

Abstract
SARS-CoV-2 continues to evolve, causing waves of the pandemic. Up to May 2022, 10 million genome sequences have accumulated, which are classified into five major variants of concern. With the growing number of sequenced genomes, analysis of the big dataset has become increasingly challenging. Here we developed systematic approaches based on sets of correlated single nucleotide variations (SNVs) for comprehensive subtyping and pattern recognition of transmission dynamics. The approach outperformed single-SNV and spike-centric scans. Moreover, the derived subtypes elucidate the relationship of signature SNVs and transmission dynamics. We found that different subtypes of the same variant, including Delta and Omicron exhibited distinct temporal trajectories. For example, some Delta and Omicron subtypes did not spread rapidly, while others did. We identified sets of characteristic SNVs that appeared to enhance transmission or decrease efficacy of antibodies for some subtypes. We also identified a set of SNVs that appeared to suppress transmission or increase viral sensitivity to antibodies. For the Omicron variant, the dominant type in the world, we identified the subtypes with enhanced and suppressed transmission in an analysis of eight million genomes as of March 2022 and further confirmed the findings in a later analysis of ten million genomes as of May 2022. While the “enhancer” SNVs exhibited an enriched presence on the spike protein, the “suppressor” SNVs are mainly elsewhere. Disruption of the SNV correlation largely destroyed the enhancer-suppressor phenomena. These results suggest the importance of fine subtyping of variants, and point to potential complex interactions among SNVs.
Keywords: COVID-19, SARS-CoV-2 variants, viral subtype, single nucleotide variation, allelic association
Significance Statement.
In this study, we develop a dimension reduction method for a viral subtyping and pattern recognition for transmission dynamics. This new method is computationally efficient and allows to analyze millions of genome sequences. Multiple analyses of two—ten million SARS-CoV-2 genomes identify viral subtypes for the contagious variants Omicron and Delta. The analysis also identifies the transmission enhancer SNVs associated with temporal rise in proportion and transmission suppressor SNVs associated with the suppression of the temporal rise. The finding is further confirmed with larger sample sizes and in more countries. The transmission enhancer-repressor hypothesis may explain a significant part of the current situations in the pandemic. This study improves our understanding of SARS-CoV-2 and controlling for the COVID-19 pandemic.
Introduction
Relative to the original Wuhan strain, SARS-CoV-2 Variants of Concern (Alpha, Beta, Gamma, Delta, and Omicron) and other known variants (e.g. Eta, Iota, Kappa, Lambda, Epsilon, Zeta, Theta, and Mu) have been identified and caused multiple waves of the pandemic. These variants are reported to confer high transmissibility and possible antibody escape, thus posing challenges to the pandemic control measures. Therefore, tracking variants and predicting their risks are crucially important for pandemic control and the development of pharmacological treatments.
Analysis of the SARS-CoV-2 genome sequences has provided unprecedented opportunities for tracking variants (1–3), characterizing the viral genomes (4–6), investigating molecular and cellular mechanisms (7–9), understanding the viral origin and evolution (2, 10–17), and scrutinizing many other aspects related to the pandemic. These utilities demonstrate the importance of analyzing the viral genome database (18).
Since the beginning of the pandemic, Global Initiative on Sharing Avian Influenza Data (GISAID) (https://www.gisaid.org/) has provided a data depository for viral genome sequences from confirmed cases. As of May 2022, ten million genome sequences have been reported, which provide an opportunity for fine subtyping of SARS-CoV-2 variants. However, as the size of the dataset grows, analysis of the big data have become increasingly challenging. Model- or likelihood-based subtyping approaches such as a phylogenetic classification are popular but require more model assumptions and intensive computation compared to a model-free approach.
We develop a model-free approach for using correlated SNV sets (CSSs) with allelic association (i.e. covariance structure) for dimension reduction of the large collection of genomes. The CSSs allow a computationally tractable ways for viral subtyping and pattern recognition of transmission dynamics. The results elucidate the relationship of signature single nucleotide variations (SNVs) of CSSs and transmission dynamics of viral strains that the relationship may be obscure in a hierarchical lineage/sublineage structures by using a phylogenetic classification.
Using this method, we found that within the commonly identified Delta (aka B.1.617.2) and Omicron (aka B.1.1.529) variants, the temporal trajectories (i.e. the frequency over time) differ significantly among their subtypes. We further identified sets of SNVs that behave as transmission “enhancers,” which are associated with increased temporal trajectory of the Delta and Omicron variants, respectively, and sets of transmission suppressors, which are associated with the “suppression” of the variants with the transmission enhancers. The results for Alpha variant are provided in the preprint of this work (19). These findings suggest the importance of fine subtyping and possible SNV interactions that be important determinants of viral fitness in the context of public health measures.
Results
Using CSSs for dimension reduction
Allelic association of SNVs is a hallmark of rising variants (Supplementary Text 1, Figs. S1 to S4, and Table S1). This characteristic allows us to use allelic association as a way to reduce the dimension of the big data and subtype the variants. We grouped SNVs with pairwise associations R2 > 0.5, and used an exponential weighted moving average (EWMA) to detect CSSs, while ignoring SNV sets with occurrence frequency lower than 20 (Refer to the “Materials and Methods” section and Fig. S5A). The sensitivity, specificity, and robustness of CSS detection are discussed in Supplementary information (Fig. S5A).
The genome of viral strains can then be represented by a combination of SNVs in CSSs with a residual term (Fig. S6). Through a three-stage dimension reduction, a 29,409 by 2,119,724 matrix of genome sequence is reduced to a 1,366 by 9,848 matrix of CSS (Fig. S6). Note that the definition of CSSs can change depending on the purpose of analysis to include any subset of the genomic database, for example, strains identified in different time span, different countries, or different segments of the genome. Additionally, the thresholds for allelic association can also vary to highlight the features of interest.
We identified a total of 1,057 CSSs, each containing 4 to 33 SNVs with a total of 1,366 signature SNVs. We found that 1,053 of 1,057 CSSs can characterize > 99.9% of the dominant strain type [Type VI defined in our previous work (5)], which accounts for 2,000,622 (94.38%) of the strains since March, 2020. The statistics are provided (Table S2).
Fig. 1A shows that the frequency of strains represented by CSSs increased with time, and CSSs almost completely represent the genome variations after July 2020. Fig. 1B shows that the residual SNVs became insignificant. Temporal change of the numbers of CSSs provides information for the dynamic evolutionary processes. We analyzed four datasets that were collected from December 2019 to 23 June and 15 December in 2021 and 23 February and 27 April in 2022 with a sample size of 2,119 K, 6,166 K, 8,475 K, and 10,089 K, respectively, to infer the number of CSSs (Fig. S7). The Omicron variant exhibited an increase in number of CSSs and superseded the other variants, indicating a rapid transmission and continued evolution of Omicron. Apart from the Alpha, Delta, and Omicron, other variants have a limited change and less evidence of ongoing host adaptation. If we use each CSS to define a subtype, these subtypes can effectively represent the whole population in recent dates (Supplementary Text 2 and Fig. S8). Therefore, CSSs can serve as a basis for both dimension reduction and subtyping, which captures the genome evolution in a computationally tractable manner. We evaluated the relationship between the number of genomes (n) and computation time (h) in our CSS analysis (Fig. S9). Our CSS analysis consisting of variation frequency calculation and CSS construction has a linear-time computational complexity. This highlights the computational efficiency of our CSS analysis.
Fig. 1.

High proportions of viral strains and SNVs can be represented by correlated SNV sets (CSSs) (n = 2,119 K genomes as of 2021 June 23). (A) Majority of viral strains can be represented by CSSs. The temporal proportions of viral strains represented (blue curve) and under-represented (red curve) by CSSs are displayed. The number of total strains (gray bar) and the number of under-represented stains (red bar) are displayed with the two histograms in the background. (B) Only a small number of SNVs cannot be represented by CSSs. Distributions of the number of residual SNVs (i.e. the SNVs under represented by any of CSSs, green bar), and the number of entire SNVs in a strain (i.e. union of SNVs represented and under represented by any of CSSs, red bar) are displayed. The median number of residual SNVs is 4. The median number of entire SNVs is 29.
