

Highlights
-
•
Two transgenic rhesus monkeys expressing an Alzheimer-mutant gene were created.
-
•
By age 10 years, one monkey had developed Aβ-amyloid in blood vessels and plaques.
-
•
This is the first report of accelerated Aβ pathology in a transgenic primate.
Abbreviations: APP, Aβ-precursor protein; APPSWE/IND, Gene coding for the Aβ-precursor protein with the Swedish (K670N/M671L) and Indiana (V717F) mutations; CAA, cerebral amyloid angiopathy; LCOs, luminescent conjugated oligothiophenes; pTau, Tau phosphorylated at threonine 181
Keywords: Alzheimer's disease, Amyloid precursor protein, Animal model, Cerebral amyloid angiopathy, Tau
Abstract
With the ultimate goal of developing a more representative animal model of Alzheimer's disease (AD), two female amyloid-β-(Aβ) precursor protein-transgenic (APPtg) rhesus monkeys were generated by lentiviral transduction of the APP gene into rhesus oocytes, followed by in vitro fertilization and embryo transfer. The APP-transgene included the AD-associated Swedish K670N/M671L and Indiana V717F mutations (APPSWE/IND) regulated by the human polyubiquitin-C promoter. Overexpression of APP was confirmed in lymphocytes and brain tissue. Upon sacrifice at 10 years of age, one of the monkeys had developed Aβ plaques and cerebral Aβ-amyloid angiopathy in the occipital, parietal, and caudal temporal neocortices. The induction of Aβ deposition more than a decade prior to its usual emergence in the rhesus monkey supports the feasibility of creating a transgenic nonhuman primate model for mechanistic analyses and preclinical testing of treatments for Alzheimer's disease and cerebrovascular amyloidosis.
1. Introduction
The defining pathologic signature of Alzheimer's disease (AD) is the accumulation of multimeric assemblies of amyloid-β (Aβ) in Aβ- ('senile') plaques along with hyperphosphorylated Tau in neurofibrillary tangles [1], [2], [3], [4]. Genetic, pathologic and biomarker findings implicate the misfolding and seeded aggregation of Aβ as an essential early step in this pathogenic process, followed by tauopathy and numerous nonspecific neurodegenerative changes [2], [5], [6], [7]. Genetically modified rodent models have yielded important insights into cellular and molecular mechanisms driving the pathogenesis of AD, but the development of effective treatments has been hampered by the lack of an animal model that fully manifests all features of the disorder [8], [9], [10], [11], [12], [13], [14], [15]. Owing to their relative longevity and their phylogenetic proximity to humans, nonhuman primates have the potential to more faithfully replicate the pathobiology of AD [9], [14], [15], [16], [17], [18], [19], [20], [21].
Aβ is enzymatically released from the Aβ-precursor protein (APP) [22], [23], a 695–770 amino acid protein that is highly homologous among primates [19], [24]. All primate species analyzed to date express human-sequence Aβ [19], [25]. As they age, nonhuman primates naturally exhibit cerebral Aβ-amyloidosis (see [13], [14], [15], [19], [20], [25], [26] for reviews). Tauopathy, while occasionally present, is generally mild or absent, and a dementia-like behavioral state involving profound impairments in multiple cognitive domains has not been demonstrated in an aged nonhuman primate [14].
One possible reason for the resistance of nonhuman primates to AD is chronological age; in most humans, clinical dementia does not set in until after the age of 65 years [27], and the accumulation of Aβ begins 20 years or more prior to the first clear signs of dementia [28], [29]. Rhesus monkeys (Macaca mulatta) have a known maximum lifespan of over 40 years [30]. Assuming a maximum lifespan in humans of just over 120 years [31], rhesus monkeys thus can be considered to age at approximately three times the rate of humans. The monkeys begin to deposit Aβ in the brain (as Aβ plaques and cerebral Aβ-amyloid angiopathy [Aβ-CAA]) in their mid-20s [19]. In old age, some rhesus monkeys (and the closely related cynomolgus monkey [Macaca fascicularis]) show signs of incipient tauopathy [26], [32], [33], [34], [35], but this does not resemble in quantity or anatomical distribution the tauopathy of advanced AD [14], [16], [35]. It is possible that two or more decades are required for cerebral Aβ-proteopathy to incite AD-like tauopathy (profuse and widespread neurofibrillary tangles) and other neurodegenerative sequelae [14]. If so, it may be necessary to induce Aβ aggregation earlier in life to fully model AD-like pathology in a nonhuman primate. As a first step toward this goal, we generated transgenic rhesus monkeys expressing human APP with the AD-associated 'Swedish' and 'Indiana' mutations. By 10 years of age, one of the two transgenic monkeys had developed neocortical Aβ plaques and cerebral Aβ-amyloid angiopathy.
2. Materials and methods
2.1. Subjects
Two female rhesus monkeys (Macaca mulatta) were generated via lentiviral gene delivery into mature oocytes, followed by in vitro fertilization and transfer of embryos into surrogate female monkeys, as previously described [36]. Using established assisted reproductive techniques in rhesus monkeys [36], [37], [38], oocytes were recovered from adult, hormone-stimulated females for in vitro fertilization and culture. The lentiviral solution was injected into the perivitelline space of metaphase-II-arrested oocytes. The oocytes then were fertilized by the intracytoplasmic injection of sperm, followed by in vitro culture until the embryos were transferred to the surrogate females at the 4–8-cell stage [36], [37], [38]. The surrogates were chosen based on hormonal evidence that their reproductive stage was compatible with the embryos. (For details of our lentiviral transgenesis methodology for nonhuman primates, see [36], [37], [38], [39]).
After delivery, the monkeys were reared in the primate nursery of the Emory National Primate Research Center according to procedures developed by Sackett and colleagues [40] to allow normal growth and the development of species-specific social skills. These procedures included daily social interactions with peers and intensive human contact (for specifics of the rearing conditions, see [41]). The animals were exposed to a 12 h/12 h light/dark cycle from birth. They were housed in quads (4 animals per large enclosure) starting at 6–7 months of age, and in pairs after approximately 12 months of age.
The primary control monkeys for the study were two age-matched, non-transgenic (wild-type) females that were nursery-reared in the same way as the APPtg monkeys, and who also served as controls for a parallel study of huntingtin-transgenic monkeys [42], [43], [44]. These animals (RCk12 and RFk12) were the controls for all imaging analyses and behavioral tests through 5 years of age, for longitudinal CSF measurement of Aβ and Tau through 9 years, for analysis of transgene expression in lymphocytes and neocortex, and for histological analysis at the end of the study. After 5 years of age, no further behavioral testing was undertaken with these monkeys, so three 6-year-old, nontransgenic nursery-reared females (designated Neo-C1, Neo-C3 and Neo-C5), which were controls for an investigation of early hippocampal lesions [45], served as controls on the Emotional Reactivity and Visual Paired Comparison tasks.
At the end of the study, the APPtg animals were humanely sacrificed (below) at the ages of 10 years, 45 weeks (APP1) and 10 years, 2 weeks (APP2). Control monkeys for histopathology (RCk12 and RFk12) both were sacrificed at 10 years, 4 weeks of age. All procedures were approved by the Emory University Institutional Animal Care and Use Committee and were performed in accordance with the Animal Welfare Act and the U.S. Department of Health and Human Services “Guide for the Care and Use of Laboratory Animals” (National Research Council, 2011). The Emory National Primate Research Center is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International.
2.2. Construction and preparation of lentiviruses carrying the APPSWE/IND gene
The APP gene coding for human-sequence APP with the Swedish K670N/M671L + Indiana V717F (APPSWE/IND) mutations, regulated by the human polyubiquitin-C promoter, was inserted into a lentiviral vector. High titer lentiviruses were generated by co-transfection of the lentiviral vector coding for APPSWE/IND (pLV-APPSWE/IND), pΔ8.9, and pVSV-G encoding the vesicular stomatitis virus envelope protein (Invitrogen, now part of ThermoFisher Scientific, Waltham, Massachusetts) into a 293FT packaging cell (Invitrogen). Viruses were then concentrated by ultracentrifugation using a previously described method [36].
2.3. Quantitative (real time) reverse transcription-polymerase chain reaction (qRT-PCR) in lymphocytes and brain tissue
For analysis of APP expression in lymphocytes, peripheral blood was collected from the two APPtg monkeys and two age- and sex-matched control monkeys (RCk12 and RFk12) at 9–10 years of age. APP expression also was analyzed in samples of neocortex collected and frozen at necropsy (see Supplementary Material for methodological details).
2.4. Magnetic resonance imaging (MRI)
MRI data were acquired with a Siemens 3-Tesla (3 T) Trio clinical scanner (Siemens Medical Solutions USA, Malvern, PA) using the Siemens CP extremity volume coil at two years of age and with the Siemens 8-channel knee array coil thereafter (when the scanner was upgraded to the Siemens Total Imaging Matrix [TIM] Trio system). Scans were performed when the transgenic animals were approximately 2, 5, and 8 years of age. The wild-type female rhesus monkeys RCk12 and RFk12 served as controls for the MRI analysis at approximately 2 years and 5 years of age. We also measured the volumes of the hippocampus (a structure affected early in AD [46], [47]) and the cerebellum (a structure affected later in AD) in the APPtg and control monkeys at 5 years of age, at which point the monkeys had reached full adulthood. At the 8-year timepoint only the transgenic monkeys were scanned in order to seek potential transgene-related structural abnormalities in the brain, of which none were found.
