

Abstract

Controlling chiral recognition and chiral information transfer has major implications in areas ranging from drug design and asymmetric catalysis to supra- and macromolecular chemistry. Especially intriguing are phenomena associated with chiral self-recognition. The design of systems that show self-induced recognition of enantiomers, i.e., involving homochiral versus heterochiral dimers, is particularly challenging. Here, we report the chiral self-recognition of α-ureidophosphonates and its application as both a powerful analytical tool for enantiomeric ratio determination by NMR and as a convenient way to increase their enantiomeric purity by simple achiral column chromatography or fractional precipitation. A combination of NMR, X-ray, and DFT studies indicates that the formation of homo- and heterochiral dimers involving self-complementary intermolecular hydrogen bonds is responsible for their self-resolving properties. It is also shown that these often unnoticed chiral recognition phenomena can facilitate the stereochemical analysis during the development of new asymmetric transformations. As a proof of concept, the enantioselective organocatalytic hydrophosphonylation of alkylidene ureas toward self-resolving α-ureidophosphonates is presented, which also led us to the discovery of the largest family of self-resolving compounds reported to date.
Introduction
Since the discovery of molecular chirality by Pasteur in the 19th century1 and the introduction of the tetrahedral carbon by van ’t Hoff2 and Le Bel,3 the chemical community has been fascinated by the myriad of phenomena associated with stereochemistry. The homochiral nature of the essential building blocks of biological systems, often denoted as “a signature of life,”4 shows the key role of chiral information transfer, which is far from being fully understood. Especially intriguing is chiral self-recognition of enantiomers, i.e., homochiral versus heterochiral dimer (and higher order aggregates) formation, governing phenomena ranging from conglomerate and racemate formation in crystallization5 to nonlinear effects in asymmetric (auto)catalysis.6 Among the most prominent examples are the attrition-enhanced deracemization7 and the chiral “sergeant and soldier” effect in supramolecular systems.8 However, predicting chiral recognition at the molecular level is still challenging, and transmission of chiral information along length scales is mainly based on serendipitous discovery.
A remarkable case of chiral self-recognition is the self-induced diastereomeric anisochronism effect9 in NMR (SIDA, also known as self-induced recognition of enantiomers). This rather unknown and often overlooked phenomenon arises from the spontaneous aggregation of certain chiral nonracemic molecules in solution, leading to diastereomeric homo- and heterochiral associates that exhibit different NMR signals (Figure 1a).10 Consequently, under ideal conditions,11 this effect enables the direct determination of the enantiomeric ratio in a scalemic mixture by simple NMR analysis.
Figure 1.

Concept of association of enantiomers (a) and its application to the development of an enantioselective synthesis of α-ureidophosphonates via hydrophosphonylation of alkylidene ureas (b and c).
Although it is difficult to predict whether a chiral compound will show the SIDA effect, certain families of compounds, including some chiral ureas,12 P(V)-based compounds,13 carboxylic acids,14 amides,12,15 and alcohols,10,16 have been found to exhibit this phenomenon. All of these chiral compounds display an evident structural feature, the presence of self-complementary hydrogen-bond acceptor and donor groups that lead to the formation of aggregates in solution. Furthermore, the presence of a stereogenic center next to these functionalities imposes a distinct three-dimensional (3D) arrangement of the homo- and heterochiral aggregates,17 giving rise to diastereomeric species with different physicochemical properties (NMR spectra, solubility, polarity). Additionally, these aggregates are also prone to exhibit spontaneous fractionation of enantiomers into enantiomerically enriched and depleted fractions in the absence of any chiral inductor (also denoted as self-disproportionation of enantiomers, SDE).18 Both effects have been recognized as potential sources of misinterpretation of chiral information, but they also provide unique opportunities for the convenient analysis of scalemic mixtures and, in some cases, ready access to enantiopure compounds.10,19 Clearly, exploring these effects might also greatly impact the development of new enantioselective transformations, as they can significantly accelerate the screening of reaction conditions and the analysis of the scope and provide an expedient way to increase the enantiopurity of the compounds.15k Nevertheless, to the best of our knowledge, these effects have never been exploited systematically in reaction development. Furthermore, addressing chiral self-recognition by design might also be highly valuable to rationally approach, e.g., chiral communication, asymmetric catalysis, supramolecular chirality, and the fundamental understanding of the origin of homochirality.
Based on our long-standing efforts on antipodal and nonlinear effects,20 chiral auto-amplification,21 and supramolecular chirality22 we have taken up the challenge to design molecules that would show chiral self-recognition. Encouraged by our previous findings on the ability of ureas to bind to phosphonates and phosphates through moderately strong inter- or intramolecular hydrogen bonds,23 we envisioned a system based on multiple self-complementary intermolecular hydrogen bonds combining phosphonate and urea moieties in chiral α-ureidophosphonates, potentially leading to a self-induced recognition of enantiomers (Figure 1b). Additionally, this chiral self-recognition could be used as an analytical tool to rapidly assess the stereochemical outcome of an asymmetric route toward α-ureidophosphonates,24 showcasing the principle. In that case, it would significantly accelerate the screening of the reaction conditions, opening new avenues in asymmetric synthesis. To this end, we conceived an enantioselective hydrophosphonylation25,26 of readily available alkylidene ureas as a convenient platform to test our hypothesis (Figure 1c). Here, we report the discovery of an entirely new family of so far underexplored compounds, i.e., α-ureidophosphonates, that exhibit self-resolving properties (SIDA + SDE). In addition, we demonstrate that such self-resolving properties facilitated the optimization of the reaction conditions, the analysis of the scope, and the enantiomer purification of the final products.
Results and Discussion
While the origin of the SIDA effect has been described in the literature,10b,13b,15c a succinct explanation is provided first for the sake of clarity. In the simplest case, considering a mixture of two enantiomers, R and S, that tend to reversibly form dimers in solution, the following equilibria can be proposed:
| 1 |
| 2 |
| 3 |
In a scenario where binary associations occur under fast exchange conditions between monomeric and dimeric species in the NMR scale, two sets of peaks would appear in the NMR spectra as a result of the distinct time-averaged local environments for the homochiral and heterochiral associates. Specifically, one set of peaks corresponds to the weighted average of the three species where R enantiomer is present (R + RR + RS, 4), and another set of peaks corresponds to the weighted average of the three species where S enantiomer is present (S + SS + RS, 5)
| 4 |
| 5 |
where δobs,R and δobs,S are the averaged chemical shifts for each set of signals observed in the NMR spectra corresponding to the R and S enantiomers, respectively; δR and δS are the chemical shifts of the monomeric enantiomers; δRR, δSS, and δRS are the chemical shifts of the dimeric homo- and heterochiral species; χR and χS are the molar fractions of each monomeric enantiomer; and χRR, χSS, and χRS are the molar fractions of the homo- and heterochiral dimers. From the above equations, it becomes evident that a change in the enantiomeric ratio of a scalemic mixture will alter the position of the equilibria shown in 1 and 2 and, consequently, change the chemical shift of the two sets of peaks (4 and 5). As a result, racemic and enantiopure solutions of a chiral compound show different NMR spectra and, in the case of a scalemic mixture, where 50:50 < e.r. < 100:0, two sets of peaks are observed and integration of the signals directly provides the e.r.
