

Abstract
Traditional drug discovery is very laborious, expensive, and time-consuming, due to the huge combinatorial complexity of the discrete molecular search space. Researchers have turned to machine learning methods for help to tackle this difficult problem. However, most existing methods are either virtual screening on the available database of compounds by protein–ligand affinity prediction, or unconditional molecular generation, which does not take into account the information of the protein target. In this paper, we propose a protein target-oriented de novo drug design method, called AlphaDrug. Our method is able to automatically generate molecular drug candidates in an autoregressive way, and the drug candidates can dock into the given target protein well. To fulfill this goal, we devise a modified transformer network for the joint embedding of protein target and the molecule, and a Monte Carlo tree search (MCTS) algorithm for the conditional molecular generation. In the transformer variant, we impose a hierarchy of skip connections from protein encoder to molecule decoder for efficient feature transfer. The transformer variant computes the probabilities of next atoms based on the protein target and the molecule intermediate. We use the probabilities to guide the look-ahead search by MCTS to enhance or correct the next-atom selection. Moreover, MCTS is also guided by a value function implemented by a docking program, such that the paths with many low docking values are seldom chosen. Experiments on diverse protein targets demonstrate the effectiveness of our methods, indicating that AlphaDrug is a potentially promising solution to target-specific de novo drug design.
Keywords: transformer, MCTS, docking, drug design
Significance Statement.
One core goal of AI-driven drug design is to generate molecules that can bind to its protein target well. However, most existing deep learning methods seldom consider the protein target, and thus the generated molecules have a poor performance on binding affinity. Here, we propose a protein target specific de novo molecular generation method, called AlphaDrug. We trained a Lmser Transformer (LT) network to learn the joint distributions of protein targets and molecules efficiently. Then a value function implemented by a docking program and the LT network is utilized to guide the Monte Carlo tree search (MCTS) for the conditional molecular generation. Experiments show that our method can generate novel molecules with high binding affinities.
Introduction
The expenditure of a new drug from research and development to the market was estimated to be between 314 million and 2.8 billion US dollars (1,2), and the development period takes more than 10 y on average (3). Computer-aided drug design utilized in-silico computational methods to accelerate the process and reduce the development cost, by modeling the interactions between small molecules and targets.
Drug design is a challenging computational problem due to the complex drug–target interaction and the vast chemical space of estimated 1060 compounds with drug-like characteristics, which is more than the number of atoms in the solar system (4).
In recent years, deep learning methods have made promising progress on the problems of molecule design, e.g. machine learning-accelerated ab-initio simulation (5,6), molecular property prediction (7,8), receptor-ligand binding affinity prediction (9,10), and so on. These methods may be used in virtual screening through the available database of drug-like compounds for drug candidates. However, such virtual screening relies on the coverage and diversity of the compound database, and is very computational intensive.
Another stream of research is de novo molecular generation by deep generative models. For example, molecules can be generated to satisfy certain physicochemical or customized properties under Variational AutoEncoder (VAE) or Generated Adversarial Networks (GAN), in terms of Simplified Molecular-Input Line-Entry System (SMILES) or molecular graph (11–15). These methods are able to generate novel compounds as drug candidates, but ignore the protein target in the generation process. Thus, the obtained molecules usually do not have good binding affinity with the target. Although there exist a few efforts (16,17) in taking the protein target into account to realize target-specific molecular generation, the performance is still far from satisfactory.
In this paper, we propose a novel method called AlphaDrug to generate molecules that can have good binding affinity with a given protein target. To enhance representation learning of drug–target interactions, we present a transformer variant by devising a hierarchy of skip connections from protein encoder to molecule decoder for improved feature transfer. We further model the molecular generation process by Monte Carlo tree search (MCTS) in a one-by-one symbol construction manner. We use not only the probability of selecting the next symbol predicted by the transformer variant but also the docking simulations computed by an external widely used program to effectively guide MCTS. Experiments verify the effectiveness of our method.
Our contributions are briefly summarized as follows:
We propose a model called AlphaDrug for de novo molecular generation given a protein target. The model is featured by a transformer variant, which enhances the efficiency of learning protein information, and an efficient MCTS guided by the transformer’s prediction and docking values.
Experiments and ablation studies demonstrate that our method outperforms the existing ones to generate molecules of binding affinity scores even higher than known ligands or drugs.
Related work
Molecular generation without considering target
Deep generative models have been developed to generate and optimize molecules with certain physicochemical and pharmacological properties. Reinforcement learning (RL) has been adopted to train the generative models to produce molecules with desired properties. The de novo drug generator regarded as the RL agent, and the agent takes actions of choosing the next SMILES symbol during molecular string generation to maximize the reward, which is computed after SMILES string completion. For string-based methods, REINVENT algorithm (18), which combines Recurrent Neural Network (RNN) with RL algorithms, was proposed to optimize the score of the generated molecules by fine-tuning the model parameters of RNN, and it is capable of controlling molecule structures. The ReLeaSE algorithm (19) integrates two separately trained generative deep neural networks to jointly generate novel chemical libraries. RL approach was introduced to adjust the generation toward those with the desired physical and/or biological properties. The ChemTS model (20) takes advantages of RNN to learn the patterns of the next symbol conditional on the intermediate molecular SMILES string, and uses the learned connection rules to guide the molecular generation by MCTS.
Efforts have also been made on graph representation of molecules. JT-VAE (11) generates valid tree-structured molecules by firstly generating a tree-structured scaffold of chemical substructures and assembling substructures according to the generated scaffold. GraphAF (14) is an autoregressive flow-based model and generates molecular graphs in a sequential manner, where the validity is checked when adding new atoms or bonds. GCPN (15) is a general graph convolutional network based model for goal directed graph generation through RL. It is trained to optimize domain-specific rewards and adversarial loss through policy gradient, and acts in an environment that incorporates domain-specific rules.
Target-specific molecular generation
Recently, a few researchers have begun to pay attention to generating molecules that bind to specific binding pockets. In LiGANN model (16), the structure of the protein pocket was mapped into the shape of the ligand through the BicycleGAN, and then the shape of the ligand was decoded into SMILES through the captioning network. In order to use 3D information of proteins to control the generation of drug-like molecules, Coulomb matrix of the coarse-grained atoms was utilized to train conditional RNN models (21). The representation of each atom in the context of the binding site was learned via graph neural networks in (22), and an autoregressive sampling scheme was developed to generate 3D molecules in 3D space. Although the above methods all considered the 3D structure of the binding site, their performances are still far from satisfactory because it is challenging to learn how molecules interact geometrically and chemically with their pockets in 3D space.
Another stream of efforts are string-based methods. The targeted drug generation problem was formulated as a translational task, and the transformer network was applied to capture the long-range dependencies (17). On the base of ChemTS (20), which was designed for sequential unconditional molecular generation, SBMolGen (23) was further developed by imposing the target docking score constraints into the molecule distributions during the process of MCTS. Our method also falls into the string-based paradigm, which has an advantage that the molecular generation is naturally formulated as a sequential decision-making problem. Our results suggest that the string-based method can be very effective in target-specific de novo molecular generation, if we can properly learn the protein target information and efficiently search in the vast space of possible drug molecules.