The anesthetized monkeys were immobilized with a custom-made head-holder and placed in the supine position. Anesthesia was maintained with 1–1.5% isoflurane mixed with O2. End-tidal CO2 (Et-CO2), inhaled CO2, O2 saturation, blood pressure, heart rate, respiration rate, and body temperature were monitored continuously and maintained within normal ranges. 3D T1-weighted images with isotropic resolution were acquired using the magnetization-prepared rapid acquisition with gradient echo (MP-RAGE) sequence with the following parameters: repetition time (TR) = 2500 ms, echo time (TE) = 3.48 ms, inversion time (TI) = 950 ms, field-of-view (FOV) = 96 mm × 96 mm, data matrix = 192 × 192, flip-angle = 8°, slice thickness = 0.5 mm, 208 slices, 6 averages.
2.4.1. Brain volume calculation
3D T1-weighted images were used for calculation of the brain volumes. Bias-field correction and brain-extraction (plus manual adjustment to exclude the non-brain tissues on T1-weighted images) were carried out using FMRIB Software Library (FSL) software (Oxford University). The volumes of the whole brain (excluding the cerebrospinal fluid), the hippocampus (bilateral) and cerebellum were calculated by multiplication of the voxel number by the voxel size (0.5 mm X 0.5 mm X 0.5 mm).
2.5. Behavioral testing
Behavioral testing was undertaken from birth to 5 years of age to assess the early developmental trajectory of the monkeys relative to wild-type controls. At 6 years of age, the monkeys were assessed for emotional reactivity, and at 9 years they were tested for recognition memory. The tests included the Infant Neurobehavioral Assessment Scale (INAS), a series of four discrimination tasks (Simple Object Discrimination (SOD), Pattern Discrimination (PD), Concurrent Discrimination (CD), and Object Discrimination Reversal (ODR)), a Barrier Detour task, Emotional Reactivity task, and Visual Paired Comparison (VPC) tasks (object recognition memory after increasing delays (VPC-Delays) and memory for spatial locations (VPC-Spatial) (Table 1). For details of the behavioral tasks, see Supplementary Material.
Table 1.
Behavioral evaluation of the APPtg and wild-type control monkeys. INAS: Infant Neurobehavioral Assessment Scale; SOD: Simple Object Discrimination; PD: Pattern Discrimination; CD: Concurrent Discrimination; ODR: Object Discrimination Reversal; VPC: Visual Paired Comparison. *VPC-Spatial includes VPC-Location, Object-in-Place, and Object Replace variants. mo = month(s), yr = years.
| Tasks |
Age |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 mo | 2 mo | 3 mo | 4 mo | 5 mo | 6 mo | 8 mo | 16 mo | 5 yr | 6 yr | 9 yr | |
| INAS | + | + | + | + | + | + | |||||
| SOD | + | + | |||||||||
| PD | + | + | |||||||||
| CD | + | ||||||||||
| ODR | + | ||||||||||
| Barrier | |||||||||||
| Detour | + | + | |||||||||
| Emotional | |||||||||||
| Reactivity | + | ||||||||||
| VPC-Delays | + | + | + | ||||||||
| VPC-Spatial* | + | + | |||||||||
2.6. Biomarkers: Aβ and Tau in the cerebrospinal fluid
Cerebrospinal fluid (CSF) samples were collected from the two APPtg monkeys and two wild-type female control monkeys at the ages of 2, 4, 6, 8, and 9 years. CSF was withdrawn from the cisterna magna under general anesthesia between the hours of 10 AM and noon. The levels of Aβ42, total Tau, and Tau phosphorylated at threonine 181 (pTau) were measured in a Luminex 200 platform using Alzbio3 (Fujirebio Diagnostics, Malvern, PA), a standard diagnostic instrument for the assessment of AD biomarkers in humans [48].
2.7. Sacrifice and tissue preparation
The animals were humanely sacrificed after an overdose of sodium pentobarbital (100 mg/kg, i.v.). The brains were rapidly removed, and the two hemispheres separated along the midline. One hemisphere was immersion-fixed in a solution of 4% paraformaldehyde in phosphate-buffered saline (PBS, 0.1 M, pH 7.4) for 2–3 weeks at 4 °C. The other hemisphere was sub-dissected into regions of interest that were placed in Eppendorf tubes, quickly frozen on dry ice, and stored at −80 °C until analysis.
The paraformaldehyde-fixed hemisphere was used for histological investigation. In brief, the hemisphere was cut into 2–3 cm-thick coronal slabs that were then cryoprotected in a solution of 30% sucrose (in 0.1 M phosphate buffer, pH7.4) for at least 1 week before being frozen and serially sectioned at a thickness of 50 μm on an HM450 freezing-sliding microtome (Thermo Fisher). The sections were placed in an antifreeze solution consisting of 30% glycerol and 30% ethylene glycol in 0.1 M phosphate buffer, and stored at −20 °C until further processing.
2.8. Histopathology
2.8.1. Antibodies
The following antibodies were used for immunohistochemistry: 6E10 (1:5000) mouse IgG1 monoclonal antibody (Covance, Princeton, NJ; Antibody Registry # AB_662798) to an epitope at residues 3–8 of Aβ [49], 4G8 (1:5000) mouse IgG2b monoclonal antibody (Covance; Antibody Registry AB_662812) to an epitope at residues 18–22 of Aβ [49]; rabbit polyclonal antibodies R361 and R398 (both at 1:2,000), provided by Dr. Pankaj Mehta (Institute for Basic Research on Developmental Disabilities, Staten Island, NY; Antibody Registry AB_2315240 and AB_2315241, respectively), raised against synthetic Aβ32-40 and Aβ33-42, respectively; CP13 (1:5,000) mouse IgG1 monoclonal antibody, a gift from Dr. Peter Davies (The Feinstein Institute for Medical Research, Manhasset, NY; Antibody Registry AB_2314223), raised against a synthetic peptide representing the region around phosphoserine residue 202 of the Tau protein [50], PHF1 (1:5,000) mouse IgG1 monoclonal antibody, also from Dr. Davies (Antibody Registry AB_2315150), raised against detergent-extracted PHF preparations, with an epitope around phosphoserine 396/404 [51], anti-glial fibrillary acidic protein (GFAP) (1:5000) purified immunoglobulin fraction of rabbit antiserum from Dako (Carpinteria, CA; Antibody Registry AB_10013382), raised against glial fibrillary acidic protein isolated from cow spinal cord; anti-Iba1 (1:1000) rabbit polyclonal antibody (Wako, Osaka, Japan; Antibody Registry # AB_839504), raised against the C-terminal region of the ionized calcium-binding adaptor molecule 1 (Iba1), a protein that is expressed by macrophages/microglia.
2.8.2. Immunohistochemistry
Endogenous peroxidase in tissues for immunostaining was inactivated with 3–10% H2O2, and nonspecific reagent-binding was blocked with 1–2% normal serum in 0.2% Tween, each for one hour at room temperature. For Aβ-immunodetection, sections were pretreated for 3–10 min in concentrated formic acid to expose antigenic sites. Tissues stained for comparison of antibodies R398 and R361 were incubated in 1% sodium borohydride in buffer following the H2O2 step to optimize antigen retrieval. Sections were incubated in primary antibody (diluted in buffer with blocking serum) overnight at 4 °C. Vectastain kits (Vector Laboratories, Burlingame, CA) were used for immunodetection of antigen–antibody complexes via the avidin–biotin complex (ABC) method. After rinsing, sections were incubated for one hour at room temperature in biotinylated secondary antibody, rinsed, immersed for 30 min in avidin–biotin complex, and then developed with diaminobenzidine (DAB) (Vector Laboratories). Tissue from human Alzheimer's disease cases was used as positive control material, and non-immune mouse IgG or rabbit sera were used in place of the primary antibodies as negative controls. In some instances, a light hematoxylin counterstain was applied after immunostaining.
2.8.3. Other stains and non-nervous tissues
Some sections were stained with Congo Red, Thioflavin-S, luminescent conjugated oligothiophenes (LCOs) [52] (kindly provided by Dr. K.P.R. Nilsson, Linköping University), Prussian Blue (Perls stain), Perls-DAB (an enhanced iron stain that detects microglia [53]), or hematoxylin and eosin (H&E).
Tissue samples also were taken from the heart, liver, spleen, and kidney for routine pathologic analysis. These samples were fixed in 4% paraformaldehyde in PBS, embedded in paraffin wax, sectioned at 8–10 um thickness, and stained with H&E and with each of the anti-Aβ antibodies 6E10, 4G8 and R398.
Photomicrographs were taken with a SPOT FLEX digital camera (Diagnostic Instruments, Sterling Heights, MI) on a Leica DMLB microscope (Wetzlar, Germany), or with a Moticam 5+ digital camera (Motic, Hong Kong) on a Leica DMLS microscope.
