

Abstract
Rationale
Autopsy and biomarker studies suggest that endotheliopathy contributes to coronavirus disease (COVID-19)-associated acute respiratory distress syndrome. However, the effects of COVID-19 on the lung endothelium are not well defined. We hypothesized that the lung endotheliopathy of COVID-19 is caused by circulating host factors and direct endothelial infection by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Objectives
We aimed to determine the effects of SARS-CoV-2 or sera from patients with COVID-19 on the permeability and inflammatory activation of lung microvascular endothelial cells.
Methods
Human lung microvascular endothelial cells were treated with live SARS-CoV-2; inactivated viral particles; or sera from patients with COVID-19, patients without COVID-19, and healthy volunteers. Permeability was determined by measuring transendothelial resistance to electrical current flow, where decreased resistance signifies increased permeability. Inflammatory mediators were quantified in culture supernatants. Endothelial biomarkers were quantified in patient sera.
Measurements and Main Results
Viral PCR confirmed that SARS-CoV-2 enters and replicates in endothelial cells. Live SARS-CoV-2, but not dead virus or spike protein, induces endothelial permeability and secretion of plasminogen activator inhibitor 1 and vascular endothelial growth factor. There was substantial variability in the effects of SARS-CoV-2 on endothelial cells from different donors. Sera from patients with COVID-19 induced endothelial permeability, which correlated with disease severity. Serum levels of endothelial activation and injury biomarkers were increased in patients with COVID-19 and correlated with severity of illness.
Conclusions
SARS-CoV-2 infects and dysregulates endothelial cell functions. Circulating factors in patients with COVID-19 also induce endothelial cell dysfunction. Our data point to roles for both systemic factors acting on lung endothelial cells and viral infection of endothelial cells in COVID-19–associated endotheliopathy.
Keywords: COVID-19, acute respiratory distress syndrome, endothelial permeability, lung endothelial injury
At a Glance Commentary
Scientific Knowledge on the Subject
The clinical course, autopsy studies, and available data on immune cell functions and circulating concentrations of inflammatory and endothelial biomarkers in patients with coronavirus disease (COVID-19) all point to the endothelium as a target organ in severe COVID-19 disease. Endothelial cell activation and dysregulation (“endotheliopathy”) are pathogenically involved in acute respiratory distress syndrome caused by sepsis and tissue injury. Currently, the understanding of the endotheliopathy of COVID-19, including its underlying mechanisms and role in driving acute respiratory distress syndrome, is rudimentary.
What This Study Adds to the Field
This translational-clinical study demonstrates that live severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) can enter primary human lung microvascular endothelial cells (HMVECs) and induce hyperpermeability and a proadhesive and proinflammatory phenotype. Analogous to the heterogeneous course of humans with COVID-19, we noted substantial variability in the responses of HMVECs from different donors. Ex vivo treatment with sera from patients with COVID-19 induces HMVEC permeability. Sera from moderately and critically ill patients with COVID-19 had a biomarker signature that was consistent with endothelial activation and dysfunction. Our data, in conjunction with other studies, suggest that COVID-19– associated lung endotheliopathy is caused by the actions of systemic and localized cells and inflammatory factors on endothelial cells, as well as SARS-CoV-2 infection of endothelial cells.
Endothelial cell dysfunction, or “endotheliopathy,” is believed to contribute to the pathogenesis of acute respiratory distress syndrome (ARDS) in severe coronavirus disease (COVID-19), and the endothelium has been proposed as a therapeutic target in severe COVID-19 disease (1, 2). However, despite an unprecedented focus on defining COVID-19 mechanisms (3), published data on the direct endothelial effects of COVID-19 are minimal (4, 5). Endothelial cells regulate vascular barrier function, vasomotor tone, vasculogenesis, coagulation and fibrinolysis, and trafficking of cells and substrates. Infection and injury induce a proinflammatory program in endothelial cells with upregulated production of cytokines, chemokines, procoagulant factors, and proadhesive proteins, as well as glycocalyx damage and vascular hyperpermeability with resultant tissue edema (6, 7). Elevated circulating endothelial cell biomarkers have been reported to associate with increased disease severity and risk of death in COVID-19 (8–12).
The radiographic features of bilateral ground-glass opacities in patients with COVID-19–associated ARDS are compatible with endothelial hyperpermeability (13). Furthermore, autopsies of patients who died of COVID-19 have revealed diffuse alveolar damage with perivascular T cell infiltration, virus within endothelial cells, endothelial injury, capillary microthrombi, and microvascular angiogenesis (14). These findings alongside the high incidence of thrombotic complications (15) implicate endotheliopathy in COVID-19–associated ARDS. The inaccessibility of endothelial cells to direct sampling in real time in humans has impeded progress toward understanding the cause(s) of COVID-19–associated endotheliopathy and its role in driving ARDS.
We hypothesized that COVID-19– associated endotheliopathy results from both viral infection of endothelial cells and secondary endothelial effects of factors in the blood of patients with COVID-19. We therefore tested the effects of live and inactivated virus, as well as sera from patients with and without COVID-19, on the activation and permeability of human lung microvascular endothelial cells (HMVECs), and we quantified serum endothelial biomarkers in patients with and without COVID-19.
Methods
HMVEC Culture, Agonists, and Antagonists
HMVECs (passages 3–6; Lonza) from male and female cadavers deceased of nonseptic causes were cultured (37°C, 5% CO2) in endothelial culture medium (EGMTM-2 EC Growth Medium). Confluent monolayers were treated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), spike protein, or sera from patients with and without COVID-19 or healthy volunteers. Some experiments used agonists, antagonists, or inhibitors, including the ACE2 agonist Diminazene aceturate (DMZ, 20 μM; Tocris), angiotensin-converting enzyme (ACE) inhibitor (CAS35115-60-7, 20 μM; Sigma-Aldrich), Toll-like receptor 4 (TLR4) antagonist LPS-RS ultrapure (1 μg/ml; InvivoGen), and TLR3 blocker CU-CPT 4a (10 μM; Tocris).
Viral Reagents
Experiments with live SARS-CoV-2 were performed in a biosafety level 3 laboratory. Live SARS-CoV-2 (strain nCoV/USA-WA1/2020) was provided by Dr. Melanie Ott. To generate viral stocks, Vero E6 cells were exposed to SARS-CoV-2 for 72 hours, and supernatants were stored at −80°C (16). Plaque assays were performed using Vero E6 cells to establish viral stock titers. We purchased inactivated SARS-CoV-2 (strain nCoV/USA-WA1/2019), produced by γ-irradiation or by heating (65°C, 30 min), and recombinant spike glycoprotein (stabilized) from SARS-CoV-2, Wuhan-Hu-1, produced in a baculovirus expression system (American Type Culture Collection).
Patient Enrollment Criteria and Serum Sample Collection
All human studies were approved by the University of California, San Francisco Institutional Review Board (IRB). Sera were collected from acutely ill patients with COVID-19 (n = 99) and patients without COVID-19 (n = 43) in the emergency department or early after admission to wards or ICUs at the Zuckerberg San Francisco General Hospital and the University of California, San Francisco Medical Center as part of two separate clinical studies: the Co-ACIT study (COVID-19 Associated Coagulopathy, Inflammation, and Thrombosis) (IRB 20-30895; n = 130) and the COMET study (COVID-19 Multi-Phenotyping for Effective Therapies; www.comet-study.org/team; IRB 20-30497; n = 12). The diagnosis of COVID-19 was based on a positive SARS-CoV-2 PCR test. The Co-ACIT study includes adults (aged >18 yr) undergoing evaluation for possible COVID-19 in the Zuckerberg San Francisco General Hospital emergency department based on fever, respiratory symptoms (cough, shortness of breath, wheezing), gastrointestinal symptoms (diarrhea, nausea/vomiting), and/or changes in taste or smell. The COMET study includes adults admitted to the wards and ICUs with symptomatic COVID-19. Subjects in both studies were followed until discharge from the emergency department or hospital, and a comprehensive collection of patient characteristics, physiological and laboratory parameters, and clinical outcomes was documented. Patients were subclassified into three disease severity categories at Day 1 based on their level of care: mild (ambulatory mild disease: discharged after evaluation in the emergency department), moderate (hospitalized: moderate disease, admitted to the floor), and critical (hospitalized: severe disease, admitted to the ICU) according to guidelines from the World Health Organization Working Group on the Clinical Characterization and Management of COVID-19 (17, 18). Table 1 includes the baseline demographics and characteristics of patients. Sera were also collected from healthy volunteers without fever or respiratory or gastrointestinal symptoms (IRB 10-01981 and 20-30895).
Table 1.
