

Abstract
Catenanes are a well-known class of mechanically interlocked molecules that possess chain-like architectures and have been investigated for decades as molecular machines and switches. However, the synthesis of higher-order catenanes with multiple, linearly interlocked molecular rings has been greatly impeded by the generation of unwanted oligomeric byproducts and figure-of-eight topologies that compete with productive ring closings. Here, we report two general strategies for the synthesis of oligo[n]catenanes that rely on a molecular “zip-tie” strategy, where the “zip-tie” is a central core macrocycle precursor bearing two phenanthroline (phen) ligands to make odd-numbered oligo[n]catenanes, or a preformed asymmetric iron(II) complex consisting of two macrocycle precursors bearing phen and terpyridine ligands to make even-numbered oligo[n]catenanes. In either case, preformed macrocycles or [2]catenanes are threaded onto the central “zip-tie” core using metal templation prior to ring-closing metathesis (RCM) reactions that generate several mechanical bonds in one pot. Using these synthetic strategies, a family of well-defined linear oligo[n]catenanes were synthesized, where n = 2, 3, 4, 5, or 6 interlocked molecular rings, and n = 6 represents the highest number of linearly interlocked rings reported to date for any isolated unimolecular oligo[n]catenane.
Short abstract
The synthesis of a series of oligo[n]catenanes (n = 2, 3, 4, 5, and 6) was achieved using a molecular “zip-tie” strategy involving orthogonal metal templation and ring-closing metathesis chemistry.
Introduction
In comparison to the most well-studied types of chemical bonds—covalent, ionic, and metallic—the mechanical bond remains relatively underexplored. Mechanical bonding occurs when two or more molecular species are interlocked via a physical entanglement in space, such that they cannot be separated without breaking covalent bonds.1 The two main classes of mechanically interlocked molecules (MIMs)2 are rotaxanes3 and catenanes,4 the former consisting of a molecularly shaped dumbbell threaded through one or more macrocycles, and the latter being constructed from two or more linearly, radially, or cyclically interlocked macrocycles. Of the two types of MIMs, catenanes5 are generally more challenging to synthesize because the ring-closing reactions required to establish the mechanical bond are notorious for generating non-interlocked byproducts6 and so-called figure-of-eight topologies.7 Linear catenanes are of particular interest because this topology maximizes the conformational degrees of freedom through rotations, translations, and rocking motions of the molecular rings.6 To facilitate catenation, template-directed syntheses operating under thermodynamic control have been pursued. These include metal-coordination,8 donor–acceptor,9 hydrogen-bonding,10 and anion–dipole11 interactions, to name some. For example, Stoddart and co-workers capitalized on charge-transfer interactions between π-electron-deficient and -rich macrocycles to successfully synthesize a linear [5]catenane they called “Olympiadane”.12,13 More recently, Iwamoto and co-workers used hydrogen bonding to template the synthesis of another linear [5]catenane.14 To date, both of these examples have stood as the record for the total number of interlocked macrocycles isolated as a linear, unimolecular oligo[n]catenane, even though Stoddart and co-workers did report by mass spectrometry a linear [7]catenane that was never isolated.15
Out of all the template-directed strategies, however, metal-coordination is usually the most efficient for assembling molecular precursors. Sauvage and co-workers pioneered16 the use of orthogonal metal templation to selectively assemble and ring-close macrocyclic precursors to make functional asymmetric catenanes. The selective orthogonal metalation arises from extended phenanthroline (phen) ligands that favor the formation of tetracoordinate complexes with monovalent metals (e.g., Cu+), and terpyridine (terpy) ligands that favor the corresponding hexacoordinate complexes with bivalent metals (e.g., Fe2+). Metal-based templation has also been used to construct catenanes of complex topology, such as Leigh and co-workers’ interwoven Star of David [2]catenane17 and Nitschke and co-workers’ cyclic [3]catenane18—both of which were preassembled using Fe2+-based helicates. Outside of well-defined unimolecular catenanes, there has also been interest in using [2]catenanes as building blocks in poly[2]catenanes,19 or as cross-linkers in polymer networks.20 Moreover, Di Stefano and co-workers demonstrated21 a fast route to synthesize a broad mixture of poly[n]catenates by performing ring-opening metathesis polymerizations on a copper(I) complex of a phen-based [2]catenane. Rowan and co-workers22,23 also adopted a one-pot approach, preassembling polydisperse metallosupramolecular polymers (MSPs) via metal coordination of open and closed macrocycles with zinc ions (i.e., Zn(II)), which were subjected to ring-closing metathesis (RCM) that produced a mixture of mostly linear and some branched and circular poly[n]catenanes. Lastly, Yagai and co-workers24 investigated the formation of supramolecular poly[n]catenanes through the self-assembly of kinetically produced toroids, where the latter catenane structures can disassemble at elevated temperatures.
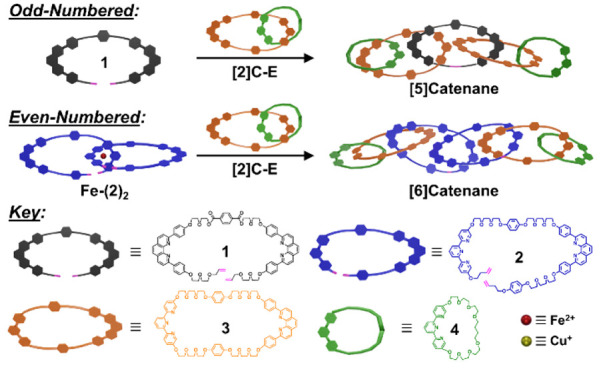
Even with these examples, there remains a fundamental synthetic gap between well-defined oligo[n]catenanes and the more broadly dispersed poly[n]catenanes. Oligo[n]catenanes, for example, can be made on a larger scale through moderately high-yielding reactions; however, they are usually of low molecular weight and possess “dead end” macrocycles that cannot be extended further (Figure 1a, previous work25 and others26,27). Poly[n]catenane syntheses may require fewer synthetic steps (i.e., one-pot reactions) and result in higher molecular weight polymers; yet, their syntheses lack control and generate product mixtures that are often difficult to purify. Here, we describe two molecular “zip-tie” synthetic strategies (Figure 1b) that build off our previous efforts to bridge this fundamental gap. The molecular “zip-tie” utilizes an open macrocycle with two phen ligands that are threaded through preformed macrocycles or [2]catenanes via coordination with copper(I), followed by a RCM step to produce odd-numbered linear oligo[2n+1]catenanes. Even-numbered [n]catenanes were synthesized as well via a double “zip-tie” approach by implementing sequential, orthogonal metal templation steps of dual-ligand macrocyclic precursors that were complexed with two transition metal ions (iron(II) and copper(I)) and preformed macrocycles or [2]catenanes. In the final step, two simultaneous RCM reactions on the precatenate metal complexes yielded well-defined, oligo[2n+2]catenanes. The sequential metal-coordinated assembly of the precursors prevented the formation of unwanted figure-of-eight topologies that often arise during simultaneous ring-closing reactions,7,28−31 a critical outcome that was proven by comparing the products generated from the syntheses of [2]- and [4]catenanes. The full utility of the double “zip-tie” strategy was demonstrated by synthesizing a linear [6]catenane, which, to the best of our knowledge, represents the highest number of linearly interlocked macrocycles ever isolated for a unimolecular catenane. This work sets the stage for future efforts to expand the syntheses toward the preparation of poly[n]catenanes, either with all mechanical bonds, or through the polymerization of terminal functional groups.32
Figure 1.