2.9. Statistical analyses
Simple group comparisons were undertaken statistically with either t-tests or analysis of variance (ANOVA) tests. For cognitive tasks administered at only one age, group differences were analyzed with t-tests, but for cognitive tasks administered at different time points, multivariate analysis of variance (MANOVA) (Group X Age) with repeated measures for the last factor were used. For the Human Intruder task, data were analyzed using a MANOVA (Group X Condition) with repeated measures for the last factor, followed by post-hoc t-tests when appropriate. (Further details regarding analyses of behavioral data are in the Supplementary Material). In all analyses, significance levels are based on two-tailed distributions. Given the small group sizes, the statistical results overall should be interpreted cautiously.
3. Results
3.1. Genotyping
The genotype of the APPtg monkeys was examined in tissues derived from the ectoderm (frontal cortex), mesoderm (peripheral blood cells), and endoderm (lung) using PCR analysis followed by sequencing to confirm the presence of the Swedish (GA → TC) and Indiana (G → T) mutations. The mutant APP transgene was detected in tissues of the three germ layers of both transgenic monkeys (Fig. 1).
Fig. 1.
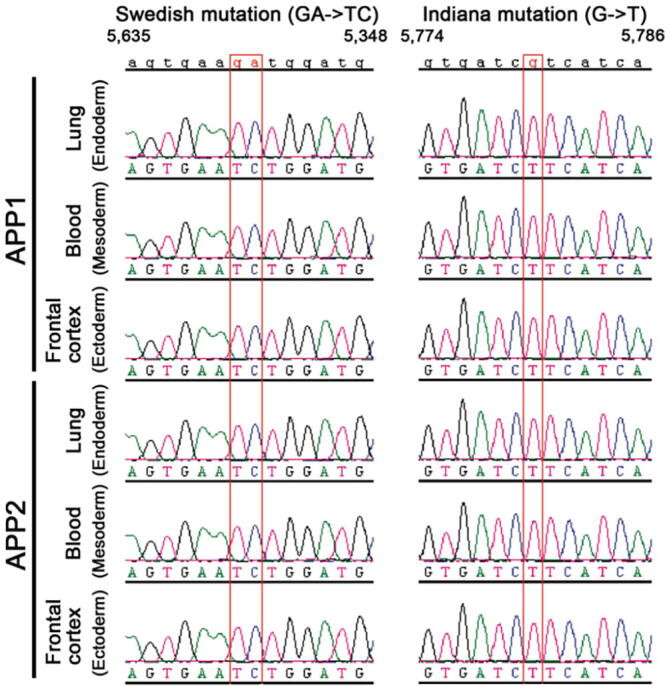
Genotyping confirmed the presence of the mutant APPSWE/IND transgene in tissues derived from the three germ layers of monkeys APP1 and APP2.
3.2. Transgene expression
Using qRT-PCR, the estimated normalized expression of APP mRNA in lymphocytes of the transgenic monkeys was 7.3 X control levels in APP1 and 5.6 X control levels in APP2. The estimated normalized neocortical expression of APP mRNA was 2.1 X control levels in APP1 and 2.3 X control levels in APP2.
3.3. Magnetic resonance imaging
The brains of the APPtg monkeys were comparable in size to those of age- and sex-matched control monkeys at 2 years and 5 years of age, with the possible exception of the somewhat smaller brain of APP1 (Fig. 2). There were no significant differences between the transgenic and control groups at 2 years (t = 0.25, p = 0.41) or 5 years of age (t = 0.64, p = 0.64). The volumes of the hippocampus and cerebellum generally corresponded to the volume of the whole brain in each animal, and neither structure showed a statistically significant difference between the transgenic and control monkeys (Supplementary Fig. 1). In the APPtg monkeys at 8 years of age, there was no evidence of hemorrhage, white matter lesions or overt structural abnormalities in the brain (Fig. 3 and Supplementary Fig. 2), nor were anomalies present in any monkey at younger ages (Supplementary Fig. 3).
Fig. 2.

Brain volumes (in mm3, not including the CSF) of the APPtg monkeys (APP1 and APP2) at 2, 5 and 8 years of age, and two age- and sex-matched wild-type control monkeys (RCk12 and RFk12) at 2 years and 5 years of age.
Fig. 3.

Horizontal MRI images of monkey APP1 at 8.75 years of age. No structural abnormalities were evident in T1-weighted images (top row), nor was there evidence of past hemorrhage or white matter lesions in T2-weighted images (bottom row). Based on the atlas of Saleem and Logothetis [94], the images (from left to right) are approximately 15 mm, 18 mm and 24 mm dorsal to the horizontal plane at the level of the interaural line.
3.4. CSF biomarkers
The CSF biomarker data are summarized in Table 2. The small n relative to the number of measures per subject precluded the use of ANOVA for repeated measures; therefore we computed t-tests (two-tailed) for each time point to highlight potentially meaningful differences between the APPtg and control monkeys.
Table 2.
The levels of Aβ42, total Tau and pTau (phosphorylated at threonine 181), and the ratios of total Tau and pTau to Aβ42 in the CSF of APPtg monkeys and non-transgenic control monkeys at 2, 4, 6, 8 and 9 years of age. Significant differences as determined by t-tests are indicated in bold.
| CSF Biomarkers | ||||||
|---|---|---|---|---|---|---|
| Group |
Monkey |
2 years |
4 years |
6 years |
8 years |
9 years |
| CSF Aβ42 (pg/ml) | ||||||
| APPtg | APP1 | 608.61 | 938.60 | 408.67 | 370.12 | 422.34 |
| APP2 | 1039.37 | 948.34 | 566.30 | 251.29 | 360.13 | |
| Control | RCk12 | 535.72 | 450.89 | 370.57 | 451.99 | 189.00 |
| RFk12 | 483.45 | 324.09 | 456.15 | 407.41 | 353.49 | |
| t (probability) | 1.45 (0.28) | 8.74 (0.01) | 0.83 (0.50) | 1.88 (0.20) | 1.36 (0.31) | |
| CSF Total Tau (pg/ml) | ||||||
| APPtg | APP1 | 27.15 | 26.18 | 23.71 | 17.81 | 26.07 |
| APP2 | 28.88 | 26.48 | 22.6 | 20.74 | 28.8 | |
| Control | RCk12 | 19.18 | 16.13 | 22.24 | 13.27 | 15.26 |
| RFk12 | 18.72 | 19 | 26.56 | 19.82 | 20.79 | |
| t (probability) | 10.13 (0.01) | 6.07 (0.03) | 0.56 (0.63) | 0.76 (0.53) | 3.05 (0.09) | |
| CSF Total Tau/Aβ42 | ||||||
| APPtg | APP1 | 0.044 | 0.028 | 0.058 | 0.048 | 0.062 |
| APP2 | 0.028 | 0.028 | 0.040 | 0.083 | 0.080 | |
| Control | RCk12 | 0.036 | 0.036 | 0.060 | 0.029 | 0.081 |
| RFk12 | 0.039 | 0.059 | 0.058 | 0.049 | 0.059 | |
| t (probability) | 0.12 (0.91) | 1.69 (0.23) | 1.12 (0.38) | 1.33 (0.31) | 0.08 (0.95) | |
| CSF pTau (pg/ml) | ||||||
| APPtg | APP1 | 44.64 | 48.16 | 37.27 | 38.95 | 36.46 |
| APP2 | 31.29 | 42.68 | 29.78 | 30.25 | 45.99 | |
| Control | RCk12 | 92.31 | 36.53 | 31.5 | 42.88 | 35.81 |
| RFk12 | 25.13 | 23.82 | 26 | 26.08 | 29.49 | |
| t (probability) | 0.61 (0.61) | 2.20 (0.16) | 1.03 (0.41) | 0.01 (0.99) | 1.50 (0.27) | |
| CSF pTau/Aβ42 | ||||||
| APPtg | APP1 | 0.073 | 0.051 | 0.091 | 0.105 | 0.086 |
| APP2 | 0.030 | 0.045 | 0.053 | 0.120 | 0.128 | |
| Control | RCk12 | 0.172 | 0.081 | 0.085 | 0.095 | 0.189 |
| RFk12 | 0.052 | 0.073 | 0.057 | 0.064 | 0.083 | |
| t (probability) | 0.95 (0.44) | 5.93 (0.03) | 0.04 (0.97) | 1.94 (0.19) | 0.52 (0.66) | |
The highest peak CSF Aβ42 levels were seen in APP1 and APP2 at the 2- and 4-year timepoints, whereas thereafter the levels were similar to those in the two control monkeys (Fig. 4). Of these timepoints, the only statistically significant difference was at 4 years of age (t = 8.74, p = 0.01). At the two youngest ages (2 and 4 years), when Aβ levels are most likely to reflect Aβ expression levels prior to the onset of deposition in the brain, the CSF Aβ42 was approximately 2 times higher in the transgenic monkeys than in the non-transgenic controls, similar to the estimated APP mRNA expression levels in the brain (see the transgene expression levels, Section 3.2).
Fig. 4.
