Kinetics of covalent inactivation of SmPutAΔα2. (A) Time-dependent inhibition of SmPutAΔα2 by 2. The colored symbols are experimental progress curves monitoring the production of o-AB-P5C in the presence of 35 mM proline, 4 mM o-AB, 0.15 mM menadione and various concentrations of 2. The black curves represent the fits to equation (1). The replot on the right shows kobs as a function of concentration of 2. Error bars come from the fits of the experimental progress curves to equation (1). The slope of the line gives an estimate of the apparent second-order rate constant (kinact/KI) as 0.19 ± 0.02 M−1 s−1. (B) Kitz and Wilson analysis of the kinetics of photoinduced covalent inactivation of SmPutAΔα2 by 3. Using the o-AB assay, PRODH activity remaining after incubation of enzyme and varying concentrations of 3 is plotted as percent activity as a function of time for four inactivator concentrations (1.25, 2.5, 5, and 10 μM). The inset shows the replot of the time constant of inactivation (t1/2) as a function of the reciprocal concentration of 3. Error bars come from the fit of the experimental data to an exponential decay function. The following inactivation parameters were obtained from the fitting: kinact = 0.76 ± 0.25 min−1 and KI = 6.22 ± 2.58 μM. The apparent second-order rate constant (kinact/KI) is 2035 ± 1078 M−1 s−1.
The photoinduced covalent inactivator 3 was tested using the approach of Kitz and Wilson in which the enzyme is incubated with the inactivator for various times and then the remaining activity is measured. 3 elicited time-dependent inhibition when incubated with SmPutAΔα2 under illumination by white light, as expected (Fig. 6B). The replot of the observed inactivation rate constant vs the reciprocal of the inactivator concentration was used to estimate the apparent kinetic parameters for enzyme inactivation of kinact = 0.76 ± 0.25 min−1 and KI = 6.22 ± 2.58 μM (Fig. 6B, inset). The apparent second-order rate constant (kinact/KI) is 2035 ± 1078 M−1 s−1. The corresponding values for other PRODHs have not been reported.
Photoinduced covalent inactivation of SmPutAΔα2 by 3 was attempted in crystallo as we had done previously with crystals of full-length SmPutA (Campbell et al., 2021b). SmPutAΔα2 was co-crystallized with 3 under low-light conditions, resulting in yellow crystals consistent with 3 being noncovalently bound in the active site. Exposure of the crystals to white light bleached the yellow color, indicating that covalent inactivation occurred. The bleached crystals were harvested and flash cooled in liquid nitrogen. The diffraction resolution was markedly degraded by covalent inactivation (Wilson B-factor of 65 Å2) and a 2.6 Å data set was obtained (Supplementary Table SII, available at PEDS online). The electron density was very weak for several sections of the polypeptide chain and many side chains, and only about 80% of the structure could be modeled. Also, the global refinement indicators of Rcryst = 25% and Rfree = 30% are high compared to typical 2.6 Å crystal structures in the PDB. Nevertheless, the electron density did exhibit a feature consistent with 1,3-dithiolane covalently bonded to the N5 of the FAD, as expected (Fig. 5D).
Oligomeric structure of SmPutAΔα2 in solution
The oligomeric structure of SmPutAΔα2 in solution was studied with SAXS. SAXS data were obtained at three protein concentrations in the range of 1.1–3.4 mg/ml (Fig. 7A). Inspection of the SAXS curves after scaling for differences in protein concentration revealed an increase in the intensity at q < 0.1 Å−1 with increasing protein concentration, suggesting self-association (Fig. 7A, inset). Likewise, Guinier analysis revealed a consistent increase in the radius of gyration (Rg) from 27 Å at the lowest protein concentration to 30 Å at the highest concentration (Fig. 7B). Calculations of the distance distribution function yielded Rg values within ~1 Å of the Guinier values (Supplementary Table SIV, available at PEDS online). For reference, the Rg values of the SmPutAΔα2 monomer and dimer in the asymmetric unit are 19.5 and 28.1 Å, respectively. Thus, the Rg data suggest that the protein is mainly dimeric in the concentration range of 1–3 mg/ml.
Fig. 7.