Characteristics at Inclusion of 142-Patient Cohort Whose Sera Were Used in Ex Vivo Endothelial Cell Stimulation Assays
| Variable | Total Cohort (N = 142) | Non–COVID-19 (n = 43) | COVID-19 (n = 99) | P Value |
|---|---|---|---|---|
| Age, yr, mean ± SD | 58 ± 16 | 59 ± 16 | 55 ± 19 | 0.24 |
| Male, n (%) | 88 (62) | 28 (65) | 60 (60.6) | 0.7 |
| Duration of symptoms before admission, d | 6.6 ± 7 | 5.3 ± 9.6 | 7.2 ± 5.3 | 0.14 |
| Ethnicity, n (%) | ||||
| White | 17 (12) | 11 (25.6) | 6 (6.1) | 0.005 |
| Black or African American | 17 (12) | 10 (23.3) | 7 (7.1) | 0.01 |
| Hispanic/Latino | 71 (50) | 12 (28) | 59 (59.6) | 0.0009 |
| Asian | 19 (13.4) | 4 (9.3) | 15 (15.1) | 0.43 |
| Native American | 1 (0.7) | 1 (2.3) | 0 (0) | 0.3 |
| Other/not reported | 17 (12) | 5 (11.6) | 12 (12.1) | 0.99 |
| Comorbidities, n (%) | ||||
| Obesity | 49 (34.5) | 10 (23.3) | 39 (39.4) | 0.08 |
| Diabetes | 41 (28.9) | 9 (20.9) | 32 (32.3) | 0.23 |
| COPD/asthma/CRD | 16 (11.3) | 9 (20.9) | 7 (7.1) | 0.02 |
| CKD | 14 (9.8) | 3 (7) | 11 (11.1) | 0.55 |
| CAD/CHF | 15 (10.6) | 6 (14) | 9 (9.1) | 0.38 |
| Stroke | 8 (5.6) | 6 (14) | 2 (2) | 0.009 |
| Liver disease | 12 (8.4) | 6 (14) | 6 (6) | 0.18 |
| HIV/AIDS | 6 (4.2) | 5 (11.6) | 1 (1) | 0.009 |
| Cancer | 13 (9.1) | 5 (11.6) | 8 (8.1) | 0.53 |
| Admission on Day 1, n (%) | ||||
| ED only/outpatient: mild | 42 (29.6) | 14 (32.6) | 28 (28.3) | 0.55 |
| Floor: moderate | 60 (43.7) | 19 (44.2) | 41 (43.4) | 0.85 |
| ICU: critical | 40 (26.7) | 10 (23.3) | 30 (28.3) | 0.42 |
| Respiratory support on Day 1, n (%) | ||||
| None | 61 (43) | 22 (51.2) | 39 (39.4) | 0.2 |
| NC/NRB | 48 (33.8) | 15 (34.8) | 33 (33.3) | 0.84 |
| HFNC | 18 (12.7) | 3 (7) | 15 (15.1) | 0.27 |
| CPAP/BiPAP | 1 (0.7) | 1 (2.3) | 0 (0) | 0.3 |
| Intubated | 14 (9.9) | 2 (4.7) | 12 (12.1) | 0.23 |
| Severity-of-illness scores | ||||
| SOFA score, mean ± SD | 1.8 ± 3 | 0.9 ± 1.2 | 2.4 ± 3.4 | 0.01 |
| Respiratory SOFA score, mean ± SD | 0.63 ± 1.1 | 0.21 ± 0.47 | 0.82 ± 1.2 | 0.02 |
| Diagnosis at admission, n (%) | ||||
| COVID-19 | 99 (69.7) | 0 (0) | 99 (100) | NA |
| Sepsis (of origin) | 18 (12.7) | 18 (41.8) | 0 (0) | NA |
| Pneumonia/URI | 12 (8.4) | 12 (27.9) | 0 (0) | NA |
| UTI | 1 (0.7) | 1 (2.3) | 0 (0) | NA |
| SSTI | 2 (1.4) | 2 (4.7) | 0 (0) | NA |
| Other/not reported | 3 (2.1) | 3 (7) | 0 (0) | NA |
| Asthma/COPD exacerbation | 6 (4.2) | 6 (14) | 0 (0) | NA |
| Heart failure | 8 (5.6) | 8 (18.6) | 0 (0) | NA |
| Pulmonary embolism | 1 (0.7) | 1 (2.3) | 0 (0) | NA |
| Gastrointestinal symptoms | 4 (2.8) | 4 (9.3) | 0 (0) | NA |
| Other/not reported | 6 (4.2) | 6 (14) | 0 (0) | NA |
Definition of abbreviations: BiPAP = bilevel positive airway pressure; CAD = coronary artery disease; CHF = chronic heart failure; CKD = chronic kidney disease; COPD = chronic obstructive pulmonary disease; COVID-19 = coronavirus disease; CPAP = continuous positive airway pressure; CRD = chronic respiratory disease; ED = emergency department; HFNC = high-flow nasal cannula; NA = not applicable; NC = nasal cannula; NRB = non-rebreather mask; SOFA = Sequential Organ Failure Assessment; SSTI = skin and soft tissue infections; URI = upper respiratory infection; UTI = urinary tract infection.
Assessment of Effects of Viral Particles, Spike Protein, and Human Sera on HMVEC Permeability and Biomarker Production
HMVECs were treated with live or inactivated SARS-CoV-2, spike protein, or human sera. Permeability was inferred from transendothelial resistance (TER), which was measured using electric cell substrate impedance sensing (Applied Biophysics), which quantifies the resistance of a cell monolayer to electrical current flow in real time (19, 20). Decreased TER reflects increased permeability. HMVECs were cultured to confluence based on reaching a stable plateau in TER measured at a frequency of 4,000 Hz (20). HMVECs were then incubated with virus or spike protein (four to eight replicates per condition) or 10% sera from patients with COVID-19 and patients without COVID-19 or healthy humans (two replicates per sample). TER was measured repeatedly over 24 hours. Data were quantitatively analyzed by calculating the area between the control curve (normalized resistance, 1) and the experimental condition, called the area under the curve, as an integrative marker of the permeability (21). At 24 hours, IL-6, IL-8, plasminogen activator inhibitor 1 (PAI-1), vascular endothelial growth factor (VEGF), and CCL-2 were quantified in supernatants by ELISA (R&D Systems). Viral PCR was performed at 24 and 72 hours in some experiments. Escherichia coli 0111:B4 LPS (1 μg/ml) served as a positive control that reliably induces HMVEC permeability across donors.
Quantification of Biomarkers in Sera from Cohort of 74 Patients
Endothelial biomarkers were quantified in sera from patients with and without COVID-19 collected in the Co-ACIT study during their first medical evaluation in the emergency department before receiving fluids, dexamethasone, remdesivir, or plasma therapy. The specific biomarkers were chosen because they are produced by or directly act on the endothelium, or, as in the case of syndecan-1, reflect endothelial glycocalyx injury. IL-6, IL-8, VEGF (R&D Systems), and syndecan-1 (Abcam) were quantified by ELISA. Other endothelial biomarkers were quantified using multiplex fluorescence-based panels (Eve Technology), including the cardiovascular disease panel (HCVD4-07-0; follistatin, platelet endothelial cell adhesion molecule 1 [PECAM-1], pentraxin-3, sE-selectin, tissue factor, s-thrombomodulin, troponin T) and the endothelial biomarker array (HDHSB10; brain-derived neurotrophic factor [BDNF], cathepsin D, myeloperoxidase, neural cell adhesion molecule [NCAM], PAI-1, platelet-derived growth factor [PDGF]-AA, PDGF-AB/BB, regulated upon activation, normal T cell expressed and secreted [RANTES], soluble intercellular adhesion molecule 1 [sICAM-1], soluble vascular cell adhesion molecule 1 [sVCAM-1]). ACE and ACE2 were quantified by ELISA (R&D Systems).
Flow Cytometry
HMVECs were detached with Accutase, and cells were labeled with fluorescein isothiocyanate CD102 (clone CBR-IC1/2), phycoerythrin-cyanine 7 CD106 (clone 429 MVCAM.A) (BioLegend), eFluor 450 CD31 (clone WM59), allophycocyanin CD62P (clone Psel.KO2.3) (eBioscience), and phycoerythrin CD144 (clone 16B-1) (Invitrogen). A fixation step was used to inactivate live virus before HMVECs underwent staining. Single-cell suspensions underwent flow cytometry the same day (LSRII Fortessa flow cytometer). The data were analyzed with FlowJo software. Endothelial cells were defined as CD31+CD102+.
Statistics
Statistics were performed using R software (www.R-project.org). Graphs show means and SDs. Nonparametric Mann-Whitney U tests were used for continuous variables; the chi-square test was used for discrete variables; or ANOVA with multiple Tukey’s post hoc tests were used for comparisons between multiple groups. For analysis of biomarkers in patient sera, to limit multiple testing–related biases, we applied a two-step-up method with a false discovery rate (q) of 5% (22). We also performed a principal component analysis based on 10 endothelial activation and injury biomarkers to assess the main relationships between the pattern of biomarkers, COVID-19 status, and disease severity. All tests were two-sided, and P values less than 0.05 were considered significant.
Results
SARS-CoV-2 Enters and Replicates in HMVECs
Using PCR, we detected SARS-CoV-2 in lysates of HMVECs after 24 and 72 hours of exposure to live SARS-CoV-2 at a multiplicity of infection (MOI) of 0.1 and 1 viral particle per cell (Figure 1A). Intracellular viral mRNA concentrations were higher at 72 hours than at 24 hours, indicating viral replication within endothelial cells. Viral mRNA was not detected in lysates of medium-treated HMVECs.
Figure 1.