Methods
Overview of our method
We propose a novel method, called AlphaDrug, for target-specific de novo molecular generation. The AlphaDrug takes a protein target as input and generates ligand molecules that have strong binding affinities with the target. The generated molecules are promising drug candidates for the given protein target. An overview of AlphaDrug is given in Fig. 1.
Fig. 1.

An overview of AlphaDrug. (A) The computational flow of AlphaDrug. Molecules are generated in an autoregressive way based on the MCTS growing policy Π, which is computed from the protein sequence and the current state of the intermediate ligand string. (B) The left figure demonstrates the structure of the original transformer implemented in (24); the right figure is the Lmser Transformer (LT) in the paper.
Specifically, the ligand generation process is modeled in a step-by-step growing way. AlphaDrug is featured by a context-embedding component and a searching component. The context Cτ at step τ is defined as the set of the target protein in terms of amino acid sequence S and the intermediate ligand string in the form of SMILES a1a2⋅⋅⋅aτ, i.e. Cτ = {S, a1a2⋅⋅⋅aτ}, where ai is a SMILES symbol, i = 1, ..., τ. We devise a deep transformer network for the context embedding. Inspired from the least mean squared error reconstruction (Lmser) network (25), we modify the standard transformer by adding a hierarchy of skip connections from protein encoder to drug decoder as in Fig. 1, so that the decoder receives different levels of features of the protein and calculates an accurate probability P(aτ + 1|Cτ) of properly selecting the next symbol aτ + 1 to be appended to the intermediate ligand string. Then, we compute an improved ligand growing policy Π (aτ + 1|Cτ) by MCTS, which takes the probability P(aτ + 1|Cτ) as the prior growing policy. We resort to an external docking software to calculate the values for the paths in a fast rollout, which is implemented by a greedy policy using P(aτ + 1|Cτ). The docking value is able to effectively assess the quality of look-ahead simulations, and control the generation from the aspect of binding affinity with the protein target. The generation stops after the intermediate ligand string reaches an end symbol. The policy network is trained on a dataset of known ligand-protein pairs. It should be noted that MCTS is not involved in the training process of the policy network, but it is activated when generating molecules for the testing protein targets.
Lmser Transformer (LT) for context embedding
Learning the structural context of the target protein and the intermediate ligand is challenging due to the following reasons. First, the structures of the proteins and the involved binding sites are very complex. Second, the protein–ligand interaction patterns are complicated. Third, proteins and ligands are different molecules with unequal sequence lengths and consist of different atom sets. A protein sequence is often over 10 times longer than that of a ligand molecule. Finally, it requires good generalization to new proteins for de novo drug design, after training on the limited amount of available experimental data. We represent proteins as sequences of amino acids and ligands as SMILES strings, and consider both as biochemical languages. We use the transformer network to translate amino acid sequences into ligand SMILES strings, where the encoder takes the protein amino acid sequence as input, the decoder is fed with the intermediate ligand string. Although the original transformer (24) has been adopted in (17) for the same task, we find that it is not efficient to involve the protein information in molecular generation. The inefficiency is due to the information transfer bottleneck from the encoder top-layer to various levels of decoder layers.
We propose a transformer variant called LT to tackle this issue. Proposed in (25), Lmser network was developed by folding autoencoder (AE) along the central hidden layer. Such folding equivalently builds forward skip connections and feedback connections between paired layers of encoder and decoder (26). Inspired by this idea, we impose a hierarchy of skip connections from protein encoder to ligand decoder, as illustrated in the right figure of Fig. 1. The hierarchical skip connections pass various levels of features of the input protein to the corresponding levels of decoder, and fuse with the features of the intermediate ligand. The decoder is filled with more details about the protein information in all levels, and leverage them to obtain accurate prediction of the next symbol for the intermediate ligand string.
The hierarchical skip connections are implemented via a cross-attention mechanism. The details are illustrated in Fig. 2. Mathematically, the cross-attention block maps a query Qm from the ligand molecule decoder and a key-value pair (KS, VS), which is passed through the skip connections from the protein encoder, into an output as a weighted sum of the values
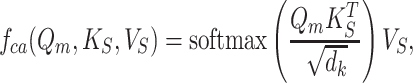 |
(1) |
where 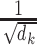 is a scaling factor, and the softmax function is used to normalized the attention scores. The information from the protein is merged with the molecule via a product in Eq. 1.
is a scaling factor, and the softmax function is used to normalized the attention scores. The information from the protein is merged with the molecule via a product in Eq. 1.
Fig. 2.

Detailed structure of LT layer. Cross attention block in decoder layer captures protein features and integrates them with molecule features.
The self-attention in both encoder and decoder is adopted, the same as the original attention block in (24) to learn the dependence within the sequences. Multihead attention mechanism is used to perform different linear projections on the queries, keys, and values for h times, allowing the model to attend information from various chemical perspectives
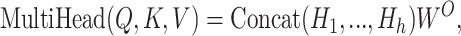 |
(2) |
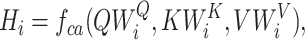 |
(3) |
where WO, 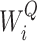 ,
, 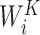 ,
, 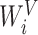 are parameters for linear projections, i = 1, …, h. Since the attention mechanism itself lacks information about the order of the sequence, we similarly add the additional positional encoding (PE) (24) to the embedding layer as follows:
are parameters for linear projections, i = 1, …, h. Since the attention mechanism itself lacks information about the order of the sequence, we similarly add the additional positional encoding (PE) (24) to the embedding layer as follows:
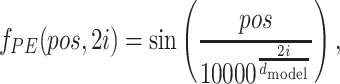 |
(4) |
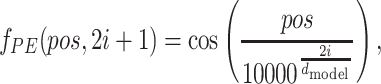 |
(5) |
where pos denotes the position of a symbol in protein sequence or SMILES, i is the dimension, and dmodel is the dimension of the embeddings. That is, each dimension of the PE corresponds to a sinusoid. Detailed information about PE is included in Supplementary Material (Fig. S4).
MCTS for molecular generation
The goal of AlphaDrug is to search the space of all possible molecules for drug candidates, such that the drug candidates can be properly docked into the protein target in an exclusive way. Although we may use LT network prediction to construct the ligand in a greedy manner, it is easy to stuck in a local optimum because the searching space is so huge. We further model the ligand growing procedure in MCTS, which is a heuristic search algorithm widely used in sequential decision-making problems.
As shown in Fig. 1, each node in the tree represents a SMILES symbol of the molecule and the path from the first root node a1 to the current one aτ forms an intermediate molecule a1a2⋅⋅⋅aτ.
For each step in the de novo molecular generation process, simulations are conducted S times and the child node with maximum number of visits is chosen as the new root node aτ + 1. As given in Fig. 3, each simulation consists of four steps: select, expand, rollout, and backup.
Fig. 3.

Detailed process of an MCTS simulation. A simulation consists of four steps: select, expand, rollout, and backup.