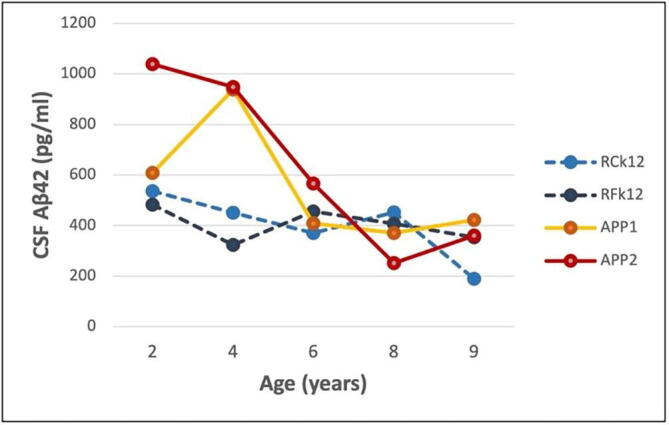
CSF Aβ42 levels in the two APPtg monkeys (APP1 and APP2) and two control monkeys (RCk12 and RFk12) from 2 to 9 years of age. The difference between the groups was statistically significant only at 4 years of age (t = 8.74, p = 0.01). See also Table 2.
The CSF total Tau levels were significantly higher in the APPtg monkeys than in control monkeys at 2 years (t = 10.13, p = 0.01) and 4 years (t = 6.07, p = 0.03) of age (Fig. 5). This difference was not significant at later ages, although there was a trend for the total Tau levels to increase in the APPtg monkeys at 9 years of age (t = 3.05, p = 0.09).
Fig. 5.
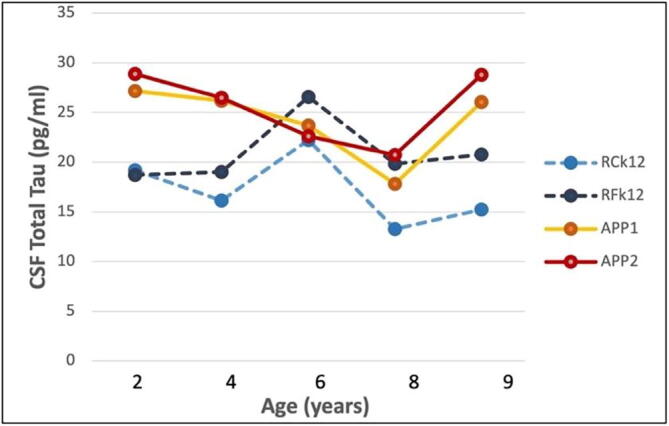
CSF total Tau levels in the two APPtg monkeys (APP1 and APP2) and two control monkeys (RCk12 and RFk12) from 2 to 9 years of age. The group difference was significant at 2 years (t = 10.13, p = 0.01) and 4 years (t = 6.07, p = 0.03) of age. See also Table 2.
Neither the total Tau/Aβ42 ratio nor the pTau levels differed significantly between the transgenic and nontransgenic monkeys at any age. The pTau/Aβ42 ratio differed significantly only at 4 years of age (t = 5.93, p = 0.03) (Table 2).
3.5. Behavior
On most behavioral tasks conducted from infancy through 16 months of age - INAS (Supplementary Table 1), VPC-Delays, VPC-Location, SOD, PD, CD, and ODR - the APPtg monkeys as a group did not differ significantly from the control monkeys. The APPtg monkeys tended to perform more poorly on several components of the Barrier Detour task at 16 months of age (Supplementary Table 2), but the only statistically significant difference was in the percent retrieval of rewards (t = 7.22, p = 0.02). At 5 years of age, this difference was no longer evident, and the only significant group difference was in perseveration following the reversal (RP Reaches) (t = 6.55, p = 0.02). APP1 in particular had a stronger tendency to perseverate (TP Reaches and RP Reaches) than did the control monkeys at 16 months and at 5 years, i.e., repeatedly attempting to reach through the transparent barrier rather than reaching around it (Supplementary Table 2).
When tested in the Emotional Reactivity task at 6 years of age, the wild-type controls exhibited the species-typical response to the human intruder, in that they primarily froze during the Profile condition and primarily expressed hostile and anxious behaviors during the Stare condition. The APPtg monkeys presented an atypical pattern of behavior, with more freezing during the Stare condition than in the Profile condition (Group X Condition: F(2,6) = 15.47, p = 0.025; Fig. 6a.). Unlike wild-type controls, the APPtg monkeys displayed more hostility during the Alone condition (Group X Condition: F(2,6) = 21.68, p = 0.003; Fig. 6b) and more displacement behaviors during the Alone and Profile conditions (Group X Condition: F(2,6) = 8.72, p = 0.017; Fig. 6c). In addition, the APPtg monkeys demonstrated significantly elevated motor stereotypies across all three conditions compared to the controls (Group: F(1,3) = 24.19, p = 0.016; Fig. 6d).
Fig. 6.

Altered emotional responses in the 6-year-old APPtg monkeys (grey bars) compared to three non-transgenic, 6-year-old female controls (white bars). Bars represent the means for (a) freezing, (b) hostility, (c) displacement behaviors, and (d) motor stereotypies during the Alone, Profile, and Stare conditions for APPtg monkeys (the yellow square represents APP1, and the red square represents APP2) and wild-type controls (blue circles represent the three individual control monkeys). *Indicates significant group differences (p < 0.05). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
At 9 years of age, the novelty preference of the APPtg group did not differ significantly from that of the wild-type control group on the VPC-Location and VPC-Replace tasks (t = 0.71, p = 0.53, and t = 2.27, p = 0.11, respectively). However, the APPtg monkeys showed significantly less preference for the novel image than did control monkeys on the Object-in-Place task (t = 3.61, p = 0.04) (Supplementary Table 4).
3.6. Histopathology
3.6.1. Brain
In transgenic monkey APP1, Aβ deposition was present primarily in association with cerebral blood vessels (Aβ-CAA) and as mostly diffuse or small, clustered parenchymal deposits (Fig. 7 and Supplementary Fig. 7). The Aβ deposits were most abundant in caudal (parietal, occipital, and caudal temporal) neocortical regions, whereas more rostral brain regions were devoid of lesions (Fig. 8). No Aβ deposition was present in non-neocortical structures, including the diencephalon, basal ganglia, hippocampal formation, amygdala, brainstem, and cerebellum. No Aβ-plaques or Aβ-CAA were present in the second APP-transgenic monkey (APP2) (Supplementary Fig. 4) or in the non-transgenic control monkeys (Supplementary Fig. 5).
Fig. 7.
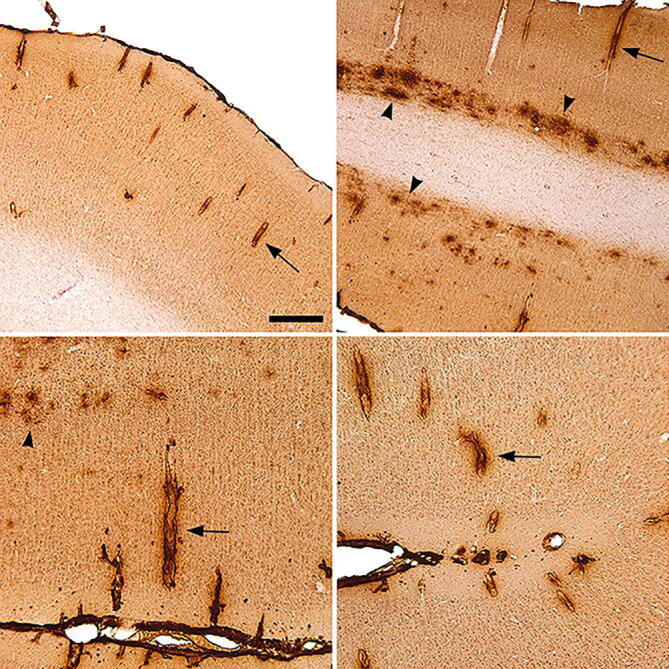
Aβ immunoreactivity in the caudal neocortex of transgenic monkey APP1 immunostained using antibody 6E10. Note the prominent Aβ-CAA (selected vessels are indicated by arrows) and diffuse parenchymal deposits (arrowheads). Light hematoxylin counterstain. Bar in the top left panel = 400 μm in the top two images, and 200 μm in the bottom two images.
Fig. 8.

Schematic diagram showing the location of Aβ deposits at three coronal brain levels in one hemisphere of monkey APP1. The lesions were restricted to the occipital, parietal, and caudal temporal neocortices. Each blue circle represents 3–5 Aβ-positive vessel profiles; each large brown square represents 3–5 plaques or a cluster of small, parenchymal Aβ puncta, and the small brown squares depict either a single plaque or a confluent patch of diffuse Aβ. Note the poor spatial overlap of plaques and Aβ-CAA. CaS: calcarine sulcus; CiS: cingulate sulcus; IOS: inferior occipital sulcus; IPS: intraparietal sulcus; LatS: lateral sulcus; LuS: lunate sulcus; MOS: medial orbital sulcus; OTS: occipitotemporal sulcus; PS: principal sulcus; STS: superior temporal sulcus. Bar = 1 cm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The Aβ-CAA affected both large vessels and capillaries, and it was immunoreactive with antibodies 6E10 (Fig. 7), 4G8, R361 and R398 (Supplementary Figs. 6 and 7). CAA and dense parenchymal deposits often stained with both antibodies R398 and R361, whereas R398 was more sensitive to diffuse deposits (Supplementary Fig. 7). Some vessels showed birefringence after staining with Congo red (Fig. 9A) and some were strongly fluorescent upon staining with Thioflavin-S (Fig. 9C) and luminescent conjugated oligothiophenes (Supplementary Fig. 8). Cored plaques were very rare, but a few parenchymal deposits were congophilic (Fig. 9B) and Thioflavin-S-positive (Fig. 9D). Antibody CP13 to Tau protein did not reveal tauopathy in any brain region (Supplementary Fig. 9). Consistent with the MRI findings (Section 3.3, above), there was no histologic evidence of overt hemorrhage, siderosis, or white matter abnormalities. There was little histological evidence of overt inflammation such as reactive microgliosis in association with the Aβ deposits (Supplementary Figure 10).