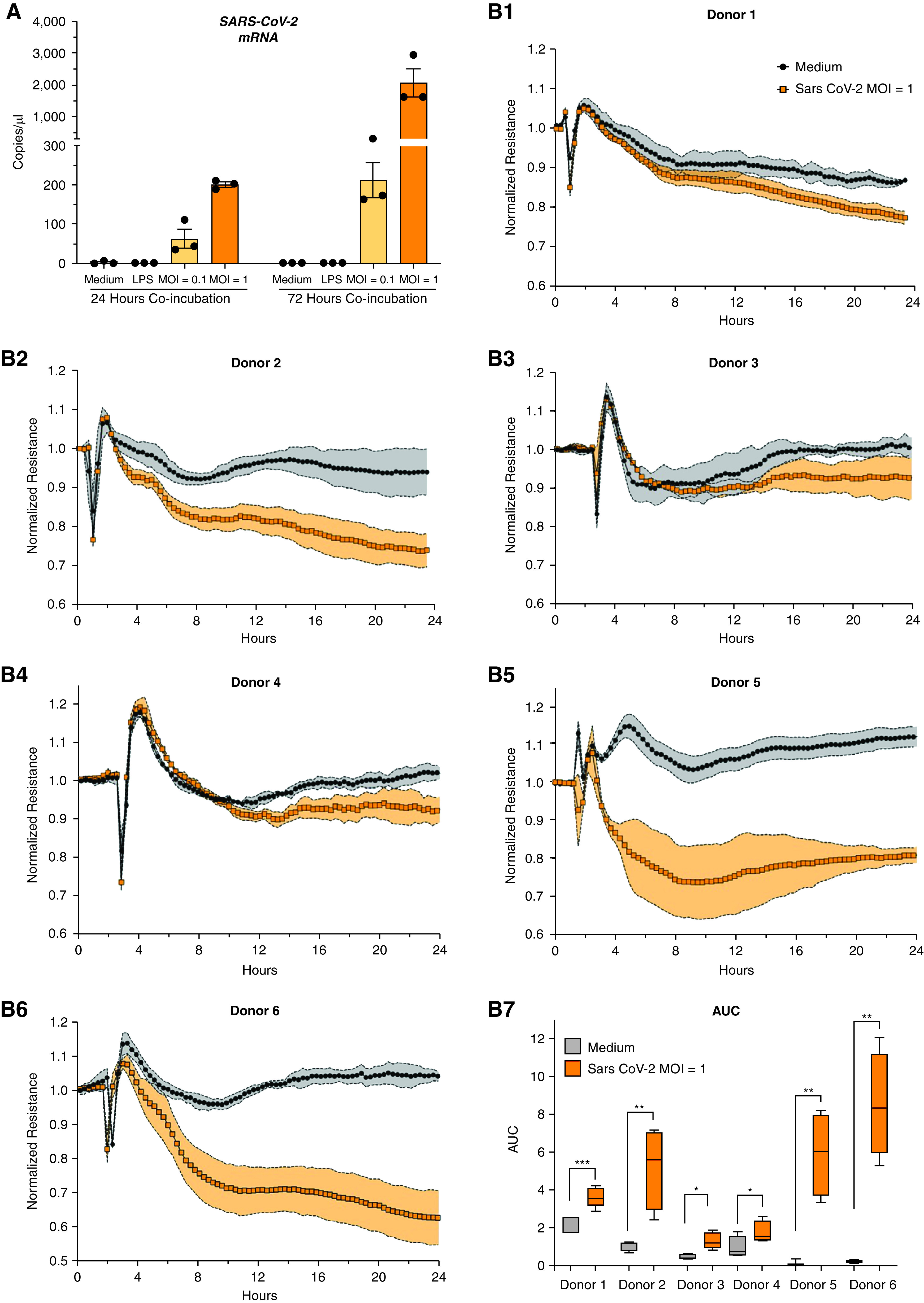
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) human lung microvascular endothelial cell (HMVEC) infection and endothelial permeability. (A) SARS-CoV-2 mRNA was detected and quantified in HMVEC lysates after 24 and 72 hours of exposure to live SARS-CoV-2 virus (multiplicity of infection [MOI] of 1; orange, strain nCoV/USA-WA1/2020) using quantitative PCR (TaqMan 2019-nCoV assay). This example shows cells from a single donor; dots are biological replicates. (B1–B6) Transendothelial resistance during treatment with medium (black line) or live virus (MOI of 1; orange) in six HMVEC donors. (B7) The corresponding area under the curve (AUC) for HMVECs on each of the six donors. *P < 0.05, **P < 0.01, ***P < 0.001 by two-tailed Mann-Whitney U test.
Exposure to Live SARS-CoV-2 Induces HMVEC Permeability
Exposure of HMVECs to SARS-CoV-2 at an MOI of 1, but not at an MOI of 0.01 or 0.1, led to decreased TER, consistent with increased permeability (Figure 1B1–6). Although the magnitude of the permeability induced by SARS-CoV-2 varied between HMVECs from different donors, there were significant differences between the areas under the curve of SARS-CoV-2 at an MOI of 1 versus medium for all six donors (Figure 1B7) (P < 0.05).
Effects of ACE and ACE2 Modulation and Innate Immune Receptor Blockade on SARS-CoV-2–induced HMVEC Permeability
Treatment with an ACE inhibitor did not affect SARS-CoV-2–induced permeability (Figure 2A1–2). Conversely, the ACE2 agonist DMZ reversed the virus-induced permeability of HMVECs from three of six donors (Figure 2A1–2; see Figure E1 in the online supplement) (P < 0.05). DMZ did not reduce intracellular SARS-CoV-2 concentrations (data not shown), which suggests that it reduces permeability independently of viral entry. Also, DMZ reduced LPS-induced permeability (Figure 2B) (P < 0.01), suggesting that its effects are not specific to SARS-CoV-2. Neither TLR4 antagonism using LPS-RS nor TLR3 inhibition using CU-CPT 4a affected virus-induced permeability (data not shown).
Figure 2.
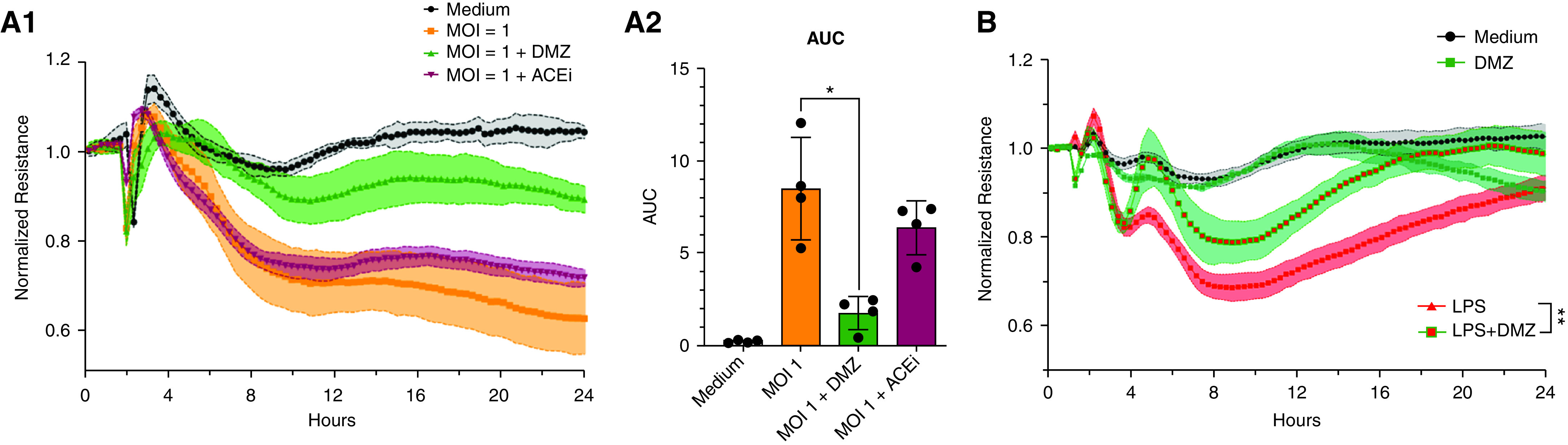
Pharmacological modulation of angiotensin-converting enzyme 2 receptor (ACE2R) pathway and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-induced human lung microvascular endothelial cell (HMVEC) permeability. (A1 and A2) Representative example of ACE2 agonist (Diminazene aceturate [DMZ] 20 μM; green) and ACE inhibitor (ACEi) effect (20 μM; claret red) on the transendothelial resistance and area under the curve (AUC) of HMVECs treated simultaneously with ACE2 agonists/antagonists and live SARS-CoV-2 (multiplicity of infection [MOI] of 1; orange) infection. (B) The stabilizing effects of DMZ on permeability induced by LPS (1 μg/ml). *P < 0.05, **P < 0.01 by two-tailed Mann-Whitney U test (AUC LPS vs. LPS + DMZ).
SARS-CoV-2 Effects on HMVEC Inflammatory Mediator and Adhesion Molecule Expression
Exposure to live SARS-CoV-2 significantly upregulated IL-6, PAI-1, and VEGF secretion by HMVECs from three of the six donors (donors 2, 5, and 6) and increased IL-8 and CCL-2 secretion by HMVECs from one donor (donor 6) (Figure 3A). On the basis of flow cytometry, there was high variability in the baseline expression of multiple adhesion molecules (VCAM-1 [CD106], ICAM-2 [CD102], P-selectin [CD62P], PECAM-1 [CD31], and VE-cadherin [CD144]) between HMVECs from different donors (Figure 3B). As shown in the heat map in Figure 3B2, exposure to SARS-CoV-2 significantly upregulated surface expression of VCAM-1 by two donors (donors 2 and 4), P-selectin by two donors (donors 2 and 5), and ICAM-2 by one donor (donor 2).
Figure 3.