SAXS analysis of SmPutAΔα2. (A) SAXS curves for three samples at protein concentrations of 1.1 mg/ml (black), 2.3 mg/ml (red) and 3.4 mg/ml (blue) mg/ml. The inset shows the low q region after scaling the data for differences in protein concentration. (B) Guinier plots. (C) SAXS Similarity heatmap based on the volatility of ratio (VR) calculated in the default range of q = 0.015–0.2 Å−1. Each matrix element represents VR for a pair of SAXS curves. The experimental curves are in rows/columns 1–3. The theoretical curves calculated from 10 models of the dimer are in rows/column 4–13. Dark blue corresponds the highest similarity (lowest VR). Dark red corresponds to the lowest similarity (highest VR). (D) Comparison of the experimental SAXS curve (1.1 mg/ml) with theoretical curves calculated from the dimer of SmPutAΔα2 in the asymmetric unit (solid red) and a monomer of SmPutAΔα2 (dotted blue). The inset shows the dimer in the asymmetric unit docked into the shape reconstructions from DENSS and DAMMIF.
The SAXS data were used to determine the quaternary structure of the dimer in solution. Analysis of crystal packing with PDBePISA (Krissinel and Henrick, 2007) revealed 10 two-body assemblies but only one was predicted to be stable in solution: the dimer in the asymmetric unit (Fig. 4B). This dimer has an Rg of 28.1 Å, interface surface area of 782 Å2 and Complexation Significance Score of 0.35. The other nine two-body assemblies span an Rg range of 29–34 Å, have lower interface areas of 100–500 Å2 and Complexation Significance Scores of 0.0. The volatility of ratio metric (VR) was used to compare the theoretical SAXS curves calculated from the 10 dimer models with the experimental data (Hura et al., 2013). The heatmap of VR shows that all three experimental curves are more similar to the dimer in the asymmetric unit than to any of the other nine dimers (Fig. 7C, matrix elements labeled with *). These results support the prediction of PDBePISA that the dimer in the asymmetric is stable in solution.
FoXS was used to compare the experimental SAXS data to the theoretical scattering curves calculated from atomic models of the monomer and the dimer in the asymmetric unit. These calculations yielded goodness-of-fit parameters (χ2) of 2–18 for the monomer and 0.1–0.5 for the dimer, indicating a much better fit for the dimer (Supplementary Table SIV, available at PEDS online). Visual inspection confirmed the superior fit for the dimer (Fig. 7D). Multi-state analysis with MultiFoXS did not improve the fits. Finally, shape reconstruction calculations returned a prolate ellipsoid with dimensions similar to the dimer in the asymmetric unit (Fig. 7D, inset). Altogether, these results are consistent with the dimer in the asymmetric unit being stable in solution and likely the predominant oligomeric form present at the concentrations used for crystallization (8–10 mg/ml).
Discussion
Protein engineering is a common technique in aiding the efforts of crystallography. Removing flexible portions of proteins and generating a more rigid and stable unit can result in crystallographic success (Derewenda, 2010; Ruggiero et al., 2012). It has become common practice to truncate the N- and C-terminal ends of proteins; however, internal insertions and deletions are less standard due to the assumption that the changes would drastically affect protein stability and folding (Dale et al., 2003). Here, minimal PRODH enzymes were generated by removing several structural domains and other elements from the bifunctional PRODH-GSALDH enzyme SmPutA, a 1233-residue protein. The best designs contain only about one-third of SmPutA and lack the α-domain, the entire GSALDH catalytic apparatus, and the ALDHSF domain. Expression and purification of the SmPutAΔα constructs were straightforward and routinely yielded tens of milligrams of highly pure, catalytically active, and crystallizable enzyme from a 1-l E. coli culture. Moreover, the minimal PRODHs are susceptible to both reversible and irreversible inhibitors.
Protein engineering of monofunctional PRODH has been investigated previously. Tallarita et al. reported a study of N- and C-truncation variants of human PRODH, an inner mitochondrial membrane protein (Tallarita et al., 2012). A total of seven constructs were generated, and one was about 50% soluble in E. coli. The soluble construct exhibited a typical flavin spectrum but has a relatively high Km for proline (0.5 M). The crystallizability of this protein was not described and a structure of the enzyme has not appeared in the PDB.