Select
Each simulation starts from the current root node aτ and selects its successive child node until a leaf node 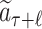 is reached after ℓ selections. For each selection t ∈ [1, ℓ], the node is obtained by the action of choosing symbol
is reached after ℓ selections. For each selection t ∈ [1, ℓ], the node is obtained by the action of choosing symbol 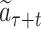 according to a variant form of the PUCT (Predictor with Upper Confidence bounds applied to Trees)algorithm (27)
according to a variant form of the PUCT (Predictor with Upper Confidence bounds applied to Trees)algorithm (27)
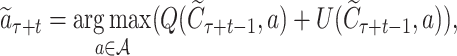 |
(6) |
where 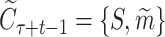 is the context with the target protein S and the current simulated intermediate molecule
is the context with the target protein S and the current simulated intermediate molecule 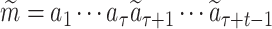 , and
, and  is the legal action space under the context, i.e. the SMILES vocabulary of molecules.
is the legal action space under the context, i.e. the SMILES vocabulary of molecules. 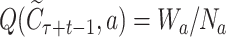 represents the average rewards selecting the symbol a in the context
represents the average rewards selecting the symbol a in the context 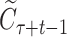 , with Wa and Na being the accumulated reward and visiting times, respectively, for the node;
, with Wa and Na being the accumulated reward and visiting times, respectively, for the node; 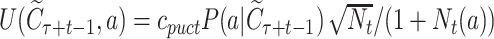 , and cpuct is a constant that controls the degree of exploration. The accumulated reward is computed as the sum of the scores docking the set
, and cpuct is a constant that controls the degree of exploration. The accumulated reward is computed as the sum of the scores docking the set 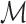 of ligands into the target protein S, i.e.
of ligands into the target protein S, i.e. 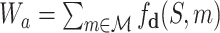 , where
, where 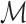 collects the valid simulation paths passing through the node, and fd is implemented by the SMINA program (28). To control the scale of the SMINA docking values, we normalize the range of
collects the valid simulation paths passing through the node, and fd is implemented by the SMINA program (28). To control the scale of the SMINA docking values, we normalize the range of 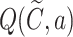 into the interval [0, 1], using the maximum docking values observed in the search tree as follows:
into the interval [0, 1], using the maximum docking values observed in the search tree as follows:
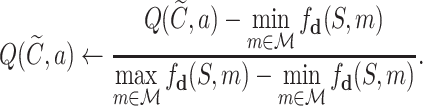 |
(7) |
This search strategy guides MCTS to initially prefer to visit the nodes of SMILES symbols with high prior probability and low number of visits, but asymptotically tend to visit the symbols that are potentially to produce a molecule of strong binding affinity with the target protein.
Expand
Given a selected leaf node 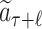 , the probability
, the probability 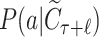 for each expandable symbol
for each expandable symbol 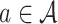 is computed by the LT network. The children nodes of
is computed by the LT network. The children nodes of 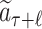 are added to the tree and each node is initialized to
are added to the tree and each node is initialized to 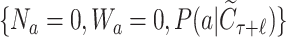 .
.
Rollout
The value, i.e. the expected return of accumulated rewards, of the reached leaf node 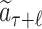 is evaluated by a fast rollout. Each SMILES symbol is selected in a greedy manner according to the probability computed by LT until a terminal symbol
is evaluated by a fast rollout. Each SMILES symbol is selected in a greedy manner according to the probability computed by LT until a terminal symbol 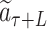 is generated or the tree reaches a maximum depth. The path from the initial symbol to the terminal forms a complete molecule
is generated or the tree reaches a maximum depth. The path from the initial symbol to the terminal forms a complete molecule 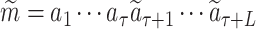 . RDKit (29) is used to validate the legitimacy of molecule. If the molecule is invalid, the value of node xl is set to be
. RDKit (29) is used to validate the legitimacy of molecule. If the molecule is invalid, the value of node xl is set to be 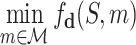 . Otherwise, a docking score is computed by the SMINA program
. Otherwise, a docking score is computed by the SMINA program 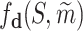 as the value of the leaf node
as the value of the leaf node 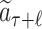 .
.
Backup
For each selection t ∈ [1, ℓ], the nodes’ statistics are updated by adding the rollout value of 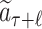 to the cumulative reward W and increasing the visiting times N by 1.
to the cumulative reward W and increasing the visiting times N by 1.
MCTS was firstly adopted in ChemTS (20) for molecular generations. Our method differs from ChemTS in several aspects. First of all, ChemTS was not developed for generating molecules that can be bound with the target protein, but for optimizing the octanol–water partition coefficient logP, synthetic accessibility (SA), and penalizing unrealistically large rings. Second, we model the molecular generation by MCTS in a more efficient and accurate way. (a) As illustrated in Fig. 4, at rollout step of AlphaDrug, only the docking score of the molecule, which is generated by the fast rollout is set as the value of the node 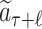 . But in ChemTS, the value of the node
. But in ChemTS, the value of the node 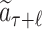 is evaluated by uniformly accumulating values of each child node, which indicates the transition probabilities from the node
is evaluated by uniformly accumulating values of each child node, which indicates the transition probabilities from the node 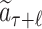 to all its children nodes are the same. The value computed by this strategy may mislead the search process. (b) When the simulation times S reach the preset integer, the most visited child node is selected by AlphaDrug as new root, and the old root is kept in memory. In contrast, the root node in ChemTS is fixed at a1 for all simulations. AlphaDrug is more efficient than ChemTS, because it balances well between the searching width and depth by pruning the unlikely branches in the selection step, while ChemTS wastes too much effort on the first part of the molecule and cares less about the later part. (c) It should be noted that AlphaDrug makes use of protein–ligand interaction patterns by the docking evaluation of the rollout molecule into the target. ChemTS does not take any information about the protein target into account to guide the molecular generation, and thus in general the generated ligands cannot be properly docked into a specified target.
to all its children nodes are the same. The value computed by this strategy may mislead the search process. (b) When the simulation times S reach the preset integer, the most visited child node is selected by AlphaDrug as new root, and the old root is kept in memory. In contrast, the root node in ChemTS is fixed at a1 for all simulations. AlphaDrug is more efficient than ChemTS, because it balances well between the searching width and depth by pruning the unlikely branches in the selection step, while ChemTS wastes too much effort on the first part of the molecule and cares less about the later part. (c) It should be noted that AlphaDrug makes use of protein–ligand interaction patterns by the docking evaluation of the rollout molecule into the target. ChemTS does not take any information about the protein target into account to guide the molecular generation, and thus in general the generated ligands cannot be properly docked into a specified target.
Fig. 4.
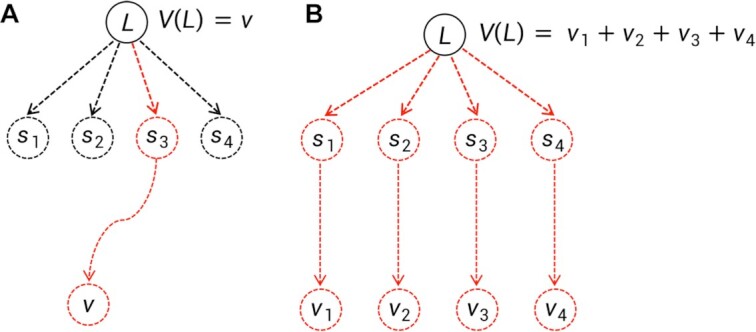
Differences between AlphaDrug and ChemTS at rollout step. (A) Our rollout. (B) Rollout in ChemTS.