Importantly, as previously pointed out by Harger,13b if the exchange rate between the species in equilibria was slow, the position of the two sets of signals would be independent of the enantiomeric ratio, and their relative intensities would be equal to the ratio of diastereoisomers rather than the ratio of enantiomers.
On the Formation of Self-Resolving Homo- and Heterochiral Aggregates Derived from α-Ureidophosphonates
To determine whether α-ureidophosphonates can form homo- and heterochiral aggregates, the preparation of a nonracemic α-ureidophosphonate was required. However, to the best of our knowledge, such compounds have never been prepared in an enantioenriched form. Consequently, we conceived a preliminary multistep sequence to access to enantioenriched α-ureidophosphonate 3 (Scheme 1a), which involved an enantioselective hydrophosphonylation of Boc-protected imine 1,27 followed by deprotection of the α-amino phosphonate ester 2 and coupling with phenyl isocyanate. Upon analysis by 1H-NMR and 31P{1H}-NMR of the crude mixture of 3 in CDCl3, we clearly observed the formation of two sets of peaks whose integration matched the expected enantiomeric ratio (90:10 of precursor 2 vs 91:9 of α-ureidophosphonate 3). Further analysis by chiral HPLC confirmed that the enantiomeric ratio of 3 was 91:9 (Scheme 1b). These results indicated that α-ureidophosphonates might be aggregating in solution and, indeed, show the SIDA effect.
Scheme 1. (a) Preliminary Access to Scalemic 3 and (b) Comparison of the 31P{1H}-NMR (Left) and Chiral HPLC Chromatogram (right) of Compound 3 Showcasing the SIDA Effect.

It is known that SIDA strongly depends on the dielectric constant of the medium, and solvents capable of disrupting intermolecular hydrogen-bonding interactions can lead to its disappearance.10a Therefore, we performed a systematic study to disclose in which solvents compound 3 showed splitting of the NMR signals. While highly polar solvents such as DMSO-d6, MeOD, or DMF-d7 led to the disappearance of the SIDA effect, lower-polarity solvents (CDCl3, CD2Cl2, toluene-d8, acetone-d6) gave rise to two sets of signals both in 1H-NMR and 31P{1H}-NMR whose relative ratio matched the expected enantiomeric ratio (see the Supporting Information). 31P{1H}-NMR spectroscopy proved particularly suitable for the determination of the optical purity of compound 3 thanks to the excellent separation of the two sets of peaks for a wide range of enantiomeric ratios (Figure 2).
Figure 2.

Comparison of the enantiomeric ratios of 3 determined by 1H-NMR (left, 400 MHz, 0.05 M, CDCl3, 25 °C), 31P{1H}-NMR (middle, 162 MHz, 0.05 M, CDCl3, 25 °C), and chiral HPLC (right table). See the Supporting Information for details.
Evidence for the Formation of Dimers
The nonequivalency of NMR spectra between enantiomers of 3 in low-polarity solvents pointed to the formation of diastereomeric aggregates. While the presence of self-complementary binding sites in α-ureidophosphonates suggested a dimeric structure of the aggregate28,29 (Figure 1b), a combined experimental and theoretical study was performed to confirm this assumption. First, diffusion-ordered spectroscopy (DOSY) measurements allowed us to calculate the diffusion coefficients of 5 in different solvents and, eventually, estimate the molecular weight of the aggregates.30 Thus, in CDCl3, compound 5 (92:8 e.r.) showed a diffusion coefficient of D = 7.0 × 10–10 m2/s, which corresponds to an estimated molecular weight of MW (measured, CDCl3) = 865 g/mol (Figure 3a). Given that the molecular weight of monomeric 5 is MW (calculated) = 334 g/mol, the estimation of 865 g/mol suggests the formation of a dimeric structure in CDCl3. In contrast, the measurement of the diffusion coefficient of 5 in DMSO-d6 gave a value of D = 2.4 × 10–10 m2/s, corresponding to an estimated molecular weight of MW (measured, DMSO-d6) = 390 g/mol, indicative of the formation of a monomeric species in this solvent (Figure 3b). Overall, this data is in excellent agreement with the observation of the SIDA effect in CDCl3, where aggregation is expected, and not in DMSO-d6.31
Figure 3.

Two-dimensional (2D)-DOSY analysis of 5 in CDCl3 (a) and DMSO-d6 (b).
Single crystals suitable for X-ray diffraction analysis could be grown for the heterochiral aggregate (racemic) of the related compound 4 (Figure 4). Its structure in the solid state revealed a clear intermolecular association between two α-ureidophosphonates with opposite chirality in an antiparallel orientation (head-to-tail), where both N–H groups of the urea functionality of one molecule are engaged in a hydrogen-bonding interaction with the phosphonate group of its partner (N–H···O=P bond distances between 2.14 and 2.15 Å).
Figure 4.

X-ray diffraction analysis of racemic 4.
Additionally, DFT calculations at the ωb97XD/6–311++G(d,p)-SMD(PhMe)//ωb97XD/6–31+G(d,p) level32 indicated that such antiparallel disposition of α-ureidophosphonates is greatly preferred over other possible (parallel) dimeric structures (Figure 5). Remarkably, such an antiparallel arrangement stabilizes the dimeric structure up to 12.9 kcal/mol for the heterochiral aggregate and 11.3 kcal/mol for the homochiral associate, as compared to the monomeric form.
Figure 5.

DFT-optimized structures of dimeric 5. See the Supporting Information for details.
The analysis of the binding isotherms for compound 5, both in racemic and enantioenriched form, by 1H-NMR titration33 allowed us to quantify the magnitude of the dimerization (see the Supporting Information). Both homochiral and heterochiral dimers showed association constants (Ka) in the range of 103 M–1, (3.3 ± 0.8) × 103 M–1 for the former and (8.3 ± 0.2) × 103 M–1 for the latter. While the SIDA effect is concentration-dependent, these relatively high association constants ensured the observation of the SIDA effect in a broad range of concentrations (0.2–0.01 M, in this study).
SDE by Achiral Column Chromatography and Fractional Precipitation
The results shown above clearly suggest that α-ureidophosphonates form dimers in solution, leading to diastereomeric aggregates. As a result of this dimerization, the phenomenon of SDE by achiral column chromatography was also investigated with α-ureidophosphonate 5, having an initial 92:8 e.r. of the crude material. A single chromatographic run using n-hexane/iPrOH 9:1 as eluent afforded 5 in 96% combined yield, where the first fraction provided a highly enantioenriched sample (98.5:1.5 e.r.) accounting for 31% yield (Table 1). The majority of the product (48%) eluted with the initial 92:8 e.r. The final fractions showed strong depletion of enantiopurity (58.5:41.5 and 52.5:47.5 e.r.) with 17% of the overall yield.34
Table 1. Achiral Phase Flash Column Chromatography of 5 with an Initial 92:8 e.r.