The engineering of the bacterial monofunctional PRODH from Thermus thermophilus (TtPRODH) has also been reported. Bacterial monofunctional PRODHs resemble the PRODH domain of PutA in having a (βα)8 barrel fold and conserved α8 helix (Supplementary Fig. S8, available at PEDS online). However, they differ from PutA in lacking both the arm that wraps around the PutA barrel and an α-helix known as α5a between PutA strands β5 and β6 (Supplementary Fig. S8, available at PEDS online). Huijbers and coworkers showed that fusion to maltose-binding protein improved the solubility and expression of TtPRODH (Huijbers and van Berkel, 2015), and the removal of α-helices from the N-terminus alters oligomerization and activity (Huijbers et al., 2017, 2018, 2019). These α-helices in TtPRODH, known as αA, αB, and αC, are located in the same region of space as the PutA α-domain and seem to be an abbreviated α-domain (Supplementary Fig. S8, available at PEDS online). Their work also showed importantly that FMN, rather than FAD, is the likely cofactor of bacterial monofunctional PRODH, which further distinguishes this class of PRODH from PutA and human PRODH (Huijbers et al., 2017).
SmPutA proved to be remarkably tolerant to molecular dissection. Especially notable is that excision of the α-domain—an internal domain of SmPutA—did not compromise PRODH activity. This result suggests that it may be possible to create bifunctional SmPutA constructs that retain both catalytic activities and lack only the α-domain. Such proteins could be useful for studying the function of the α-domain in PutA, which has been hypothesized to be membrane association and/or substrate channeling (Christgen et al., 2017; Tanner, 2019). Biosensing of l-proline is another potential application for minimal PRODHs. Electrodes containing immobilized PRODH have been investigated for measuring l-proline in cells by differential-pulse voltammetry (Hasanzadeh et al., 2017). The robustness and small size of SmPutAΔα constructs may be useful in this application.
Removing the α-domain was essential for crystallization. Whereas none of the N- and C-terminal truncation constructs were crystallizable, all four SmPutAΔα constructs produced positive results in crystal screening trials. Also, trimming the C-terminus of SmPutAΔα1 by 16 residues to SmPutAΔα2 appeared to facilitate crystallization. Initial testing suggests that SmPutAΔα2 may yield the best crystals; a 1.23 Å resolution structure was obtained from a crystal harvested directly from a crystal screen. However, it is possible that further optimization of the other SmPutAΔα constructs could yield equally high-quality crystals.
The current P1 crystal form of SmPutAΔα2 appears to be well suited for co-crystallization with noncovalent inhibitors yet has some drawbacks. For example, soaking ligand-free crystals with reversible inhibitors was not successful. Although the crystals were undamaged by soaking, electron density for the inhibitor was absent. Thus, the P1 crystal form is not optimal for fragment-based inhibitor discovery in which crystal soaking is used as the primary screen to identify active site binders. Nevertheless, it may be possible to use soaking to screen for fragments that bind outside of the active site, as can occur in fragment-based inhibitor discovery (Schiebel et al., 2016). Also, crystals of SmPutAΔα2 covalently inactivated by 2 diffracted poorly, and the photoinduced covalent inactivation of SmPutAΔα2 in crystallo degraded the crystal quality. These limitations may be due to crystal packing. The P1 asymmetric unit contains two proteins arranged such that their active sites contact each. In particular, the α8 helices of the active sites contact each other in the dimer interface (Fig. 4B). This is notable because the α8 helix is critical for ligand recognition by contributing conserved residues that contact proline analog inhibitors (Fig. 5A and B), and it is known to be a dynamic element that moves in response to inhibitor binding and covalent inactivation by 2 and 3 (Luo et al., 2012; Singh et al., 2014). It may be that the P1 crystal lattice prohibits the conformational changes associated with ligand entry in crystallo and covalent inactivation. Although the P1 crystal form appears to be suboptimal for studying covalent inactivation of PRODH, it may be possible to find alternative crystal forms by performing crystal screening of minimal PRODHs inactivated in solution by 2 and 3. Curiously, the dimer chosen for the asymmetric unit is likely present in solution based on SAXS data and the analysis of protein-protein interfaces in the crystal lattice with PDBePISA. Since co-crystallization with inhibitors was successful, it appears that the active site of the dimer in solution is flexible enough to allow inhibitor binding.
Supplementary Material
Acknowledgements
We thank Jonathan Schuermann and Igor Kourinov for help with X-ray diffraction data collection and processing. We thank Greg Hura and Kathryn Burnett for assistance with SAXS data collection and processing. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16 M detector on 24-ID-E is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. SAXS experiments were conducted at the Advanced Light Source (ALS), a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, supported by DOE Office of Biological and Environmental Research. Additional support comes from the National Institute of Health project ALS-ENABLE (P30 GM124169) and a High-End Instrumentation Grant S10OD018483.
