

Abstract
DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid)-based chelates that give only a single isomer in solution when complexed with lanthanide (Ln3+) ions is of value for studying protein dynamics and interactions via NMR. Herein, we have investigated the geometries, energetics, and electrostatic potentials of Lu complexed with DOTA (1), ring methylated M4DOTA (2), and arm methylated R-DOTMA (3) and S-DOTMA (4), as well as, both ring and arm methylated 4S-4S-M4DOTMA (5) and 4S-4R-M4DOTMA (6) at the level of M06-L/6–31+G(d)-SDD, to elucidate the origin of the isomer stability. These analyses indicate that the electrostatic repulsion between the arm methyl and the neighboring carboxylate significantly destabilizes the square antiprism (SAP) isomer of Lu-5 and the twisted square antiprism (TSAP) isomer of Lu-6, while the steric repulsion between the ring and arm methyl groups attenuates the stability of both TSAP of Lu-5 and SAP of Lu-6. To rationalize the variable temperature proton NMR spectra, the energy barriers for the inter-conversion in Lu-5 and Lu-6 via arm rotation were also calculated. The modulation of the stability and rigidity of Ln complexes via a modification of DOTA is also discussed. Our investigation will aid to design better chelates for the Ln3+ ions for its use in molecular medicine.
Graphical Abstract
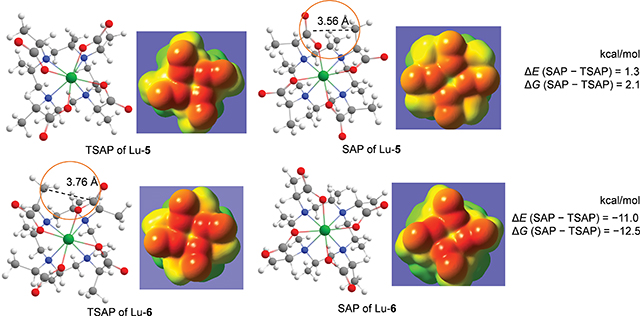
The geometries, energetics, and electrostatic potentials of Lu complexed with the polymethylated 4S-4S-M4DOTMA (5) and 4S-4R-M4DOTMA (6) were investigated with DFT to elucidate the origin of the isomer stability. The modulation of the stability and rigidity of Ln complexes is also discussed with the aim of designing better chelates for the Ln3+ ions for its use in molecular medicine.
Introduction
The magnetic and optical properties of lanthanide ions (Ln3+) find their use in nuclear magnetic resonance (NMR), paramagnetic chemical exchange saturation transfer (PARACEST)[1], protein structural NMR analysis[2], and luminescence[3]. In nuclear medicine, 177Lu can be used to selectively destroy cancer cells via radioimmunotherapy.[4] Lanthanide ions are toxic and must be administered in the form of a complex with high thermodynamic stability and kinetic inertness. In this context, the 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) ligand offers an excellent match between the cavity of the ligand and the size of the Ln3+, making it an ideal chelator. The DOTA ligand binds Ln3+ octadentately through four nitrogen atoms (N4) of the macrocycle and four oxygen atoms (O4) of the carboxylate groups. The ethylene groups of the macrocycle can adopt two different gauche orientations (λ and δ)[5] while the acetate arms are twisted around the Ln3+ in either Δ or Λ orientation (Figure 1). Two diastereomeric forms Λ(δδδδ)/Δ(λλλλ) and Δ(δδδδ)/Λ(λλλλ) are present in solution, differing in the orientation of the parallel planes formed by the Ln3+-bound nitrogen and oxygen atoms. In the Λ(δδδδ)/Δ(λλλλ), the average angle of rotation between the N4 and O4 planes is ∼40°, which leads to the square antiprism (SAP) isomer; in the Δ(δδδδ)/Λ(λλλλ), an angle of ~24° corresponds to the twisted square antiprism (TSAP) isomer. The TSAP and SAP isomers can undergo an exchange process either by ring inversion (δδδδ) ↔ (λλλλ) or arm rotation Δ ↔ Λ (Figure 1). The interconversion is slow on the NMR time scale and can therefore be seen as two sets of peaks in the 1H or 13C NMR spectra. The major isomer of the large Ln3+ (La3+-Nd3+) DOTA complexes adopts the TSAP conformation, while the major isomer for the smaller Ln3+ (Sm3+-Lu3+) DOTA adopts the SAP conformation. The ninth coordination site of the Ln3+ in the complex is usually occupied by a labile ligand, e.g., apical water/bound water molecule in aqueous solutions. The Ln-OH2 bond length is known to play an essential role in the exchange rate between bound and bulk water, a process central for its effectiveness as MRI contrasting agents.[6]
Figure 1.