Remarkably, the CSSs for the variants with the same Pango nomenclature exhibited different temporal trajectories. These subtypes carried similar core SNVs (defining SNVs), but some additional SNVs may be different and influence the fitness and transmission of the CSSs. Examples for the Delta variant (aka B.1.617.2) and Omicron variant (aka B.1.1.529) are given in Fig. 2A and Fig. 3A, respectively. Detailed composition of the Delta CSSs and Omicron CSSs are provided in Table 1 and Table S3, respectively.
Fig. 2.

The CSS analysis identifies multiple Delta subtypes with differential temporal trajectories (n = 2,119 K genomes as of 2021 June 23). (A) Eleven Delta (aka B.1.617.2) CSSs are identified in India. Three CSSs (Delta-01 to 03) have an increasing temporal trajectory and eight CSSs (Delta-04 to 11) have a much lower temporal trajectory, revealing that CSSs provide a more detailed information for the subtypes and their transmission patterns for Delta. (B to D) Missing transmission enhancer SNVs causes a dramatically decrease in the temporal trajectory in the enhanced CSS Delta-01. The curve with a symbol o indicates the temporal trajectory of Delta-01. The curve with a symbol x indicates the Delta-01 variant with the maximum temporal trajectory among the different Delta-01 variants without or with a missing of specific SNVs. Nmax indicates the maximum number of strains in a temporal trajectory. Delta-01 carries 8 transmission enhancer SNVs (Table 1), where there are 3 SNVs in the spike protein and 5 SNVs in the non-spike proteins. In (B), it shows that missing any of 8 transmission enhancer SNVs causes a decrease in the temporal trajectory in Delta-01. In (C), it shows that missing any of 3 transmission enhancer SNVs in the spike protein causes a decrease in the temporal trajectory in Delta-01. In (D), it shows that missing any of 5 transmission enhancer SNVs in the non-spike proteins causes a decrease in the temporal trajectory in Delta-01. When any of the transmission enhancer SNVs are missing (the curve without a symbol x), the temporal trajectories are dramatically reduced. This phenomenon explains that the transmission enhancer SNVs work cooperatively. (E to G) Missing all of the transmission suppressor SNVs causes an increase in the temporal trajectory in the suppressed CSS Delta-04. The curve with a symbol o indicates the temporal trajectory of Delta-04. The curve with a symbol x indicates the Delta-04 variant with the maximum temporal trajectory among the different Delta-04 variants without or with a missing of specific SNVs. Nmax indicates the maximum number of strains in a temporal trajectory. Delta-04 carries five transmission enhancer SNVs and four transmission suppressor SNVs (Table 1). Among the four suppressor SNVs, one is located in the spike protein and the other three are located in the non-spike proteins. In (E), it shows that missing all of the four transmission suppressor SNVs causes an increase in the temporal trajectory. In (F), it shows that missing the only transmission suppressor SNV in the spike protein (conditional on that the three non-spike suppressor SNVs are remained) does not cause an increase in the temporal trajectory. In (G), it shows that missing any of the transmission suppressor SNVs in the non-spike proteins (conditional on that the spike suppressor SNV is remained) does not cause an increase in the temporal trajectory. When all of the four transmission suppressor SNVs are remained, the temporal trajectory of Delta-04 is dramatically reduced. This phenomenon illustrates that transmission suppression can be contributed by a single spike SNV or a set of transmission suppressor SNVs.
Fig. 3.

The CSS analysis identifies multiple Omicron subtypes with differential temporal trajectories (n = 8,475 K genomes as of 2022 February 23). (A) Seven Omicron (aka B.1.1.529) CSSs are identified in the United Kingdom. Six CSSs (Omicron-01 to 06) have an increasing temporal trajectory and one CSS (Omicron-07) has a much lower temporal trajectory, revealing that CSSs provide a more detailed information for the subtypes and their transmission patterns for Omicron. (B to D) Missing transmission enhancer SNVs causes a dramatically decrease in the temporal trajectory in Omicron-01. The curve with a symbol o indicates the temporal trajectory of Omicron-01. The curve with a symbol x indicates the Omicron-01 variant with the maximum temporal trajectory among the different Omicron-01 variants without or with a missing of specific SNVs. Nmax indicates the maximum number of strains in a temporal trajectory. Omicron-01 carries 44 transmission enhancer SNVs, where there are 24 SNVs in the spike protein and 20 SNVs in the non-spike proteins (Table S3). In (B), it shows that missing any of 44 transmission enhancer SNVs causes a decrease in the temporal trajectory in Omicron-01. In (C), it shows that missing any of 24 transmission enhancer SNVs in the spike protein causes a decrease in the temporal trajectory in Omicron-01. In (D), it shows that missing any of 20 transmission enhancer SNVs in the non-spike proteins causes a decrease in the temporal trajectory in Omicron-01. When any of the transmission enhancer SNVs are missing (the curve without a symbol x), the temporal trajectories are dramatically reduced. This phenomenon explains that the transmission enhancer SNVs work cooperatively. (E to G) Missing all of the transmission suppressor SNVs causes an increase in the temporal trajectory in Omicron-07. The curve with a symbol o indicates the temporal trajectory of Omicron-07. The curve with a symbol x indicates the Omicron-07 variant with the maximum temporal trajectory among the different Omicron-07 variants without or with a missing of specific SNVs. Nmax indicates the maximum number of strains in a temporal trajectory. Omicron-07 carries five transmission enhancer SNVs and four transmission suppressor SNVs. Among the four suppressor SNVs, one is located in the spike protein and the other three are located in the non-spike proteins. In (E), it shows that missing all of the four transmission suppressor SNVs causes an increase in the temporal trajectory. In (F), it shows that missing the only transmission suppressor SNV in the spike protein (conditional on that the three non-spike suppressor SNVs are remained) cause a slightly increase in the temporal trajectory. In (G), it shows that missing any of the transmission suppressor SNVs in the non-spike proteins (conditional on that the spike suppressor SNV is remained) does not cause an increase in the temporal trajectory. When all of the four transmission suppressor SNVs are remained, the temporal trajectory of Omicron-07 is dramatically reduced. This phenomenon illustrates that transmission suppression can be contributed by a single spike SNV or a set of transmission suppressor SNVs.
Table 1.
Transmission enhancer and suppressor SNVs for the Delta CSSs (subtypes) with differential transmission dynamics (n = 2,119 K genomes as of 2021 June 23).