Contributor Information
Alexandra N Bogner, Department of Biochemistry, University of Missouri, Columbia, MO 65211, USA.
Juan Ji, Department of Biochemistry, University of Missouri, Columbia, MO 65211, USA.
John J Tanner, Department of Biochemistry, University of Missouri, Columbia, MO 65211, USA; Department of Chemistry, University of Missouri, Columbia, MO 65211, USA.
Conflict of Interest
The authors have no conflict of interest to declare.
Funding
This work was supported by the National Institute of General Medical Sciences, National Institutes of Health (R01GM132640).
Authors' Contributions
A.N.B.: Conceptualization, Methodology, Investigation, Writing-Original Draft, Writing-Review & Editing and Visualization. J.J.: Investigation. J.J.T.: Conceptualization, Writing-Original Draft, Writing-Review & Editing, Visualization, Validation, Supervision, Project administration and Funding acquisition.
Abbreviations
ALDHSF, aldehyde dehydrogenase superfamily; EcPutA86–630, variant of proline utilization A from E. coli containing residues 86–630; GSALDH, l-glutamate-γ-semialdehyde dehydrogenase; o-AB, ortho-aminobenzaldehyde; P5C, Δ1-pyrroline-5-carboxylate; PRODH, proline dehydrogenase; PutA, proline utilization A; SAXS, small-angle X-ray scattering; SmPutA, proline utilization A from S. meliloti; TtPRODH, monofunctional proline dehydrogenase from T. thermophilus.
Data availability
The Materials and Methods section contains statements about depositions to the Protein Data Bank (PDB) and the Small-Angle Scattering Biological Data Bank (SASBDB). The depoistion IDs are also listed in Supplementary Tables SII and SIV.
References
- Afonine, P.V., Grosse-Kunstleve, R.W., Echols, N., Headd, J.J., Moriarty, N.W., Mustyakimov, M., Terwilliger, T.C., Urzhumtsev, A., Zwart, P.H. and Adams, P.D. (2012) Acta Crystallogr. D Biol. Crystallogr., 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, F.H., Kennard, O., Watson, D.G., Brammer, L., Orpen, A.G. and Taylor, R. (1987) J Chem Soc. Perkin Trans., 2, S1–S19. [Google Scholar]
- Arnold, K., Bordoli, L., Kopp, J. and Schwede, T. (2006) Bioinformatics, 22, 195–201. [DOI] [PubMed] [Google Scholar]
- Bergers, G. and Fendt, S.M. (2021) Nat. Rev. Cancer, 21, 162–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogner, A.N. and Tanner, J.J. (2022) Org. Biomol. Chem., 20, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, A.C., Becker, D.F., Gates, K.S. and Tanner, J.J. (2020) ACS Chem. Biol., 15, 936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, A.C., Bogner, A.N., Mao, Y., Becker, D.F. and Tanner, J.J. (2021a) Arch. Biochem. Biophys., 698, 108727. 10.1016/j.abb.2020.108727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, A.C., Prater, A.R., Bogner, A.N., Quinn, T.P., Gates, K.S., Becker, D.F. and Tanner, J.J. (2021b) ACS Chem. Biol., 16, 2268–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, V.B., Arendall, W.B.3rd, Headd, J.J., Keedy, D.A., Immormino, R.M., Kapral, G.J., Murray, L.W., Richardson, J.S. and Richardson, D.C. (2010) Acta Crystallogr. D Biol. Crystallogr., D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christgen, S.L., Zhu, W., Sanyal, N., Bibi, B., Tanner, J.J. and Becker, D.F. (2017) Biochemistry, 56, 6292–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Aniello, C., Patriarca, E.J., Phang, J.M. and Minchiotti, G. (2020) Front. Oncol., 10, 776. 10.3389/fonc.2020.00776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale, G.E., Oefner, C. and D'Arcy, A. (2003) J. Struct. Biol., 142, 88–97. [DOI] [PubMed] [Google Scholar]
- Derewenda, Z.S. (2010) Acta Crystallogr. D Biol. Crystallogr., 66, 604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald, S.P., Sun, X.Y., Hu, C.A., Yu, J., Mei, J.M., Valle, D. and Phang, J.M. (2001) Cancer Res., 61, 1810–1815. [PubMed] [Google Scholar]