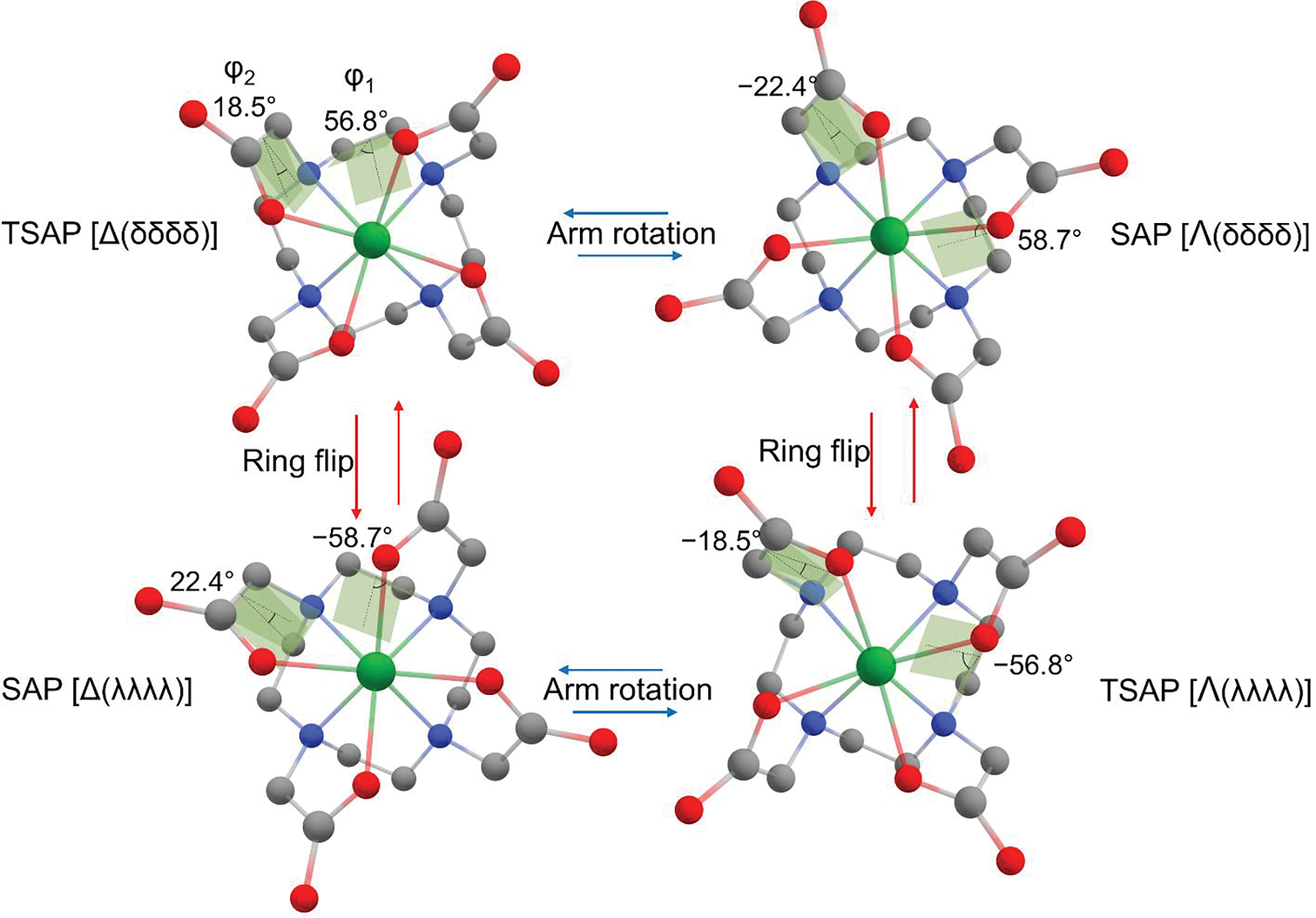
Interconversion of the conformers of Lu-1. Shades depict the dihedral angles of N−C−C−N (ϕ1) and N−C−C−O (ϕ2). The conformers having the same and opposite sign of the dihedral angles are designated as TSAP and SAP, respectively. The structures of TSAP [Δ(δδδδ)] and SAP [Λ(δδδδ)] were obtained via geometry optimization at the level of M06-L/6–31+G(d)-SDD in the water reaction field. Their respective enantiomers were obtained by inversion. The ring flip of TSAP [Δ(δδδδ)] transforms the dihedral angle of ϕ1 from positive (δ) to negative (λ) resulting in SAP [Δ(λλλλ)]; the arm rotation of TSAP [Δ(δδδδ)] transforms the dihedral angle of ϕ2 from positive (Δ) into a negative (Λ) resulting in SAP [Λ(δδδδ)]. Atoms are colored as follows: gray, carbon; blue, nitrogen; red, oxygen; green, Lu3+. No hydrogens are shown.
Lanthanide complexes can also be used to tag proteins for structural NMR studies due to their ability to induce paramagnetic relaxation enhancement and pseudocontact shifts (PCS). While the development of Ln3+ tags that induce only one set of resonances for protein analysis is desirable, this effort has been hampered due to the presence of the TSAP and SAP isomers that produce additional resonances. One way of eliminating these undesirable peaks is by introducing structural rigidity in the DOTA chelates via methylation of the ring or the arm or both.[7]