| Defining SNVs | Transmission | Transmission | Signature | High-frequency | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| for Delta | enhancer SNV | suppressor SNV | SNVs in CSSs | SNVs in CSSs | ||||||||||
| Delta variant | Enhanced CSSs | Suppressed CSSs | ||||||||||||
| Nucleotide change | Protein region | Amino acid change | Type of SNVs | Delta-01 | Delta-02 | Delta-03 | Delta-04 | Delta-05 | Delta-06 | Delta-07 | Delta-08 | Delta-09 | Delta-10 | Delta-11 |
| G29742T | 3'UTR | – | Enhancer | – | – | – | – | – | – | – | – | – | – | – |
| T22917G | S (RBD, RBM) | L452R | Defining | R | R | R | R | R | R | R | R | R | R | R |
| C23604G | S (S1) | P681R | Defining | R | R | R | R | R | R | R | R | R | R | R |
| C25469T | ORF3a | S26L | Defining | L | L | L | L | L | L | L | L | L | L | L |
| T27638C | ORF7a | V82A | Defining | A | A | A | A | A | A | A | A | A | A | A |
| G28881T | N | R203M | Defining | M | M | M | M | M | M | M | M | M | M | M |
| G29402T | N | D377Y | Defining | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| A23403G | S (S1) | D614G | Defining | G | G | G | G | G | G | G | G | G | G | G |
| C21618G | S (NTD) | T19R | Defining | R | R | R | R | R | R | R | R | R | R | R |
| C22995A | S (RBD, RBM) | T478K | Defining | K | K | K | K | K | K | K | K | K | K | K |
| G24410A | S (HR1) | D950N | Defining | N | N | N | N | N | N | N | N | N | N | N |
| T26767C | M | I82T | Defining | T | T | T | T | T | T | T | T | T | T | T |
| C27752T | ORF7a | T120I | Defining | I | I | I | I | I | I | I | I | I | I | I |
| A28461G | N | D63G | Defining | G | G | G | G | G | G | G | G | G | G | G |
| G15451A | nsp12 | G5063S | Enhancer | S | S | S | S | S | S | S | S | |||
| C16466T | nsp13 | P5401L | Enhancer | L | L | L | L | L | L | L | L | |||
| G4181T | nsp3 | A1306S | Enhancer | S | S | S | ||||||||
| C6402T | nsp3 | P2046L | Enhancer | L | L | L | ||||||||
| C7124T | nsp3 | P2287S | Enhancer | S | S | S | ||||||||
| C8986T | nsp4 | D2907D | Enhancer | D | D | D | ||||||||
| G9053T | nsp4 | V2930L | Enhancer | L | L | L | ||||||||
| C10029T | nsp4 | T3255I | Enhancer | I | I | I | ||||||||
| A11332G | nsp6 | V3689V | Enhancer | V | V | V | ||||||||
| C19220T | nsp14 | A6319V | Enhancer | V | V | V | ||||||||
| C27874T | ORF7b | T40I | Enhancer | I | I | I | ||||||||
| G28916T | N | G215C | Enhancer | C | C | C | ||||||||
| C20320T | nsp15 | H6686Y | Suppressor | Y | Y | Y | Y | |||||||
| G29427A | N | R385K | Suppressor | K | K | K | K | |||||||
| C24745T | S (S2) | V1061V | Suppressor | V | V | V | ||||||||
| C6539T | nsp3 | H2092Y | Suppressor | Y | Y | Y | ||||||||
| T12946C | nsp9 | Y4227Y | Suppressor | Y | Y | Y | Y | |||||||
| A20262G | nsp15 | L6666L | Suppressor | L | L | L | L | |||||||
| C1191T | nsp2 | P309L | Suppressor | L | L | L | L | L | L | L | ||||
| C1267T | nsp2 | G334G | Suppressor | G | G | G | G | G | G | G | ||||
| C27739T | ORF7a | L116F | Suppressor | F | F | F | F | |||||||
| G9203A | nsp4 | D2980N | Suppressor | N | N | N | ||||||||
| T9678C | nsp4 | F3138S | Suppressor | S | S | S | ||||||||
| A17496G | nsp13 | E5744E | Suppressor | E | E | E | ||||||||
| A21792C | S (NTD) | K77T | Suppressor | T | T | T | ||||||||
| C28253T | ORF8 | F120F | Suppressor | F | F | F | ||||||||
| C11005A | nsp6 | H3580Q | Suppressor | Q | Q | Q | ||||||||
| A5584G | nsp3 | T1773T | Suppressor | T | ||||||||||
| C11514T | nsp6 | T3750I | Suppressor | I | ||||||||||
| C13019T | nsp9 | L4252L | Suppressor | L | ||||||||||
| C22227T | S (NTD) | A222V | Suppressor | V | ||||||||||
| N: # of strains in each CSS in the world | 102,529 | 95,969 | 81,013 | 3,481 | 3,990 | 4,216 | 7,583 | 1,297 | 1,849 | 1,684 | 11,059 | |||
| n: # of strains in each CSS in India | 9,212 | 5,227 | 2,350 | 1,209 | 1,493 | 1,563 | 2,333 | 501 | 816 | 674 | 976 | |||
Eleven CSSs (subtypes) were identified in India. These CSSs can be separated into two categories: (1) Enhanced CSSs (Delta-01 to 03) that they have a rising temporal trajectory; (2) Suppressed CSSs (Delta-04 to 11) that they have a much lower temporal trajectory relative to the trajectories of the enhanced CSSs. In the first sub-table entitled “Delta variant,” the columns are “nucleotide change,” “protein region,” “amino acid change,” and “Type of SNVs” for the signature SNVs in CSSs. Types of SNVs include: (a) Defining SNVs for Delta from the Pango classification (green); (b) Transmission enhancer SNVs” (red); (c) Transmission suppressor SNVs (cyan). The second and third sub-tables entitled “Enhanced CSSs” and “Suppressed CSSs” show the alternate alleles of the signature SNVs in the three enhanced CSSs and seven suppressed CSSs, respectively. SNVs located on the spike protein are bolded. If 100% of the strains in a CSS carry the alternate allele, then the cells are highlighted with a yellow color; If ≥ 50% of the strains in the CSS carry the alternate allele, then the cells are marked with a gray color; If < 50% of the strains in the CSS carry the alternate allele, then the cells are empty. The number of strains in each of the eleven CSSs in the world (N) and in the original country that the Delta variant was discovered (n) are listed in the bottom of this table.
The Delta transmission enhancer and suppressor SNVs
To characterize and subtype strain variations in more detail, the CSS approach can be applied to individual countries. As the highly contagious variant Delta was first discovered in India, we further subtyped the Delta strain sequenced in India using the proposed CSS approach, which resulted in eleven subtypes. For illustration, we focus on the first six subtypes (Delta-01 to 06). The strains in the six Delta subtypes carry all or the majority of the signature SNVs defined for the Delta variant (T19R, T478K, D950N, D614G, L452R, P681R in the spike protein, and 7 SNVs in other proteins) (Green cells in column “SNV” in Table 1). Yet, the subtypes exhibited distinct temporal trajectories (Fig. 2A).
The first three subtypes “enhanced CSSs” (Delta-01 to 03) exhibited increasing temporal trajectories and the other three subtypes “suppressed CSSs” (Delta-04 to 06) had much lower temporal trajectories (Fig. 2A). Remarkably, the same pattern of the differential temporal trajectories for the subtypes were found in many other countries consistently (Fig. S10), indicating that the subtypes and their differences are reproducible.
The first three subtypes with rising temporal trajectories (Delta-01 to 03 in Fig. 2A and Table 1) are defined by eight, nine, and eleven signature SNVs, respectively, with 100% allelic associations (Yellow cells in column “Delta-01 to 03” in Table 1). Excluding the Delta defining SNVs, we define the remaining signature SNVs as “transmission enhancers” (Red cells in column “SNV” in Table 1), since they are strongly associated with the rapid rise in proportion. The remaining three subtypes with lower temporal trajectories (Delta-04 to 06 in Fig. 2A and Table 1) all contain a set of 100% associated signature SNVs, in addition to the Delta defining SNVs and CSS enhancer SNVs in some strains. It appears that these signature SNVs “suppressed” the rise of the temporal trajectories. Thus, we define them as “transmission suppressors” (Cyan cells in column “SNV” in Table 1).