Overview of the “zip-tie” strategies to synthesize linear oligo[n]catenanes (n is the number of interlocked macrocycles). (a) Previous synthetic approach to prepare a metalated [4]catenane, [4]C-M-PM, in 55% yield. (b) This work, where single and double “zip-tie” synthetic strategies allowed for the synthesis of odd- and even-numbered [n]catenanes, respectively.
Results and Discussion
The synthesis of linear [3]catenanes often proceeds by the coupling of two [2]pseudorotaxanes33−36 or through the ring-closing of a [3]pseudorotaxane,37−39 the latter of which was employed here for the synthesis of the linear [3]catenane [3]C (Figure 2a, Scheme S18). However, many [3]catenanes have been constructed using small monofunctionalized terminal macrocycles, often resulting in low molecular weight products with limited opportunity for further catenation. In order to quickly achieve higher molecular weight MIMs while retaining the potential to synthesize higher order catenanes, a “zip-tie” approach was implemented with open phen-based macrocycle 1 (Figure 1b and Schemes S1–S3) and a 73-membered dual-ligand molecular ring 3 (Figure 1b, and Schemes S10–S11) to synthesize [3]C. Initial efforts to directly complex macrocycle 1 with 3 were unsuccessful because 3 collapses during monometalation with Cu+, producing a heteroleptic Cu+-phen-terpy complex.40 This unwanted complexation was circumvented by first “blocking” the terpy ligands with Fe2+ to give the dimeric macrocycle complex Fe-(3)2 (Figure 2a, Scheme S14). With the terpy ligands occupied, the phen ligands of Fe-(3)2 were readily monometalated with CuI to give Cu·Fe-(3)2. The pre-[3]catenate complex was formed by the addition of 1 in CH2Cl2, which then underwent a single RCM reaction with Grubbs' catalyst. In order to simplify the purification, the crude, ring-closed products were demetalated in a two-step process (Figure 2a, Scheme S18) using K2CO3 in DMF to remove Fe2+, followed by KCN in acetonitrile (MeCN)/H2O to remove Cu+. After an aqueous workup, the demetalated mixture was purified via recycling prep-GPC (Figure 3a, Figure S57) to afford [3]C, which was isolated in a 36% yield over the final three steps (i.e., complexation, ring-closing, and demetalation). The lower yield is due to the formation of [2]catenane byproduct and higher molecular weight oligomers which were observed during purification by recycling prep-GPC. Moreover, although this yield is lower than that (55%) from our previously reported synthesis to make the monophen-macrocycle-terminated [4]catenate (Figure 1a, [4]C-M-PM), the terminal rings of [3]C can be used for further catenation reactions and growth toward higher molecular weight oligo[n]catenanes.
Figure 2.

| “Zip-tie” strategies to synthesize odd-numbered linear oligo[n]catenanes. (a) (i) CuI, MeCN, 1, 25 °C, 2 days; (ii) Grubbs’ second-generation catalyst, CH2Cl2, 35 °C, 4 days; (iii) K2CO3, DMF, 75 °C, 1 day. (iv) KCN, MeCN/H2O, 25 °C, 2 h. (b) (v) FeCl2, CH2Cl2, DMF, 25 °C, 1 h; (vi) CuI, MeCN, 1, 25 °C, 18 h; (vii) Grubbs’ second-generation catalyst, CH2Cl2, 35 °C, 1 day; (iii) K2CO3, DMF, 75 °C, 1 day. (iv) KCN, MeCN/H2O, 25 °C, 2 h.
Figure 3.
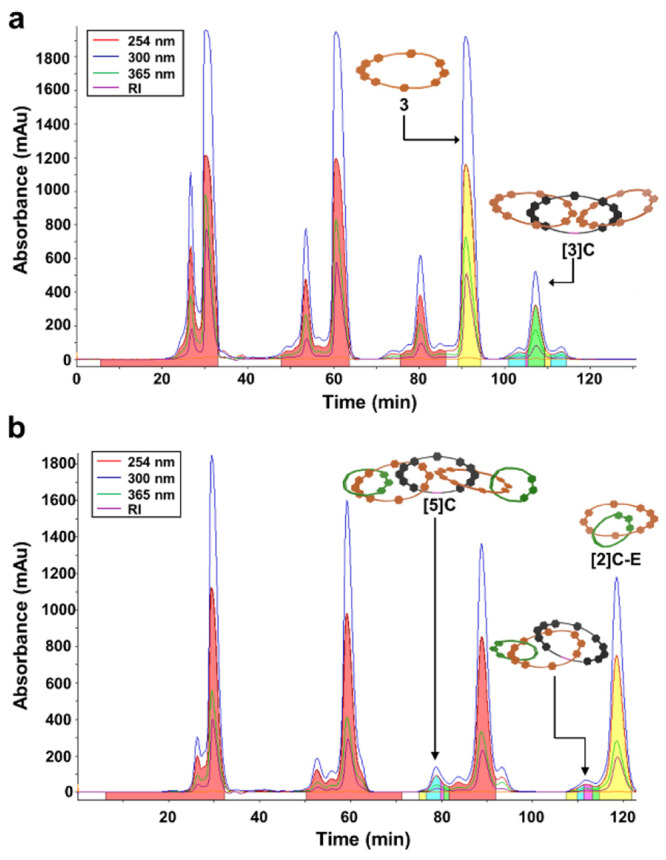
Recycling prep-GPC data for [3]C (a) and [5]C (b) collected in a DMF mobile phase at 8 mL·min–1. Pure [3]C was isolated in the green fraction at 108 min. Pure [5]C was isolated in the blue fraction at 75 min.