- Dyer K.N., Hammel M., Rambo R.P., Tsutakawa S.E., Rodic I., Classen S., Tainer J.A. and Hura G.L. (2014) High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. In: Methods in Molecular Biology. Clifton, Totowa, NJ: Humana Press, 1091, 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia, I., Broekaert, D., Christen, S., Boon, R., Radaelli, E., Orth, M.F., Verfaillie, C., Grunewald, T.G.P. and Fendt, S.M. (2017) Nat. Commun., 8, 15267. 10.1038/ncomms15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley, P., Lohkamp, B., Scott, W.G. and Cowtan, K. (2010) Acta Crystallogr. D Biol. Crystallogr., 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, P.R. (2011) Acta Crystallogr. D Biol. Crystallogr., 67, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke, D. and Svergun, D.I. (2009) J. Appl. Cryst., 42, 342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore, S., Sanz, G.E., Hendrickx, P.M.S.et al. (2017) Structure, 25, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant, T.D. (2018) Nat. Methods, 15, 191–193. [DOI] [PubMed] [Google Scholar]
- Hasanzadeh, M., Nahar, A.S., Hassanpour, S., Shadjou, N., Mokhtarzadeh, A. and Mohammadi, J. (2017) Enzyme Microb. Technol., 105, 64–76. [DOI] [PubMed] [Google Scholar]
- Huijbers, M.M., Martinez-Julvez, M., Westphal, A.H., Delgado-Arciniega, E., Medina, M. and vanBerkel, W.J. (2017) Sci. Rep., 7, 43880. 10.1038/srep43880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers, M.M. and vanBerkel, W.J. (2015) Biotechnol. J., 10, 395–403. [DOI] [PubMed] [Google Scholar]
- Huijbers, M.M.E., vanAlen, I., Wu, J.W., Barendregt, A., Heck, A.J.R. and vanBerkel, W.J.H. (2018) Molecules, 23, 184–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers, M.M.E., Wu, J.W., Westphal, A.H. and vanBerkel, W.J.H. (2019) Biotechnol. J., 14, e1800540. 10.1002/biot.201800540. [DOI] [PubMed] [Google Scholar]
- Hura, G.L., Budworth, H., Dyer, K.N., Rambo, R.P., Hammel, M., McMurray, C.T. and Tainer, J.A. (2013) Nat. Methods, 10, 453–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch, W. (2010) Acta Crystallogr. D Biol. Crystallogr., 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikhney, A.G., Borges, C.R., Molodenskiy, D.S., Jeffries, C.M. and Svergun, D.I. (2020) Protein Sci., 29, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitz, R. and Wilson, I.B. (1962) J. Biol. Chem., 237, 3245–3249. [PubMed] [Google Scholar]
- Konarev, P.V., Volkov, V.V., Sokolova, A.V., Koch, M.H.J. and Svergun, D.I. (2003) J. Appl. Cryst., 36, 1277–1282. [Google Scholar]
- Krissinel, E. and Henrick, K. (2007) J. Mol. Biol., 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Liebschner, D., Afonine, P.V., Moriarty, N.W., Poon, B.K., Sobolev, O.V., Terwilliger, T.C. and Adams, P.D. (2017) Acta Crystallogr D Struct Biol, 73, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L.K., Becker, D.F. and Tanner, J.J. (2017) Arch. Biochem. Biophys., 632, 142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. and Phang, J.M. (2012a) Biofactors, 38, 398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W. and Phang, J.M. (2012b) Autophagy, 8, 1407–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, M., Arentson, B.W., Srivastava, D., Becker, D.F. and Tanner, J.J. (2012) Biochemistry, 51, 10099–10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, M., Gamage, T.T., Arentson, B.W., Schlasner, K.N., Becker, D.F. and Tanner, J.J. (2016) J. Biol. Chem., 291, 24065–24075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, B.W. (1968) J. Mol. Biol., 33, 491–497. [DOI] [PubMed] [Google Scholar]
- Maxwell, S.A. and Davis, G.E. (2000) Proc. Natl. Acad. Sci. U. S. A., 97, 13009–13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell, S.A. and Rivera, A. (2003) J. Biol. Chem., 278, 9784–9789. [DOI] [PubMed] [Google Scholar]