| fraction | e.r. | % yield |
|---|---|---|
| 1 | 98.5:1.5 | 31 |
| 2 | 92:8 | 48 |
| 3 | 58.5:41.5 | 9 |
| 4 | 52.5:47.5 | 8 |
Alternatively, the spontaneous separation of enantiomers can also be achieved by harnessing the different solubility of the homo- and heterochiral dimers in organic solvents. Thus, when a saturated solution of compound 3 (135.5 mg) with an initial 79:21 e.r. in hexane/iPrOH 8:2 was cooled to 2 °C for 24–40 h, a white solid precipitated (Figure 6). The analysis of the supernatant solution and the solid fraction revealed a remarkable fractionation of enantiomers. While the precipitate consisted of nearly racemic 3 (42.2 mg, 53:47 e.r.), the supernatant contained compound 3 with significantly increased enantiopurity (92.1 mg, 92.5:7.5 e.r.). These experiments showcase the potential of SDE to obtain highly enantioenriched compounds by employing inexpensive purification methods.
Figure 6.

Spontaneous fractionation of enantiomers based on the different solubility of homo- and heterochiral dimers of 3.
Application of the Self-Resolving Properties of α-Ureidophosphonates to the Development of an Enantioselective Hydrophosphonylation Reaction
At the onset of our studies, we selected the reaction comprising the readily available (E)-1-benzylidene-3-phenylurea356 and the commercially available diethyl phosphite as our model system. In the past decades, a wide range of catalysts have been used in enantioselective hydrophosphonylations of imines,25,26 although, to the best of our knowledge, a direct comparison of different catalyst classes is missing, and asymmetric hydrophosphonylation of alkylidene ureas is unprecedented. Motivated by addressing this gap, Lewis acidic metal complexes (A, B), cinchona alkaloids (C, D), thiourea-derived catalysts (E, F),36 chiral cyclopentadienyl-based Brønsted acid catalysts (G, H),37 and chiral phosphoric acids (I–L) were examined (Table 2). A model reaction between alkylidene urea 6 and diethyl phosphite was carried out in the presence of 10 mol % of a chiral catalyst. Al-salen complex A showed high catalytic activity; however, the hydrophosphonylation resulted in a nearly racemic product 3. In comparison, Sharpless-catalyst B gave the product with better enantioselectivity (36:64 e.r.), albeit in lower conversion. Cinchona alkaloids C and D (entry 2), thiourea-based catalysts E and F (entry 3), and cyclopentadienyl Brønsted acids G and H (entry 4) did not prove to be suitable for our transformation, giving the product with low to good conversions and <55:45 e.r. Finally, using the simple (S)-BINOL-derived chiral phosphoric acid I, product 3 was obtained in a promising 82% conversion and 62.5:37.5 e.r. Encouraged by this result, we further explored the catalytic activity of other chiral phosphoric acids J–L (entries 5 and 6). Among them, the commercially available MacMillan TiPSY catalyst L afforded the desired product with 87:13 e.r. while maintaining a conversion of 79% (entry 6).
Table 2. Catalyst Screeninga.

| entry | catalyst | conversion (%)b | e.r.c |
|---|---|---|---|
| 1 | metal complexes A, B | 88/26 | 49:51/36:64 |
| 2 | Cinchona alkaloids C, D | 31–43 | <52.5:47.5 |
| 3 | thioureas E, F | <10 | <55:45 |
| 4 | cyclopentadienyl-based Brønsted acids G, H | 56–60 | <52.5:47.5 |
| 5 | phosphoric acids I–K | 61–83 | 62.5:37.5–78:22 |
| 6 | phosphoric acid L | 79 | 87:13 |
| 7 | phosphoric acid Ld | 88 (63)e | 92.5:7.5 |
General reaction conditions: alkylidene urea 6 (0.2 mmol), the indicated catalyst (0.02 mmol, 10 mol%), and diethyl phosphite (0.24 mmol) in PhMe (1 mL) at room temperature for 18 h.
Determined by 31P{1H}-NMR.
Enantiomeric ratios were determined by 1H-NMR/31P{1H}-NMR analysis, benefiting from the SIDA effect, and/or chiral HPLC analysis (see the Supporting Information for details).
Under optimal conditions: Alkylidene urea 6 (0.2 mmol), L catalyst (0.01 mmol, 5 mol%), and diethyl phosphite (0.24 mmol) in deoxygenated PhMe (2 mL) at 30 °C for 48 h.
Isolated yield in brackets.
Taking advantage of the SIDA effect enabling efficient reaction analysis, we further conducted an extensive reaction optimization, including an investigation of the solvent effect, temperature, catalyst loading, stoichiometry, and concentration (see the Supporting Information for details). We found that the best results were obtained using 1.2 equiv of diethyl phosphite and 5 mol % of TiPSY catalyst L in deoxygenated toluene (0.1 m) at 25 °C. Under these conditions, compound 3 was obtained in 63% isolated yield and 92.5:7.5 e.r. (entry 7).
Having identified the optimal reaction conditions, we explored the scope of phosphites and alkylidene ureas with various substitution patterns (Scheme 2).38 Initially, we tested a broad variety of phosphites in the hydrophosphonylation reaction of 6. While diethyl phosphite and dimethyl phosphite afforded the corresponding α-ureidophosphonates 3 and 5, respectively, in good yields and enantioselectivities, other bulkier phosphites led to the desired products with lower stereoselectivities (4, 7–11). Importantly, we observed that for the more reactive diphenyl and neopentylene phosphites, lower temperatures (6 °C) were required to achieve better stereocontrol (9 and 10, 87:13 e.r. and 76:24 e.r., respectively). Remarkably, the hydrophosphonylation reaction using dimethyl phosphite could be performed in a 2 mmol scale using 2 mol% of catalyst L, resulting in α-ureidophosphonate 5 in 96% isolated yield and 92:8 e.r.