After the successful synthesis of [3]C, the “zip-tie” strategy was used to evaluate the efficacy of synthesizing a higher order linear [5]catenane [5]C (Figure 2b, Scheme S20) by replacing macrocycle 3 with a preformed [2]catenane [2]C-E (Schemes S15–S16), the latter of which bears phen and terpy coordination sites. Compound [2]C-E was first metalated with FeCl2 to occupy the terpy ligands, followed by monometalation of the unobstructed phen with CuI in the same reaction flask. Addition of 1 to this bimetallic intermediate yielded the pre-[5]catenate complex, which was subjected to the same RCM reaction and demetalation conditions as [3]C. Purification of the crude material via recycling prep-GPC (Figure 3b, Figure S59) produced [5]C in 15% yield (vide infra) over the final four steps. The purity of [3]C and [5]C was confirmed by narrow and unimodal analytical GPC traces (Figure 6c, Figure S51) and by 1H NMR (Figures S17–S18, S21–S22). Additionally, MALDI-TOF-MS of [5]C (Figure 4a, Figure S73) exhibited a well-defined parent molecular ion [M + H]+ with m/z = 5045 Da, as well as macrocyclic and [n]catenane fragments that are indicative of the linear [5]catenane topology. A similar fragmentation pattern was observed during MS/MS of [5]C (Figure 4b, Figures S67–S68), in which the [M + 3H]3+ adduct was isolated and fragmented, which produced distinct [3]- and [4]catenane fragments that could only be derived from [5]C. All other observable peaks by prep-GPC were isolated and subjected to MS/MS analysis. None showed any ring-closed products. Thus, the tandem MS data confirmed only one ring-closing reaction occurred during the synthesis of [5]C instead of the formation of deleterious figure-of-eight intermediates6,22−25 which have been reported previously for multiple, concurrent ring-closures (e.g., Figure 5).
Figure 6.

Double “zip-tie” strategies to synthesize even-numbered oligo[n]catenanes. (a) (i) in MeCN, added to Cu(MeCN)4PF6 (solid), 25 °C, 30 min; (ii) Fe-(2)2, 50% CH2Cl2/MeCN, 25 °C, 1 day; (iii) Grubbs’ second-generation catalyst, CH2Cl2, 35 °C, 2 day; (iv) Cs2CO3, DMF, 75 °C, 1 day; (v) KCN, MeCN/H2O, 25 °C, 30 min (b) “Zip-tie” synthesis of [4]C under topological control favors intramolecular ring closing and thus the catenane topology. (c) (vi) Cu(MeCN)4PF6 in MeCN, 25 °C, 30 min; (vii) Fe-(2)2, MeCN, 25 °C, 2.5 day; (iii) Grubbs’ second-generation catalyst, CH2Cl2, 35 °C, 18 h. (viii) K2CO3, DMF, 75 °C, 1 day; (v) KCN, MeCN/H2O, 25 °C, 30 min.
Figure 4.
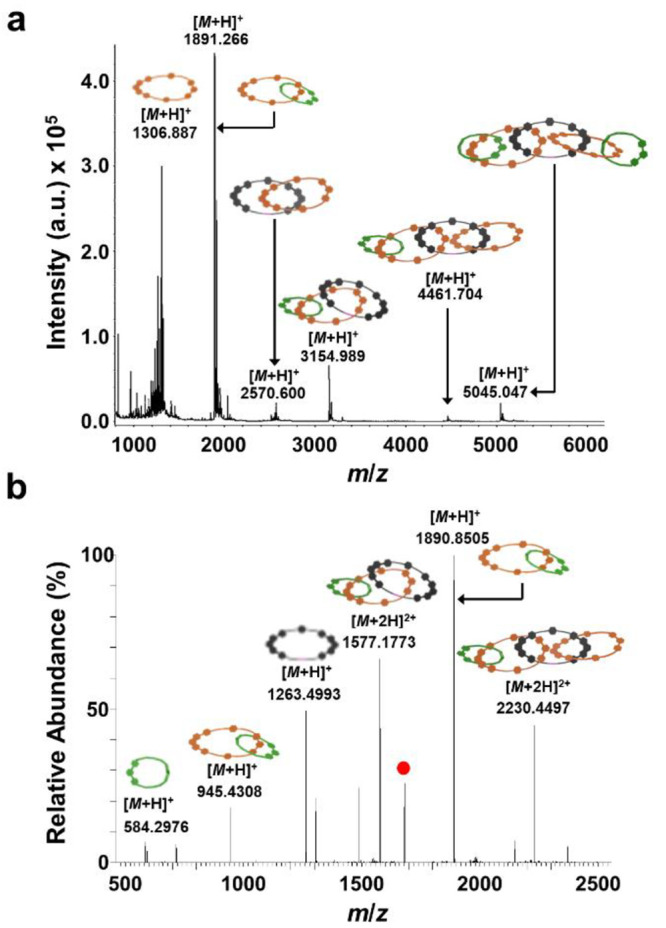
(a) MALDI-TOF-MS of [5]C with α-cyano-4-hydroxycinnamic acid matrix. (b) THRMS-ESI (i.e., MS/MS) of [5]C; the [M + 3H]3+ = 1682.06 Da peak (red dot) was isolated and fragmented.
Figure 5.

(a) Synthesis of topological products [2]C and I. (i) Grubbs’ second-generation catalyst, CH2Cl2, 35 °C, 1 day; (ii) Cs2CO3, DMF, 75 °C, 1 day. (b) Tandem high-resolution mass spectrometry–electrospray ionization (THRMS-ESI, i.e., MS/MS) of [2]C; the [M + 3H]3+ peak was isolated, and its fragmentation data are shown in the bottom spectrum. (c) THRMS-ESI of I; the [M + 3H]3+ peak was isolated, and its fragmentation data are shown in the bottom spectrum.