Loss function
We train the LT in a supervised way on the available ligand–protein pairs in the public database  . For each pair
. For each pair 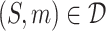 , S denotes the target protein while m is the molecule in the SMILES format
, S denotes the target protein while m is the molecule in the SMILES format 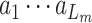 . We construct the a sequence of contexts Cτ = {S, a1⋅⋅⋅aτ}, τ = 1, …, Lm, and feed them into LT network to compute the probability P (a|Cτ) of predicting the next symbol aτ + 1. The objective is to minimize the following cross-entropy loss:
. We construct the a sequence of contexts Cτ = {S, a1⋅⋅⋅aτ}, τ = 1, …, Lm, and feed them into LT network to compute the probability P (a|Cτ) of predicting the next symbol aτ + 1. The objective is to minimize the following cross-entropy loss:
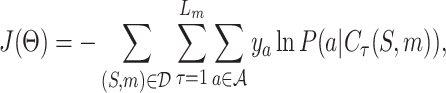 |
(8) |
where Θ denotes the set of parameters in the LT network and 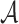 is the vocabulary set of ligand SMILES. The binary label ya indicates whether a is the next symbol.
is the vocabulary set of ligand SMILES. The binary label ya indicates whether a is the next symbol.
Experiments
Datasets
BindingDB (30) is a public database of protein–ligand pairs where the proteins are considered to be drug–targets, and the ligands are small, drug-like molecules. We downloaded the latest version that contains over 2.3 million binding records. Protein–ligand pairs were filtered from the crude database using the following criteria as similarly in (17).
Proteins only belong to “Homo sapiens”.
The IC50 value is less than 100 nM; if the IC50 is missing, then Kd is less than 100 nM; if both are missing, then EC50 is less than 100 nM.
The record has a chemical identifer (PubChem CID).
The record has SMILES representations.
The molecular weight is less than 1000 Da.
The record has a protein identifer (Uniprot ID).
Protein sequence length is greater than 79 and lower than 1001.
Number of protein chains in target equals to 1 (>1 implies a multichain complex).
After filtering, we built a dataset with 239,455 protein–ligand pairs among which there are 981 unique protein sequences. Then, we utilize Mmseqs2 (31) to yield sequence clusters at a sequence identity level of 30%, which is similarly used to build the Big Fantastic Database in AlphaFold2 (32) and randomly select 25 clusters as test set. We select 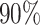 of the remaining clusters as the training set and the other
of the remaining clusters as the training set and the other 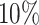 for validation. In summary, our dataset contains 192,712 protein–ligand pairs for training, 17,049 pairs for validation, and 100 proteins from the 25 clusters for testing. Since MCTS is activated in the testing and the 3D structure of the testing protein is required for the docking process, we download the corresponding 3D protein structures from PDB bind website (http://www.pdbbind.org.cn/index.php). We utilize the Clustal Omega tool (33) provided by EMBL-EBI’s website (https://www.ebi.ac.uk/Tools/msa/clustalo/) to compute similarities between different proteins. The distributions of pairwise sequence similarities between different subsets are given in Fig. 5. All of the protein sequences share less than
for validation. In summary, our dataset contains 192,712 protein–ligand pairs for training, 17,049 pairs for validation, and 100 proteins from the 25 clusters for testing. Since MCTS is activated in the testing and the 3D structure of the testing protein is required for the docking process, we download the corresponding 3D protein structures from PDB bind website (http://www.pdbbind.org.cn/index.php). We utilize the Clustal Omega tool (33) provided by EMBL-EBI’s website (https://www.ebi.ac.uk/Tools/msa/clustalo/) to compute similarities between different proteins. The distributions of pairwise sequence similarities between different subsets are given in Fig. 5. All of the protein sequences share less than 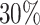 similarity. Detailed information about Similarity Matrix of Proteins is included in Supplementary Material (Fig. S3).
similarity. Detailed information about Similarity Matrix of Proteins is included in Supplementary Material (Fig. S3).
Fig. 5.
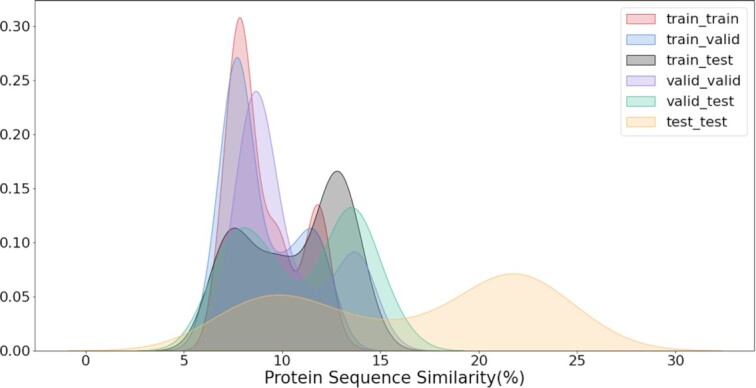
Kernel density estimates for the distributions of protein sequence similarity between different subsets.
Training and validation instances are constructed by shifting right the input ligand string as the ground-truth output. For convenience, we add a start symbol “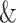 ” in front of all protein sequences and ligand strings and a terminal symbol “$” at the end.
” in front of all protein sequences and ligand strings and a terminal symbol “$” at the end.
Evaluation metrics
We use the following criteria to evaluate the generated molecules using the testing proteins as targets. It is noted that the docking score is the main metric because the task of this paper requires the generated molecules to bind with the protein target well.
Docking score. Generally, drugs should be docked well to a binding site of its protein target. Binding energy is regarded as a general indicator to describe the binding affinity between molecules and proteins. SMINA (28) is a free, widely used program to compute the binding affinity. We use the negative value of the output by SMINA as the docking score. The higher the docking score, the better the molecule is docked into the protein.
- Uniqueness. Drug design models should be able to generate different molecules according to different proteins. The higher the uniqueness value is, the more sensitive the model is to the protein. This metric is computed as follows:
where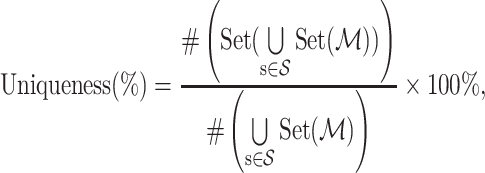
(9) 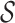 indicates the set of testing proteins,
indicates the set of testing proteins, 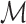 denotes the collection of molecules generated by a method for the target protein
denotes the collection of molecules generated by a method for the target protein 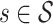 ,
, 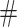 counts the number of molecules, and Set is an operator to remove the repetitive molecules.
counts the number of molecules, and Set is an operator to remove the repetitive molecules. LogP (the water–octanol partition coefficient). Large LogP value indicates the substance is lipophilic, while small LogP means it is easy to dissolve in water. According to Ghose filter (34), the LogP value of a drug should range from −0.4 to + 5.6.
Quantitative estimate of drug-likeness (QED). The score ranges from 0 to 1. A higher QED indicates that the molecule is more likely to be a potential drug-like compound, with the desired molecular properties such as hydrogen bond acceptor, hydrogen bond donor, and polar molecular surface area.