Scheme 2. Scope of the Enantioselective Hydrophosphonylation of Alkylidene Ureas with Phosphites,,
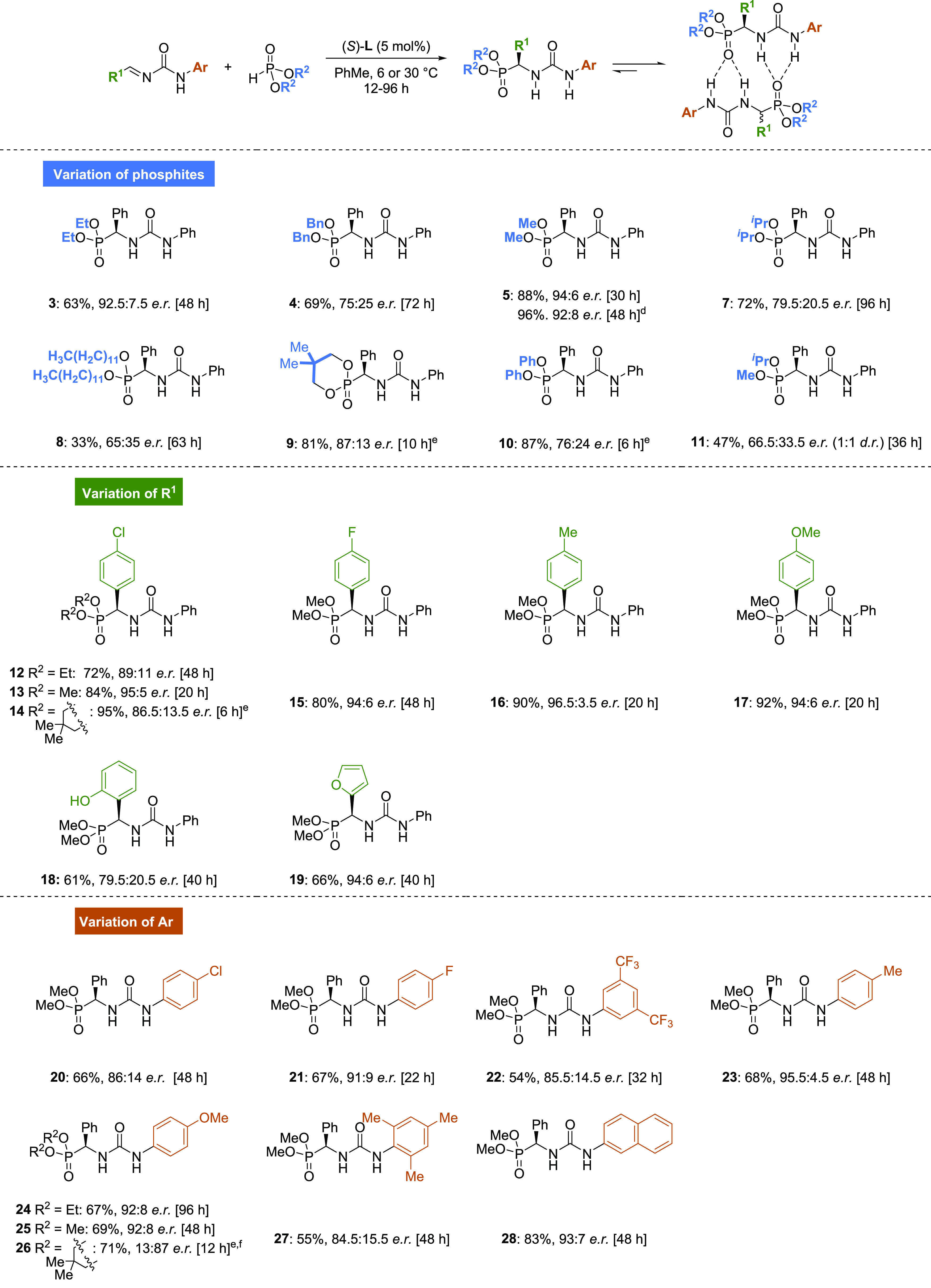
General reaction conditions: alkylidene urea (0.2 mmol), the corresponding alkyl/aryl phosphite (0.24 mmol), and catalyst (S)-L (0.01 mmol, 5 mol %) in deoxygenated PhMe (2 mL) at 30 °C for the indicated time.
Yields of isolated products are given.
Enantiomeric ratios were determined by 1H-NMR/31P{1H}-NMR analysis, benefiting from the SIDA effect, and chiral HPLC analysis for selected examples as controls (see the Supporting Information for details).
Performed in a 2 mmol scale.
Reaction performed at 6 °C.
Using (R)-L as the catalyst.
Next, we analyzed the variations on the alkylidene urea backbone by introducing both electron-donating and electron-withdrawing groups in ortho-, meta-, or para-positions. Substitution in R1 was well tolerated, and α-ureidophosphonates 12–18 were obtained in good to excellent yields and enantiomeric ratios ranging from 79.5:20.5 to 96.5:3.5. We were pleased to observe that the reaction tolerated not only electron-poor (12–15) and electron-rich aromatic groups (16–19) but also the presence an unprotected phenol (18) and a heteroaromatic group (19).
The substitution effect was more pronounced in the case of variation on the Ar group. The introduction of electron-withdrawing substituents led to a slight reduction of the enantioselectivity. Thus, products 20 and 21, bearing halogen groups in the para-position, and 22, possessing two trifluoromethyl groups in meta-positions, were obtained in moderate yields and enantioselectivities. In contrast, higher isolated yields and enantioselectivities were obtained for α-ureidophosphonates bearing electron-rich aryl rings (23–28). An exception to this observation was noted for α-ureidophosphonate 27 having a bulky mesitylene group. We reasoned that such bulky substituent might lead to an unfavorable conformation of the starting alkylidene urea, as the urea group and mesitylene moiety cannot adopt a coplanar conformation, giving rise to a diminished reactivity and stereoselectivity.
It is important to remark that all α-ureidophosphonates shown in Scheme 2 form dimers in low-polarity solvents, which enabled the facile analysis of their enantiomeric purity by simple NMR techniques, as shown earlier for compound 3. The analysis of the scope of the hydrophosphonylation reaction provided us with a library of up to 25 examples of compounds exhibiting chiral self-recognition properties (SIDA and SDE), which, to the best of our knowledge, represents the largest family of self-resolving compounds reported to date.
Conclusions
To conclude, we have described an entirely new family of compounds, α-ureidophosphonates, that show self-resolving properties, namely, their enantiomeric purity can be directly determined by simple NMR techniques (SIDA effect) and their optical purity can be easily increased by inexpensive physicochemical techniques (achiral column chromatography and fractional precipitation). A combined experimental and computational work enabled us to determine that the formation of stable homo- and heterochiral dimers anchored through multiple intermolecular hydrogen bonds between the urea moieties and phosphonate groups is, ultimately, responsible for their self-resolving properties. Moreover, such self-resolving properties were systematically applied to the development of their first enantioselective synthesis via an unprecedented organocatalytic hydrophosphonylation of alkylidene ureas. Remarkably, the hydrophosphonylation reaction proved to be general and provided the desired α-ureidophosphonates in good yields and good enantiomeric excesses.
The fact that aggregation of enantiomers can transform an enantiomeric relationship into a diastereomeric one has important implications in the development of new asymmetric transformations and chiral supramolecular systems. Its potential to significantly simplify reaction analysis and be explored in phenomena ranging from chiral recognition to self-assembly and control of dynamic molecular systems might provide ample opportunities in future molecular design.
Acknowledgments
This work was supported financially by the Netherlands Organization for Scientific Research (NWO-CW), the European Research Council (ERC, Advanced Grant No. 694345 to B.L.F.), and the Ministry of Education and Culture and Science (Gravitation Program No. 024.001.035). The COFUND project oLife has received funding from the European Union′s Horizon 2020 research and innovation programme under Grant Agreement No. 847675 (oLife postdoctoral fellowship). The authors are grateful to the CESGA (Xunta de Galicia) for computational time. The authors are also grateful to J. L. Sneep for collecting high-resolution mass spectrometry data for all newly reported compounds. The authors thank R. Toyoda for assistance in measuring the X-ray crystal structure.
Glossary
Abbreviations
- SIDA
self-induced diastereomeric anisochronism
- SDE
self-disproportionation of enantiomers
- DOSY
diffusion-ordered spectroscopy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c10911.
Experimental procedures, characterization of new compounds, computational details, NMR experiments, and chromatograms (PDF)
Author Contributions
‡ V.D. and D.P. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Any funds used to support the research of the manuscript should be placed here (per journal style).
The authors declare no competing financial interest.