Since the “zip-tie” approach with a single central ring combined with preformed macrocycles or [2]catenanes is designed to minimize the number of necessary ring-closing events, and thus reduce the likelihood of generating unwanted byproducts, higher yielding syntheses were anticipated. However, an obvious dichotomy exists between the combined overall yields obtained for catenanes [5]C (15%) and [3]C (36%), where each yield includes metalation, self-assembly, RCM, and demetalation but excludes the steps required to make the premade macrocycle and [2]catenane end-caps. We speculate the difference in yields for this head-to-head comparison largely stems from the relative stability of the respective precatenate complexes during the RCM step, which in principle should be the same for each mechanical bond-forming reaction due to the commonality of the approach and central macrocycle 1. This notion is supported by comparing the recycling prep-GPC traces obtained during the purification of [3]C versus [5]C (Figure 3a,b). For the former, the largest peak outside of the starting macrocycle 3 (which was added in excess) relates to the [3]C product, whereas for the latter, there is a substantial peak for the incomplete [3]catenane byproduct that results from ring closing of a partial precatenate complex. This partial instability may also lead to the production of other unwanted kinetic byproducts, such as disperse oligomers, which may further decrease the overall yield for [5]C.
Although the single “zip-tie” reaction with 1 was successfully implemented to produce [3]C and [5]C, it has thus far been limited to the synthesis of lower molecular weight [2n+1]catenanes, where n is the number of preformed interlocked macrocycles. Inspired by the double ring-closing approach established by Sauvage over three decades ago,41 we envisioned a process to achieve higher-order even-numbered linear catenanes by using a preformed [2]catenane building block that can undergo further catenation. We previously reported25 the synthesis of the dual-ligand open macrocycle 2 (Figure 1, and Schemes S7–S9) and its iron(II) complex Fe-(2)2 (Figure 5a and Scheme S12). Open macrocycle 2 contains tridentate terpy and bidentate phen ligands, which preferentially bind bivalent and monovalent metals, respectively. Asymmetric short and long olefin-functionalized linkers were also implemented in an attempt to mitigate the intermolecular metathesis reaction that leads to topologically trivial figure-of-eights, while still promoting intramolecular ring-closing to afford the desired [2]catenane product. Despite these efforts, the double-RCM reactions of Fe-(2)2 with the second-generation Grubbs’ catalyst (Figure 5a, Scheme S13) afforded the figure-of-eight product I and the [2]catenane product [2]C in a nearly 2:1 ratio. Moreover, due to the asymmetric nature of 2, compound [2]C was isolated as a racemic mixture (Figure S1). To simplify the purification process, the mixture of ring-closed products was demetalated with a suspension of Cs2CO3 in DMF at 75 °C. The metal-free products were separated via recycling prep-GPC (Figure S56). The identity of the isolated fractions was determined by MS/MS experiments (Figure 5b,c, Figures S62–S63), where in each case, the [M + 3H]3+ adduct was isolated and fragmented until the parent adduct was consumed. The cleavage of any bond in one of the macrocycles that make up [2]C yields an intact macrocycle, which is what was observed (Figure 5b, lower spectrum) as the major species in the MS/MS experiment. However, the fragmentation of the larger macrocycle I generated a complex mass spectrum with a wide range of fragments (Figure 5c, lower spectrum). These fragmentation patterns are consistent with those observed by Au-Yeung and co-workers31 who also differentiated linear oligo[n]catenanes from their figure-of-eight counterparts using MS/MS.
The predominate isolation of I over [2]C led to the hypothesis that the flexible glycol-based linkers in Fe-(2)2 readily allowed for intermolecular ring-closings instead of the desired intramolecular reaction (Figure 5a), ultimately yielding the figure-of-eight product as the major topological intermediate product. We speculated that if the flexible linkers could be “rigidified” (Figure 6),26,27 the intermolecular RCM reaction would become less favorable. As an alternative to making chemical modifications of 2 to accomplish this goal, we envisaged complex Fe-(2)2 could be “rigidified” by noncovalently threading on the iron(II)-macrocycle dimer Fe-(3)2 before ring closing to afford a pre-[4]catenate complex (Figure 6a,b), in a manner similar to the synthesis of [3]C. Thus, under topological control, the resultant pre-[4]catenate complex (Figure 6b) can spatially arrange the olefin linkers to favor intramolecular RCM reactions over unproductive intermolecular pathways.
We previously demonstrated (Figure 1a) a similar double “zip-tie” strategy to synthesize a linear [4]catenane after two simultaneous one-pot RCM reactions; however, in that case the smaller, monofunctionalized phen-based macrocycles at each terminus produced lower molecular weight products and prevented further syntheses of higher-order catenanes.25 Additionally, the asymmetric nature of the previous end-cap macrocycles led to difficult purification, as well as a mixture of six topologically diastereomeric linear [4]catenanes that gave rise to complicated NMR spectra. In order to alleviate these issues, the double “zip-tie” strategy was adapted using the larger and more symmetric macrocycle 3 as its Fe2+ dimer Fe-(3)2 to synthesize the linear [4]catenane [4]C, which is composed of four large dual-ligand terpy-phen-based macrocycles (Figure 1, Figure 6a,b, and Scheme S19). With the terpy ligands “rigidified” as octahedral Fe2+ complexes, the phen ligands were readily monometalated with Cu(MeCN)4PF6 to give Cu·Fe-(3)2 as an air-sensitive intermediate, which was stirred under N2 atmosphere at 25 °C for 15 min before a solution of Fe-(2)2 was added to form the pre-[4]catenate complex (Figure 6b). Double RCM reactions were carried out next with Grubbs' catalyst, and the conversion to [4]C was confirmed by 1H NMR. After sequential demetalation steps with Cs2CO3 and KCN (Figure 6a, Scheme S19), the demetalated mixture was purified via recycling prep-GPC (Figure 7a, Figure S58) to afford [4]C in an 11% yield over the final three steps. Once again, the isolated yield to make the larger-ringed [4]C was lower presumably because of complex instability during the RCM step. The prep-GPC trace reveals substantial peaks for a [2]catenane byproduct, as well as unidentified higher and lower molecular weight byproducts, which were purified out. All other minor peaks by prep-GPC were isolated and subjected to MS/MS analysis. None showed any ring-closed products. This result is consistent with the aforementioned synthesis to make [5]C, except in this instance two RCM steps were required to establish the desired number of mechanical bonds.
Figure 7.

Recycling pre-GPC data for [4]C (a) (second injection) and [6]C (b) were collected in a DMF mobile phase at 8 mL·min–1. Pure [4]C was isolated in the blue fraction at 235 min, whereas pure [6]C was isolated in the green fraction at 125 min. (c) Overlay of analytical GPC traces (normalized dRI) of [3]C, [4]C, [5]C, and [6]C in DMF with 0.025 M LiBr at 60 °C. (d) Overlay of analytical HPLC traces (normalized absorbance at 254 nm) of [3]C, [4]C, [5]C, and [6]C with a gradient mobile phase of MeCN/H2O (0.1% TFA): 5 to 100% in 10 min and 100% MeCN for 15 min at 40 °C. Note: the solubility of the catenane products in the HPLC mobile phase was poor until the end of the gradient, i.e., closer to 95% MeCN. This caused the catenane products to streak on the column. Additionally, see Figures S75–S78 for corresponding LRMS-ESI spectra.