The octamethylated 4S-4S-M4DOTMA (5 in Scheme 1) bears four S-methyl groups in the ring and another four S-methyl groups in the pendent arm. We have shown the unusual preference of the larger lanthanides (Ce-Nd3+) complexes of 4S-4S-M4DOTMA to form the SAP isomer; smaller lanthanide complexes of 4S-4S-M4DOTMA predominantly exist as TSAP in solution at room temperature.[8] This observation is in contrast to Ln-DOTA, where the larger lanthanide complexes adopt the TSAP conformation. [9] Interestingly, the inversion of the pendent arm of 4S-4S-M4DOTMA to 4S-4R-M4DOTMA (6) resulted in a single isomer SAP across the Ln3+ series (Pr3+-Yb3+).[10] In line with this finding, the Dy-, Tm-, Tb- and Lu-complexes of (4R4S)-M8-Spy was shown to favor the SAP conformation thermodynamically via DFT calculations.[7e] The synthesis of the ligands such as 4S-4S- or 4S-4R-M4DOTMA is laborious, and the isomer composition can be assessed only after the preparation of the complex. The prediction/modulation of the isomer stability of a Ln3+ complex via quantum mechanical (QM) studies thus can be of significant value in the synthesis of the rigidified Ln-DOTA-like complexes.
Scheme 1.