SA score. Low SA score is preferred such that the molecules are easy to synthesize.
Natural products likeness(NP-likeness). Natural products play an important role in the history of drug discovery. Many drugs are natural products and their derivatives. The higher the score is, the more likely the molecule is to be a natural product.
Implementation details
We include the existing methods, i.e. original Transformer with Beam Search (T+BS) in (17), LiGANN (16), SBMolGen (23), which is developed from ChemTS (20) for target-specific molecular generation, and SBDD-3D (22), for comparisons. In (17), when each time a symbol is decoded, the candidates with top K highest probabilities computed by the transformer are selected, where K is the beam size. Here, we use K = 10 as in (17) to generate 10 ligand candidates for each testing protein, and denote it as “T+BS10”. Moreover, we implement LT with BS (LT+BS10) as a baseline to compare LT with the original transformer. We directly use the web-based application (https://www.playmolecule.org/LiGANN) from the original authors to implement LiGANN on the testing proteins. For SBDD-3D, we utilize the trained model published by the authors. For a fair comparison, we both collect 10 generated molecules for each testing protein from LiGANN and SBDD-3D.
In AlphaDrug, we set the constant cpuct in Eq. 6 to 1.5 for balancing between exploitation and exploration. The simulation times S affect the performance and computational burden of MCTS. For a reasonable trade-off, we set S = 50. We provide two versions of AlphaDrug for molecular generation on a given testing protein. First, we execute MCTS by selecting the next symbol with the maximum visited times of simulations, and denote this version as AlphaDrug(max). Second, we execute MCTS by randomly selecting the next symbol according to the frequency of visited times, and denote this version as AlphaDrug(freq). For fair comparisons, we also use AlphaDrug to generate 10 molecules for each testing protein.
Results
The average scores of the metrics on all testing proteins are reported in Table 1. Ten candidate molecules are generated for each testing protein. All 1000 candidates are evaluated by the metrics, and the scores are averaged for an overall comparison. The “decoys” in the first row is implemented by randomly selecting 10 compounds from ZINC database (https://zinc.docking.org/), a free database of commercially available compounds for virtual screening of drugs. The “known ligands” in the second row indicates the original molecules binding to proteins in the database.
Table 1.
Average scores of the metrics of all generated molecules on 100 testing proteins.
| Methods | Docking | Uniqueness | LogP | QED | SA | NP |
|---|---|---|---|---|---|---|
| Decoys | 7.3 | - | 2.4 | 0.8 | 2.4 | −1.2 |
| Known ligands | 9.8 | - | 2.2 | 0.5 | 3.3 | −1.0 |
| LiGANN (16) | 6.7 | 94.7% | 2.9 | 0.6 | 3.0 | −1.1 |
| SBMolGen (23) | 7.7 | 100% | 2.6 | 0.7 | 2.8 | −1.2 |
| SBDD-3D (22) | 7.7 | 99.3% | 1.5 | 0.6 | 4.0 | 0.3 |
| T + BS10 (17) | 8.5 | 90.6% | 3.8 | 0.5 | 2.8 | −0.8 |
| LT + BS10 | 8.5 | 98.1% | 4.0 | 0.5 | 2.7 | −1.0 |
| AlphaDrug (freq) | 10.8 | 99.5% | 4.9 | 0.4 | 2.9 | −1.0 |
| AlphaDrug (max) | 11.6 | 100% | 5.2 | 0.4 | 2.7 | −0.8 |
We observe that, in terms of docking score, the decoys baseline is obviously worse than most other methods because the randomly selected molecules are usually not good candidates for a specific target. Notice that although LiGANN takes the 3D structure information of the protein target into account, its docking score is even lower than the decoys baseline. LiGANN would produce some molecules to have relatively small molecular mass (usually containing only one ring and quite short side chains). These molecules are too small to bind well to the protein, dragging down the docking scores. The results of SBMolGen is even worse than the “T+BS10” (17) using BS, because the context of the target is not properly learned by SBMolGen and the adopted MCTS from ChemTS is not efficient, as illustrated in Fig. 4. Although SBDD-3D has used 3D coordinates of atoms of protein pockets as chemical contexts to coach the model to generate molecules, the resulted docking score is still far from satisfactory. SBDD-3D, which also used BS, is even inferior to “LT+BS10”. It seems that the sequence-based representation learning by the vanilla transformer or LT may be more effective than the context learning on the 3D protein by SBDD-3D. It is noted that the uniqueness percentage is further improved when replacing the vanilla transformer with LT based on BS.
The average docking scores in Table 1 demonstrate that AlphaDrug outperforms other methods, and AlphaDrug (max) achieves the highest average docking score. AlphaDrug (max) may output the same molecule for each protein because its decision-making of next symbol in MCTS is deterministic according to the maximum visited times and the fast rollout for docking values is made by choosing the child nodes of the highest probability. AlphaDrug (max) is better than AlphaDrug (freq) in terms of the average docking score, but AlphaDrug (freq) allows to search more paths for top candidates. The frequency-based random decision-making of next symbol gives more chances to explore better possibilities, at the cost of lowering the mean performance.
We further investigate the distributions of docking scores between the generated molecules and the target protein. As given in Fig. 6, all distributions are approximately unimodal in bell-shapes, and they are consistent with the observations from Table 1. We assess the statistical significance of the improvement by AlphaDrug (freq) over the state-of-the-art methods, i.e. “T+BS10” (17), SBDD-3D (22), and LiGANN (16). We compute P-values of two tailed t-test for all testing proteins and report them in Table 2. The results suggest that the improvements by AlphaDrug (freq) are all significant (P < 0.01) for all targets, verifying the generalization performance of AlphaDrug for generating promising drug candidates.
Fig. 6.

Box plots of docking scores between the target proteins and their corresponding generated molecules by different methods.
Table 2.
P-values of two tailed t-test for molecules generated by different methods for all testing proteins.
Finally, we check the physicochemical properties of the generated ligand candidates. The results in Table 1 indicate that the produced molecules by all methods have comparable QED, SA, NP-likeness scores, and suitable range of LogP for drugs. Detailed distributions of each property are in Supplementary Material (Figs. S1 and S2).
Case study
According to the docking scores between molecules generated by AlphaDrug and known ligands in the testing set, we find that 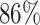 of the new molecules have higher scores than the corresponding known ligands. In this section, we will visualize examples of generated molecules of top binding affinities and study their details. We consider the following three target proteins:
of the new molecules have higher scores than the corresponding known ligands. In this section, we will visualize examples of generated molecules of top binding affinities and study their details. We consider the following three target proteins:
Protein 3gcs is a human P38 MAP kinase in complex with Sorafenib (a novel multitargeted oral drug for the treatment of tumors).
Protein 3eig is a crystal structure of a methotrexate-resistant mutant of human dihydrofolate reductase. Its known ligand is MTX, the main drug for the treatment of rheumatoid arthritis.
Protein 4o28 is a structural basis for resistance to diverse classes of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors, a bottleneck enzyme that plays a key role in recycling nicotinamide to maintain the adequate nicotinamide adenine dinucleotide (NAD+) level inside the cell (35).