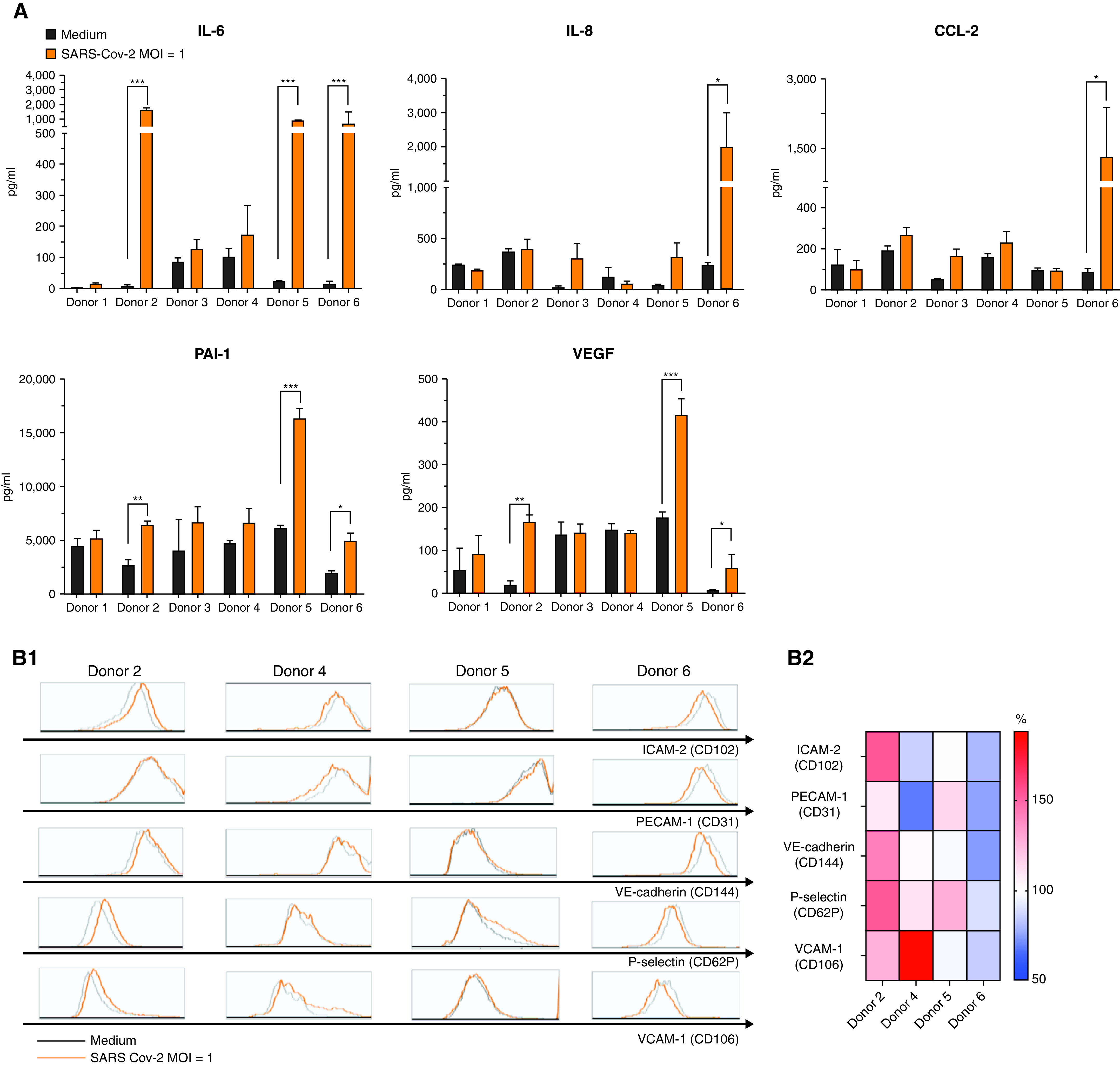
Effect of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection on human lung microvascular endothelial cell (HMVEC) mediator secretion and proadhesive phenotype. (A) Concentrations of IL-6, IL-8, CCL-2, plasminogen activator inhibitor 1 (PAI-1), and vascular endothelial growth factor (VEGF) in supernatants of HMVECs from six different donors treated with medium or SARS-CoV-2 (multiplicity of infection [MOI] of 1; orange, strain nCoV/USA-WA1/2020). *P < 0.05, **P < 0.01, ***P < 0.001 by two-tailed Mann-Whitney U test. (B1) Histograms for CD102 (ICAM-2), CD31 (platelet endothelial cell adhesion molecule 1 [PECAM-1]), CD106 (vascular cell adhesion molecule 1 [VCAM-1]), CD144 (VE-cadherin), and CD62P (P-selectin) expression on HMVECs from four different donors after 24 hours of treatment with medium (gray) or infection with SARS-CoV-2 (MOI of 1; orange, strain nCoV/USA-WA1/2020). (B2) A double-gradient heat map of the relative mean fluorescence intensity on endothelial cells from four different donors after 24 hours of treatment with medium versus virus.
Spike Protein and Inactivated Virus Do Not Induce HMVEC Permeability or Inflammation
There were no significant effects of spike protein (0.1–1,000 ng/ml) or inactivated virus (104, 105, or 106 copies/ml) on permeability (Figure E2A), nor did spike protein or inactivated virus induce IL-6, IL-8, or CCL-2 secretion by HMVECs from any of the three donors tested (Figure E2B). Moreover, on the basis of flow cytometry, neither spike protein nor inactivated virus affected the surface expression of endothelial cell proteins, including CD31 (PECAM-1), CD102 (ICAM-2), CD106 (VCAM-1), CD144 (VE-cadherin), and CD62P (P-selectin) (Figure E2C). These data suggest that spike protein and viral particles do not activate endothelial cell surface innate immune receptors.
Effects of Sera from Patients with and without COVID-19 on HMVEC Permeability and Inflammation
We exposed HMVECs to sera from patients with COVID-19 (n = 99) and patients without COVID-19 (n = 43) and from healthy volunteers (n = 18). Table 1 includes baseline demographics, level of care, and comorbidities of the cohort. Hispanic/Latino individuals were substantially overrepresented, whereas Black or African American and White individuals were substantially underrepresented, among patients with COVID-19 versus patients without COVID-19. Compared with sera from healthy subjects, sera from patients with COVID-19 with mild (P < 0.05), moderate (P < 0.001), or critical (P < 0.001) disease induced reductions in TER, reflecting increased permeability (Figure 4). Sera from critical patients without COVID-19 also induced HMVEC permeability (P < 0.001), whereas healthy donor sera did not (Figure 4). There were no differences in permeability induced by sera from patients without and with COVID-19 with comparable illness severities (Figure 4B).
Figure 4.
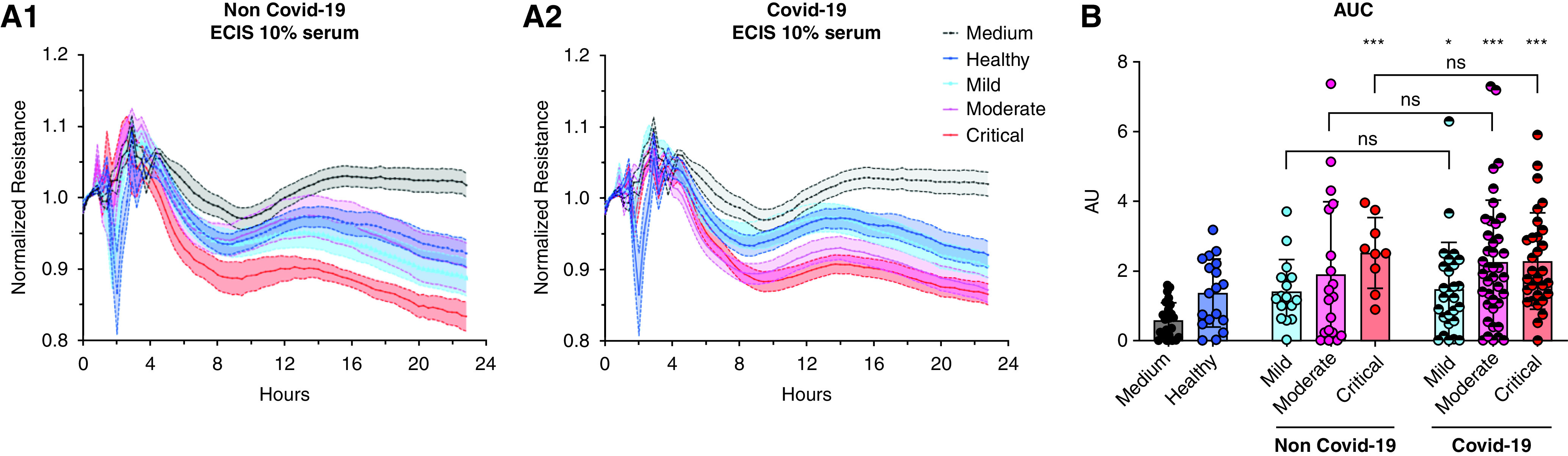
Sera from patients with and without coronavirus disease (COVID-19) induce human lung microvascular endothelial cell (HMVEC) permeability proportionate to their clinical severity. (A1 and A2) Transendothelial resistance tracings of HMVECs during treatment with medium or with sera from healthy volunteers (n = 18); patients with COVID-19 with mild, moderate, or critical illness (n = 99; mild [n = 28], moderate [n = 41], critical [n = 30]); or patients without COVID-19 with mild, moderate, or critical illness (n = 43; mild [n = 14], moderate [n = 19], critical [n = 10]). Each curve represents the mean value of the group ± SD. (B) The corresponding electric cell substrate impedance sensing (ECIS) area under the curve (AUC) represents the intensity of sera-induced HMVEC permeability. *P < 0.05, ***P < 0.001 versus healthy donors by two-tailed Mann-Whitney U test.
There was considerable variability in the effects of COVID-19 sera on HMVEC secretion of different inflammatory mediators and in the pattern of upregulation of mediators induced by COVID-19 and non–COVID-19 sera (Figure E3). Compared with healthy human sera, sera from critical but not mild or moderate COVID-19 disease increased HMVEC secretion of IL-6 (P < 0.05), whereas sera from patients without COVID-19 did not. Exposure to sera from patients with COVID-19 of any disease severity and from mild and moderate patients without COVID-19 significantly upregulated CCL-2 secretion compared with healthy human serum. COVID-19 and non-COVID-19 sera did not affect IL-8 or PAI-1 secretion by HMVECs. Finally, sera from moderate and critical patients with COVID-19, but not patients without COVID-19, significantly increased VEGF secretion (Figure E3).