Supplementary Material
References
- Pasteur L. Mémoire sur la relation qui peut exister entre la forme crystalline et la composition chimique, et sur la cause de la polarisation rotatoire. C. R. Acad. Sci., Paris 1848, 26, 535–538. [Google Scholar]
- van ’t Hoff J. H. A suggestion looking to the extension into space of the structural formulas at present used in chemistry. And a note upon the relation between the optical activity and the chemical constitution of organic compounds. (English translation from the original pamphlet). Arch. Neerl. Sci. Exactes Nat. 1874, 9, 445–454. [Google Scholar]
- Le Bel J. A. Sur les relations qui existent entre les formules atomiques des corps organiques et le pouvoir rotatoire de leurs dissolutions. Bull. Soc. Chim. Fr. 1874, 22, 337–347. [Google Scholar]
- a Feringa B. L.; van Delden R. A. Absolute Asymmetric Synthesis: The Origin, Control, and Amplification of Chirality. Angew. Chem., Int. Ed. 1999, 38, 3418–3438. . [DOI] [PubMed] [Google Scholar]; b Blackmond D. G. The Origin of Biological Homochirality. Cold Spring Harb. Perspect. Biol. 2019, 11, a032540 10.1101/cshperspect.a032540. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Blackmond D. G. Autocatalytic Models for the Origin of Biological Homochirality. Chem. Rev. 2020, 120, 4831–4847. 10.1021/acs.chemrev.9b00557. [DOI] [PubMed] [Google Scholar]
- Buhse T.; Cruz J.-M.; Noble-Terán M. E.; Hochberg D.; Ribó J. M.; Crusats J.; Micheau J.-C. Spontaneous Deracemizations. Chem. Rev. 2021, 121, 2147–2229. 10.1021/acs.chemrev.0c00819. [DOI] [PubMed] [Google Scholar]
- a Puchot C.; Samuel O.; Dunach E.; Zhao S.; Agami C.; Kagan H. B. Nonlinear effects in asymmetric synthesis. Examples in asymmetric oxidations and aldolization reactions. J. Am. Chem. Soc. 1986, 108, 2353–2357. 10.1021/ja00269a036. [DOI] [PubMed] [Google Scholar]; b Guillaneux D.; Zhao S.-H.; Samuel O.; Rainford D.; Kagan H. B. Nonlinear Effects in Asymmetric Catalysis. J. Am. Chem. Soc. 1994, 116, 9430–9439. 10.1021/ja00100a004. [DOI] [Google Scholar]; c Satyanarayana T.; Abraham S.; Kagan H. B. Nonlinear Effects in Asymmetric Catalysis. Angew. Chem., Int. Ed. 2009, 48, 456–494. 10.1002/anie.200705241. [DOI] [PubMed] [Google Scholar]; d Bryliakov K. P. Dynamic Nonlinear Effects in Asymmetric Catalysis. ACS Catal. 2019, 9, 5418–5438. 10.1021/acscatal.9b00697. [DOI] [Google Scholar]; e Soai K.; Shibata T.; Morioka H.; Choji K. Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature 1995, 378, 767–768. 10.1038/378767a0. [DOI] [Google Scholar]; f Athavale S. V.; Simon A.; Houk K. N.; Denmark S. E. Demystifying the asymmetry-amplifying, autocatalytic behaviour of the Soai reaction through structural, mechanistic and computational studies. Nat. Chem. 2020, 12, 412–423. 10.1038/s41557-020-0421-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Trapp O.; Lamour S.; Maier F.; Siegle A. F.; Zawatzky K.; Straub B. F. In Situ Mass Spectrometric and Kinetic Investigations of Soai’s Asymmetric Autocatalysis. Chem. - Eur. J. 2020, 26, 15871–15880. 10.1002/chem.202003260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorduin W. L.; Vlieg E.; Kellogg R. M.; Kaptein B. From Ostwald Ripening to Single Chirality. Angew. Chem., Int. Ed. 2009, 48, 9600–9606. 10.1002/anie.200905215. [DOI] [PubMed] [Google Scholar]
- a Green M. M.; Reidy M. P.; Johnson R. D.; Darling G.; O’Leary D. J.; Willson G. Macromolecular stereochemistry: the out-of-proportion influence of optically active comonomers on the conformational characteristics of polyisocyanates. The sergeants and soldiers experiment. J. Am. Chem. Soc. 1989, 111, 6452–6454. 10.1021/ja00198a084. [DOI] [Google Scholar]; b Yashima E.; Ousaka N.; Taura D.; Shimomura K.; Ikai T.; Maeda K. Supramolecular Helical Systems: Helical Assemblies of Small Molecules, Foldamers, and Polymers with Chiral Amplification and Their Functions. Chem. Rev. 2016, 116, 13752–13990. 10.1021/acs.chemrev.6b00354. [DOI] [PubMed] [Google Scholar]
- Williams T.; Pitcher R. G.; Bommer P.; Gutzwiller J.; Uskokovic M. Diastereomeric solute-solute interactions of enantiomers in achiral solvents. Nonequivalence of the nuclear magnetic resonance spectra of racemic and optically active dihydroquinine. J. Am. Chem. Soc. 1969, 91, 1871–1872. 10.1021/ja01035a060. [DOI] [Google Scholar]
- a Szakács Z.; Sánta Z.; Lomoschitz A.; Szántay C. Self-induced recognition of enantiomers (SIRE) and its application in chiral NMR analysis. Trends Anal. Chem. 2018, 109, 180–197. 10.1016/j.trac.2018.07.020. [DOI] [Google Scholar]; b Baumann A.; Wzorek A.; Soloshonok V. A.; Klika K. D.; Miller A. K. Potentially Mistaking Enantiomers for Different Compounds Due to the Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon. Symmetry 2020, 12, 1106 10.3390/sym12071106. [DOI] [Google Scholar]
- The SIDA effect strongly depends on the concentration, temperature and solvent. The magnetic field of the instrument used is related to the magnitude of the splitting of the signals.