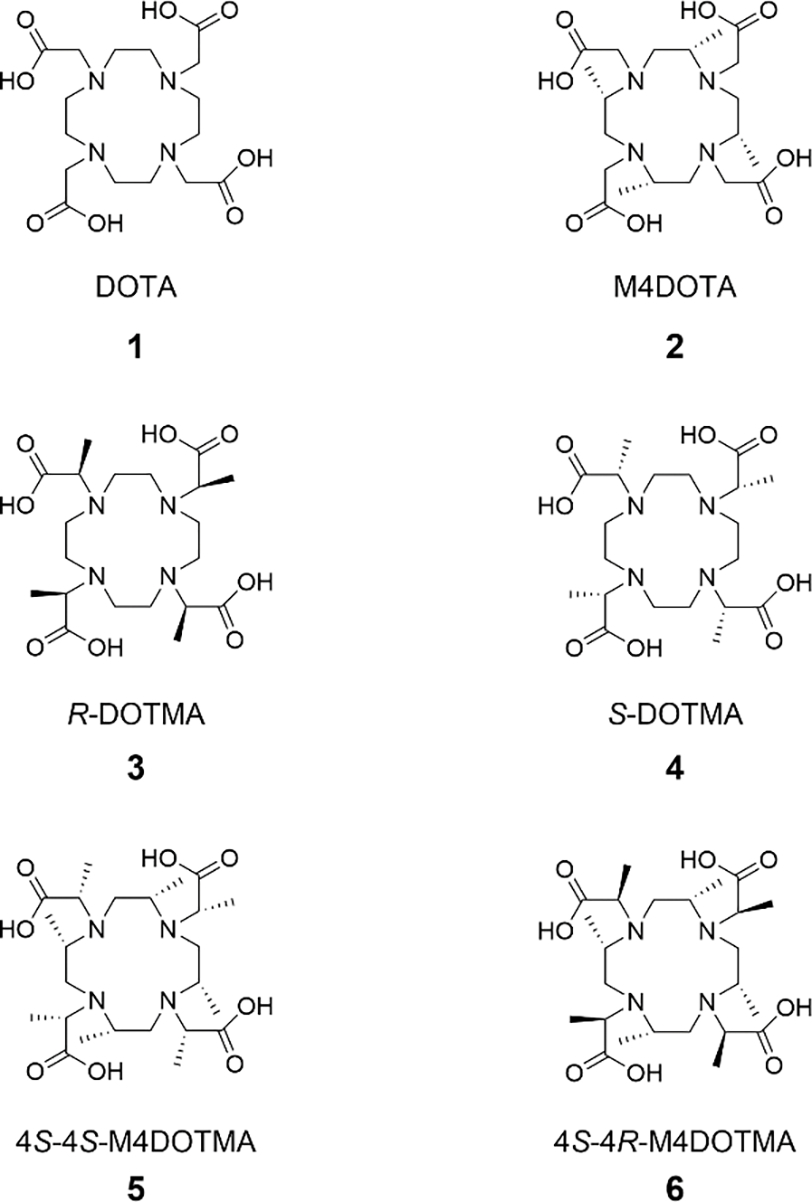
Structures of DOTA and methylated DOTA ligands.
In the present study, the structures and energetics of the isomers of both Lu-5 and Lu-6 were calculated with density functional theory (DFT) at the level of M06-L/6–31+G(d)-SDD. The proton chemical shifts of these structures were also calculated at the level of B3LYP/6–311+G(2d,p)-SDD//B3LYP/6–31+G(d)-SDD to characterize the isomers in solution. The calculated structures were used to assign the nuclear Overhauser effect (NOE) cross peaks to identify the respective isomers. To elucidate the origin of the isomer stability of Ln3+ complexed with DOTA-like chelates, we have systematically introduced methyl groups into the ring and arm to DOTA (1), as depicted in Scheme 1. Their structures and energetics, as well as electrostatic potentials (EPs), were then compared to identify the structural motifs that dictate the stability of the isomers of Lu3+ complexes. The interconversion energy barriers for the isomers of 1, 4, and 5-6 bound to Lu3+ were calculated to estimate the isomer energy barrier. These were, in turn, used to provide a rationale for the variable temperature proton NMR spectra of Lu-5 and Lu-6.