Patients with COVID-19 Have Elevated Serum Endothelial Activation and Injury Biomarkers
Concentrations of endothelial biomarkers in sera from patients with and without COVID-19 are shown in Table E1. IL-6 (P = 0.001), IL-8 (P = 0.003), VEGF (P = 0.01), follistatin (P = 0.04), tissue factor (P = 0.02), serum thrombomodulin (P = 0.03), sVCAM-1 (P = 0.03), and syndecan-1 were positively associated with the level of care in COVID-19 and non–COVID-19 groups. Conversely, BDNF (P = 0.04) and PDGF-AA (P = 0.03) concentrations were negatively associated with level of care (Table E2). Disregarding level of care, patients with COVID-19 had higher sVCAM-1 (P < 0.0001), sICAM-1 (P = 0.006), NCAM (P = 0.009), cathepsin (P = 0.002), PAI-1 (P = 0.0005), and syndecan-1 (P = 0.017) and lower BDNF (P = 0.03) than patients without COVID-19 (Figure 5A). ACE and ACE2 serum concentrations were similar between patients without and with COVID-19 (Figure 5A, Table E3). After stratification by level of care, patients with COVID-19 had significantly higher circulating concentrations of sVCAM-1, cathepsin D, sICAM-1, NCAM, PAI-1, and syndecan-1 than patients without COVID-19 (Figure 5B, Table E2). Applying a two-step-up method with a false discovery rate (q) of 5% (22), only sVCAM-1 (q = 0.0006), NCAM (q = 0.015), sICAM-1 (q = 0.03), cathepsin D (q = 0.003), syndecan-1 (q = 0.02), PAI-1 (total) (q = 0.003), and IL-8 (q = 0.015) were significantly different between patients with and without COVID-19. An exploratory multivariate analysis by a principal component analysis based on 10 endothelium-related biomarkers and the level of care showed that patients with COVID-19 have increased sVCAM-1, NCAM, sICAM-1, cathepsin D, and PAI-1 but lower BDNF and RANTES in comparison to patients without COVID-19 (Figure 5C).
Figure 5.
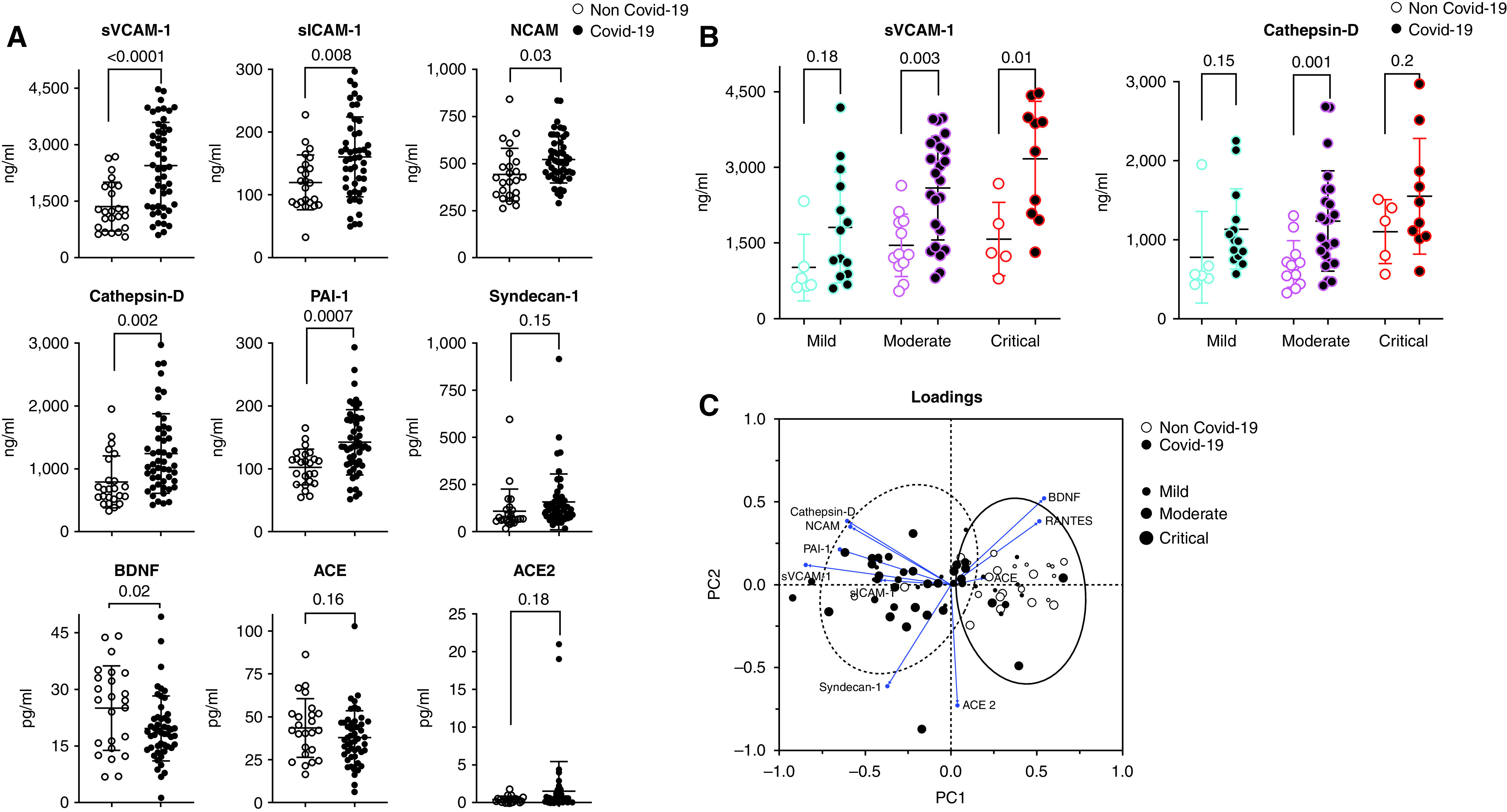
Admission serum endothelial biomarkers in patients with versus without coronavirus disease (COVID-19). (A) Comparison of endothelium-related biomarkers in serum from patients with COVID-19 (n = 50) and patients without COVID-19 (n = 24). P values are based on two-sided Mann-Whitney U tests. (B) Comparison of endothelium-related biomarkers between patients with COVID-19 (n = 50) and patients without COVID-19 (n = 24), stratified according to the severity of illness. (C) Principal component analysis showing representation of COVID-19 and non-COVID-19 populations in the two dimensions (score plot). Scores and loadings are presented in a scatterplot of one principal component (PC) against another. The loadings are represented in a circle of correlations: The closer the arrow of a loading is to the circle (shown in blue), the more the variable is well represented in the space of the two plotted PCs and contributed to the building of these PCs. The first two PCs allowed the discrimination of patients with and without COVID-19, consolidating results of the univariate analysis. Patients with COVID-19 (dotted line ellipse) were characterized by increased cathepsin D, neural cell adhesion molecule (NCAM), soluble vascular cell adhesion molecule 1 (sVCAM-1), soluble intercellular adhesion molecule 1 (sICAM-1), plasminogen activator inhibitor 1 (PAI-1), and syndecan-1. Patients without COVID-19 (solid line ellipse) were characterized by increased brain-derived neurotrophic factor (BDNF), RANTES (regulated upon activation, normal T cell expressed and secreted), and angiotensin-converting enzyme (ACE).
Discussion
This study highlights the complexity of the endotheliopathy associated with SARS-CoV-2 infection. We found that SARS-CoV-2 infects and induces permeability of HMVECs from all donors, and it variably affects inflammatory mediator secretion by HMVECs from different donors. We observed that ex vivo exposure of HMVECs to sera of patients with COVID-19 induces endothelial permeability and variable inflammatory activation. At the time of presentation to the emergency department, enrolled patients with COVID-19 had elevated serum endothelial biomarkers that correlated with their severity of illness. Our data, in conjunction with autopsy findings of SARS-CoV-2 within lung endothelial cells (14) and reports of elevated circulating endothelial biomarkers (8–12), suggest that both viral infection of endothelial cells and secondary effects of circulating factors and intrapulmonary leukocytes mediate COVID-19–associated lung endotheliopathy and, by extension, ARDS. Figure 6 shows our hypothetical model of endothelial cells as targets for SARS-CoV-2 infection and secondary injury.
Figure 6.