The purity of [4]C was confirmed by the narrow and unimodal analytical GPC traces (Figure 7c, Figure S51) and HPLC traces (Figure 7d, Figure S61), as well as by 1H NMR (Figures S19–S20). The topology of [4]C was confirmed using spectrometric methods, whereby MALDI-TOF-MS (Figure 8a, Figure S72) showed a prominent peak for the corresponding parent molecular ion [M + H]+, as well as the macrocycle and catenane fragments. The asymmetric [2]catenane fragment at m/z = 2579 Da (Figure 8a) could have only originated from the linear [4]catenane, as opposed to the hypothetical [3]catenane derived from the figure-of-eight topological product. A similar fragmentation pattern was observed during MS/MS of [4]C (Figure 8c, Figures S65–S66), in which the [M + 3H]3+ adduct was isolated and fragmented. In addition to the asymmetric [2]catenane, the ring-closed product of 2 was also observed, both of which can only originate from the pathway to [4]C. In contrast with the RCM reaction of complex Fe-(2)2, in which intramolecular and intermolecular pathways occur concurrently to produce a 2:1 ratio of figure-of-eight to catenane products, the double “zip-tie” approach involving two Cu·Fe-(3)2 threaded onto Fe-(2)2 exclusively produced the catenane product [4]C as no figure-of-eight impurities were ever isolated.
Figure 8.

MALDI-TOF-MS of [4]C (a) and [6]C (b) with α-cyano-4-hydroxycinnamic acid matrix. (c) THRMS-ESI (i.e., MS/MS) of [4]C; the [M + 3H]3+ = 1720.06 Da peak (red dot) was isolated and fragmented. (d) THRMS-ESI of [6]C; the [M + 3Na]3+ = 2130 Da peak (red dot) was isolated and fragmented. Each peak labeled with a red dot represents remaining unfragmented catenane.
The complete versatility of the double “zip-tie” approach was demonstrated by substituting the end-cap macrocycles 3 that were used to prepare [4]C with preformed [2]catenanes [2]C-E to synthesize a linear [6]catenane [6]C (Figure 6c, Scheme S21). A slight modification of the synthetic protocol was made between [5]C and [6]C, namely, the preformed [2]catenane was metalated with Fe2+ in a separate step producing [2]catenate Fe-[2]C-E (Scheme S17). The open phen-coordination site of Fe-[2]C-E was then monometalated with Cu(MeCN)4PF6, analogous to the synthesis of [4]C. A solution of Fe-(2)2 was added to Cu·Fe-[2]C-E via syringe, and the dark red solution was allowed to stir under N2 atmosphere for 2.5 days to ensure formation of the pre-[6]catenate complex consisting of two Cu·Fe-[2]C-E catenates threaded onto complex Fe-(2)2. Next, RCM with Grubbs' catalyst was carried out on the pre-[6]catenate complex in anhydrous CH2Cl2 under mild heating. The crude ring-closed products were demetalated (Figure 6c, Scheme S21) and purified via recycling prep-GPC (Figure 7b, Figure S60) to afford [6]C, which was isolated in a 25% yield over the final three steps. It is important to note that the yield here was higher than that for [4]C, which we surmise is due to the [2]catenane end-cap [2]C-E being relatively more stable than the iron(II)-macrocycle dimer complex Fe-(3)2 during the course of two RCM reactions. Also, the use of complex Fe-(2)2 as the key “zip-tie” core seems to be more compatible with the end-cap [2]catenane [2]C-E versus that when macrocycle 1 was used to synthesize [5]C in 15% yield.
The purity of [6]C was verified using analytical GPC (Figure 7c, Figure S51) and HPLC (Figure 7d, Figure S61), as well as 1H NMR (Figures S23–S24, S26). Due to its higher molecular weight, [6]C exhibited a shorter retention time by GPC compared to [3]C, [4]C, and [5]C. MALDI-TOF-MS of [6]C (Figure 8b, Figure S74) displayed clear peaks for the parent molecular ion [M + H]+ and the various [n]catenane fragments. The asymmetric [3]catenane fragment at m/z = 3163 Da is distinctive to the linear [6]catenane topology because a hypothetical [5]catenane derived from the figure-of-eight topological product could not fragment in this manner. Further evidence for the topology of [6]C was obtained via MS/MS experiments (Figure 8d, Figures S69–S70), in which the [M + 3H]3+ adduct was isolated and fragmented. The asymmetric [2]catenane fragment at m/z = 2579 Da and the ring-closed macrocycle from 2 at m/z = 1272 Da could only be observed by fragmentation of [6]C. The detection of these fragmented species by MALDI-TOF-MS and MS/MS, combined with analytical chromatography methods, provide definitive evidence for the successful synthesis and isolation of the unimolecular, linear [6]catenane. Moreover, all other observable peaks by prep-GPC were isolated and subjected to MS/MS analysis. None showed any ring-closed products. As was observed during the synthesis of [4]C, the topological control afforded by the double “zip-tie” method again prevented intermolecular ring-closing and yielded only the expected catenane product [6]C and no figure-of-eights (based on characterization of all isolated GPC fractions).
Conclusion
Two general synthetic blueprints have been developed to make higher-order, linear [2n+1]catenanes and [2n+2]catenanes under topological control using orthogonal metal templation to generate precatenate complexes, followed by RCM reactions in a single step. In the case of the even-numbered catenane, the so-called molecular “zip-tie” approach eliminated the formation of figure-of-eight topological products during the critical ring-closing steps that lead to mechanical bond formation. These methodologies were applied to synthesize a family of linear oligo[n]catenanes, where n = 2, 3, 4, 5, or 6, the latter of which representing the highest order unimolecular and linear catenane ever isolated. We offer these synthetic approaches as a way to bridge the gap between the synthesis of lower molecular weight oligo[n]catenanes and true linear poly[n]catenanes, making their study in materials a realistic proposition. Future endeavors in this research area relate to the synthesis of catenanes with terminal rings that bear polymerizable functional groups,32 such that poly[n]catenanes can be generated through traditional polymerization methods (e.g., step-growth), in addition to refining the synthetic approach toward true poly[n]catenanes consisting exclusively of mechanically bonded molecular rings.