Fig. 9.
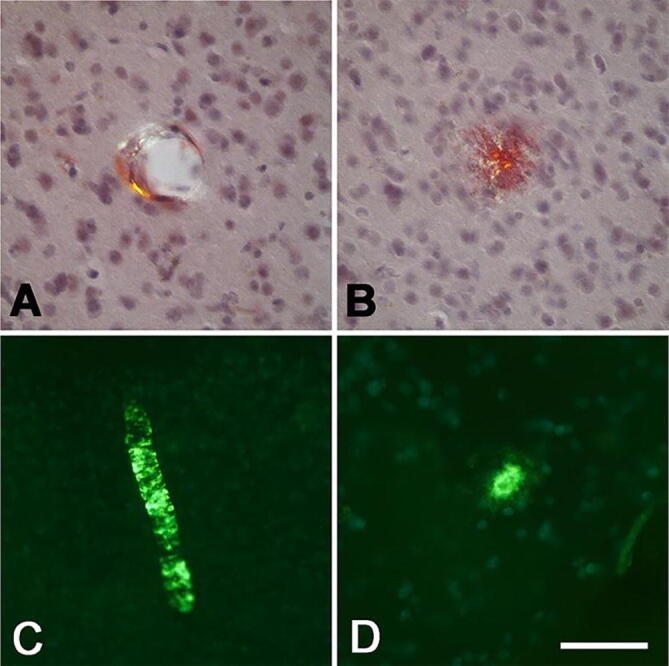
Congo Red- and Thioflavin-S-stained blood vessels (A,C) and parenchymal plaques (B,D) in the caudal neocortex of monkey APP1. The amyloid was birefringent under crossed polarizing filters when stained with Congo Red (A,B), and yielded a fluorescent signal when stained with Thioflavin-S (C,D). Hematoxylin counterstain in A and B. Bar in D = 50 μm in A, B and D, and 100 μm in C. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.6.2. Other organs
Histological examination of the APPtg monkeys revealed no abnormalities in heart muscle, liver, spleen or kidney, and no Aβ deposition was identified in these organs when immunostained with antibodies 6E10, 4G8 or R398.
4. Discussion
The presence of Aβ-plaques and Aβ-CAA more than a decade before their usual appearance in rhesus monkeys indicates that the intracerebral deposition of Aβ can be accelerated in a nonhuman primate by the overexpression of transgenic APP bearing AD-associated mutations. The resulting lesions were located only in more posterior regions of the neocortex (occipital, parietal, and caudal temporal neocortices), whereas no Aβ deposition was present in the frontal lobe, medial temporal lobe, or in non-neocortical structures. This exclusively neocortical pattern of Aβ deposition approximately corresponds to Thal Phase 1 of AD [54], [55], and to Stage 1 Aβ-CAA [56] in humans.
No Aβ deposition was detected in a second APPtg rhesus monkey (APP2) in which the estimated APP expression was comparable to that in APP1. Although the unaffected monkey was slightly younger at the time of death (10 years, 2 weeks) than was monkey APP1 (10 years, 45 weeks), it is likely that other, still unknown factors, interact with APP expression to incite the aggregation of Aβ. Studies of APP duplication [57] and triplication [58] in humans have noted variability in the age of disease onset and clinical features in patients with the same genetic anomaly, so the absence of obvious lesions in one of the monkeys is not unexpected at this young stage of life. As in humans, phenotypic variability is likely to be a characteristic of outbred animal models such as monkeys.
Analysis of biomarkers in the CSF found high levels of Aβ42 in the APPtg monkeys early in life, followed by a relatively steep decline from 2 to 6 years of age (Fig. 4). This temporal pattern is reminiscent of that in humans at preclinical stages of dominantly inherited AD (i.e., high levels of Aβ initially, followed by a decline as the disease progresses [59]). However, in a cross-sectional study of wild-type cynomolgus monkeys, CSF Aβ levels were shown to decrease with age [60]. Thus, the longitudinal decline of Aβ in the present study may reflect normal, age-associated changes in CSF levels of the protein. The amount of total Tau in CSF was greater in the transgenic monkeys at 2 years, 4 years, and (non-significantly) at 9 years, but the levels of phospho-Tau (which is thought to be a relatively specific biomarker of AD-type pathology [61]) were similar in the two groups. The ratios of Tau and pTau to Aβ42 (except for a [possibly anomalous] spike in pTau/Aβ42 at 4 years of age) were largely similar in APPtg and control monkeys, consistent with the absence of tauopathy histologically.
Behavioral tests found that the APPtg monkeys were hyperactive and irritable as young adults compared to control animals that were similarly reared in the nursery. Neuropsychiatric symptoms have been reported to predict progression to mild cognitive impairment (MCI) among cognitively normal patients [62], to predict progression to AD among prodromal patients [63], and to be prognostic of earlier progression to severe AD dementia [64], [65], [66]. When tested at 9 years of age, the overall performance of the transgenic monkeys on visual paired comparison tasks was generally similar to that of wild-type control monkeys. A possible exception was the diminished preference of the APPtg monkeys for the novel images in the relatively difficult Object-in-Place task. In humans, the VPC task has been shown to predict the progression from an unimpaired state to mild cognitive impairment (MCI) and from MCI to AD [67].
Despite somewhat deficient performance on some behavioral tasks, particularly for monkey APP1, there was no evidence of a dementia-like cognitive state, i.e., extreme impairments in multiple cognitive domains [14] in either transgenic monkey. This is not unexpected, given the limited degree of Aβ deposition and the absence of tauopathy, along with the paucity of inflammation and other degenerative changes that characterize the later stages of AD. Monkey APP2, which lacked histologically detectable Aβ deposition, also displayed some behavioral differences from the control animals. However, owing to the small number of subjects and the somewhat younger age of the control monkeys (6 years) for the VPC assessment, the behavioral findings should be interpreted guardedly. Furthermore, while the behavioral anomalies of the APPtg monkeys may indicate the initial effects of increased APP and/or Aβ (possibly in oligomeric form), we cannot rule out potential non-specific effects such as those that might result from the unpredictable genomic integration sites of the transgene.
Transgene-induced premature Aβ-amyloidogenesis, combined with a potential lifespan of 40+ years [30], could favor the eventual emergence of profuse, human-like neurofibrillary tangles in transgenic rhesus monkeys. Studies of very old wild-type monkeys of several species have found evidence of incipient tauopathy [9], [26], [32], [33], [34], [35], [68], indicating that the complete pathologic phenotype of AD might be achievable in nonhuman primates if the disease process is initiated early in life. Since tauopathy follows Aβ deposition in human AD [2], [5], [6], [7], and in light of the early stage of Aβ deposition in the affected monkey, the absence of tauopathy is not unexpected. We should note that non-fibrillar tau is rapidly de-phosphorylated postmortem, and that by quickly inactivating phosphatases, perfusion-fixation can enable the detection of non-fibrillar phosphorylated tau [26]. Because our histologic analysis used immersion-fixed tissue, the immunohistochemical methods may not have detected a very early stage of aberrant tau phosphorylation. Future studies are needed to determine if fully human-like AD tauopathy can be modeled in transgenic monkeys.
There was little histopathological evidence of reactive gliosis in the monkeys, even in areas with evident Aβ deposition. However, histology may fail to detect early biochemical changes associated with inflammation. In humans with AD, reactive astrocytes [69] and microglia [70], [71], [72] are most strongly linked to the later stages of AD pathogenesis, and they appear to emerge subsequent to Aβ deposition [69]. The microenvironment of Aβ plaques in nonhuman primates is similar to that in humans [21]. Thus, a similar glial response might be expected in APP-transgenic monkeys as the pathogenic process progresses, a possibility that must be addressed in longer-term analyses.