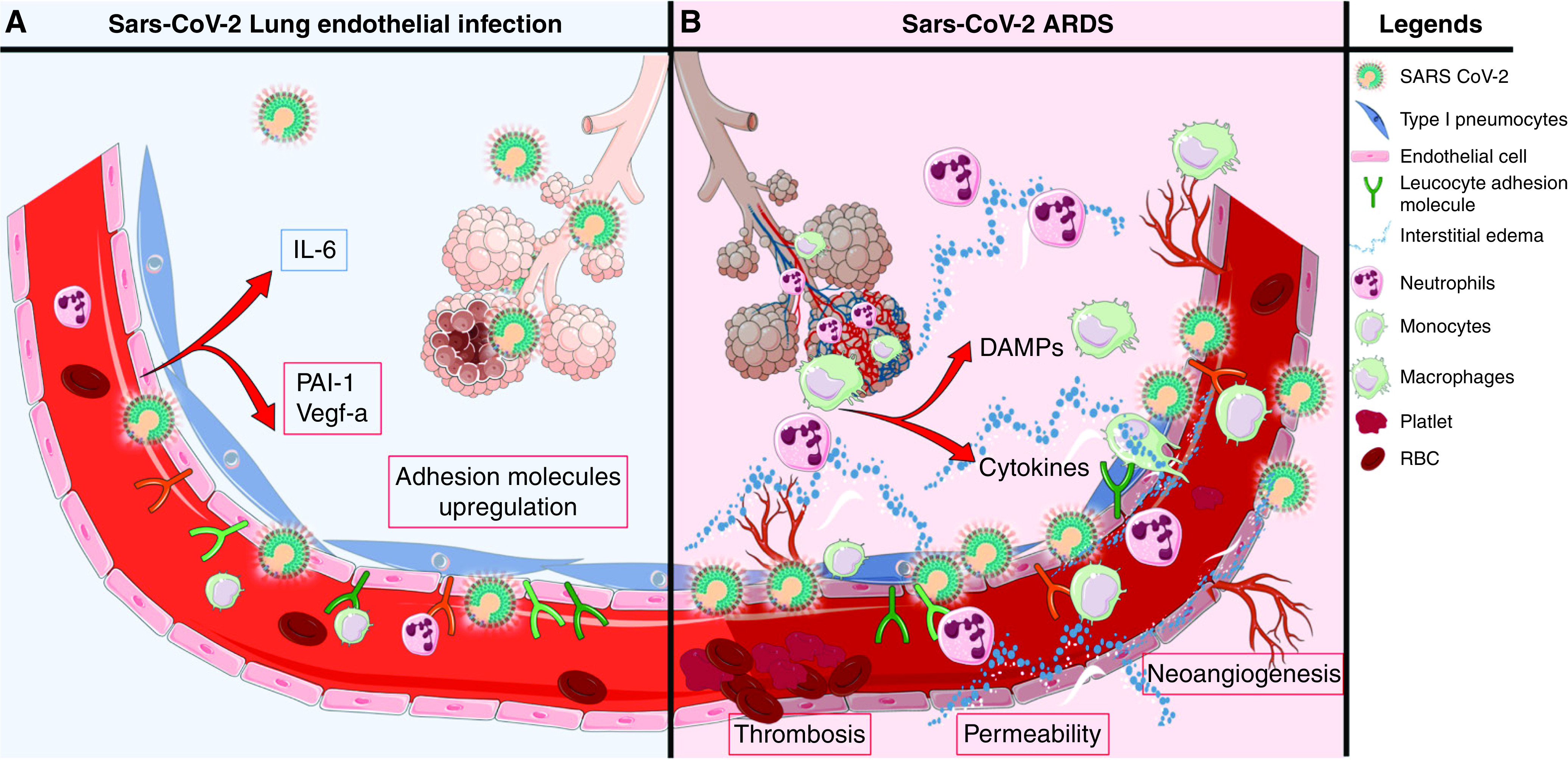
Hypothetical model of coronavirus disease (COVID-19)-associated lung microvascular endotheliopathy. On the basis of our experimental results, we speculate that viral infection and secondary injury of the endothelium by inflammatory mediators and immune cells contribute to the lung endotheliopathy of COVID-19 and, in turn, COVID-19–associated acute respiratory distress syndrome (ARDS). (A) Endothelial cell exposure to live severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) could occur early or later in the course of the disease if there is persistence of virus in the host, including in alveolar epithelial cells, which are in proximity to microvascular endothelial cells. In susceptible individuals, immune dysfunction could lead to increased viral load and release by alveolar epithelial cells. In this speculative model, SARS-CoV-2 infection of lung microvascular endothelial cells induces permeability, upregulates surface expression of leukocyte adhesion molecules and secretion of plasminogen activator inhibitor 1 (PAI-1) and vascular endothelial growth factor (VEGF) secretion, and induces low levels of secretion of cytokines and chemokines. In this context, infection of endothelial cells may promote and/or amplify endotheliopathy and ARDS. (B) Secondary injury to the lung endothelium is likely to be a principal determinant of COVID-19–induced endotheliopathy. Secondary injury may result from the actions of circulating inflammatory mediators and toxins and of activated pulmonary leukocytes and damaged alveolar epithelial cells. In susceptible individuals, impaired viral clearance and persistent viral replication in epithelial or endothelial cells may promote leukocyte recruitment to the lung and cell death and the release of damage-associated molecular patterns (DAMPs). These DAMPs could, analogously to their roles in sepsis and injury, further exacerbate the endotheliopathy, as well as acute lung injury and ARDS. RBC = red blood cells.
We found that exposure to sera from patients with mild, moderate, or critical COVID-19 significantly increased HMVEC permeability. This indicates the presence of circulating factors capable of dysregulating endothelial function early during symptomatic COVID-19. We observed differences in the effects of sera from patients with mild, moderate, and severe COVID-19 on HMVEC production of specific proinflammatory mediators. Sera from patients with critical COVID-19 significantly upregulated IL-6 production by HMVECs. We also observed that live SARS-CoV-2 significantly upregulated production of IL-6, as well as PAI-1 and VEGF, by HMVECs from half of the donors. Notably, live SARS-CoV-2 induced the highest degree of permeability in HMVECs from the same donors that produced the most IL-6. Circulating IL-6 levels are predictive of nonsurvival in COVID-19 (9). Our data suggest that enhanced IL-6 secretion by endothelial cells is linked to endothelial dysfunction and is associated with increased endothelial permeability.
We observed large differences in live SARS-CoV-2 effects on permeability and activation of HMVECs from different donors. We hypothesize that intrinsic differences in lung endothelial cell susceptibility to SARS-CoV-2 infection and to the actions of systemic and localized immune factors underlie the variability in the outcomes of COVID-19. Possible sources of the heterogeneity in the responses of different donor HMVECs include genetics, epigenetics, age, concomitant diseases, recent infection or injury, or the cause of the donor’s death. Interindividual differences in the ACE/ACE2 system, such as ACE2 polymorphisms or cell surface expression of ACE2 (23–26), could also contribute to variability in the susceptibility to HMVEC infection and downstream inflammation and permeability (27). One study examining SNPs in ACE2 and ACE genes in 297 patients with COVID-19 and 253 patients without COVID-19 reported that the ACE2 rs2285666, the GG genotype, or the G allele was significantly associated with a twofold increased risk of SARS-CoV-2 infection and a threefold increased risk of severe or lethal COVID-19 (28). However, it remains unclear whether endothelial cells express ACE2 (29–31). In our study, the ACE2 agonist DMZ reduced SARS-CoV-2– induced permeability, without reducing viral entry into HMVECs. DMZ also reduced LPS-induced permeability. This suggests that activation of ACE2 signaling pathways helps to stabilize the endothelial barrier during acute inflammation caused by multiple processes rather than being specific to COVID-19 (32, 33).
We observed that sera from patients with COVID-19 and patients without COVID-19 with comparable severities of illness induced similar levels of HMVEC permeability. This finding is not surprising, given the known role of endothelial injury and dysfunction in sepsis and ARDS, but it does not answer the question whether the pathogenesis of COVID-19–associated endotheliopathy is analogous to that of sepsis or ARDS. There are a number of reports of elevated blood endothelial biomarkers in COVID-19, and some studies have shown associations between elevated serum endothelial biomarkers, such as PAI-1 and syndecan-1, with organ failure and death (8–12, 34–39). Despite similarities between serum endothelial biomarkers and the degree of HMVEC permeability induced by sera from patients with and without COVID-19, our data suggest that there is a serum endothelial biomarker signature of COVID-19 disease that differs from that of non–COVID-19 disease and includes increased levels of NCAM, sVCAM-1, and sICAM-1. The upregulation of these adhesion molecules may reflect the intensity of lung leukocyte infiltration and the angiocentric alveolar inflammation in COVID-19 ARDS, analogous to sepsis-induced ARDS (40–42). We found that cathepsin D levels were positively associated with the severity of illness and were higher in patients with COVID-19 than in patients without COVID-19. Cathepsin D increases vascular permeability and can activate VEGF (43) and could play a role in the lung edema and neoangiogenesis in COVID-19 (14). Finally, serum BDNF levels were lower in patients with COVID-19 than in patients without COVID-19. BDNF is involved in neuronal survival and differentiation (44) and in vascular stability (45). We speculate that lower BDNF levels may contribute to COVID-19–associated endotheliopathy and neurological dysfunction (46–48).
Our study has certain limitations. First, we used patient sera that were collected within the first 24 hours of hospitalization. This limits the ability to define any associated dynamic relationship with outcomes. However, an advantage to using these early serum samples is that none of the patients received fluids, dexamethasone, remdesivir, or plasma before sample collection, which should have minimized biases resulting from dilution or the administration of plasma, immunomodulatory drugs, or antiviral therapy. Second, the study was designed to include symptomatic patients with mild, moderate, and severe COVID-19 to reflect the broader spectrum of the disease, but the low mortality in the COVID-19 group (3 of 99 patients with COVID-19 deceased) does not allow analysis of the predictive value of our measured endpoints on clinical outcome. Third, we observed considerable heterogeneity in the effects of live virus on HMVECs from different donors. This finding limits our ability to draw conclusions about the lung endothelial cells that are generalizable to all individuals. Also, for the serum exposure experiments, we used a single donor’s HMVECs because of limitations in the quantity of sera available for testing. This could have created a potential “donor-related bias.” This might have been avoided by using an immortalized cell line, but using primary human cells is more clinically relevant. Fourth, we used only one strain of virus for our experiments and cannot rule out the possibility that other variants would have different effects. Last, we tested effects of sera and virus on HMVECs under static conditions. However, COVID-19–induced lung injury undoubtedly involves a complicated and dynamic interplay among multiple cell types, including alveolar epithelial cells, endothelial cells, and leukocytes (12).