- a Hong C. Y.; Kishi Y. Enantioselective total synthesis of (-)-decarbamoylsaxitoxin. J. Am. Chem. Soc. 1992, 114, 7001–7006. 10.1021/ja00044a008. [DOI] [Google Scholar]; b Huang S.-H.; Bai Z.-W.; Feng J.-W. Chiral self-discrimination of the enantiomers of α-phenylethylamine derivatives in proton NMR. Magn. Reson. Chem. 2009, 47, 423–427. 10.1002/mrc.2406. [DOI] [PubMed] [Google Scholar]
- a Harger M. J. P. Nuclear magnetic resonance non-equivalence of the enantiomers in optically active samples of phosphinic amides. J. Chem. Soc., Chem. Commun. 1976, 555–556. 10.1039/c39760000555. [DOI] [Google Scholar]; b Harger M. J. P. Proton magnetic resonance non-equivalence of the enantiomers of alkylphenylphosphinic amides. J. Chem. Soc., Perkin Trans. 2 1977, 1882–1887. 10.1039/p29770001882. [DOI] [Google Scholar]; c Harger M. J. P. Chemical shift non-equivalence of enantiomers in the proton magnetic resonance spectra of partly resolved phosphinothioic acids. J. Chem. Soc., Perkin Trans. 2 1978, 326–331. 10.1039/p29780000326. [DOI] [Google Scholar]; d Ouryupin A. B.; Kadyko M. I.; Petrovskii P. V.; Fedin E. I. Chiral discrimination in nonracemic mixtures of methanephosphonic acid, N,N′-bis(1-phenylethyl)diamides. Chirality 1994, 6, 1–4. 10.1002/chir.530060103. [DOI] [Google Scholar]; e Ouryupin A. B.; Kadyko M. I.; Petrovskii P. V.; Fedin E. I.; Okruszek A.; Kinas R.; Stec W. J. Enantiomeric 2-anilino-2-oxo-1,3,2-oxazaphosphorinanes: Synthesis and NMR-investigation of their non-racemic mixtures. Tetrahedron: Asymmetry 1995, 6, 1813–1824. 10.1016/0957-4166(95)00227-G. [DOI] [Google Scholar]
- Horeau A.; Guetté J. P. Interactions diastereoisomeres d’antipodes en phase liquide. Tetrahedron 1974, 30, 1923–1931. 10.1016/S0040-4020(01)97323-5. [DOI] [Google Scholar]
- a Cung M. T.; Marraud M.; Neel J. Etude experimentale de L′auto-association des molécules modèles dipeptidiques. III. Influence de la dimerisation stéréosélective sur les spectres de résonance magnétique protonique. Biopolymers 1977, 16, 715–729. 10.1002/bip.1977.360160403. [DOI] [PubMed] [Google Scholar]; b Cung M. T.; Marraud M.; Neel J.; Aubry A. Experimental study on aggregation of model dipeptide molecules. V. Stereoselective association of leucine dipeptides. Biopolymers 1978, 17, 1149–1173. 10.1002/bip.1978.360170505. [DOI] [Google Scholar]; c Dobashi A.; Saito N.; Motoyama Y.; Hara S. Self-induced nonequivalence in the association of D- and L-amino acid derivatives. J. Am. Chem. Soc. 1986, 108, 307–308. 10.1021/ja00262a027. [DOI] [Google Scholar]; d Giordano C.; Restelli A.; Villa M.; Annunziata R. A case of self-induced anisochrony in the proton NMR spectra of 1,5-benzothiazepines. J. Org. Chem. 1991, 56, 2270–2272. 10.1021/jo00006a064. [DOI] [Google Scholar]; e Jursic B. S.; Goldberg S. I. Enantiomer discrimination arising from solute-solute interactions in partially resolved chloroform solutions of chiral carboxamides. J. Org. Chem. 1992, 57, 7172–7174. 10.1021/jo00052a035. [DOI] [Google Scholar]; f Ghosh S. K. Chiral recognition in dipeptides containing 1-aminocyclopropane carboxylic acid or α-aminoisobutyric acid: NMR studies in solution. Pept. Res. 1999, 53, 261–274. 10.1034/j.1399-3011.1999.00030.x. [DOI] [PubMed] [Google Scholar]; g Wagger J.; Grdadolnik S. G.; Grošelj U.; Meden A.; Stanovnik B.; Svete J. Chiral solvating properties of (S)-1-benzyl-6-methylpiperazine-2,5-dione. Tetrahedron: Asymmetry 2007, 18, 464–475. 10.1016/j.tetasy.2007.02.010. [DOI] [Google Scholar]; h Klika K. D.; Budovská M.; Kutschy P. NMR spectral enantioresolution of spirobrassinin and 1-methoxyspirobrassinin enantiomers using (S)-(−)-ethyl lactate and modeling of spirobrassinin self-association for rationalization of its self-induced diastereomeric anisochromism (SIDA) and enantiomer self-disproportionation on achiral-phase chromatography (ESDAC) phenomena. J. Fluor. Chem. 2010, 131, 467–476. [Google Scholar]; i Klika K. D.; Budovská M.; Kutschy P. Enantiodifferentiation of phytoalexin spirobrassinin derivatives using the chiral solvating agent (R)-(+)-1,1′-bi-2-naphthol in conjunction with molecular modeling. Tetrahedron: Asymmetry 2010, 21, 647–658. 10.1016/j.tetasy.2010.03.035. [DOI] [Google Scholar]; j Szántay C.; Demeter Á.. Chapter 14 - Self-Induced Recognition of Enantiomers (SIRE) in NMR Spectroscopy. In Anthropic Awareness; Szántay C., Ed.; Elsevier: Boston, 2015; pp 401–415. [Google Scholar]; k Storch G.; Haas M.; Trapp O. Attracting Enantiomers: Chiral Analytes That Are Simultaneously Shift Reagents Allow Rapid Screening of Enantiomeric Ratios by NMR Spectroscopy. Chem. - Eur. J. 2017, 23, 5414–5418. 10.1002/chem.201700461. [DOI] [PubMed] [Google Scholar]; l Budovská M.; Tischlerová V.; Mojžiš J.; Harvanová M.; Kozlov O.; Gondová T.; Tomášková N. 2′-Aminoanalogues of the cruciferous phytoalexins spirobrassinin, 1-methoxyspirobrassinin and 1-methoxyspirobrassinol methyl ether: Synthesis and anticancer properties. Tetrahedron 2017, 73, 6356–6371. 10.1016/j.tet.2017.09.033. [DOI] [Google Scholar]
- a Arnold W.; Daly J. J.; Imhof R.; Kyburz E. An efficient resolution of 3-PPP and assignment of the absolute configuration. Tetrahedron Lett. 1983, 24, 343–346. 10.1016/S0040-4039(00)81402-1. [DOI] [Google Scholar]; b Luchinat C.; Roelens S.; Group I. V. A. organometallic reagents. 2. Enantiomeric purity determination of 1,2-diols through NMR spectroscopy without chiral auxiliaries. J. Am. Chem. Soc. 1986, 108, 4873–4878. 10.1021/ja00276a027. [DOI] [Google Scholar]; c Hui R. A. H. F.; Salamone S.; Williams T. H. Self-induced nonequivalence in the 1H-NMR spectra of the (+)- and (−)-isomers of a cannabinoid ketone intermediate. Pharm. Biochem. Behav. 1991, 40, 491–496. 10.1016/0091-3057(91)90352-3. [DOI] [PubMed] [Google Scholar]; d Navrátilová H.; Potácek M. Enantiomeric enrichment of (+)-(3R,4S)-4-(4-fluorophenyl)-3-hydroxymethyl-1-methylpiperidine by crystallization. Enantiomer 2001, 6, 333–337. [PubMed] [Google Scholar]; e Nieminen V.; Murzin D. Y.; Klika K. D. NMR and molecular modeling of the dimeric self-association of the enantiomers of 1,1′-bi-2-naphthol and 1-phenyl-2,2,2-trifluoroethanol in the solution state and their relevance to enantiomer self-disproportionation on achiral-phase chromatography (ESDAC). Org. Biomol. Chem. 2009, 7, 537–542. 10.1039/B814905D. [DOI] [PubMed] [Google Scholar]; f Charpentier M.; Hans M.; Jauch J. Enantioselective Synthesis of Myrtucommulone A. Eur. J. Org. Chem. 2013, 2013, 4078–4084. 10.1002/ejoc.201300179. [DOI] [Google Scholar]; g Xu Z.; Wang Q.; Zhu J. Total Syntheses of (−)-Mersicarpine, (−)-Scholarisine G, (+)-Melodinine E, (−)-Leuconoxine, (−)-Leuconolam, (−)-Leuconodine A, (+)-Leuconodine F, and (−)-Leuconodine C: Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon for Scholarisine G and Leuconodines A and C. J. Am. Chem. Soc. 2015, 137, 6712–6724. 10.1021/jacs.5b03619. [DOI] [PubMed] [Google Scholar]