As the enhancer SNVs are 100% associated in Delta-01 to 03, we looked for similar strains without the complete set of the enhancer SNVs. The result showed that strains missing any one of the 8 signature SNVs in Delta-01 dramatically reduced the temporal trajectory (Fig. 2B). This phenomenon suggests that the signature SNVs work cooperatively to gain viral fitness (i.e. a synergistic or positive cooperativity effect), and disfavors the possible hitchhiking passenger roles played by some SNVs. We also evaluated the effects of the signature SNVs in the spike protein and non-spike proteins, separately. Compared to Delta-01, the strains that they had the five non-spike SNVs [G29742Del (3’UTR), S26L (ORF3a), V82A (ORF7a), R203M (N), and D377Y (N)] but missed any one of the 3 signature SNVs in the spike protein had a dramatically reduced temporal trajectory (Fig. 2C). Similarly, the strains that they had the three spike SNVs [L452R (S), P681R (S), and D614G (S)] but missed any one of 5 SNVs in the non-spike proteins also had a dramatically reduced temporal trajectory (Fig. 2D). The results suggest that both the spike and non-spike protein SNVs play a critical role in transmission dynamics. Similar patterns were also found in Delta-02 and Delta-03.
In contrast to the first three subtypes, the other three subtypes (Delta-04 to 06) exhibit different temporal patterns (Fig. 2A). The majority of the strains in these three subtypes also carried the defining SNVs in the spike protein for the Delta variant (i.e. the T19R-L452R-T478K-D614G-P681R-D950N haplotype) (Table 1). Nevertheless, the temporal trajectories of the three subtypes are suppressed dramatically after acquiring the suppressor SNVs located mainly on the non-spike proteins (Fig. 2A and Table 1). At most one suppressor SNV is located in the spike protein in each of the suppressed CSSs.
Delta-04 carries the transmission suppressors H6686Y (nsp15), R385K (N), V1061V (S), and H2092Y (nsp3) (Table 1), and exhibits the low temporal trajectory (Fig 2A). Delta-05 and Delta-06 carry overlapping but different sets of transmission suppressors with some synonymous SNVs (Table 1). These two subtypes also generally suppressed the rise of the temporal proportion, although not as effective as Delta-04 (Fig. 2A).
We examined the influence of missing suppressor SNVs. Compared to the Delta-04 strains, the strains containing only the 5 Delta defining SNVs but missing all transmission suppressor SNVs of Delta-04 exhibited a rising temporal frequency (Orange line in Fig. 2E). We further evaluated the effects of missing suppressor SNVs in the spike and non-spike proteins, separately. Compared to the Delta-04 strains, when the Delta strains missed the only suppressor SNV in the spike protein [i.e. V1061V (S)], the temporal trajectory was not increased (Fig. 2F). When the Delta strains missed any of the three transmission suppressors in the non-spike proteins, the temporal trajectory was not increased either (Fig. 2G). The potential mechanism of transmission suppressor SNVs is discussed in the subsection “Suppression effect” below.
Remarkably, the identified enhancer and suppressor SNVs can be further confirmed in an analysis of six million SARS-CoV-2 genomes (Fig. S11). This reflects the robustness of the identified transmission enhancer and suppressor SNVs.
The Omicron transmission enhancer and suppressor SNVs
As of 2022 February 23 (n = 8,475 K), we applied the proposed CSS-based approach to subtype the Omicron variants in the United Kingdom with a larger sample size compared to other countries (Fig. 3A). Omicron is known as the variant with a large number of SNVs especially in the spike protein. The first six subtypes (Omicron-01 to 06) present the much higher temporal trajectory than the seventh subtype (Omicron-07). These Omicron subtypes were also found in many other countries, and showed the consistent temporal trajectory patterns (Fig. S12). Excluding the Omicron defining SNVs, we defined the signature SNVs of Omicron-01 to 06 as transmission enhancers, as they enhanced the transmission of the variant (Red cells in column “SNV” in Table S3). Reduced transmission may be found after a viral competition with other subtypes or the newly emergent variants. Omicron-03 and Omicron-06 exhibited a stronger viral competition among the enhanced CSSs. In addition to the Omicron defining SNVs (Green cells in column “SNV” in Table S3), Omicron-07 contained some of, but most importantly, they acquired a set of suppressors that appeared to suppress the rise of the temporal trajectory (Cyan cells in column “SNV” in Table S3). Furthermore, these findings were successfully confirmed in an analysis of ten million SARS-CoV-2 genomes accumulated as of 2022 April 27 (n = 10,089 K) (Fig. S13).
The proportion of the strains pertaining to the first six subtypes were reduced dramatically if any of the transmission enhancers were missing (e.g. the pattern of Omicron-01 in Fig. 3B). This finding suggests that the enhancers work cooperatively. We also evaluated the effects of the signature SNVs in the spike and non-spike proteins in Omicron-01, separately. Missing any of the spike signature SNVs (Fig. 3C) or non-spike signature SNVs (Fig. 3D) resulted in a dramatically reduced temporal trajectory. The results suggest that both the spike and non-spike protein SNVs play a critical role in transmission dynamics.
Omicron-07 carries the transmission suppressors L3290L (nsp5), I1081V (S), L106F (ORF3a), and D343G (N) (Table S3), and exhibits the low temporal trajectory (Fig. 3A). Missing some number of transmission suppressor SNVs (Fig. 3E) or any suppressors in the non-spike proteins (Fig. 3G) do not cause a large rising temporal trajectory, but missing the suppressor in the spike protein (Fig. 3F) causes a slightly increase, suggesting that transmission suppression can be contributed by a single spike SNV or a set of transmission suppressor SNVs. The mechanism of transmission suppressor SNVs is discussed in the subsection “ Suppression effect” below.
We looked for the composition of the seven identified Omicron CSSs (Table 2). Omicron sublineages such as BA.1 from the Pango nomenclature can be classified into several CSSs that they had respective sets of signature SNVs and exhibited different transmission dynamics. Therefore, CSSs provide a fine subtyping for the lineages and sublineages of Omicron. In addition, a CSS can be composed of multiple lineages and/or sublineages. For example, Omicron-03 consisted of part of BA.1, BA.1.1, and BA.2 strains that they carried the common transmission enhancer SNVs with high allelic association. Therefore, CSSs provide a subtyping system alternative to the SARS-CoV-2 nomenclature system from a phylogenetic classification. Remarkably, the viral subtyping by using CSSs found important signature SNVs directly related to transmission dynamics. The results pave a way for a better understanding about the viral transmission in the pandemic.
Table 2.
Differential strain compositions of in the Omicron CSSs (subtypes).
| Omicron variant | Enhanced CSSs | Suppressed CSS | |||||
|---|---|---|---|---|---|---|---|
| Pango lineage | Omicron-01 | Omicron-02 | Omicron-03 | Omicron-04 | Omicron-05 | Omicron-06 | Omicron-07 |
| BA.1 | 62.36% | 65.08% | 46.88% | 57.07% | 57.22% | 0.00% | 99.77% |
| BA.1.1 | 37.64% | 34.89% | 35.31% | 42.87% | 42.71% | 0.00% | 0.19% |
| BA.2 | 0.00% | 0.00% | 17.60% | 0.00% | 0.00% | 100.00% | 0.00% |
| BA.3 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| None | 0.00% | 0.03% | 0.21% | 0.06% | 0.07% | 0.00% | 0.04% |
Six enhanced CSSs and one suppressed CSS were identified in the United Kingdom. In these CSSs with differential transmission dynamics, their strain compositions are different. For examples, Omicron-03 consists of 46.88% BA.1 strains, 35.31% of BA.1.1, and 17.60% of BA.2 strains, and 0.21% of other Omicron strains.
Suppression effect
Transmission suppression can be contributed by a single spike SNV or a set of suppressor SNVs. Delta-11 (Fig. 2 and Table 1) and Omicron-07 (Fig. 3 and Table S3) are illustrated as examples. Compared to the strains without A222V (S), the strains with A222V (S) exhibited a lower temporal trajectory in the world and many countries for all variants (Fig. S14A), Delta (Fig. S14B), and Omicron (Fig. S14C), illustrating that A222V (S) indeed has a suppressor effect. In addition, Delta with A222V (S) has a higher temporal trajectory than Delta‐11, which carries a full set of transmission suppressor SNVs: T1773T(nsp3)–T3750I(nsp6)–L4252L(nsp9)–A222V(S), indicating that the set of four transmission suppressor SNVs work cooperatively and have a stronger synergistic effect in suppressing transmission suppression than a single suppressor A222V (S) (Fig. 4). Since some of the suppressor SNVs in Delta-04 to 06 are synonymous SNVs, the effect may come from codon usage or RNA-level interactions, although the hitchhiking effect cannot be ruled out. The result also explains the necessity of a CSS analysis compared to a SNV by SNV analysis that it fails to account for genetic epistasis.