In summary, these studies indicate that live SARS-CoV-2 can infect and induce permeability and a proadhesive phenotype of HMVECs and that sera from patients with COVID-19 induce permeability and mild inflammatory activation of HMVECs. Our data, alongside other published studies on circulating biomarkers in COVID-19 and autopsy studies, suggest that both viral infection of endothelial cells and secondary injury and activation of endothelial cells by circulating and localized cells and mediators contribute to COVID-19–associated lung endotheliopathy, as depicted in our hypothetical model in Figure 6. We speculate that live SARS-CoV-2 may have the opportunity to interact with and infect lung endothelial cells longitudinally over the course of the disease, facilitated by persistence of live virus in nearby alveolar epithelial cells (Figure 6A). We further speculate that circulating and local tissue leukocytes, inflammatory mediators, and damage-associated molecular patterns contribute to COVID-19–associated lung microvascular endotheliopathy (Figure 6B). In some patients, impaired viral clearance and viral replication in epithelial or endothelial cells may exacerbate the endotheliopathy by promoting leukocyte recruitment to the lung and cell death with release of more damage-associated molecular patterns. Further studies on direct endothelial infection by SARS-CoV-2 and on the secondary actions of systemic factors and pulmonary leukocytes in COVID-19– associated endotheliopathy could lead to the identification of endothelial cell–targeted approaches to treat COVID-19 disease.
Acknowledgments
Acknowledgment
The authors thank Melanie Ott, M.D., Ph.D., University of California, San Francisco, and J. David Gladstone Institutes, for providing us with live SARS-CoV-2 USA-WA1/2020 strain and Vero E6 cells for viral propagation and titer determination. The authors acknowledge the Co-ACIT Study Group for enrolling patients and the COMET Consortium (immunox.ucsf.edu/covid19-research) for providing 12 additional human serum samples.
COMET Consortium members: Sharvari Bhide1,2, Carolyn S. Calfee2,3, Sidney A. Carrillo2, Suzanna Chak2, David J. Erle2,3,4, Gabriela K. Fragiadakis4,5,6, Rajani Ghale2, Jeremy Giberson1,2, Pat Glenn7, Ana Gonzalez1,2, Carolyn Hendrickson2,3, Alejandra Jauregui2, Norman Jones8, Kirsten Kangelaris9, Serena Ke1,2, Matthew F. Krummel5,10, Charles R. Langelier11,12, Tasha Lea10, Deanna Lee1,2, Aleskandra Lelidowicz2,13, Raphael Lota6, Michael Matthay2,3, Jeff Milush8, Viet Nguyen1,2, Ahmad Sadeed Rashid6, Nicklaus Rodriguez6, Austin Sigman2, Kevin Tang6, Luz Torres Altamirano6, Alyssa Ward1, Andrew Willmore2, Michael Wilson1,3, Prescott G. Woodruff2,3,5
Co-ACIT Study Group members: Biniam Ambachew1, Sarah Cary1, Lauren Chalwell3, Christopher Colwell14, Chayse Jones2, Clayton Josephy2, Carolyn Hendrickson2,3, Deanna Lee1, Matthieu LeGrand15, Juan Carlos Montoy3, Aaron E. Kornblith14, Philip Kurien15, Brenda Nunez-Garcia1, Viet Nguyen1, John J. Park1, Arun Prakash15, Brittany Robinson1, India Shelley1, Katherine D. Wick2
1Division of General Surgery, Trauma and Surgical Critical Care, Zuckerberg San Francisco General Hospital, 2Division of Pulmonary, Critical Care, Allergy and Sleep Medicine, Department of Medicine, 3Cardiovascular Research Institute, 4CoLabs, 5ImmunoX Initiative, 6Division of Rheumatology, Department of Medicine, 7Helen Diller Family Comprehensive Cancer Center, 8Core Immunology Laboratory, Department of Experimental Medicine, Zuckerberg San Francisco General Hospital, 9Division of Hospital Medicine, 10Department of Pathology, 11Division of Infectious Disease, Department of Medicine, 14Department of Emergency Medicine, and 15Department of Anesthesia and Perioperative Care, School of Medicine, University of California, San Francisco, San Francisco, California; 12Chan Zuckerberg Biohub, San Francisco, California; and 13Interdepartmental Division of Critical Care Medicine, University of Toronto, Toronto, Ontario, Canada
Footnotes
A complete list of COMET Consortium and Co-ACIT Study Group members may be found before the beginning of the References.
Supported in part by awards from the University of California, San Francisco (UCSF): the UCSF COVID-19 Rapid Response Pilot Grant Program, supported by the National Center for Advancing Translational Sciences and the National Institutes of Health (sponsored by the UCSF Academic Senate, Clinical and Translational Science Institute, and the Research Development Office [M.A.M., L.Z.K.]), the San Francisco Foundation (J.H.), and the UCSF Department of Anesthesia and Perioperative Care (J.H.). The COMET (COVID-19 Multi-Phenotyping for Effective Therapies) study was funded in part by the National Institute of Allergy and Infectious Diseases IMPACC (Immunophenotyping Assessment in a COVID-19 Cohort) Network and the National Institutes of Health (3U19AI077439-13S2). J.J. was supported by a fellowship from Société de Réanimation de Langue Française and Amicale des Anciens Internes en medicine des Hopitaux de Paris and Assistance Publique – Hopitaux de Paris. C.S.C. was funded by National Institutes of Health grant R35 HL140026.
Author Contributions: J.J. and J.H. conceptualized and designed the study. J.J. acquired and analyzed the data and drafted and revised the manuscript. J.H. provided critical edits to the manuscript. L.R. was responsible for virus propagation and viral titer determination. L.R. and A.S. supervised biosafety level 3 (BSL-3) working conditions. E.L. participated in cell culture, stimulation, and immunoassays. The other authors (Z.A.M., A.T.F., R.J.B., P.K., C.S.C., P.G.W., D.J.E., C.H., M.F.K., C.R.L, M.A.M., and L.Z.K.) were part of patient enrollment in the COMET (COVID-19 Multi-Phenotyping for Effective Therapies) and Co-ACIT (COVID-19 Associated Coagulopathy, Inflammation, and Thrombosis) studies or members of the COMET executive committees and provided critical edits to the manuscript. Studies not requiring BSL-3 laboratory precautions were performed in J.H.’s laboratory, and those requiring BSL-3 laboratory precautions were performed in A.S.’s laboratory.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202107-1774OC on June 1, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
Contributor Information
on behalf of the COVID-19 Multi-Phenotyping for Effective Therapies (COMET) ConsortiumCOVID-19 Associated Coagulopathy, Inflammation, and Thrombosis (Co-ACIT) Study Group:
Sharvari Bhide, Carolyn S. Calfee, Sidney A. Carrillo, Suzanna Chak, David J. Erle, Gabriela K. Fragiadakis, Rajani Ghale, Jeremy Giberson, Pat Glenn, Ana Gonzalez, Carolyn Hendrickson, Alejandra Jauregui, Norman Jones, Kirsten Kangelaris, Serena Ke, Matthew F. Krummel, Charles R. Langelier, Tasha Lea, Deanna Lee, Aleskandra Lelidowicz, Raphael Lota, Michael Matthay, Jeff Milush, Viet Nguyen, Ahmad Sadeed Rashid, Nicklaus Rodriguez, Austin Sigman, Kevin Tang, Luz Torres Altamirano, Alyssa Ward, Andrew Willmore, Michael Wilson, Prescott G. Woodruff, Biniam Ambachew, Sarah Cary, Lauren Chalwell, Christopher Colwell, Chayse Jones, Clayton Josephy, Carolyn Hendrickson, Deanna Lee, Matthieu LeGrand, Juan Carlos Montoy, Aaron E. Kornblith, Philip Kurien, Brenda Nunez-Garcia, Viet Nguyen, John J. Park, Arun Prakash, Brittany Robinson, India Shelley, and Katherine D. Wick
References
- 1. Jin Y, Ji W, Yang H, Chen S, Zhang W, Duan G. Endothelial activation and dysfunction in COVID-19: from basic mechanisms to potential therapeutic approaches. Signal Transduct Target Ther . 2020;5:293. doi: 10.1038/s41392-020-00454-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Teuwen LA, Geldhof V, Pasut A, Carmeliet P. COVID-19: the vasculature unleashed. Nat Rev Immunol . 2020;20:389–391. doi: 10.1038/s41577-020-0343-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matthay MA, Leligdowicz A, Liu KD. Biological mechanisms of COVID-19 acute respiratory distress syndrome. Am J Respir Crit Care Med . 2020;202:1489–1491. doi: 10.1164/rccm.202009-3629ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Okada H, Yoshida S, Hara A, Ogura S, Tomita H. Vascular endothelial injury exacerbates coronavirus disease 2019: the role of endothelial glycocalyx protection. Microcirculation . 2021;28:e12654. doi: 10.1111/micc.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Osuchowski MF, Winkler MS, Skirecki T, Cajander S, Shankar-Hari M, Lachmann G, et al. The COVID-19 puzzle: deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir Med . 2021;9:622–642. doi: 10.1016/S2213-2600(21)00218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ait-Oufella H, Maury E, Lehoux S, Guidet B, Offenstadt G. The endothelium: physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med . 2010;36:1286–1298. doi: 10.1007/s00134-010-1893-6. [DOI] [PubMed] [Google Scholar]
- 7. Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial responses in sepsis. Am J Respir Crit Care Med . 2020;202:361–370. doi: 10.1164/rccm.201910-1911TR. [DOI] [PubMed] [Google Scholar]
- 8. Yao Y, Cao J, Wang Q, Shi Q, Liu K, Luo Z, et al. D-dimer as a biomarker for disease severity and mortality in COVID-19 patients: a case control study. J Intensive Care . 2020;8:49. doi: 10.1186/s40560-020-00466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lavillegrand JR, Garnier M, Spaeth A, Mario N, Hariri G, Pilon A, et al. Elevated plasma IL-6 and CRP levels are associated with adverse clinical outcomes and death in critically ill SARS-CoV-2 patients: inflammatory response of SARS-CoV-2 patients. Ann Intensive Care . 2021;11:9. doi: 10.1186/s13613-020-00798-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Philippe A, Chocron R, Gendron N, Bory O, Beauvais A, Peron N, et al. Circulating von Willebrand factor and high molecular weight multimers as markers of endothelial injury predict COVID-19 in-hospital mortality. Angiogenesis . 2021;24:505–517. doi: 10.1007/s10456-020-09762-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhatraju PK, Morrell ED, Zelnick L, Sathe NA, Chai XY, Sakr SS, et al. Comparison of host endothelial, epithelial and inflammatory response in ICU patients with and without COVID-19: a prospective observational cohort study. Crit Care . 2021;25:148. doi: 10.1186/s13054-021-03547-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leisman DE, Mehta A, Thompson BT, Charland NC, Gonye ALK, Gushterova I, et al. Alveolar, endothelial, and organ injury marker dynamics in severe COVID-19. Am J Respir Crit Care Med . 2022;205:507–519. doi: 10.1164/rccm.202106-1514OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hani C, Trieu NH, Saab I, Dangeard S, Bennani S, Chassagnon G, et al. COVID-19 pneumonia: a review of typical CT findings and differential diagnosis. Diagn Interv Imaging . 2020;101:263–268. doi: 10.1016/j.diii.2020.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med . 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, et al. CRICS TRIGGERSEP Group (Clinical Research in Intensive Care and Sepsis Trial Group for Global Evaluation and Research in Sepsis) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med . 2020;46:1089–1098. doi: 10.1007/s00134-020-06062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wurtz N, Penant G, Jardot P, Duclos N, La Scola B. Culture of SARS-CoV-2 in a panel of laboratory cell lines, permissivity, and differences in growth profile. Eur J Clin Microbiol Infect Dis . 2021;40:477–484. doi: 10.1007/s10096-020-04106-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.World Health Organization (WHO) 2020.