- Alkorta I.; Elguero J. Self-Discrimination of Enantiomers in Hydrogen-Bonded Dimers. J. Am. Chem. Soc. 2002, 124, 1488–1493. 10.1021/ja011348c. [DOI] [PubMed] [Google Scholar]
- a Cundy K. C.; Crooks P. A. Unexpected pehnomenon in the high-performance liquid chromatographic anlaysis of racemic 14C-labelled nicotine: Separation of enantiomers in a totally achiral system. J. Chromatogr. A 1983, 281, 17–33. 10.1016/S0021-9673(01)87863-8. [DOI] [Google Scholar]; b Trapp O.; Schurig V. Nonlinear effects in enantioselective chromatography: prediction of unusual elution profiles of enantiomers in non-racemic mixtures on an achiral stationary phase doped with small amounts of a chiral selector. Tetrahedron: Asymmetry 2010, 21, 1334–1340. 10.1016/j.tetasy.2010.04.027. [DOI] [Google Scholar]
- a Soloshonok V. A.; Roussel C.; Kitagawa O.; Sorochinsky A. E. Self-disproportionation of enantiomers via achiral chromatography: a warning and an extra dimension in optical purifications. Chem. Soc. Rev. 2012, 41, 4180–4188. 10.1039/c2cs35006h. [DOI] [PubMed] [Google Scholar]; b Han J.; Kitagawa O.; Wzorek A.; Klika K. D.; Soloshonok V. A. The self-disproportionation of enantiomers (SDE): a menace or an opportunity?. Chem. Sci. 2018, 9, 1718–1739. 10.1039/C7SC05138G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynberg H.; Feringa B. Enantiomeric recognition and interactions. Tetrahedron 1976, 32, 2831–2834. 10.1016/0040-4020(76)80131-7. [DOI] [Google Scholar]
- a Fletcher S. P.; Jagt R. B. C.; Feringa B. L. An astrophysically-relevant mechanism for amino acid enantiomer enrichment. Chem. Commun. 2007, 2578–2580. 10.1039/b702882b. [DOI] [PubMed] [Google Scholar]; b Dašková V.; Buter J.; Schoonen A. K.; Lutz M.; de Vries F.; Feringa B. L. Chiral Amplification of Phosphoramidates of Amines and Amino Acids in Water. Angew. Chem., Int. Ed. 2021, 60, 11120–11126. 10.1002/anie.202014955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a van Dijken D. J.; Beierle J. M.; Stuart M. C. A.; Szymański W.; Browne W. R.; Feringa B. L. Autoamplification of Molecular Chirality through the Induction of Supramolecular Chirality. Angew. Chem., Int. Ed. 2014, 53, 5073–5077. 10.1002/anie.201311160. [DOI] [PubMed] [Google Scholar]; b Zhang Q.; Crespi S.; Toyoda R.; Costil R.; Browne W. R.; Qu D.-H.; Tian H.; Feringa B. L. Stereodivergent Chirality Transfer by Noncovalent Control of Disulfide Bonds. J. Am. Chem. Soc. 2022, 144, 4376–4382. 10.1021/jacs.1c10000. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Xu F.; Crespi S.; Pacella G.; Fu Y.; Stuart M. C. A.; Zhang Q.; Portale G.; Feringa B. L. Dynamic Control of a Multistate Chiral Supramolecular Polymer in Water. J. Am. Chem. Soc. 2022, 144, 6019–6027. 10.1021/jacs.2c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Vlatković M.; Feringa B. L.; Wezenberg S. J. Dynamic Inversion of Stereoselective Phosphate Binding to a Bisurea Receptor Controlled by Light and Heat. Angew. Chem., Int. Ed. 2016, 55, 1001–1004. 10.1002/anie.201509479. [DOI] [PubMed] [Google Scholar]; b Wezenberg S. J.; Feringa B. L. Photocontrol of Anion Binding Affinity to a Bis-urea Receptor Derived from Stiff-Stilbene. Org. Lett. 2017, 19, 324–327. 10.1021/acs.orglett.6b03423. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jong J.; Feringa B. L.; Wezenberg S. J. Light-Modulated Self-Blockage of a Urea Binding Site in a Stiff-Stilbene Based Anion Receptor. ChemPhysChem 2019, 20, 3306–3310. 10.1002/cphc.201900917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For previous methods to access to achiral or racemic α-ureidophosphonates see:; a Birum G. H. Urylenediphosphonates. General method for the synthesis of.alpha.-ureidophosphonates and related structures. J. Org. Chem. 1974, 39, 209–213. 10.1021/jo00916a019. [DOI] [Google Scholar]; b Huber J. W.; Gilmore W. F. Optically active α-aminophosphonic acids from ureidophosphonates. Tetrahedron Lett. 1979, 20, 3049–3052. 10.1016/S0040-4039(01)95316-X. [DOI] [Google Scholar]; c Li Z.-G.; Wang Q.-M.; Huang R.-Q.; Cheng J.-R.; Ma J.-A. New and effective synthesis of unsymmetrical α-ureidophosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2000, 160, 51–59. 10.1080/10426500008043670. [DOI] [Google Scholar]; d Li Z.-G.; Huang R.-Q. A Convenient Synthesis of α-Ureidomethylphosphonates with Heterocycle Moiety. J. Chem. Res. 2001, 2001, 470–471. 10.3184/030823401103168712. [DOI] [Google Scholar]; e Kaboudin B.; Afsharinezhad M. B.; Yokomatsu T. A convenient and general procedure for the synthesis of α-ureidophosphonates under catalyst-free conditions. Arkivoc 2011, 2012, 44–53. 10.3998/ark.5550190.0013.405. [DOI] [Google Scholar]; f Kaboudin B.; Arefi M.; Emadi S.; Sheikh-Hasani V. Synthesis and inhibitory activity of ureidophosphonates, against acetylcholinesterase: Pharmacological assay and molecular modeling. Bioorg. Chem. 2012, 41-42, 22–27. 10.1016/j.bioorg.2012.01.003. [DOI] [PubMed] [Google Scholar]; g Bouzina A.; Berredjem M.; Bouacida S.; Merazig H.; Aouf N.-E. A greener procedure for the synthesis of α-ureidophosphonates under ultrasound irradiation. An X-ray crystallographic study. RSC Adv. 2015, 5, 99775–99780. 10.1039/C5RA19886K. [DOI] [Google Scholar]