Fig. 4.

Set of transmission suppressor SNVs work cooperatively and has a stronger synergistic effect in suppressing transmission suppression than a single suppressor in Delta-11 (n = 6,166 K genomes as of 2021 December 15 ). We analyzed the effects of transmission suppression contributed by a single SNV and a set of SNVs. Delta-11 (Fig. 2 and Table 1) is illustrated as an example. In each subfigure, three curves indicate the temporal trajectories for three subgroups: (i) “Delta-11” carries a full set of transmission suppressor SNVs: T1773T(nsp3)–T3750I(nsp6)–L4252L(nsp9)–A222V(S) (green color and circle symbol); (ii) “Delta-11 with T1773T(nsp3)–T3750I(nsp6)–L4252L(nsp9)” indicates the Delta strains that they carry the signature SNVs similar to Delta-11 but miss a suppressor SNV A222V(S), i.e. the Delta strains, which carry only the suppressor triplet T1773T(nsp3)–T3750I(nsp6)–L4252L(nsp9) (blue color and x symbol); (iii) “Delta with A222V(S)” indicates the Delta strains with a transmission suppressor SNV in the spike protein A222V (red color and triangle symbol). The number of total strains per date (gray bar) is displayed with the histogram in the background. “Delta with A222V(S)” has a higher temporal trajectory than “Delta-11,” indicating that the set of four transmission suppressor SNVs work cooperatively and have a stronger synergistic effect in suppressing transmission suppression than a single SNV A222V(S). The curves for “Delta-11” and “Delta-11 with T1773T(nsp3)–T3750I(nsp6)–L4252L(nsp9)” are very close, reflecting that the four suppressor SNVs T1773T(nsp3)–T3750I(nsp6)–L4252L(nsp9)–A222V(S) have a high allelic association, and therefore it's hard to observe any missing suppressor SNVs from the set of transmission suppressor SNVs. (A) The World; (B) The United Kingdom; (C) Mexico; (D) Spain.
Similarly, both Omicron-07 and Omicron with I1081V (S) have a lower temporal trajectory compared to the one in Omicron with L3290L(nsp5)–L106F(ORF3a)–D343G(N), indicating that I1081V (S) and/or L3290L(nsp5)–I1081V(S)–L106F(ORF3a)–D343G(N) have an effect in suppressing a viral transmission of Omicron (Fig. 5). Because of a high allelic association in the transmission suppressor SNVs L3290L(nsp5)–I1081V(S)–L106F(ORF3a)–D343G(N), more data are needed to distinguish that the suppressing effect is contributed by I1081V solely and/or the full set of transmission suppressor SNVs.
Fig. 5.

Single spike suppressor SNV and set of transmission suppressor SNVs contributes to transmission suppression in Omicron-07 (n = 8,475 K genomes as of 2022 February 23). We analyzed transmission suppression contributed by a single suppressor SNV and a set of suppressor SNVs. Omicron-07 (Fig. 3 and Table S3) is illustrated as an example. In each subfigure, three curves indicate the temporal trajectories for the following three subgroups: (i) “Omicron-07” carries a full set of four transmission suppressor SNVs: L3290L(nsp5)–I1081V(S)–L106F(ORF3a)–D343G(N) (green color and circle symbol); (ii) “Omicron-07 with L3290L(nsp5)–L106F(ORF3a)–D343G(N)” indicates the Omicron strains that they carry the signature SNVs similar to Omicron-07 but miss a transmission suppressor SNV I1081V in the spike protein, i.e. it only carries the suppressor triplet L3290L(nsp5)–L106F(ORF3a)–D343G(N) (blue color and x symbol); (iii) “Omicron with I1081V(S)” indicates the Omicron strains carrying a transmission suppressor SNV in the spike protein I1081V (red color and triangle symbol). The number of total strains per date (gray bar) is displayed with the histogram in the background. “Omicron-07” and “Omicron with I1081V(S)” have very close temporal trajectories, representing that the suppressor SNV I1081V(S) and triplet L3290L(nsp5)–L106F(ORF3a)–D343G(N) co-appeared in the Omicron variants. They have a lower temporal trajectory compared to the one in “Omicron-07 with L3290L(nsp5)–L106F(ORF3a)–D343G(N),” indicating that I1081V(S) and/or L3290L(nsp5)–I1081V(S)–L106F(ORF3a)–D343G(N) have an effect in suppressing a viral transmission of Omicron. More data are needed to distinguish that the suppressing effect is contributed by I1081V solely and/or the full set of transmission suppressor SNVs: L3290L(nsp5)–I1081V(S)–L106F(ORF3a)–D343G(N). (A) The World; (B) The United Kingdom; (C) The United States; (D) Canada.
Single-SNV scan and spike-centric CSS scan
A genome-wide single-SNV scan examines the temporal trajectories SNV by SNV. In an analysis of two million SARS-CoV-2 genomes accumulated as of 2021 June 23 (n = 2,119 K) and an analysis of eight million SARS-CoV-2 genomes accumulated as of 2022 February 23 (n = 8,475 K), the single-SNV scans can identify the transmission enhancer SNVs for Delta (Fig. S15A) and Omicron (Fig. S15B), respectively. However, single SNVs fail to provide a reasonable viral subtyping because almost all viral subtypes carry multiple signature SNVs with allelic association. In addition, a single-SNV scan also identified a large number of false-positive transmission suppressors. The results illustrate the limitation of a single-SNV scan compared to a genome-wide multilocus CSS scan. A spike-centric CSS scan can also identify the transmission enhancer SNVs on the spike protein for Omicron (Table S3). However, the transmission suppressors SNVs cannot be detected. The set of transmission suppressor SNVs L3290L(nsp5)–I1081V(S)–L106F(ORF3a)–D343G(N) contained at most one SNV on the spike protein (Table S3). No other SNVs on the spike protein had an allelic association with I1081V (S). The result illustrates a genome-wide multilocus CSS scan provides a more intact transmission enhancer and suppressor detection than a spike-centric CSS scan.
Discussion
SARS-CoV-2 is an RNA virus and can readily acquire mutations during the replication process and generate new variants and subtypes. The subtypes arising from the recent common ancestral variant may have highly correlated SNVs but remarkably different genomic sequences and transmission patterns. In this study, we developed a systematic dimension reduction approach to characterizing the viral subtypes based on the correlated SNV sets with allelic association and monitoring their emergence and growth. We also developed a pattern recognition approach to grouping CSSs and detecting the sets of transmission enhancers and suppressors. By analyzing 10 million genome sequences of SARS-CoV-2, we provided real-world evidence for the viral subtypes. The identified subtypes exhibit differential temporal trajectories. The patterns can be characterized by the sets of transmission enhancers and suppressors located both on the spike protein and elsewhere. This highlights the importance of SNVs in both spike and non-spike proteins.