Methods
The synthesis and spectroscopic characterization of all precursors and catenanes are described in full detail in Schemes S1–S21 and Figures S1–S78.
Synthesis of Linear [6]catenane [6]C
A solution of end-cap [2]catenate Fe-[2]C-E (0.0601 g, 0.0284 mmol, 4.0 equiv) was prepared in 10 mL of N2-purged anhydrous MeCN in an oven-dried 50 mL round-bottom (RB) flask. To this was added a solution of Cu(MeCN)4PF6 (0.0105 g, 0.028 mmol, 4.0 equiv) in 3 mL of N2-purged anhydrous MeCN. The solution was stirred at room temperature for 0.5 h under N2 atmosphere before the addition of a solution of Fe-(2)2 (0.022 g, 0.0077 mmol, 1.0 equiv) in 7 mL of N2-purged anhydrous MeCN via syringe. The dark red solution was stirred under N2 atmosphere for 2.5 days. The solvent was removed via rotary evaporator, and the crude was taken up in 100 mL of CH2Cl2. The organics were washed with 3 × 50 mL of DI H2O and dried over Na2SO4. The dark red solution was filtered, and the filtrate was concentrated via rotary evaporator to afford the pre-[6]catenate complex as a dark red film, which was used without further purification. The crude pre-[6]catenate complex was redissolved in 25 mL of anhydrous CH2Cl2 in a 100 mL RB flask, and a solution of Grubbs’ second-generation catalyst (0.0013 g, 0.0015 mmol, 0.2 equiv) in 1 mL CH2Cl2 was added. The flask was fitted with a Vigreux column and was heated to 35 °C while stirring under N2 atmosphere. After 18 h, an aliquot was taken and quenched with ethyl vinyl ether (EVE). The reaction was deemed complete by 1H NMR, and the remaining reaction mixture was quenched with 1 mL of EVE and 5 mL of MeCN. The solvent was then removed via rotary evaporator to afford the crude [6]catenate mixture as a dark red film. In order to remove noninterlocked structures and simplify the purification process, the Fe2+ and Cu+ templates were removed. The Fe2+ ion was removed from the crude mixture with addition of a weak inorganic base and moderate heating, while the Cu+ ion was removed in a second step by the addition of a strongly competing ligand. The crude film was redissolved in 25 mL of DMF in a 100 mL RB flask. Solid K2CO3 (1.0 g, 7.23 mmol, 1000 equiv) was added, and the suspension was heated to 75 °C for 1 day while stirring open to air. The solvent was removed via rotary evaporator, and 50 mL of MeCN was added to the reaction mixture. A solution of KCN (0.2 g, 3.07 mmol, 400 equiv) in 10 mL of H2O was added via syringe. The suspension was stirred open to air at room temperature for 0.5 h. The suspension was diluted with 300 mL of CH2Cl2. The organics were washed with 3 × 100 mL DI H2O, dried over Na2SO4, and filtered. The solvent was removed via rotary evaporator, and the crude was redissolved in 4 mL of GPC grade DMF. The solution was filtered via syringe filter and was purified via recycling preparative GPC with DMF mobile phase. The mixed fractions were also collected and repurified. The linear [6]catenane [6]C was isolated as a yellow/orange film (0.0122 g, 25% based on Fe-(2)2).
Gel Permeation Chromatography (GPC)
Recycling preparative gel permeation chromatography (prep-GPC) was performed on a Japan Analytical Industry LaboAce instrument with one JAIGEL-2HR column and one JIAGEL-2.5HR column in sequence, running with DMF at 8 mL·min–1 as the mobile phase. Analytical GPC analyses were performed on an Agilent 1260 Infinity setup with two Shodex GPC KD-8060 columns in sequence, running with DMF (0.025 M LiBr) at 1 mL·min–1 as the mobile phase. The differential refractive index (dRI) of each compound was monitored using a Wyatt Optilab T-rEX detector.
High-Pressure Liquid Chromatography (HPLC)
Analytical HPLC analyses were performed on an Avant 2000 HPLC with a Shodex Asahipak ODP-50-2D reverse-phase column with a gradient mobile phase of H2O with 0.1% trifluoracetic acid (TFA) and MeCN with 0.1% TFA running at 0.2 mL·min–1, which was in series with an Advion Expression-L Compact Mass Spectrometer. UV–vis absorbance was recorded at 254 nm.
Tandem High-Resolution Mass Spectrometry Electrospray Ionization (THRMS-ESI)
THRMS-ESI mass spectra were recorded on a Bruker maXis 4G Q-TOF mass spectrometer.
MALDI-Time-of-Flight (MALDI-TOF)
MALDI-TOF mass spectra were recorded on a Bruker Solaris 12T FT-MS; samples were prepared using 2,5-dihydroxybenzoic or α-cyano-4-hydroxycinnamic acid matrices.
Acknowledgments
Funding support was provided by the David and Lucile Packard Foundation through J.C.B.’s Packard Fellowship for Science and Engineering. This research is also supported by the National Science Foundation under Grant CHE-2204184. A.O.D. acknowledges support from the National Science Foundation Graduate Research Fellowship Program (NSF GRFP; DGE-1745038) and the PEO International Scholar Award.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications Web site as a single PDF. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.2c00697.
Synthesis and characterization data (i.e., nuclear magnetic resonance (NMR), mass spectrometry (MS), gel permeation chromatography (GPC), and liquid chromatography (LC), etc.) (PDF)
The authors declare the following competing financial interest(s): The authors filed a non-provisional patent containing part of the work described in this manuscript.