Results and Discussion
Structures and energetics of the polymethylated DOTA complexed with Lu3+
The SAP isomer of Lu-1 was first geometry optimized at the level of M06L/6–31+G(d)-SDD (Figure 1) for comparison with the X-ray structure of the SAP of Lu-1 with a bound water. The average Lu−N distances calculated with and without a bound water were, 2.605 Å and 2.553 Å respectively, comparable to that of the X-ray structure (2.614 Å).[11] The calculated average Lu−O distances with and without a water molecule were 2.279 Å and 2.250 Å, respectively, which are also close to the experimental value of 2.279 Å. Whereas the experiment[9b] indicates that the SAP isomer of Lu-1, unlike TSAP, has a coordinated water molecule in solution, our energetics calculations for both isomers were done in the reaction field of water without the coordinated water molecule. The calculations nonetheless show that the SAP isomer is slightly more stable (0.3 kcal/mol) than TSAP in terms of relative Gibbs free energy (ΔG) at 298.15 K, as compared to the experimental ΔG of 1.0 kcal/mol favoring the SAP conformation[12a]. These agreements between theory and the experiments validate that our quantum chemical approach, which is now extended to investigate the geometry and energetics of other methylated DOTA chelates (Scheme 1) complexed with Lu3+.
Figure 2 shows the geometry optimized structures of both 5 and 6 complexed with Lu3+. The rings all have the δδδδ conformation, and the isomer interconversion occurs only through the arm rotation Δ(δδδδ) ↔ Λ(δδδδ).[8, 13] The conformers with the axial methyl groups were not considered due to their high energy, as discussed under the section of Conformers of Lu-1 and Lu-2. Our calculations indicate that the TSAP isomer of Lu-5 is more stable than SAP by both the relative electronic energy (ΔE) = −1.3 kcal/mol and ΔG = −2.1 kcal/mol at 298.15 K, consistent with our previous HPLC and NMR findings.[8] When the chirality of the methyl group on the α position of the acetate arm was inverted, the SAP isomer of Lu-6 becomes more stable (ΔE = −11.0 kcal/mol) relative to TSAP. Whereas these calculated energies are consistent with the single isomer appearance in solution across the Ln3+ series when complexed with 6 [10], it still remains unclear as to the origin of the switch in the conformer stability of Lu-6 along with its magnitude with respect to Lu-5.