We consider two groups of generated molecules based on the similarity with the known ligands. We compute the Tanimoto coefficients implemented by RDKit (29) between the molecules and known ligands. Tanimoto coefficient is a popular similarity measure for comparing chemical structures represented by means of fingerprints (36). In the first group, we select three molecules with high Tanimoto coefficients to the known ligands and visualize them in the third column of Fig. 7. To facilitate the comparisons, we highlight in green the maximum common substructure shared between molecules and known ligands. For protein 3gcs, the generated molecule contains 1-ethyl-4-methyltetrahydroquinoxaline instead of N-methylpicolinamide in the known ligand. For protein 3eig, the generated one can be derived from the known ligand by replacing the glutamic acid part with aspartic acid part. The only difference between them is that the side chain of glutamic acid has one more methylene group than that of aspartic acid. For protein 4o28, the (R)-3-hydroxy-N-benzoyl-pyrrolidine is generated instead of imidazo[1,2-a]pyridine in left part, and the carboxyl group instead of 3,5-difluorophenylsulfonyl group in right part. The results indicate that our method is reliable and consistent with known experimental evidence, and is able to suggest possible modifications to optimize the known ligands.
Fig. 7.

Protein pockets and co-crystal structures of protein with generated molecule. Shared maximum common substructure are colored in green or blue. The numbers under molecular pictures are docking scores computed by SMINA.
In the second group, we pick three novel top-scored molecules, which are very different from the known ligands. They are listed in the last column of Fig. 7, where the common part is highlighted in blue. For the three proteins, the common parts are only an aniline, or a pyrimidine-2,4-diamine, or a phenylmethanamine in both molecules. Notice that the two groups of newly designed molecules, all have higher docking scores than the known ligands. Therefore, AlphaDrug is a promising method for de novo drug design.
The effect of MCTS
MCTS is a tree search process guided by a trade-off between exploration and exploitation via Eq. 6, making the search to focus on high-reward nodes.
We take “LT + Greedy”, which is realized via a greedy policy based on the probability computed by LT, as a baseline for evaluating the improvements on the docking score metric brought by MCTS. The (average) docking score metric is computed by docking the generated molecules into the target protein. The results are reported here in Table 3. Detailed trace plots of docking scores can be found in Supplementary Material (Fig. S5). We observe that MCTS brings a significant increment of the docking score metric under two settings of different simulation times (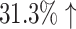 and
and 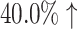 , respectively). It is expected that the version of AlphaDrug (max) with 50 MCTS simulations performs better than the one with 10 simulations, because it consumes more computational resources.
, respectively). It is expected that the version of AlphaDrug (max) with 50 MCTS simulations performs better than the one with 10 simulations, because it consumes more computational resources.
Table 3.
Statistics and analysis of docking calculations and efficiency of our methods.
| Method | Molecular length | Docking times [L × S] | Score ↑ | Validness ↑ |
|---|---|---|---|---|
| LT + Greedy (baseline) | 53.96 | - | 8.3 | 80.0% |
| AlphaDrug (max)* | 55.88 | 104.8 [559.0] (81.3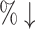 ) ) |
10.9 (31.3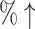 ) ) |
100% (20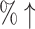 ) ) |
| AlphaDrug (max) | 56.54 | 394.3 [2825.0] (86.0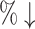 ) ) |
11.6 (40.0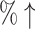 ) ) |
100% (20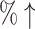 ) ) |
All values in this table are average of results on 100 testing proteins. Here, “*” denotes the MCTS simulation times S = 10, while S = 50 are set as default. “Validness” indicates the ratio of generated molecules that are chemically legal. Values in the third column, e.g. 104.8 and 559.0, are actual docking times and theoretical docking times, respectively.
We count the number of docking calculations in our method and evaluate the efficiency of our approach. In MCTS, considerable dockings are carried out for exploring the huge unknown chemical space where one docking process can even take up to 30 seconds on average. Theoretically, to generate a molecule, which has a length of L, our approach needs to utilize SMINA for L × S times, where S is the MCTS simulation times. However, in many situations, docking values of the same molecule–protein pair are computed due to the use of “LT + Greedy” at the rollout step. To improve the efficiency of MCTS, we keep a docking table 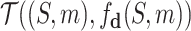 in memory. Each time a new molecule protein pair (S′, m′) is produced at the rollout step, a fresh record (S′, m′), fd (S′, m′) will be added into the docking table. As in Table 3, the actual docking times in the two AlphaDrug versions are much smaller than the theoretical docking times in square brackets (
in memory. Each time a new molecule protein pair (S′, m′) is produced at the rollout step, a fresh record (S′, m′), fd (S′, m′) will be added into the docking table. As in Table 3, the actual docking times in the two AlphaDrug versions are much smaller than the theoretical docking times in square brackets (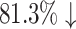 and
and 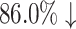 , respectively).
, respectively).
Besides, we also compare the performance of MCTS with other regular search under the same number of docking times. Here, BS method is chosen as a baseline from the regular search methods. All methods are constrained to use the same number of docking times, and use the same LT network for predicting the probability of selecting the next symbol. Specifically, we consider MCTS under three different search settings, i.e. S = 10, 50, 500, where S denotes the number of simulations in MCTS. For each S, the number of actual docking times taken in MCTS is counted, and BS is constrained to use exactly the same docking times. Table 4 reports the average over the docking scores of all generated molecules, and the average (in parenthesis) over the top-1 score among all docking scores for each of 100 testing proteins, as well as the t-test P-values between BS and MCTS.
Table 4.
Average docking scores by different search methods on 100 testing proteins.
| Docking times N \ Method | BS | MCTS | P-value |
|---|---|---|---|
| N = 105; S = 10 | 8.4 (10.9) | 10.9 (11.5) | 1.8 × 10−34 (4.5 × 10−3) |
| N = 394; S = 50 | 8.3 (11.4) | 11.6 (12.2) | 1.4 × 10−31 (1.8 × 10−3) |
| N = 1345; S = 500 | 8.4 (11.9) | 12.4 (13.2) | 2.2 × 10−39 (8.2 × 10−6) |
All methods are constrained to use the same number N of docking times, and use the same LT network. The average is computed in two ways, i.e. over the docking scores of all generated molecules or (in parenthesis) over the top-1 score among all docking scores for each testing protein. P-values are computed using t-test on the results between BS and MCTS.
It is observed that MCTS outperforms BS under the same number of docking times, in terms of the average docking scores not only on all molecules or but also on the top-1 molecules for each testing protein. The increments of MCTS over BS are all significant as the corresponding P-values are smaller than 0.05. MCTS balances well between exploitation and exploration in the selection step by the criterion in Eq. 6, which takes into account both LT’s predicted probability and the SMINA’s docking score. Moreover, it is expected that the performances of both methods become better as the number of docking times increases, where BS achieves higher average top-1 scores and MCTS improves in terms of both average docking scores.