Supplementary Material
References
- Stoddart J. F. The chemistry of the mechanical bond. Chem. Soc. Rev. 2009, 38 (6), 1802–1820. 10.1039/b819333a. [DOI] [PubMed] [Google Scholar]
- De Bo G. Mechanochemistry of the mechanical bond. Chem. Sci. 2018, 9 (1), 15–21. 10.1039/C7SC04200K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M.; Yang Y.; Chi X.; Yan X.; Huang F. Development of Pseudorotaxanes and Rotaxanes: From Synthesis to Stimuli-Responsive Motions to Applications. Chem. Rev. 2015, 115 (15), 7398–7501. 10.1021/cr5005869. [DOI] [PubMed] [Google Scholar]
- Gil-Ramírez G.; Leigh D. A.; Stephens A. J. Catenanes: Fifty Years of Molecular Links. Angew. Chem., Int. Ed. 2015, 54 (21), 6110–6150. 10.1002/anie.201411619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au-Yeung H. Y.; Deng Y. Distinctive features and challenges in catenane chemistry. Chem. Sci. 2022, 13 (12), 3315–3334. 10.1039/D1SC05391D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena-Hernando S.; Pérez E. M. Mechanically interlocked materials. Rotaxanes and catenanes beyond the small molecule. Chem. Soc. Rev. 2019, 48 (19), 5016–5032. 10.1039/C8CS00888D. [DOI] [PubMed] [Google Scholar]
- Belfrekh N.; Dietrich-Buchecker C.; Sauvage J.-P. Unexpected Synthesis of an 8-Shaped Macrocycle Instead of an Interlocking-Ring System. Inorg. Chem. 2000, 39 (22), 5169–5172. 10.1021/ic991502k. [DOI] [PubMed] [Google Scholar]
- Lewis J. E. M.; Beer P. D.; Loeb S. J.; Goldup S. M. Metal ions in the synthesis of interlocked molecules and materials. Chem. Soc. Rev. 2017, 46 (9), 2577–2591. 10.1039/C7CS00199A. [DOI] [PubMed] [Google Scholar]
- Griffiths K. E.; Stoddart J. F. Template-directed synthesis of donor/acceptor [2]catenanes and [2]rotaxanes. Pure Appl. Chem. 2008, 80 (3), 485–506. 10.1351/pac200880030485. [DOI] [Google Scholar]
- Evans N. H. Recent Advances in the Synthesis and Application of Hydrogen Bond Templated Rotaxanes and Catenanes. Eur. J. Org. Chem. 2019, 2019 (21), 3320–3343. 10.1002/ejoc.201900081. [DOI] [Google Scholar]
- Vickers M. S.; Beer P. D. Anion templated assembly of mechanically interlocked structures. Chem. Soc. Rev. 2007, 36 (2), 211–225. 10.1039/B518077P. [DOI] [PubMed] [Google Scholar]
- Amabilino D. B.; Ashton P. R.; Reder A. S.; Spencer N.; Stoddart J. F. Olympiadane. Angew. Chem., Int. Ed. Engl. 1994, 33 (12), 1286–1290. 10.1002/anie.199412861. [DOI] [Google Scholar]
- Amabilino D. B.; Ashton P. R.; Balzani V.; Boyd S. E.; Credi A.; Lee J. Y.; Menzer S.; Stoddart J. F.; Venturi M.; Williams D. J. Oligocatenanes Made to Order1. J. Am. Chem. Soc. 1998, 120 (18), 4295–4307. 10.1021/ja9720873. [DOI] [Google Scholar]
- Iwamoto H.; Tafuku S.; Sato Y.; Takizawa W.; Katagiri W.; Tayama E.; Hasegawa E.; Fukazawa Y.; Haino T. Synthesis of linear [5]catenanes via olefin metathesis dimerization of pseudorotaxanes composed of a [2]catenane and a secondary ammonium salt. Chem. Commun. 2016, 52 (2), 319–322. 10.1039/C5CC07562A. [DOI] [PubMed] [Google Scholar]
- Ashton P. R.; Baldoni V.; Balzani V.; Claessens C. G.; Credi A.; Hoffmann H. D. A.; Raymo F. M.; Stoddart J. F.; Venturi M.; White A. J. P.; Williams D. J. Template-Directed Syntheses, Spectroscopic Properties, and Electrochemical Behavior of [n]Catenanes. Eur. J. Org. Chem. 2000, 2000 (7), 1121–1130. . [DOI] [Google Scholar]
- Cárdenas D. J.; Gaviña P.; Sauvage J.-P. Construction of Interlocking and Threaded Rings Using Two Different Transition Metals as Templating and Connecting Centers: Catenanes and Rotaxanes Incorporating Ru(terpy)2-Units in Their Framework. J. Am. Chem. Soc. 1997, 119 (11), 2656–2664. 10.1021/ja963955j. [DOI] [Google Scholar]
- Leigh D. A.; Pritchard R. G.; Stephens A. J. A Star of David catenane. Nat. Chem. 2014, 6 (11), 978–982. 10.1038/nchem.2056. [DOI] [PubMed] [Google Scholar]
- Wood C. S.; Ronson T. K.; Belenguer A. M.; Holstein J. J.; Nitschke J. R. Two-stage directed self-assembly of a cyclic [3]catenane. Nat. Chem. 2015, 7 (4), 354–358. 10.1038/nchem.2205. [DOI] [PubMed] [Google Scholar]
- Weidmann J.-L.; Kern J.-M.; Sauvage J.-P.; Muscat D.; Mullins S.; Köhler W.; Rosenauer C.; Räder H. J.; Martin K.; Geerts Y. Poly[2]catenanes and Cyclic Oligo[2]catenanes Containing Alternating Topological and Covalent Bonds: Synthesis and Characterization. Chem.—Eur. J. 1999, 5 (6), 1841–1851. . [DOI] [Google Scholar]
- Xing H.; Li Z.; Wu Z. L.; Huang F. Catenane Crosslinked Mechanically Adaptive Polymer Gel. Macromol. Rapid Commun. 2018, 39 (1), 1700361. 10.1002/marc.201700361. [DOI] [PubMed] [Google Scholar]
- Berrocal J. A.; Pitet L. M.; Nieuwenhuizen M. M. L.; Mandolini L.; Meijer E. W.; Di Stefano S. Ring-Opening Metathesis Polymerization of a Diolefinic [2]-Catenane-Copper(I) Complex: An Easy Route to Polycatenanes. Macromolecules 2015, 48 (5), 1358–1363. 10.1021/ma502392v. [DOI] [Google Scholar]
- Wu Q.; Rauscher P. M.; Lang X.; Wojtecki R. J.; de Pablo J. J.; Hore M. J. A.; Rowan S. J. Poly[n]catenanes: Synthesis of molecular interlocked chains. Science 2017, 358 (6369), 1434–1439. 10.1126/science.aap7675. [DOI] [PubMed] [Google Scholar]