- McCoy, A.J., Grosse-Kunstleve, R.W., Adams, P.D., Winn, M.D., Storoni, L.C. and Read, R.J. (2007) J. Appl. Cryst., 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty, N.W., Grosse-Kunstleve, R.W. and Adams, P.D. (2009) Acta Crystallogr. D Biol. Crystallogr., 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrander, E.L., Larson, J.D., Schuermann, J.P. and Tanner, J.J. (2009) Biochemistry, 48, 951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallag, G., Nazarian, S., Ravasz, D., Bui, D., Komlodi, T., Doerrier, C., Gnaiger, E., Seyfried, T.N. and Chinopoulos, C. (2022) Int. J. Mol. Sci., 23, 5111–5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang, J.M. (2019) Antioxid. Redox Signal., 30, 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang, J.M. and Liu, W. (2012) Front. Biosci. (Landmark Ed), 17, 1835–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang, J.M., Liu, W., Hancock, C. and Christian, K.J. (2012) Front. Oncol., 2, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang, J.M., Liu, W., Hancock, C.N. and Fischer, J.W. (2015) Curr. Opin. Clin. Nutr. Metab. Care, 18, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piiadov, V., Ares de Araujo, E., Oliveira, N.M., Craievich, A.F. and Polikarpov, I. (2019) Protein Sci., 28, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak, K., Xia, Y., Zweier, J.L., Kinzler, K.W. and Vogelstein, B. (1997) Nature, 389, 300–305. [DOI] [PubMed] [Google Scholar]
- Ruggiero, A., Smaldone, G., Squeglia, F. and Berisio, R. (2012) Protein Pept. Lett., 19, 732–742. [DOI] [PubMed] [Google Scholar]
- Salminen, K.A., Leppanen, J., Venalainen, J.I., Pasanen, M., Auriola, S., Juvonen, R.O. and Raunio, H. (2011) Drug Metab. Dispos., 39, 412–418. [DOI] [PubMed] [Google Scholar]
- Schiebel, J., Radeva, N., Krimmer, S.G.et al. (2016) ACS Chem. Biol., 11, 1693–1701. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny, D., Hammel, M. and Sali, A. (2010) Nucleic Acids Res., 38, W540–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, G.K., Mahoney, S., Scott, M., Loureiro, A., Lopez-Ramirez, A., Tanner, J.J., Ellerby, L.M. and Benz, C.C. (2021) Amino Acids, 53, 1927–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, G.K., Yau, C., Becker, B.C., Khateeb, S., Mahoney, S., Jensen, M.B., Hann, B., Cowen, B.J., Pegan, S.D. and Benz, C.C. (2019) Mol. Cancer Ther., 18, 1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, D.S. and Hura, G.L. (2022) Advanced Light Source. Berkeley, CA: The SIBYLS Beamline 12.3.1 at the Advanced Light Source. [Google Scholar]
- Singh, H., Arentson, B.W., Becker, D.F. and Tanner, J.J. (2014) Proc. Natl. Acad. Sci. U. S. A., 111, 3389–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, D., Zhu, W., Johnson, W.H.Jr., Whitman, C.P., Becker, D.F. and Tanner, J.J. (2010) Biochemistry, 49, 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarita, E., Pollegioni, L., Servi, S. and Molla, G. (2012) Protein Expr. Purif., 82, 345–351. [DOI] [PubMed] [Google Scholar]
- Tanner, J.J. (2019) Antioxid. Redox Signal., 30, 650–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner, J.J., Fendt, S.M. and Becker, D.F. (2018) Biochemistry, 57, 3433–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, T.A., Johnson, W.H.Jr., Whitman, C.P. and Tanner, J.J. (2008) Biochemistry, 47, 5573–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, T.A., Krishnan, N., Becker, D.F. and Tanner, J.J. (2007) J. Biol. Chem., 282, 14316–14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M., White, T.A., Schuermann, J.P., Baban, B.A., Becker, D.F. and Tanner, J.J. (2004) Biochemistry, 43, 12539–12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, W., Haile, A.M., Singh, R.K., Larson, J.D., Smithen, D., Chan, J.Y., Tanner, J.J. and Becker, D.F. (2013) Biochemistry, 52, 4482–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Materials and Methods section contains statements about depositions to the Protein Data Bank (PDB) and the Small-Angle Scattering Biological Data Bank (SASBDB). The depoistion IDs are also listed in Supplementary Tables SII and SIV.