- For reviews on hydrophosphonylation of imines see:; a Merino P.; Marqués-López E.; Herrera R. P. Catalytic Enantioselective Hydrophosphonylation of Aldehydes and Imines. Adv. Synth. Catal. 2008, 350, 1195–1208. 10.1002/adsc.200800131. [DOI] [Google Scholar]; b Ordóñez M.; Rojas-Cabrera H.; Cativiela C. An overview of stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 2009, 65, 17–49. 10.1016/j.tet.2008.09.083. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhao D.; Wang R. Recent developments in metal catalyzed asymmetric addition of phosphorus nucleophiles. Chem. Soc. Rev. 2012, 41, 2095–2108. 10.1039/C1CS15247E. [DOI] [PubMed] [Google Scholar]; d Bhadury P. S.; Li H. Organocatalytic Asymmetric Hydrophosphonylation/Mannich Reactions Using Thiourea, Cinchona and Brønsted Acid Catalysts. Synlett 2012, 2012, 1108–1131. 10.1055/s-0031-1290193. [DOI] [Google Scholar]; e Herrera R. P. Organocatalytic Hydrophosphonylation Reaction of Carbonyl Groups. Chem. Rec. 2017, 17, 833–840. 10.1002/tcr.201600129. [DOI] [PubMed] [Google Scholar]
- For recent reports on enantioselective hydrophosphonylation of imines not included in the reviews above see:; a Nakamura S.; Hayashi M.; Hiramatsu Y.; Shibata N.; Funahashi Y.; Toru T. Catalytic Enantioselective Hydrophosphonylation of Ketimines Using Cinchona Alkaloids. J. Am. Chem. Soc. 2009, 131, 18240–18241. 10.1021/ja908940e. [DOI] [PubMed] [Google Scholar]; b George J.; Sridhar B.; Reddy B. V. S. First example of quinine-squaramide catalyzed enantioselective addition of diphenyl phosphite to ketimines derived from isatins. Org. Biomol. Chem. 2014, 12, 1595–1602. 10.1039/c3ob42026d. [DOI] [PubMed] [Google Scholar]; c Cheng M.-X.; Ma R.-S.; Yang Q.; Yang S.-D. Chiral Brønsted Acid Catalyzed Enantioselective Phosphonylation of Allylamine via Oxidative Dehydrogenation Coupling. Org. Lett. 2016, 18, 3262–3265. 10.1021/acs.orglett.6b01514. [DOI] [PubMed] [Google Scholar]; d Suneja A.; Unhale R. A.; Singh V. K. Enantioselective Hydrophosphonylation of in Situ Generated N-Acyl Ketimines Catalyzed by BINOL-Derived Phosphoric Acid. Org. Lett. 2017, 19, 476–479. 10.1021/acs.orglett.6b03623. [DOI] [PubMed] [Google Scholar]; e Chassillan L.; Yamashita Y.; Yoo W.-J.; Toffano M.; Guillot R.; Kobayashi S.; Vo-Thanh G. Enantioselective hydrophosphonylation of N-Boc imines using chiral guanidine–thiourea catalysts. Org. Biomol. Chem. 2021, 19, 10560–10564. 10.1039/D1OB01953H. [DOI] [PubMed] [Google Scholar]; f Chen D.-H.; Sun W.-T.; Zhu C.-J.; Lu G.-S.; Wu D.-P.; Wang A.-E.; Huang P.-Q. Enantioselective Reductive Cyanation and Phosphonylation of Secondary Amides by Iridium and Chiral Thiourea Sequential Catalysis. Angew. Chem., Int. Ed. 2021, 60, 8827–8831. 10.1002/anie.202015898. [DOI] [PubMed] [Google Scholar]
- Pettersen D.; Marcolini M.; Bernardi L.; Fini F.; Herrera R. P.; Sgarzani V.; Ricci A. Direct Access to Enantiomerically Enriched α-Amino Phosphonic Acid Derivatives by Organocatalytic Asymmetric Hydrophosphonylation of Imines. J. Org. Chem. 2006, 71, 6269–6272. 10.1021/jo060708h. [DOI] [PubMed] [Google Scholar]
- Similar dimeric structures have been previously observed in amido-thioureas:; a Zuend S. J.; Jacobsen E. N. Mechanism of Amido-Thiourea Catalyzed Enantioselective Imine Hydrocyanation: Transition State Stabilization via Multiple Non-Covalent Interactions. J. Am. Chem. Soc. 2009, 131, 15358–15374. 10.1021/ja9058958. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ford D. D.; Lehnherr D.; Kennedy C. R.; Jacobsen E. N. On- and Off-Cycle Catalyst Cooperativity in Anion-Binding Catalysis. J. Am. Chem. Soc. 2016, 138, 7860–7863. 10.1021/jacs.6b04686. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Strassfeld D. A.; Jacobsen E. N. The Aryl-Pyrrolidine- tert -Leucine Motif as a New Privileged Chiral Scaffold: The Role of Noncovalent Stabilizing Interactions. Supramol. Catal. 2022, 361–385. 10.1002/9783527832033.ch25. [DOI] [Google Scholar]
- For related dimeric, yet achiral, structures see:; White L. J.; Wells N. J.; Blackholly L. R.; Shepherd H. J.; Wilson B.; Bustone G. P.; Runacres T. J.; Hiscock J. R. Towards quantifying the role of hydrogen bonding within amphiphile self-association and resultant aggregate formation. Chem. Sci. 2017, 8, 7620–7630. 10.1039/C7SC03888G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R.; Dal Poggetto G.; Nilsson M.; Morris G. A. Improving the Interpretation of Small Molecule Diffusion Coefficients. Anal. Chem. 2018, 90, 3987–3994. 10.1021/acs.analchem.7b05032. [DOI] [PubMed] [Google Scholar]
- A 2D-DOSY analysis of racemic 5 was also performed, obtaining comparable results. See section 3.4 in the Supporting Information.
- See section 9 in the Supporting Information for computational details.
- a Thordarson P.Bindfit. http://www.supramolecular.org (accessed October, 2022).; b Thordarson P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. 10.1039/C0CS00062K. [DOI] [PubMed] [Google Scholar]; c Brynn Hibbert D.; Thordarson P. The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis. Chem. Commun. 2016, 52, 12792–12805. 10.1039/C6CC03888C. [DOI] [PubMed] [Google Scholar]
- For another example of spontaneous fractionation of enantiomers by achiral column chromatography see Supporting Information.
- a Lillo V. J.; Mansilla J.; Saá J. M. Organocatalysis by Networks of Cooperative Hydrogen Bonds: Enantioselective Direct Mannich Addition to Preformed Arylideneureas. Angew. Chem., Int. Ed. 2016, 55, 4312–4316. 10.1002/anie.201511555. [DOI] [PubMed] [Google Scholar]; b Lillo V. J.; Mansilla J.; Saá J. M. The role of proton shuttling mechanisms in solvent-free and catalyst-free acetalization reactions of imines. Org. Biomol. Chem. 2018, 16, 4527–4536. 10.1039/C8OB01007B. [DOI] [PubMed] [Google Scholar]
- Xie H.; Song A.; Zhang X.; Chen X.; Li H.; Sheng C.; Wang W. Quinine-thiourea catalyzed enantioselective hydrophosphonylation of trifluoromethyl 2(1H)-quinazolinones. Chem. Commun. 2013, 49, 928–930. 10.1039/C2CC38097H. [DOI] [PubMed] [Google Scholar]
- Gheewala Chirag D.; Collins Bridget E. Lambert Tristan, H. An aromatic ion platform for enantioselective Brønsted acid catalysis. Science 2016, 351, 961–965. 10.1126/science.aad0591. [DOI] [PubMed] [Google Scholar]
- Special precautions were taken for the determination of the enantiomeric ratios of the compounds described herein. Since spontaneous fractionation of enantiomers during purification may happen, we determined the enantiomeric purity of α-ureidophosphonates in the crude mixture and after column chromatography. Generally, we observed small differences between both values. See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.