Spike-protein signature SNVs are often used as a proxy for diagnosing variants and explaining increasing viral transmissibility. Our result shows that almost all CSSs contain SNVs on the spike protein (1,053/1,057 = 99.62%), and only 4 CSSs do not contain any defining SNVs on spike. In total, 37.16% of the defining SNVs of a CSS are located in the spike protein, which is significantly larger than the proportion of SNVs in the spike protein in the whole genome (3,822/29,903 = 12.78%) with p = 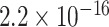 by using parametric t and Wilcoxon signed-ranked tests; the two tests yielded the same p-value. Remarkably, spike-protein is highly related to allelic association; spike-protein is characterized by the high ratio of non-synonymous SNVs vs. synonymous SNVs with allelic association, frequent intergenic allelic association (i.e. Spike-nucleocapsid association), and frequent intragenic allelic association. These results indicate that spike is constantly under selection and that it is the most important protein coded by the viral genome, which determines the overall viral fitness and transmission effectiveness.
by using parametric t and Wilcoxon signed-ranked tests; the two tests yielded the same p-value. Remarkably, spike-protein is highly related to allelic association; spike-protein is characterized by the high ratio of non-synonymous SNVs vs. synonymous SNVs with allelic association, frequent intergenic allelic association (i.e. Spike-nucleocapsid association), and frequent intragenic allelic association. These results indicate that spike is constantly under selection and that it is the most important protein coded by the viral genome, which determines the overall viral fitness and transmission effectiveness.
The non-spike protein signature SNVs have been commonly found in the important variants; however, their roles have been largely under-appreciated. Our results reveal that the non-spike-proteins signature SNVs provide subtle information for the subtypes of a variant. In addition, the non-spike-proteins signature SNVs are also relevant to the viral transmissibility. Allelic association provides a direction for investigating mechanistic interactions. The non-spike-proteins SNVs can directly elevate or reduce the viral transmissibility through a direct genetic epistasis with other SNVs (i.e. genetic buffering). On the other hand, the observed association of non-spike protein SNVs may be explainable by genetic hitchhiking. However, this explanation is disfavored because we did not find “hitchhikers” of less than the whole set of suppressors. The current vaccines have been designed targeting at the spike protein. Our results suggest that the non-spike-proteins SNVs should also be considered to improve the sensitivity of SARS-CoV-2 variants such as Omicron and Delta to pharmacological intervention. The results for Alpha are only provided in the preprint of this work (19).
Our results show that the ratio of nonsynonymous vs. synonymous amino acid change (r) is much higher in the transmission enhancer SNVs compared to the suppressor SNVs. Interestingly, the Alpha variant has a high r value (r = 4.500) for the transmission enhancer SNVs, and the Delta variant has an even higher r value (r = 11.500). Moreover, the Delta variant has a r value (r = 1.375) for the transmission suppressor SNVs much lower than the Alpha variant (r = 4.000). A higher nonsynonymous vs. synonymous proportion may suggest a possible positive selection (20). The results reveal that the Delta variant may have a higher positive selection in the transmission enhancer SNVs and a lower negative selection in the transmission suppressor SNVs. This partially reflects the dominance of Delta compared to other variants. Compared to the previous variants, Omicron is known as a variant with a significant enrichment of spike signature SNVs. SNVs in the receptor binding domain (RBD) of the spike protein (S) can alter the affinity to the angiotensin converting enzyme 2 (ACE2) receptor and potentially cause vaccine escape (7, 21, 22). Omicron has exhibited a more rapid transmission than previous Variants of Concern.
Because of a lasting evolution of SARS-CoV-2, there is an unmet need to systematically track the dynamic changes of the viral subtypes and understand their signature SNVs. However, the computation becomes a hurdle when the number of genomes exceeds a ten-million scale. We find that allelic association is a hallmark for an emergence and growth of a subtype and therefore can be employed to detect a strain subtype. Viral strains in a subtype share the signature SNVs with high allelic association (i.e. CSS). A CSS-based approach provides a multilocus analysis that it is more informative than a SNV by SNV analysis. A SNV by SNV analysis ignores allelic association and genetic epistasis, causing an increased false positive in identifying transmission suppressors and an underestimated effect of transmission suppression. In addition, a large number of SNVs are neutral and do not benefit the fitness gain of SARS-CoV-2 (23). It is redundant to consider all SNVs in the 30 K genome of SARS-CoV-2. The CSS-based analysis addresses this problem. A CSS-based analysis based on a set of correlated SNVs significantly overcomes the computational bottleneck in the strain-by-strain and SNV-by-SNV whole-genome analysis.
Overall, the proposed method allows to analyze tens of millions of genomes, identify the emerging subtypes and their transmission enhancer and suppressor SNVs, and therefore improve our understanding of SARS-CoV-2. A continuous trend monitoring of viral subtypes and their evolution as new genomes are added to the database promotes risk assessment of SARS-CoV-2 transmission and pandemic control of COVID-19. Future work is warranted to extend the current method to an online version and provide a real-time monitoring system.
Materials and Methods
Data download and preprocessing
We downloaded and preprocessed 2,215 K, 6,316 K, 8,940 K, and 10,663 K whole-genome sequences data from the Global Initiative on Sharing Avian Influenza Data (GISAID) database (https://www.gisaid.org/) on 07 July, 29 December in 2021 and 02 March and 04 May in 2022, respectively (Fig. S16). Strain information was extracted from the meta information in GISAID. After data quality control (discarding the duplicated samples, the samples with an aligned sequence of < 29 K bases, and the samples without sample recruitment date), it remained the complete sequences of 2,119 K, 6,166 K, 8,475 K, and 10,089 K genomes, respectively. Multiple sequence alignment was performed by using MAFFT v.7 (24). The Wuhan-Hu-1 that the strain was originally isolated in China and had 29,903 nucleotides (25) was employed as the reference genome. Our major sequence analysis discarded two ends (5’ leader and 3’ terminal sequences) and focused on the SNV base positions from 266 to 29,674. Nucleotides different from the Wuhan-Hu-1 strain were assigned as a SNV. Deletions were also detected. Annotation of the SNVs was collected from CNCB (https://bigd.big.ac.cn/ncov/release_genome). Statistical analyses were performed by using our self-developed R codes.
CSS analysis
CSS subtyping was established based on the proposed analysis procedures (Fig. S5A). Matrix representation for a dimension reduction of the proposed CSS analysis procedure is provided (Fig. S6). Identification of preliminary SNV groups (PSGs) based on allelic association and determination of CSSs by using an exponential weighted moving average (EWMA) (26) are explained. We calculated variation (allele) frequencies of SNVs in different countries and in the whole world by using a direct allele counting. For the SNVs with a frequency > 0.01, we calculated pairwise allelic association for any pairs of SNVs by using the square of the correlation coefficient (27) as follows:
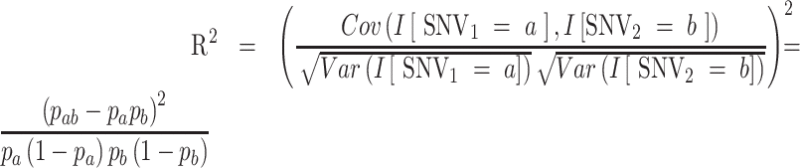 |
where Cov and Var indicate covariance and variance, respectively. 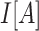 indicates an indicator variable with a value of 1 if event A holds, 0 otherwise. Frequency
indicates an indicator variable with a value of 1 if event A holds, 0 otherwise. Frequency 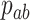 indicates occurrence frequency (haplotype frequency) with allele a at the first SNV and allele b at the second SNV.
indicates occurrence frequency (haplotype frequency) with allele a at the first SNV and allele b at the second SNV. 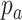 (
(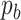 ) indicates the frequency of allele a (b) at the first (second) SNV.
) indicates the frequency of allele a (b) at the first (second) SNV.