Even if transgenic monkeys fail to display human-like AD, they might serve as more representative models of Aβ-CAA. The buildup of Aβ in the vascular wall is a feature of nearly all AD brains [73], [74], [75], although the degree of Aβ-CAA is variable, being severe in only about a quarter of the cases [76]. Cerebrovascular amyloidosis has been linked to lobar intracerebral hemorrhage, white matter lesions, cerebral ischemia, and cognitive impairment, and thus is itself a significant source of morbidity and mortality [77]. None of these sequelae were apparent in the affected APPtg monkey, although the Aβ-CAA was probably at an early stage of development. CAA-related anomalies should be possible in transgenic nonhuman primates, inasmuch as naturally occurring Aβ-CAA has been associated with white matter lesions and neuroinflammation in an aged, wild-type squirrel monkey (Saimiri sciureus) [78]. It is noteworthy that cases of APP duplication and triplication in humans are characterized by early-onset AD with prominent Aβ-CAA [58], [79], [80], [81]. Many cases of trisomy 21 in which an extra copy of the APP gene is present also manifest early-onset AD with Aβ-CAA [79], [82]. The pathologic phenotype in the APPtg rhesus monkeys thus has parallels with cases of human APP-gene multiplication, albeit at an early stage in the monkeys. As in the case of Aβ plaques, further aging will be required to evaluate the full phenotype resulting from transgene-induced Aβ-CAA in monkeys.
Rhesus monkeys are advantageous primates for modeling AD due to their extensive use in aging research and their relatively close evolutionary relationship to humans [17], [21], [83], [84]. Although the long timecourse of pathogenesis is a shortcoming of the model, it is conceivable that an extended period of pathogenesis is necessary to completely model the pathobiology of AD, including neurofibrillary tangles, neuroinflammation, and the progressive loss of neurons and their connections. Several research groups are developing primate models of AD-like pathology using germline genetic modification (e.g., [85], [86]). One such paradigm uses the marmoset (Callithrix jacchus) [85], a small, New World primate with a known maximum lifespan of around 22 years [87]. Although they are more evolutionarily distant from humans, if the accumulation of misfolded Aβ can be accelerated in these animals, which our findings indicate is likely, colonies of germline-transgenic marmosets could be developed for use in mechanistic and therapeutic investigations. In addition, the exogenous administration of various transgenes [88] or proteopathic seeds [89], [90], [91], [92], [93] might be employed to expedite a more human-like AD state in germline-transgenic monkeys. If this can be achieved, the primate paradigm could serve as a useful complement to existing animal models, none of which yet manifest AD as it occurs in humans.
Conflict of interest statement
W.T.H. has patents on CSF-based diagnosis of frontotemporal lobar degeneration (FTLD) with TDP-43 inclusions (FTLD-TDP), and prognosis of MCI due to AD and spinal muscular atrophy; has received research support from Fujirebio Diagnostics; and has consulted for Apellis Pharmaceuticals, Fujirebio Diagnostics, and Hoffman-LaRoche. The other authors declare no competing interests. This work was prepared while Anthony W.S. Chan and Rebecca F. Rosen were employed at Emory University. The opinions expressed in this article are the authors' own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
Acknowledgments
Acknowledgments
We gratefully acknowledge enlightening discussions with Mathias Jucker, Harry LeVine, Alison R. Weiss, Nicholas Seyfried and Stuart Zola. Jean-François Paré, Susan Jenkins and Jeromy Dooyema provided expert technical assistance. Drs. Jack Orkin, Doty Kempf, Ruth Connelly and Fawn Connor-Stroud contributed skilled veterinary assistance.
Funding
Portions of this work were supported by National Institutes of Health (NIH) grants P50 AG025688, ORIP/OD P51 OD011132, and the Transgenic Huntington’s Disease Monkey Resource grant awarded by the ORIP/NIH (OD010930) to A.W.S.C. Neuropsychiatric behavioral assessments were supported by generous donations of the Arthur and Sarah Merrill Foundation.
Author contributions
A.W.S.C and L.C.W. designed the study with input from J.B., J.R., S.P.M., X.Z., J.G.H., R.F.R., W.T.H., J.J.L., A.I.L. and Y.S. A.W.S.C. and S.-H.Y. created the transgenic monkeys. C-X.L. and S.P. collected (C-X.L. and S.P) and analyzed (C-X.L.) the MRI data under the supervision of X.Z. J.R. designed and interpreted the neuropsychiatric behavioral assessments, and J.B. designed and interpreted the cognitive assessments. R.R., S.P.M., T.C., K.H.C., C.E.H. and R.C.M. participated in the collection and interpretation of the behavioral data. A.W.S.C., I.K.C., H.X., S.G., Y.S. and L.C.W. oversaw the preparation and analysis of tissue samples, and W.T.H. directed and interpreted the biomarker analyses of the cerebrospinal fluid.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbas.2022.100044.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Duyckaerts C., Delatour B., Potier M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118(1):5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- 2.Jack C.R., Bennett D.A., Blennow K., Carrillo M.C., Dunn B., Haeberlein S.B., et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long J.M., Holtzman D.M. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312–339. doi: 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson P.T., Alafuzoff I., Bigio E.H., Bouras C., Braak H., Cairns N.J., et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–381. doi: 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe D.J. A is for amyloid. J Prev Alzheimers Dis. 2020;7(3):140–141. doi: 10.14283/jpad.2020.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker L.C., Lynn D.G., Chernoff Y.O. A standard model of Alzheimer's disease? Prion. 2018;12(5–6):261–265. doi: 10.1080/19336896.2018.1525256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finch C.E., Austad S.N. Commentary: is Alzheimer's disease uniquely human? Neurobiol Aging. 2015;36(2):553–555. doi: 10.1016/j.neurobiolaging.2014.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freire‐Cobo C., Edler M.K., Varghese M., Munger E., Laffey J., Raia S., et al. Comparative neuropathology in aging primates: A perspective. Am J Primatol. 2021;83(11):e23299. doi: 10.1002/ajp.23299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jucker M. The benefits and limitations of animal models for translational research in neurodegenerative diseases. Nat Med. 2010;16(11):1210–1214. doi: 10.1038/nm.2224. [DOI] [PubMed] [Google Scholar]
- 11.Philippens I.H.C.H.M., Langermans J.A.M. Preclinical marmoset model for targeting chronic inflammation as a strategy to prevent Alzheimer's disease. Vaccines (Basel) 2021;9(4):388. doi: 10.3390/vaccines9040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rapoport S.I., Nelson P.T. Biomarkers and evolution in Alzheimer disease. Prog Neurobiol. 2011;95(4):510–513. doi: 10.1016/j.pneurobio.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Dam D., De Deyn P.P. Non human primate models for Alzheimer's disease-related research and drug discovery. Expert Opin Drug Discov. 2017;12(2):187–200. doi: 10.1080/17460441.2017.1271320. [DOI] [PubMed] [Google Scholar]
- 14.Walker L.C., Jucker M. The exceptional vulnerability of humans to Alzheimer's disease. Trends Mol Med. 2017;23(6):534–545. doi: 10.1016/j.molmed.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeiss C.J. Utility of spontaneous animal models of Alzheimer's disease in preclinical efficacy studies. Cell Tissue Res. 2020;380(2):273–286. doi: 10.1007/s00441-020-03198-6. [DOI] [PubMed] [Google Scholar]
- 16.Beckman D., Morrison J.H. Towards developing a rhesus monkey model of early Alzheimer's disease focusing on women's health. Am J Primatol. 2021;83(11) doi: 10.1002/ajp.23289. [DOI] [PubMed] [Google Scholar]
- 17.Didier E.S., MacLean A.G., Mohan M., Didier P.J., Lackner A.A., Kuroda M.J. Contributions of nonhuman primates to research on aging. Vet Pathol. 2016;53(2):277–290. doi: 10.1177/0300985815622974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haque R.U., Levey A.I. Alzheimer’s disease: A clinical perspective and future nonhuman primate research opportunities. Proc Natl Acad Sci USA. 2019;116(52):26224–26229. doi: 10.1073/pnas.1912954116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heuer E., Rosen R.F., Cintron A., Walker L.C. Nonhuman primate models of Alzheimer-like cerebral proteopathy. Curr Pharm Des. 2012;18(8):1159–1169. doi: 10.2174/138161212799315885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H.W., Zhang L., Qin C. Current state of research on non-human primate models of Alzheimer's disease. Anim Model Exp Med. 2019;2(4):227–238. doi: 10.1002/ame2.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Souder D.C., Dreischmeier I.A., Smith A.B., Wright S., Martin S.A., Sagar M.A.K., et al. Rhesus monkeys as a translational model for late-onset Alzheimer's disease. Aging Cell. 2021;20(6):e13374. doi: 10.1111/acel.13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haass C., Kaether C., Thinakaran G., Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2(5):a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Brien R.J., Wong P.C. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci. 2011;34(1):185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Podlisny M.B., Tolan D.R., Selkoe D.J. Homology of the amyloid beta protein precursor in monkey and human supports a primate model for beta amyloidosis in Alzheimer's disease. Am J Pathol. 1991;138(6):1423–1435. [PMC free article] [PubMed] [Google Scholar]