The effect of protein sequence as input
We perform an experimental analysis on the role of the protein sequence jointly as input, and the results are reported in Table 5. Here, we train a transformer encoder, denoted as “TE”, to learn molecular growth strategies, without the input of protein sequences. Note that “TE” is trained similarly like “T” (vanilla transformer) and “LT” and shares the same training hyperparameters with the encoder of “T” and “LT”. The difference of “TE” from “T” and “LT” is that “TE” does not have protein sequence as input, while the latter two have. More details of the training hyperparameters can be found in Supplementary Material (Table S1). We introduce two metrics, Uniqueness and Score per Symbol (denoted as SpS) to demonstrate the effect of the protein sequence in inputs. Uniqueness is computed in Eq. 9 and SpS is given below
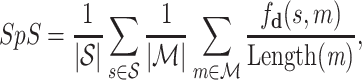 |
(10) |
where 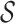 indicates the set of 100 testing proteins,
indicates the set of 100 testing proteins, 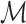 denotes the set of molecules generated by a method for the target protein
denotes the set of molecules generated by a method for the target protein 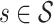 , and fd (s, m) evaluates the docking score of the generated molecule m with the target protein s.
, and fd (s, m) evaluates the docking score of the generated molecule m with the target protein s.
Table 5.
An ablation study on analysing how well input of the protein sequence information would help MCTS find high-affinity binders.
| Group | Method | Uniqueness ↑ | SpS ↑ | Molecular length |
|---|---|---|---|---|
| 1 | TE + MCTS* (baseline) | 65.0% | 0.1771 | 66.15 |
| T + MCTS* | 98.0% (33%↑) | 0.1959 (10.62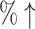 ) ) |
59.33 | |
| LT + MCTS* | 100.0% (35%↑) |
0.2025 (14.34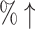 ) )
|
55.88 | |
| 2 | TE + MCTS (baseline) | 81.0% | 0.1926 | 62.93 |
| T + MCTS | 98.0% (17%↑) | 0.2149 (11.58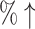 ) ) |
55.63 | |
| LT + MCTS | 100.0% (19%↑) |
0.2159 (12.10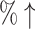 ) )
|
56.54 |
Here, “*” denotes the MCTS simulation times S = 10, while S = 50 are set as default. “TE”, “T” and “LT” denote a transformer encoder, a vanilla transformer, and our LT, respectively. In MCTS, we select the next symbol (the new root node) with the maximum visited times of simulations, which is the same with AlphaDrug (max). “SpS” denotes score per symbol.
The Uniqueness metric indicates the sensitivity of a model to the target protein. The higher the uniqueness value is, the more sensitive the model is to the protein. Besides, within a certain suitable length, the longer molecules intend to have higher scores due to more explorations in MCTS. The SpS metric is used to get rid of the influence of the length factor on the docking score.
We consider two groups of experimental settings, i.e. Group 1 and Group 2 for the MCTS simulation times being 10 and 50, respectively. The results of both groups in Table 5 demonstrate that the baseline (i.e. “TE + MCTS”) is improved in terms of Uniqueness by 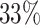 and
and 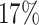 , respectively, when the protein target sequence is jointly fed into the model of “T + MCTS”. The improvement is also observed in terms of SpS, i.e. by
, respectively, when the protein target sequence is jointly fed into the model of “T + MCTS”. The improvement is also observed in terms of SpS, i.e. by 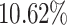 and
and 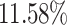 , respectively. These improvements indicate that it is better to use the protein sequence as an additional input, because our method is developed to capture the conditional distribution of the high-binding affinity molecules given the protein target.
, respectively. These improvements indicate that it is better to use the protein sequence as an additional input, because our method is developed to capture the conditional distribution of the high-binding affinity molecules given the protein target.
The effect of LT
We also observe slight improvements in Table 5 on Uniqueness and SpS in both groups when replacing the vanilla transformer (T) with our LT. The improvement becomes marginal when MCTS is growing more powerful in explorations with five times more simulations, i.e. from Group 1 to Group 2. The hierarchical skip connections employed by LT are effective in sending more information of the protein target to the molecular decoder.
According to Table 3, “LT + MCTS*” (i.e. AlphaDrug*) only takes about a quarter of the time of “LT + MCTS” (i.e. AlphaDrug) to generate molecules (52 and 197 minutes per protein on average, respectively), but their performances are comparably similar. Thus, “LT + MCTS*” is preferred in situations where computing resources are scarce.
Conclusion and discussion
We have proposed a deep learning model, AlphaDrug, for de novo molecular generation according to given protein targets. The model is featured by an effective representation learning on the protein target information, and an efficient heuristic search by MCTS to reduce the computational complexity due to the huge search space of all possible drug molecules. Specifically, we devised a variant transformer with a hierarchy of skip connections from protein encoder to molecule decoder for enhanced feature transfer. Molecular generation is modeled as searching optimal paths in MCTS, and the searching process is properly guided by not only the predicted probabilities of growing the next symbol of the molecule by the variant transformer, but also a value function of docking score. Experiments and ablation studies verify the advantages of our method over the existing methods. AlphaDrug is a promising model to speed up the drug discovery process.
There is still room to improve AlphaDrug’s performance for real world drug design applications. First, the deep representation learning of AlphaDrug is made on sequence data, i.e. SMILES strings of molecules and amino acid sequences of proteins. Although AlphaDrug has been demonstrated to be even more efficient than the existing methods that used 3D coordinates of the proteins, the 3D structure of the binding pocket is definitely a critical context to guide the molecular generation. Recent advances about 3D structure deep learning methods, such as 3D convolutional neural network, geometric deep learning, and so on, may be employed to take 3D information into account. Second, the value function of AlphaDrug is currently implemented by calling an external, fixed docking program, i.e. SMINA, which is computational expensive when the number of calls grows large in MCTS simulations. Building a learnable end-to-end deep value network is a good direction to go, because joint learning of both policy and value would benefit from each other and adapt better to the data. Third, it is not efficient to execute fast rollout in every MCTS simulations in AlphaDrug. If we want to tackle this limitation, we need to evaluate the potential binding affinity without growing a complete, valid molecule, which is extremely challenging. To summarize, it deserves more future work to improve AlphaDrug, which is potentially powerful in real applications.
Previous Presentation
The results were not presented anywhere before.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the anonymous reviewers for their valuable suggestions.
Notes
Competing Interest: The authors declare no competing interest.
Contributor Information
Hao Qian, Department of Computer Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China; Centre for Cognitive Machines and Computational Health (CMaCH), Shanghai Jiao Tong University, Shanghai 200240, China.
Cheng Lin, Department of Computer Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China; Centre for Cognitive Machines and Computational Health (CMaCH), Shanghai Jiao Tong University, Shanghai 200240, China.
Dengwei Zhao, Department of Computer Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China; Centre for Cognitive Machines and Computational Health (CMaCH), Shanghai Jiao Tong University, Shanghai 200240, China.
Shikui Tu, Department of Computer Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China; Centre for Cognitive Machines and Computational Health (CMaCH), Shanghai Jiao Tong University, Shanghai 200240, China.
Lei Xu, Department of Computer Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China; Centre for Cognitive Machines and Computational Health (CMaCH), Shanghai Jiao Tong University, Shanghai 200240, China.
Funding
This work was supported by The National Key Research and Development Program (2018AAA0100700) of the Ministry of Science and Technology of China, the National Natural Science Foundation of China (62172273), and Shanghai Municipal Science and Technology Major Project (2021SHZDZX0102).