- 18. WHO Working Group on the Clinical Characterisation and Management of COVID-19 infection. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect Dis . 2020;20:e192–e197. doi: 10.1016/S1473-3099(20)30483-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tiruppathi C, Malik AB, Del Vecchio PJ, Keese CR, Giaever I. Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc Natl Acad Sci USA . 1992;89:7919–7923. doi: 10.1073/pnas.89.17.7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Szulcek R, Bogaard HJ, van Nieuw Amerongen GP. Electric cell-substrate impedance sensing for the quantification of endothelial proliferation, barrier function, and motility. J Vis Exp . 2014;(85):51300. doi: 10.3791/51300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Joffre J, Lloyd E, Wong E, Chung-Yeh C, Nguyen N, Xu F, et al. Catecholaminergic vasopressors reduce Toll-like receptor agonist-induced microvascular endothelial cell permeability but not cytokine production. Crit Care Med . 2021;49:e315–e326. doi: 10.1097/CCM.0000000000004854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benjamini Y, Krieger AM, Yekutieli D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika . 2006;93:491–507. [Google Scholar]
- 23. Bakhshandeh B, Sorboni SG, Javanmard AR, Mottaghi SS, Mehrabi MR, Sorouri F, et al. Variants in ACE2; potential influences on virus infection and COVID-19 severity. Infect Genet Evol . 2021;90:104773. doi: 10.1016/j.meegid.2021.104773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gerard L, Lecocq M, Bouzin C, Hoton D, Schmit G, Pereira JP, et al. Increased angiotensin-converting enzyme 2 and loss of alveolar type II cells in COVID-19-related acute respiratory distress syndrome. Am J Respir Crit Care Med . 2021;204:1024–1034. doi: 10.1164/rccm.202012-4461OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gómez J, Albaiceta GM, García-Clemente M, López-Larrea C, Amado-Rodríguez L, Lopez-Alonso I, et al. Angiotensin-converting enzymes (ACE, ACE2) gene variants and COVID-19 outcome. Gene . 2020;762:145102. doi: 10.1016/j.gene.2020.145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science . 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen F, Zhang Y, Li X, Li W, Liu X, Xue X. The impact of ACE2 polymorphisms on COVID-19 disease: susceptibility, severity, and therapy. Front Cell Infect Microbiol . 2021;11:753721. doi: 10.3389/fcimb.2021.753721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Möhlendick B, Schönfelder K, Breuckmann K, Elsner C, Babel N, Balfanz P, et al. ACE2 polymorphism and susceptibility for SARS-CoV-2 infection and severity of COVID-19. Pharmacogenet Genomics . 2021;31:165–171. doi: 10.1097/FPC.0000000000000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McCracken IR, Saginc G, He L, Huseynov A, Daniels A, Fletcher S, et al. Lack of evidence of angiotensin-converting enzyme 2 expression and replicative infection by SARS-CoV-2 in human endothelial cells. Circulation . 2021;143:865–868. doi: 10.1161/CIRCULATIONAHA.120.052824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang J, Dong J, Martin M, He M, Gongol B, Marin TL, et al. AMP-activated protein kinase phosphorylation of angiotensin-converting enzyme 2 in endothelium mitigates pulmonary hypertension. Am J Respir Crit Care Med . 2018;198:509–520. doi: 10.1164/rccm.201712-2570OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Am J Respir Crit Care Med . 2020;202:756–759. doi: 10.1164/rccm.202001-0179LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gustave CA, Gossez M, Demaret J, Rimmelé T, Lepape A, Malcus C, et al. Septic shock shapes B cell response toward an exhausted-like/immunoregulatory profile in patients. J Immunol . 2018;200:2418–2425. doi: 10.4049/jimmunol.1700929. [DOI] [PubMed] [Google Scholar]
- 33. Victorino GP, Newton CR, Curran B. Effect of angiotensin II on microvascular permeability. J Surg Res . 2002;104:77–81. doi: 10.1006/jsre.2002.6412. [DOI] [PubMed] [Google Scholar]
- 34. Pine AB, Meizlish ML, Goshua G, Chang CH, Zhang H, Bishai J, et al. Circulating markers of angiogenesis and endotheliopathy in COVID-19. Pulm Circ . 2020;10:2045894020966547. doi: 10.1177/2045894020966547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zuo Y, Warnock M, Harbaugh A, Yalavarthi S, Gockman K, Zuo M, et al. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci Rep . 2021;11:1580. doi: 10.1038/s41598-020-80010-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Targosz-Korecka M, Kubisiak A, Kloska D, Kopacz A, Grochot-Przeczek A, Szymonski M. Endothelial glycocalyx shields the interaction of SARS-CoV-2 spike protein with ACE2 receptors. Sci Rep . 2021;11:12157. doi: 10.1038/s41598-021-91231-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fraser DD, Patterson EK, Slessarev M, Gill SE, Martin C, Daley M, et al. Endothelial injury and glycocalyx degradation in critically ill coronavirus disease 2019 patients: implications for microvascular platelet aggregation. Crit Care Explor . 2020;2:e0194. doi: 10.1097/CCE.0000000000000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wadowski PP, Jilma B, Kopp CW, Ertl S, Gremmel T, Koppensteiner R. Glycocalyx as possible limiting factor in COVID-19. Front Immunol . 2021;12:607306. doi: 10.3389/fimmu.2021.607306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ogawa F, Oi Y, Nakajima K, Matsumura R, Nakagawa T, Miyagawa T, et al. Temporal change in syndecan-1 as a therapeutic target and a biomarker for the severity classification of COVID-19. Thromb J . 2021;19:55. doi: 10.1186/s12959-021-00308-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Amalakuhan B, Habib SA, Mangat M, Reyes LF, Rodriguez AH, Hinojosa CA, et al. Endothelial adhesion molecules and multiple organ failure in patients with severe sepsis. Cytokine . 2016;88:267–273. doi: 10.1016/j.cyto.2016.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Whalen MJ, Doughty LA, Carlos TM, Wisniewski SR, Kochanek PM, Carcillo JA. Intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 are increased in the plasma of children with sepsis-induced multiple organ failure. Crit Care Med . 2000;28:2600–2607. doi: 10.1097/00003246-200007000-00070. [DOI] [PubMed] [Google Scholar]
- 42. Figueras-Aloy J, Gómez-López L, Rodríguez-Miguélez JM, Salvia-Roiges MD, Jordán-García I, Ferrer-Codina I, et al. Serum soluble ICAM-1, VCAM-1, L-selectin, and P-selectin levels as markers of infection and their relation to clinical severity in neonatal sepsis. Am J Perinatol . 2007;24:331–338. doi: 10.1055/s-2007-981851. [DOI] [PubMed] [Google Scholar]
- 43. Monickaraj F, McGuire P, Das A. Cathepsin D plays a role in endothelial-pericyte interactions during alteration of the blood-retinal barrier in diabetic retinopathy. FASEB J . 2018;32:2539–2548. doi: 10.1096/fj.201700781RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bathina S, Das UN. Brain-derived neurotrophic factor and its clinical implications. Arch Med Sci . 2015;11:1164–1178. doi: 10.5114/aoms.2015.56342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Donovan MJ, Lin MI, Wiegn P, Ringstedt T, Kraemer R, Hahn R, et al. Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development . 2000;127:4531–4540. doi: 10.1242/dev.127.21.4531. [DOI] [PubMed] [Google Scholar]
- 46. Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, et al. Neurological associations of COVID-19. Lancet Neurol . 2020;19:767–783. doi: 10.1016/S1474-4422(20)30221-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, et al. Post-acute COVID-19 syndrome. Nat Med . 2021;27:601–615. doi: 10.1038/s41591-021-01283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pranata R, Huang I, Lim MA, Yonas E, Vania R, Kuswardhani RAT. Delirium and mortality in coronavirus disease 2019 (COVID-19) — a systematic review and meta-analysis. Arch Gerontol Geriatr . 2021;95:104388. doi: 10.1016/j.archger.2021.104388. [DOI] [PMC free article] [PubMed] [Google Scholar]