- 25.Selkoe D.J., Bell D.S., Podlisny M.B., Price D.L., Cork L.C. Conservation of brain amyloid proteins in aged mammals and humans with Alzheimer's disease. Science. 1987;235(4791):873–877. doi: 10.1126/science.3544219. [DOI] [PubMed] [Google Scholar]
- 26.Arnsten A.F.T., Datta D., Preuss T.M. Studies of aging nonhuman primates illuminate the etiology of early-stage Alzheimer's-like neuropathology: An evolutionary perspective. Am J Primatol. 2021;83(11) doi: 10.1002/ajp.23254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holtzman D.M., Morris J.C., Goate A.M. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3(77):77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jack C.R., Jr., Holtzman D.M. Biomarker modeling of Alzheimer's disease. Neuron. 2013;80(6):1347–1358. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jack C.R., Jr., Knopman D.S., Jagust W.J., Shaw L.M., Aisen P.S., Weiner M.W., et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9(1):119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stonebarger G.A., Urbanski H.F., Woltjer R.L., Vaughan K.L., Ingram D.K., Schultz P.L., et al. Amyloidosis increase is not attenuated by long-term calorie restriction or related to neuron density in the prefrontal cortex of extremely aged rhesus macaques. Geroscience. 2020;42(6):1733–1749. doi: 10.1007/s11357-020-00259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finch C.E., Sapolsky R.M. The evolution of Alzheimer disease, the reproductive schedule, and apoE isoforms. Neurobiol Aging. 1999;20(4):407–428. doi: 10.1016/s0197-4580(99)00053-6. [DOI] [PubMed] [Google Scholar]
- 32.Kiatipattanasakul W., Nakayama H., Yongsiri S., Chotiapisitkul S., Nakamura S., Kojima H., et al. Abnormal neuronal and glial argyrophilic fibrillary structures in the brain of an aged albino cynomolgus monkey (Macaca fascicularis) Acta Neuropathol. 2000;100(5):580–586. doi: 10.1007/s004010000215. [DOI] [PubMed] [Google Scholar]
- 33.Oikawa N., Kimura N., Yanagisawa K. Alzheimer-type tau pathology in advanced aged nonhuman primate brains harboring substantial amyloid deposition. Brain Res. 2010;1315:137–149. doi: 10.1016/j.brainres.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Paspalas C.D., Carlyle B.C., Leslie S., Preuss T.M., Crimins J.L., Huttner A.J., et al. The aged rhesus macaque manifests Braak stage III/IV Alzheimer's-like pathology. Alzheimers Dement. 2018;14(5):680–691. doi: 10.1016/j.jalz.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uchihara T., Endo K., Kondo H., Okabayashi S., Shimozawa N., Yasutomi Y., et al. Tau pathology in aged cynomolgus monkeys is progressive supranuclear palsy/corticobasal degeneration- but not Alzheimer disease-like - Ultrastructural mapping of tau by EDX. Acta Neuropathol Commun. 2016;4(1):118. doi: 10.1186/s40478-016-0385-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang S.H., Cheng P.H., Banta H., Piotrowska-Nitsche K., Yang J.J., Cheng E.C., et al. Towards a transgenic model of Huntington's disease in a non-human primate. Nature. 2008;453(7197):921–924. doi: 10.1038/nature06975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan A.W.S. In: Transgenic animal technology, a laboratory handbook. Pinkert C.A., editor. Academic Press; San Diego, CA: 2014. Production of transgenic nonhuman primate models of human diseases; pp. 359–378. [Google Scholar]
- 38.Chang A.T.C., Chan A.W.S. In: Methods in molecular biology. Pelegri F., editor. Humana Press, Totowa; New Jersey: 2010. Reproductive technology in nonhuman primates. [Google Scholar]
- 39.Chan A.W., Yang S.H. Generation of transgenic monkeys with human inherited genetic disease. Methods. 2009;49(1):78–84. doi: 10.1016/j.ymeth.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sackett G., Ruppenthal G., Hewitson L., Simerly C., Schatten G. Neonatal behavior and infant cognitive development in rhesus macaques produced by assisted reproductive technologies. Dev Psychobiol. 2006;48(3):243–265. doi: 10.1002/dev.20132. [DOI] [PubMed] [Google Scholar]
- 41.Goursaud A.P., Bachevalier J. Social attachment in juvenile monkeys with neonatal lesion of the hippocampus, amygdala and orbital frontal cortex. Behav Brain Res. 2007;176(1):75–93. doi: 10.1016/j.bbr.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 42.Chan A.W.S., Jiang J., Chen Y., Li C., Prucha M.S., Hu Y., et al. Progressive cognitive deficit, motor impairment and striatal pathology in a transgenic Huntington disease monkey model from infancy to adulthood. PLoS ONE. 2015;10(5):e0122335. doi: 10.1371/journal.pone.0122335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kocerha J., Xu Y., Prucha M.S., Zhao D., Chan A.W. microRNA-128a dysregulation in transgenic Huntington's disease monkeys. Mol Brain. 2014;7:46. doi: 10.1186/1756-6606-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meng Y., Jiang J., Bachevalier J., Zhang X., Chan A.W. Developmental whole brain white matter alterations in transgenic Huntington's disease monkey. Sci Rep. 2017;7(1):379. doi: 10.1038/s41598-017-00381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blue S.N., Kazama A.M., Bachevalier J. Development of memory for spatial locations and object/place associations in infant rhesus macaques with and without neonatal hippocampal lesions. J Int Neuropsychol Soc. 2013;19(10):1053–1064. doi: 10.1017/S1355617713000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gosche K.M., Mortimer J.A., Smith C.D., Markesbery W.R., Snowdon D.A. Hippocampal volume as an index of Alzheimer neuropathology: findings from the Nun Study. Neurology. 2002;58(10):1476–1482. doi: 10.1212/wnl.58.10.1476. [DOI] [PubMed] [Google Scholar]
- 47.Serrano-Pozo A., Frosch M.P., Masliah E., Hyman B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herskovits A.Z., Locascio J.J., Peskind E.R., Li G., Hyman B.T. A Luminex assay detects amyloid beta oligomers in Alzheimer's disease cerebrospinal fluid. PLoS ONE. 2013;8(7):e67898. doi: 10.1371/journal.pone.0067898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim K.S., Miller D.L., Sapienza V.J., Chen C.M.J., Bai C., Grundke-Iqbal I., et al. Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun. 1988;2:121–130. [Google Scholar]
- 50.Duff K., Knight H., Refolo L.M., Sanders S., Yu X., Picciano M., et al. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis. 2000;7(2):87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- 51.Otvos L., Jr., Feiner L., Lang E., Szendrei G.I., Goedert M., Lee V.M. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39(6):669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- 52.Nilsson K.P., Aslund A., Berg I., Nystrom S., Konradsson P., Herland A., et al. Imaging distinct conformational states of amyloid-beta fibrils in Alzheimer's disease using novel luminescent probes. ACS Chem Biol. 2007;2(8):553–560. doi: 10.1021/cb700116u. [DOI] [PubMed] [Google Scholar]
- 53.Kenkhuis B., Somarakis A., de Haan L., Dzyubachyk O., IJsselsteijn M.E., de Miranda N.F.C.C., et al. Iron loading is a prominent feature of activated microglia in Alzheimer's disease patients. Acta Neuropathol Commun. 2021;9(1) doi: 10.1186/s40478-021-01126-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thal D.R., Capetillo-Zarate E., Del Tredici K., Braak H. The development of amyloid beta protein deposits in the aged brain. Sci Aging Knowledge Environ. 2006;2006(6):re1. doi: 10.1126/sageke.2006.6.re1. [DOI] [PubMed] [Google Scholar]
- 55.Thal D.R., Rub U., Orantes M., Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 56.Thal D.R., Ghebremedhin E., Orantes M., Wiestler O.D. Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol. 2003;62(12):1287–1301. doi: 10.1093/jnen/62.12.1287. [DOI] [PubMed] [Google Scholar]
- 57.Guyant-Marechal I., Berger E., Laquerriere A., Rovelet-Lecrux A., Viennet G., Frebourg T., et al. Intrafamilial diversity of phenotype associated with APP duplication. Neurology. 2008;71(23):1925–1926. doi: 10.1212/01.wnl.0000339400.64213.56. [DOI] [PubMed] [Google Scholar]
- 58.Grangeon L., Cassinari K., Rousseau S., Croisile B., Formaglio M., Moreaud O., et al. Early-onset cerebral amyloid angiopathy and Alzheimer disease related to an APP locus triplication. Neurology Genetics. 2021;7(5):e609. doi: 10.1212/NXG.0000000000000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDade E., Wang G., Gordon B.A., Hassenstab J., Benzinger T.L.S., Buckles V., et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology. 2018;91(14):e1295–e1306. doi: 10.1212/WNL.0000000000006277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yue F., Lu C., Ai Y., Chan P., Zhang Z. Age-associated changes of cerebrospinal fluid amyloid-beta and tau in cynomolgus monkeys. Neurobiol Aging. 2014;35(7):1656–1659. doi: 10.1016/j.neurobiolaging.2014.01.139. [DOI] [PubMed] [Google Scholar]