Authors' Contributions
S.T. and L.X. conceived the project; H.Q. and C.L. collected the data; H.Q., D.Z., and S.T. developed the model; H.Q. conducted the experiments; H.Q. and S.T. wrote and reviewed the manuscript.
Preprints
This article has not been published as a preprint anywhere.
Data Availability
The data and source codes are available in https://github.com/CMACH508/AlphaDrug.
References
- 1. DiMasi JA, Grabowski HG, Hansen RW. 2016. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 47:20–33. [DOI] [PubMed] [Google Scholar]
- 2. Wouters OJ, McKee M, Luyten J. 2020. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA. 323(9):844–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paul SM, et al. 2010. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Disc. 9(3):203–214. [DOI] [PubMed] [Google Scholar]
- 4. Polishchuk PG, Madzhidov TI, Varnek A. 2013. Estimation of the size of drug-like chemical space based on GDB-17 data. J Comput Aided Mol Des. 27(8):675–679. [DOI] [PubMed] [Google Scholar]
- 5. Zhang L, Han J, Wang H, Car R, Weinan E. 2018. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys Rev Lett. 120(14):143001. [DOI] [PubMed] [Google Scholar]
- 6. Casalino L, et al. 2021. AI-driven multiscale simulations illuminate mechanisms of SARS-CoV-2 spike dynamics. Int J High Perform Comput Appl. 35:432–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bennett WD, et al. 2020. Predicting small molecule transfer free energies by combining molecular dynamics simulations and deep learning. J Chem Inf Model. 60(11):5375–5381. [DOI] [PubMed] [Google Scholar]
- 8. Jiang S, Balaprakash P. Graph neural network architecture search for molecular property prediction. Los Alamitos, CA, USA,2020. In: Jermaine C, Xiong L, Wu X, editors. 2020 IEEE International conference on big data (big data). IEEE. p. 1346–1353. [Google Scholar]
- 9. Zhu F, Zhang X, Allen JE, Jones D, Lightstone FC. 2020. Binding affinity prediction by pairwise function based on neural network. J Chem Inf Model. 60(6):2766–2772. [DOI] [PubMed] [Google Scholar]
- 10. Li S, et al. 2020. MONN: a multi-objective neural network for predicting compound-protein interactions and affinities. Cell Syst. 10(4):308–322. [Google Scholar]
- 11. Jin W, Barzilay R, Jaakkola T. 2018. Junction tree variational autoencoder for molecular graph generation. Paper presented at: 35th International Conference on Machine Learning (ICML); Stockholm, Sweden. PMLR. p. 2323–2332. [Google Scholar]
- 12. Kusner MJ, Paige B, Hernández-Lobato JM. 2017. Grammar variational autoencoder. Paper presented at: 34th International Conference on Machine Learning (ICML); Sydney, Australia. PMLR. p. 1945–1954. [Google Scholar]
- 13. Ma T, Chen J, Xiao C. 2018. Constrained generation of semantically valid graphs via regularizing variational autoencoders. arXiv:180902630. 10.48550/arXiv.1809.02630, preprint: not peer reviewed. [DOI] [Google Scholar]
- 14. Shi C, et al. 2020. GraphAF: a flow-based autoregressive model for molecular graph generation. arXiv:200109382. 10.48550/arXiv.2001.09382, preprint: not peer reviewed. [DOI] [Google Scholar]
- 15. You J, Liu B, Ying R, Pande V, Leskovec J. 2018. Graph convolutional policy network for goal-directed molecular graph generation. arXiv:180602473. 10.48550/arXiv.1806.02473, preprint: not peer reviewed. [DOI] [Google Scholar]
- 16. Skalic M, Sabbadin D, Sattarov B, Sciabola S, De Fabritiis G. 2019. From target to drug: generative modeling for the multimodal structure-based ligand design. Mol Pharm. 16(10):4282–4291. [DOI] [PubMed] [Google Scholar]
- 17. Grechishnikova D. 2021. Transformer neural network for protein-specific de novo drug generation as a machine translation problem. Sci Rep. 11(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Olivecrona M, Blaschke T, Engkvist O, Chen H. 2017. Molecular de-novo design through deep reinforcement learning. J Cheminformatics. 9(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Popova M, Isayev O, Tropsha A. 2018. Deep reinforcement learning for de novo drug design. Sci Adv. 4(7):eaap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang X, Zhang J, Yoshizoe K, Terayama K, Tsuda K. 2017. ChemTS: an efficient python library for de novo molecular generation. Sci Technol Adv Mate. 18(1):972–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu M, Ran T, Chen H. 2021. De novo molecule design through the molecular generative model conditioned by 3D information of protein binding sites. J Chem Inf Model. 61(7):3240–3254. [DOI] [PubMed] [Google Scholar]
- 22. Luo S, Guan J, Ma J, Peng J. 2021. A 3D generative model for structure-based drug design. Adv Neural Inf Process Syst. 34:6229–6239. [Google Scholar]
- 23. Ma B, et al. 2021. Structure-based de novo molecular generator combined with artificial intelligence and docking simulations. J Chem Inf Model. 61(7):3304–3313. [DOI] [PubMed] [Google Scholar]
- 24. Vaswani A, et al. Attention is all you need. 2017. In: Guyon I, editor. Advances in neural information processing systems (NeurIPS 2017). Long Beach, CA, USA.NIPS Foundation. p. 5998–6008. [Google Scholar]
- 25. Xu L. 1993. Least mean square error reconstruction principle for self-organizing neural-nets. Neural Netw. 6(5):627–648. [Google Scholar]
- 26. Xu L. 2019. An overview and perspectives on bidirectional intelligence: Lmser duality, double IA harmony, and causal computation. IEEE/CAA J Automatica Sinica. 6(4):865–893. [Google Scholar]
- 27. Rosin CD. 2011. Multi-armed bandits with episode context. Ann Math Artif Int. 61(3):203–230. [Google Scholar]
- 28. Koes DR, Baumgartner MP, Camacho CJ. 2013. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model. 53(8):1893–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Landrum G, 2013. RDKit: a software suite for cheminformatics, computational chemistry, and predictive modeling.http://www.rdkit.org/RDKit_Overview.pdf [Google Scholar]
- 30. Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. 2007. BindingDB: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 35(1):D198–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steinegger M, Söding J. 2017. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 35(11):1026–1028. [DOI] [PubMed] [Google Scholar]
- 32. Jumper J, et al. 2021. Highly accurate protein structure prediction with AlphaFold. Nature. 596(7873):583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Madeira F, et al. 2019. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47(W1):W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ghose AK, Viswanadhan VN, Wendoloski JJ. 1999. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1(1):55–68. [DOI] [PubMed] [Google Scholar]
- 35. Panwar S, et al. 2021. Structure-based virtual screening, molecular dynamics simulation and in vitro evaluation to identify inhibitors against NAMPT. J Biomol Struct Dyn. DOI: 10.1080/07391102.2021.1943526. [DOI] [PubMed] [Google Scholar]
- 36. Bajusz D, Rácz A, Héberger K. 2015. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?. J Cheminformatics. 7(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and source codes are available in https://github.com/CMACH508/AlphaDrug.