An algorithm was developed to group SNVs as a preliminary SNV group (PSG) that a PSG contains more than 3 SNVs and all pairwise allelic associations 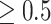 . We removed very rare PSGs, which contained less than and equal to 20 strains in the last 90 days in a country or in the world. On the one hand, signature SNVs in PSGs were extended if the SNVs on the spike protein had a very high proportion. On the other hand, a major SNV subset (i.e. a haplotype) in a PSG was also regarded as PSG if the subset had a proportion ≥ 0.1. The number of major SNV subsets corresponding to a PSG is few, particularly at the PSG, which carries SNVs with high pairwise allelic associations, because: (1) if a major SNV subset in a PSG was regarded as PSG, then the subset must contain more than 3 SNVs with all pairwise allelic associations
. We removed very rare PSGs, which contained less than and equal to 20 strains in the last 90 days in a country or in the world. On the one hand, signature SNVs in PSGs were extended if the SNVs on the spike protein had a very high proportion. On the other hand, a major SNV subset (i.e. a haplotype) in a PSG was also regarded as PSG if the subset had a proportion ≥ 0.1. The number of major SNV subsets corresponding to a PSG is few, particularly at the PSG, which carries SNVs with high pairwise allelic associations, because: (1) if a major SNV subset in a PSG was regarded as PSG, then the subset must contain more than 3 SNVs with all pairwise allelic associations 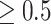 in the current analysis; (2) SNVs in a PSG have high allelic associations so that genetic diversity of SNV subsets is limited and the number of major SNV subsets with a high proportion (i.e. haplotype frequency) is few typically. In an analysis of two million SARS-CoV-2 genomes accumulated as of 2021 June 23 (n = 2,119 K), about 21.38% of 1,057 CSSs were identified through a subset of PSGs. The percentage will reduce when we increase the thresholds of pairwise allelic associations and proportion of a major SNV subset. EWMA control chart for testing
in the current analysis; (2) SNVs in a PSG have high allelic associations so that genetic diversity of SNV subsets is limited and the number of major SNV subsets with a high proportion (i.e. haplotype frequency) is few typically. In an analysis of two million SARS-CoV-2 genomes accumulated as of 2021 June 23 (n = 2,119 K), about 21.38% of 1,057 CSSs were identified through a subset of PSGs. The percentage will reduce when we increase the thresholds of pairwise allelic associations and proportion of a major SNV subset. EWMA control chart for testing 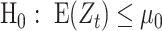 vs.
vs. 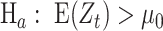 was applied to detect correlated SNV sets (CSSs) and track the growth change of a variant over time as follows: Let
was applied to detect correlated SNV sets (CSSs) and track the growth change of a variant over time as follows: Let 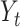 denote the temporal proportion at time point t for a PSG, where the time index t corresponds to a time window of nine dates (four dates of a specific date in each side were considered for yielding a smoothed temporal proportion) in the current analysis. The EWMA statistic
denote the temporal proportion at time point t for a PSG, where the time index t corresponds to a time window of nine dates (four dates of a specific date in each side were considered for yielding a smoothed temporal proportion) in the current analysis. The EWMA statistic
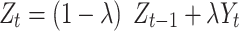 |
represents a weighted average characteristic of the past and current occurrence proportion of a variant, where 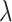 indicates a (smoothing) weight for the current temporal proportion. A PSG was identified as an emerging CSS (i.e.
indicates a (smoothing) weight for the current temporal proportion. A PSG was identified as an emerging CSS (i.e. 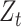 is out-of-control) if
is out-of-control) if
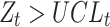 |
where the upper control limit and
and
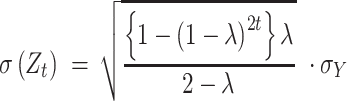 |
where 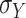 denotes the standard deviation of
denotes the standard deviation of 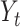 . Default
. Default 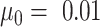 and
and 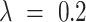 were considered. The analysis was conducted by using R package qcc (28).
were considered. The analysis was conducted by using R package qcc (28).
Once CSSs had been determined, genome sequence of a CSS was determined by substituting the genome of the Wuhan-Hu-1 strain with the signature SNVs with high allelic association of the CSS. A CSS-based phylogenetic analysis (Fig. S8) based on maximum parsimony (MP) was conducted by using MEGA X (29). Subtree-pruning-regrafting algorithm (30) was employed for a tree topology search heuristic.
Detection of transmission enhancer SNVs and transmission suppressor SNVs
Transmission enhancer SNVs and transmission suppressor SNVs were detected by using the proposed procedures (Fig. S5B). Decision rule, parameter vector, and parameter updating by using Particle Swarm Optimization (31) are explained below.
On the basis of the identified CSSs and their temporal proportions for a variant, we proposed a decision rule to classify the CSSs into enhanced, suppressed, and undetermined CSSs for a variant. First, we only included the CSSs with a temporal proportion >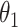 at some time (hitting time) and focused on the temporal proportions after the first hitting time. Second, CSSs were initially grouped if an average difference of their temporal proportions over time was <
at some time (hitting time) and focused on the temporal proportions after the first hitting time. Second, CSSs were initially grouped if an average difference of their temporal proportions over time was <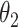 . Third, when all CSSs belong to the same group: (i) if the maximum temporal proportion was <
. Third, when all CSSs belong to the same group: (i) if the maximum temporal proportion was <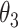 , then these CSSs were classified as the undetermined CSS group. (ii) If the maximum temporal proportion was located between
, then these CSSs were classified as the undetermined CSS group. (ii) If the maximum temporal proportion was located between 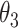 and
and 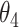 : (ii-a) we further monitored the slope of the temporal trajectory. Let D denote the increment that we subtracted the sum of negative slopes from the sum of positive slopes. If D was
: (ii-a) we further monitored the slope of the temporal trajectory. Let D denote the increment that we subtracted the sum of negative slopes from the sum of positive slopes. If D was 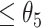 , then the CSSs were classified as the undetermined CSS group. (ii-b) If D was >
, then the CSSs were classified as the undetermined CSS group. (ii-b) If D was >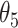 , then the CSSs were classified as the enhanced CSS group. (iii) If the maximum temporal proportion was
, then the CSSs were classified as the enhanced CSS group. (iii) If the maximum temporal proportion was 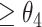 , then the CSSs were classified as the enhanced CSS group. Finally, when CSSs were grouped into multiple groups: (i) if the maximum temporal proportion was <
, then the CSSs were classified as the enhanced CSS group. Finally, when CSSs were grouped into multiple groups: (i) if the maximum temporal proportion was <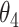 : (i-a) If D was
: (i-a) If D was 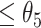 , then these CSSs were classified as the undetermined CSS group. (i-b) If D for all CSS were >
, then these CSSs were classified as the undetermined CSS group. (i-b) If D for all CSS were >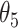 , then these CSSs were classified as the enhanced CSS group. (i-c) If D for some CSSs were >
, then these CSSs were classified as the enhanced CSS group. (i-c) If D for some CSSs were >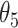 and some CSSs were
and some CSSs were 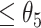 , then the former CSSs were classified as the enhanced CSS group and the latter CSSs were classified as the suppressed CSS group. (ii) if the maximum temporal proportion was
, then the former CSSs were classified as the enhanced CSS group and the latter CSSs were classified as the suppressed CSS group. (ii) if the maximum temporal proportion was 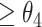 , then the CSSs were classified as the enhanced CSS group. Therefore, given a parameter vector
, then the CSSs were classified as the enhanced CSS group. Therefore, given a parameter vector 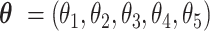 , CSSs can be classified into enhanced, suppressed, or undetermined CSSs.
, CSSs can be classified into enhanced, suppressed, or undetermined CSSs.
An optimal parameter vector is critical for the decision rule (a classifier of CSSs). The parameter vector 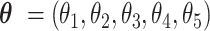 was updated and optimized by using Particle Swarm Optimization (31) as follows:
was updated and optimized by using Particle Swarm Optimization (31) as follows:
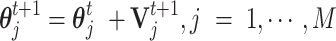 |
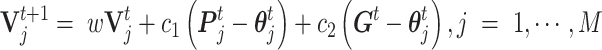 |
where w is the weight of 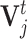 (default:
(default: 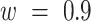 ); acceleration constant for individuals (“particle”)
); acceleration constant for individuals (“particle”) 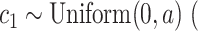 default:
default: 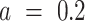 ); acceleration constant for population (“swarm”)
); acceleration constant for population (“swarm”) 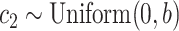 (default:
(default: 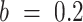 ); M is the number of initial random particles (default:
); M is the number of initial random particles (default: 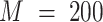 ).