- Tranquilli M. M.; Wu Q.; Rowan S. J. Effect of metallosupramolecular polymer concentration on the synthesis of poly[n]catenanes. Chem. Sci. 2021, 12 (25), 8722–8730. 10.1039/D1SC02450G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S.; Kato Y.; Higashiharaguchi S.; Aratsu K.; Isobe A.; Saito T.; Prabhu D. D.; Kitamoto Y.; Hollamby M. J.; Smith A. J.; Dalgliesh R.; Mahmoudi N.; Pesce L.; Perego C.; Pavan G. M.; Yagai S. Self-assembled poly-catenanes from supramolecular toroidal building blocks. Nature 2020, 583 (7816), 400–405. 10.1038/s41586-020-2445-z. [DOI] [PubMed] [Google Scholar]
- Colley N. D.; Nosiglia M. A.; Li L.; Amir F.; Chang C.; Greene A. F.; Fisher J. M.; Li R.; Li X.; Barnes J. C. One-Pot Synthesis of a Linear [4]Catenate Using Orthogonal Metal Templation and Ring-Closing Metathesis. Inorg. Chem. 2020, 59 (15), 10450–10460. 10.1021/acs.inorgchem.0c00735. [DOI] [PubMed] [Google Scholar]
- Wang K.; Yee C.-C.; Au-Yeung H. Y. Facile syntheses of [3]-, [4]- and [6]catenanes templated by orthogonal supramolecular interactions. Chem. Sci. 2016, 7 (4), 2787–2792. 10.1039/C5SC04774A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A. W. H.; Lai S. K.-M.; Yee C.-C.; Au-Yeung H. Y. Macrocycle Dynamics in a Branched [8]Catenane Controlled by Three Different Stimuli in Three Different Regions. Angew. Chem., Int. Ed. 2022, 61 (1), e202110200 10.1002/anie.202110200. [DOI] [PubMed] [Google Scholar]
- Fuller A.-M. L.; Leigh D. A.; Lusby P. J.; Slawin A. M.; Walker D. B. Selecting topology and connectivity through metal-directed macrocyclization reactions: a square planar palladium [2] catenate and two noninterlocked isomers. J. Am. Chem. Soc. 2005, 127 (36), 12612–12619. 10.1021/ja053005a. [DOI] [PubMed] [Google Scholar]
- Loren J. C.; Gantzel P.; Linden A.; Siegel J. S. Synthesis of achiral and racemic catenanes based on terpyridine and a directionalized terpyridine mimic, pyridyl-phenanthroline. Org. Biomol. Chem. 2005, 3 (17), 3105–3116. 10.1039/b506101f. [DOI] [PubMed] [Google Scholar]
- Leigh D. A.; Lusby P. J.; McBurney R. T.; Morelli A.; Slawin A. M.; Thomson A. R.; Walker D. B. Getting harder: cobalt (III)-template synthesis of catenanes and rotaxanes. J. Am. Chem. Soc. 2009, 131 (10), 3762–3771. 10.1021/ja809627j. [DOI] [PubMed] [Google Scholar]
- Yee C.-C.; Ng A. W. H.; Au-Yeung H. Y. Control over the macrocyclisation pathway and product topology in a copper-templated catenane synthesis. Chem. Commun. 2019, 55 (44), 6169–6172. 10.1039/C9CC02263E. [DOI] [PubMed] [Google Scholar]
- Nosiglia M. A.; Colley N. D.; Danielson M. K.; Palmquist M. S.; Delawder A. O.; Tran S. L.; Harlan G. H.; Barnes J. C. Metalation/Demetalation as a Postgelation Strategy To Tune the Mechanical Properties of Catenane-Crosslinked Gels. J. Am. Chem. Soc. 2022, 144 (22), 9990–9996. 10.1021/jacs.2c03166. [DOI] [PubMed] [Google Scholar]
- Sauvage J. P.; Weiss J. Synthesis of biscopper(I) [3]-catenates: multiring interlocked coordinating systems. J. Am. Chem. Soc. 1985, 107 (21), 6108–6110. 10.1021/ja00307a049. [DOI] [PubMed] [Google Scholar]
- Dietrich-Buchecker C. O.; Hemmert C.; Khemiss A. K.; Sauvage J. P. Synthesis of dicopper [3]-catenates and [3]-catenands by acetylenic oxidative coupling. Preparation and study of corresponding homodimetallic [3]-catenates [silver(1+), zinc(2+), cobalt(2+), and nickel(2+)]. J. Am. Chem. Soc. 1990, 112 (22), 8002–8008. 10.1021/ja00178a024. [DOI] [Google Scholar]
- Megiatto J. D.; Schuster D. I. ″Click″ Methodology for Synthesis of Functionalized [3]Catenanes: Toward Higher Interlocked Structures. Chem.—Eur. J. 2009, 15 (22), 5444–5448. 10.1002/chem.200900536. [DOI] [PubMed] [Google Scholar]
- Nguyen M. T.; Ferris D. P.; Pezzato C.; Wang Y. P.; Stoddart J. F. Densely Charged Dodecacationic [3]- and Tetracosacationic Radial [5]Catenanes. Chem-Us 2018, 4 (10), 2329–2344. 10.1016/j.chempr.2018.07.010. [DOI] [Google Scholar]
- Ashton P. R.; Brown C. L.; Chrystal E. J. T.; Goodnow T. T.; Kaifer A. E.; Parry K. P.; Slawin A. M. Z.; Spencer N.; Stoddart J. F.; Williams D. J. Self-Assembling [3]Catenanes. Angew. Chem. Int. Edit 1991, 30 (8), 1039–1042. 10.1002/anie.199110391. [DOI] [Google Scholar]
- Gupta M.; Kang S. S.; Mayer M. F. A double ring-closing olefin metathesis approach to [3]catenanes. Tetrahedron Lett. 2008, 49 (18), 2946–2950. 10.1016/j.tetlet.2008.03.012. [DOI] [Google Scholar]
- Wojtecki R. J.; Wu Q.; Johnson J. C.; Ray D. G.; Korley L. T. J.; Rowan S. J. Optimizing the formation of 2,6-bis(N-alkyl-benzimidazolyl)pyridine-containing [3]catenates through component design. Chem. Sci. 2013, 4 (12), 4440–4448. 10.1039/c3sc52082j. [DOI] [Google Scholar]
- De S.; Pramanik S.; Schmittel M. A monomer-dimer nanoswitch that mimics the working principle of the SARS-CoV 3CLpro enzyme controls copper-catalysed cyclopropanation. Dalton Trans. 2014, 43 (28), 10977–10982. 10.1039/C4DT01508H. [DOI] [PubMed] [Google Scholar]
- Dietrichbuchecker C. O.; Sauvage J. P.; Kintzinger J. P. A New Family of Molecules - Metallo-Catenanes. Tetrahedron Lett. 1983, 24 (46), 5095–5098. 10.1016/S0040-4039(00)94050-4. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.