- 61.Drummond E., Pires G., MacMurray C., Askenazi M., Nayak S., Bourdon M., et al. Phosphorylated tau interactome in the human Alzheimer's disease brain. Brain. 2020;143(9):2803–2817. doi: 10.1093/brain/awaa223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Geda Y.E., Roberts R.O., Mielke M.M., Knopman D.S., Christianson T.J., Pankratz V.S., et al. Baseline neuropsychiatric symptoms and the risk of incident mild cognitive impairment: A population-based study. Am J Psychiatry. 2014;171(5):572–581. doi: 10.1176/appi.ajp.2014.13060821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dietlin S., Soto M., Kiyasova V., Pueyo M., de Mauleon A., Delrieu J., et al. Neuropsychiatric symptoms and risk of progression to Alzheimer's disease among mild cognitive impairment subjects. J Alzheimers Dis. 2019;70(1):25–34. doi: 10.3233/JAD-190025. [DOI] [PubMed] [Google Scholar]
- 64.Burhanullah M.H., Tschanz J.T., Peters M.E., Leoutsakos J.M., Matyi J., Lyketsos C.G., et al. Neuropsychiatric symptoms as risk factors for cognitive decline in clinically normal older adults: The Cache County Study. Am J Geriatr Psychiatry. 2020;28(1):64–71. doi: 10.1016/j.jagp.2019.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leoutsakos J.M., Forrester S.N., Lyketsos C.G., Smith G.S. Latent classes of neuropsychiatric symptoms in NACC controls and conversion to mild cognitive impairment or dementia. J Alzheimers Dis. 2015;48(2):483–493. doi: 10.3233/JAD-150421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Peters M.E., Schwartz S., Han D., Rabins P.V., Steinberg M., Tschanz J.T., et al. Neuropsychiatric symptoms as predictors of progression to severe Alzheimer's dementia and death: The Cache County Dementia Progression Study. Am J Psychiatry. 2015;172(5):460–465. doi: 10.1176/appi.ajp.2014.14040480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zola S.M., Manzanares C.M., Clopton P., Lah J.J., Levey A.I. A behavioral task predicts conversion to mild cognitive impairment and Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2013;28(2):179–184. doi: 10.1177/1533317512470484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schultz C., Dehghani F., Hubbard G.B., Thal D.R., Struckhoff G., Braak E., et al. Filamentous tau pathology in nerve cells, astrocytes, and oligodendrocytes of aged baboons. J Neuropathol Exp Neurol. 2000;59(1):39–52. doi: 10.1093/jnen/59.1.39. [DOI] [PubMed] [Google Scholar]
- 69.Vehmas A.K., Kawas C.H., Stewart W.F., Troncoso J.C. Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol Aging. 2003;24(2):321–331. doi: 10.1016/s0197-4580(02)00090-8. [DOI] [PubMed] [Google Scholar]
- 70.Fukumoto H., Asami-Odaka A., Suzuki N., Iwatsubo T. Association of A beta 40-positive senile plaques with microglial cells in the brains of patients with Alzheimer's disease and in non-demented aged individuals. Neurodegeneration. 1996;5(1):13–17. doi: 10.1006/neur.1996.0002. [DOI] [PubMed] [Google Scholar]
- 71.Ohgami T., Kitamoto T., Shin R.W., Kaneko Y., Ogomori K., Tateishi J. Increased senile plaques without microglia in Alzheimer's disease. Acta Neuropathol. 1991;81(3):242–247. doi: 10.1007/BF00305864. [DOI] [PubMed] [Google Scholar]
- 72.Vogelgesang S., Schroeder E., Walker L.C., Pahnke J., Naubereit A., Walther R., et al. Activated microglia do not mediate the early deposition of Abeta in carriers of the apolipoprotein Eepsilon4 allele. Clin Neuropathol. 2002;21(3):99–106. [PubMed] [Google Scholar]
- 73.Greenberg S.M., Bacskai B.J., Hernandez-Guillamon M., Pruzin J., Sperling R., van Veluw S.J. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol. 2020;16(1):30–42. doi: 10.1038/s41582-019-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Revesz T., Ghiso J., Lashley T., Plant G., Rostagno A., Frangione B., et al. Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol. 2003;62(9):885–898. doi: 10.1093/jnen/62.9.885. [DOI] [PubMed] [Google Scholar]
- 75.Yamada M. Cerebral amyloid angiopathy: emerging concepts. J Stroke. 2015;17(1):17–30. doi: 10.5853/jos.2015.17.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kamara D.M., Gangishetti U., Gearing M., Willis-Parker M., Zhao L., Hu W.T., et al. Cerebral amyloid angiopathy: Similarity in African-Americans and Caucasians with Alzheimer's disease. J Alzheimers Dis. 2018;62(4):1815–1826. doi: 10.3233/JAD-170954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Auriel E., Greenberg S.M. The pathophysiology and clinical presentation of cerebral amyloid angiopathy. Curr Atheroscler Rep. 2012;14(4):343–350. doi: 10.1007/s11883-012-0254-z. [DOI] [PubMed] [Google Scholar]
- 78.Heuer E., Jacobs J., Du R., Wang S., Keifer O.P., Jr., Cintron A.F., et al. Amyloid-related imaging abnormalities in an aged squirrel monkey with cerebral amyloid angiopathy. J Alzheimers Dis. 2017;57(2):519–530. doi: 10.3233/JAD-160981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mann D.M.A., Davidson Y.S., Robinson A.C., Allen N., Hashimoto T., Richardson A., et al. Patterns and severity of vascular amyloid in Alzheimer's disease associated with duplications and missense mutations in APP gene, Down syndrome and sporadic Alzheimer's disease. Acta Neuropathol. 2018;136(4):569–587. doi: 10.1007/s00401-018-1866-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rovelet-Lecrux A., Hannequin D., Raux G., Le Meur N., Laquerriere A., Vital A., et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38(1):24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 81.Sleegers K., Brouwers N., Gijselinck I., Theuns J., Goossens D., Wauters J., et al. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain. 2006;129(Pt 11):2977–2983. doi: 10.1093/brain/awl203. [DOI] [PubMed] [Google Scholar]
- 82.Carmona-Iragui M., Videla L., Lleo A., Fortea J. Down syndrome, Alzheimer disease, and cerebral amyloid angiopathy: The complex triangle of brain amyloidosis. Dev Neurobiol. 2019;79(7):716–737. doi: 10.1002/dneu.22709. [DOI] [PubMed] [Google Scholar]
- 83.Perelman P., Johnson W.E., Roos C., Seuánez H.N., Horvath J.E., Moreira M.A.M., et al. A molecular phylogeny of living primates. PLoS Genet. 2011;7(3):e1001342. doi: 10.1371/journal.pgen.1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stonebarger G.A., Bimonte-Nelson H.A., Urbanski H.F. The rhesus macaque as a translational model for neurodegeneration and Alzheimer's disease. Front Aging Neurosci. 2021;13:734173. doi: 10.3389/fnagi.2021.734173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sato K, Sasaguri H, Kumita W, Inoue T, Kurotaki Y, Nagata K, Mihira N, Sato K, Sakuma T, Yamamoto T, Tagami M, Manabe R, Ozaki K, Okazaki Y, Saido TC, Sasaki E, 2020. A non-human primate model of familial Alzheimer’s disease. bioRxiv.
- 86.Seita Y., Morimura T., Watanabe N., Iwatani C., Tsuchiya H., Nakamura S., et al. Generation of transgenic cynomolgus monkeys overexpressing the gene for amyloid-beta precursor protein. J Alzheimers Dis. 2020;75(1):45–60. doi: 10.3233/JAD-191081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nishijima K., Saitoh R., Tanaka S., Ohsato-Suzuki M., Ohno T., Kitajima S. Life span of common marmoset (Callithrix jacchus) at CLEA Japan breeding colony. Biogerontology. 2012;13(4):439–443. doi: 10.1007/s10522-012-9388-1. [DOI] [PubMed] [Google Scholar]
- 88.Beckman D., Chakrabarty P., Ott S., Dao A., Zhou E., Janssen W.G., et al. A novel tau-based rhesus monkey model of Alzheimer's pathogenesis. Alzheimers Dement. 2021;17(6):933–945. doi: 10.1002/alz.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beckman D., Ott S., Donis-Cox K., Janssen W.G., Bliss-Moreau E., Rudebeck P.H., et al. Oligomeric Abeta in the monkey brain impacts synaptic integrity and induces accelerated cortical aging. Proc Natl Acad Sci U S A. 2019;116(52):26239–26246. doi: 10.1073/pnas.1902301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Forny-Germano L., Lyra e Silva N.M., Batista A.F., Brito-Moreira J., Gralle M., Boehnke S.E., Coe B.C., Lablans A., Marques S.A., Martinez A.M., Klein W.L., Houzel J.C., Ferreira S.T., Munoz D.P., De Felice F.G. Alzheimer's disease-like pathology induced by amyloid-beta oligomers in nonhuman primates. J Neurosci. 2014;34(41):13629–13643. doi: 10.1523/JNEUROSCI.1353-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jucker M., Walker L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501(7465):45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lourenco M.V., Clarke J.R., Frozza R.L., Bomfim T.R., Forny-Germano L., Batista A.F., et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer's beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013;18(6):831–843. doi: 10.1016/j.cmet.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 93.Yue F., Feng S., Lu C., Zhang T., Tao G., Liu J., et al. Synthetic amyloid-beta oligomers drive early pathological progression of Alzheimer's disease in nonhuman primates. iScience. 2021;24(10):103207. doi: 10.1016/j.isci.2021.103207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saleem K.S., Logothetis N.K. Elsevier; 2007. A combined MRI and histology atlas of the rhesus monkey brain. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.