). 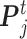 is the best state for individual j at iteration t and
is the best state for individual j at iteration t and 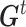 is the best state for population at iteration t. In each updating of
is the best state for population at iteration t. In each updating of 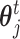 and
and 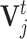 , the best states
, the best states 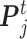 and
and 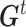 were updated simultaneously as follows:
were updated simultaneously as follows:
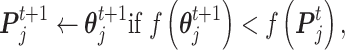 |
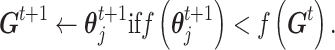 |
Here we considered an objective function (i.e. misclassification frequency in n CSSs) as follows:
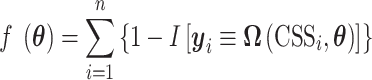 |
where 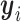 and
and 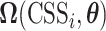 indicate the true and predicted states (i.e. “enhancer,” “suppressor,” or “undetermined”) of the i-th CSS given a parameter vector
indicate the true and predicted states (i.e. “enhancer,” “suppressor,” or “undetermined”) of the i-th CSS given a parameter vector 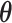 . The true state of a CSS was initially determined based on a heuristic discussion about the pattern of the temporal trajectory of the CSS in a multidisciplinary expert team. The predicted state of a CSS was obtained according to the aforementioned decision rule given a parameter vector
. The true state of a CSS was initially determined based on a heuristic discussion about the pattern of the temporal trajectory of the CSS in a multidisciplinary expert team. The predicted state of a CSS was obtained according to the aforementioned decision rule given a parameter vector 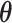 . For a CSS, if the true and predicted states are identical, then the misclassification error
. For a CSS, if the true and predicted states are identical, then the misclassification error 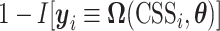 is 0, otherwise, 1. The parameter updating procedure was iterated to minimize the misclassification error (
is 0, otherwise, 1. The parameter updating procedure was iterated to minimize the misclassification error (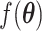 ). The iteration was stop if: 1)
). The iteration was stop if: 1) 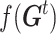 reached the minimum of f (In our case, 0 is the minimum of f, this represents every predicted state of CSSs is the true state.) or 2) it reached the maximum number of iterations (default: 25),
reached the minimum of f (In our case, 0 is the minimum of f, this represents every predicted state of CSSs is the true state.) or 2) it reached the maximum number of iterations (default: 25), 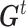 is the optimal estimator of
is the optimal estimator of 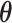 . The optimization was performed by using R package pso (32).
. The optimization was performed by using R package pso (32).
The optimal parameter vector was plugged into the decision rule to find the candidate transmission enhancers and suppressors for variant(s) and their corresponding signature SNVs. Finally, the results need to be confirmed in at least 80% of the studied countries and further confirmed in a later dataset with a larger sample size. The established decision rule can be directly apply to determine the CSS states, or serve as a good initial in an adaptive decision rule for more other variants.
Classification
Biological, Health, and Medical Science/Public Health and Epidemiology
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs. Ling-Jyh Chen, Shang-Te Danny Hsu, and Kay-Hooi Khoo for their useful discussions. We thank Global Initiative on Sharing Avian Influenza Data (GISAID) for providing the rich data for the genome sequences of SARS-CoV-2. We thank National Center for High-performance Computing of National Applied Research Laboratories of Taiwan for providing computational resources.
Notes
Competing Interest: The authors declare no competing interest.
Contributor Information
Hsin-Chou Yang, Institute of Statistical Science, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
Jen-Hung Wang, Institute of Statistical Science, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
Chih-Ting Yang, Institute of Statistical Science, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
Yin-Chun Lin, Institute of Statistical Science, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
Han-Ni Hsieh, Institute of Statistical Science, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
Po-Wen Chen, Institute of Statistical Science, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
Hsiao-Chi Liao, Institute of Statistical Science, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
Chun-houh Chen, Institute of Statistical Science, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
James C Liao, Institute of Biological Chemistry, Academia Sinica, Academia Rd, Nangang District Taipei 115, Taiwan.
Authors' Contributions
J.C.L. and H.C.Y. conceived the study, designed the research, and wrote the manuscript. J.H.W., C.T.Y., Y.C.L., H.N.H., B.W.C., H.C.L., and C.h.C. performed data analysis. J.C.L. and H.C.Y. supervised the project.
Data Availability
All data are available at the Global Initiative on Sharing Avian Influenza Data (GISAID) (https://gisaid.org/).
References
- 1. Elbe S, Buckland-Merrett G. 2017. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall. 1:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hadfield J, et al. 2018. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 34:4121–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rambaut A, et al. 2020. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol. 5:1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu B, Guo H, Zhou P, Shi Z-LJNRM. 2021. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 19:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang HC, et al. 2020. Analysis of genomic distributions of SARS-CoV-2 reveals a dominant strain type with strong allelic associations. Proc Natl Acad Sci USA. 117:30679–30686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zeng HL, Dichio V, Horta ER, Thorell K, Aurell E. 2020. Global analysis of more than 50,000 SARS-CoV-2 genomes reveals epistasis between eight viral genes. Proc Natl Acad Sci USA. 117:31519–31526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shang J, et al. 2020. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci USA. 117:11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang QH, et al. 2020. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell. 181:894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. 2021. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 19:155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang YZ, Holmes EC. 2020. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell. 181:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lu RJ, et al. 2020. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 395:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boni MF, et al. 2020. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat Microbiol. 5:1408–1417. [DOI] [PubMed] [Google Scholar]
- 13. Lam TTY, et al. 2020. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 583:282–285. [DOI] [PubMed] [Google Scholar]
- 14. Zhu N, et al. 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 382:727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Forster P, Forster L, Renfrew C, Forster M. 2020. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci USA. 117:9241–9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou P, et al. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 579:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rochman ND, et al. 2021. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc Nat Acad Sci USA. 118:e2104241118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Noorden RV. 2021. Scientists call for fully open sharing of coronavirus genome data. Nature. 590:195–196. [DOI] [PubMed] [Google Scholar]
- 19. Yang H-C, et al. 2022. Subtyping of major SARS-CoV-2 variants reveals different transmission dynamics. doi:10.1101/2022.04.10.486823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kimura M. 1977. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature. 267:275–276. [DOI] [PubMed] [Google Scholar]
- 21. Harvey WT, et al. 2021. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 19:409–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yi CY, et al. 2020. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell Mol Immunol. 17:621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Dorp L, et al. 2020. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat Commun. 11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu F, et al. 2020. A new coronavirus associated with human respiratory disease in China. Nature. 579:265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hunter JS. 1986. The exponentially weighted moving average. JQT. 18:203–210. [Google Scholar]
- 27. Hill WG, Robertson A. 1968. Linkage disequilibrium in finite populations. Theor Appl Genet. 38:226–231. [DOI] [PubMed] [Google Scholar]
- 28. Scrucca L. 2004. qcc: an R package for quality control charting and statistical process control [accessed 2022 Jun 15]. https://cran.r-project.org/web/packages/qcc/vignettes/qcc_a_quick_tour.html. [Google Scholar]
- 29. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 35:1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nei M, Kumar S. 2000. Molecular evolution and phylogenetics. New York: Oxford university press. [Google Scholar]
- 31. Kennedy J, Eberhart R. 1995. Particle Swarm Optimization. Proc IEEE Int Conf Neural Netw. 4:1942–1948. [Google Scholar]
- 32. Bendtsen C. 2015. pso: Particle Swarm Optimization. R package version 2.10.0 [accessed 2022 Jun 15]. https://cran.r-project.org/web/packages/pso/index.html. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available at the Global Initiative on Sharing Avian Influenza Data (GISAID) (https://gisaid.org/).