Figure 2.
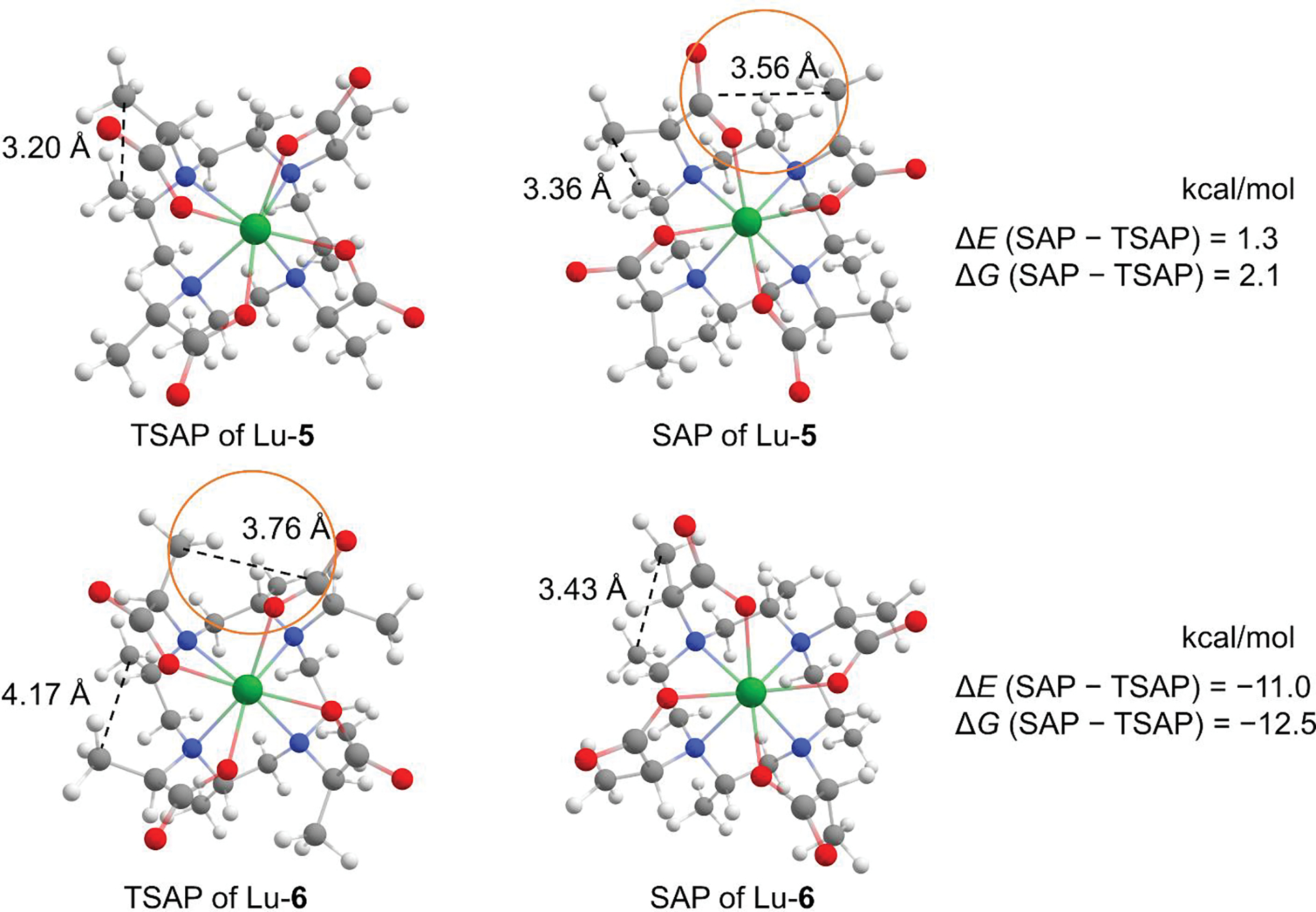
Geometry optimized conformers of Lu-5 and Lu-6. Dashed lines in the circle represent the distance between the carbons of the carboxyl and the neighboring arm methyl. Dashed lines without the circle indicate the distance between the carbons of the ring methyl and the arm methyl. Atoms are colored as follows: white, hydrogen; gray, carbon; blue, nitrogen; red, oxygen; green, Lu3+.
Characterization of the solution structure of Lu-5 and Lu-6
The proton NMR spectrum of Lu-5 (Figure 3, top) shows that the calculated chemical shifts of ring CH3 protons are found upfield (δ 1.19) with respect to the chemical shifts of the arm CH3 protons (δ 1.33); it also indicates a better shielding of the ring CH (ax2) proton (δ 3.28) when compared to the arm CH proton (δ 4.12). These differences can be ascribed to the low electron density on the Cα atom of the arm acetate, giving rise to the weaker shielding of both arm CH3 and CH protons. The chemical shifts of the equatorial proton (δ 2.66) and the axial (ax1) proton (δ 2.99) (Figure 3, top inset), as well as their spin-spin couplings, are all consistent with the calculated structures. Furthermore, the calculated chemical shifts for the ring CH3 protons (δ 1.17), the arm CH3 protons (δ 1.39), and the arm CH proton (δ 4.30) for the TSAP isomer of Lu-5 (Table 1) are in better agreement with the observed chemical shifts than those for SAP. In particular, the calculated chemical shift of the arm CH proton (δ 4.30) for the TSAP isomer is downfield compared to the SAP (δ 3.34), and comparable to the observed chemical shift (δ 4.12), suggesting that it can be a prominent marker for the characterization of isomers of comparable energetics. The proton NMR spectrum of Lu-5 also shows the presence of minor peaks arising from the SAP conformer. As listed in Table 1, the calculated chemical shifts for the ring CH3 protons (δ 1.04) and the arm CH3 protons (δ 1.68) for the SAP isomer of Lu-5 are in agreement with the observed peaks of the minor isomer. Both the proton chemical shifts and the spin-spin couplings of the Lu-6 (Figure 3, bottom inset) are comparable to those of Lu-5, except that the peaks of the ax1-H and the ax2-H of Lu-6 are merged; no peaks corresponding to the minor isomer were observed. The calculated chemical shifts for the ring CH3 (δ 1.14) and the arm CH3 (δ 1.54) protons for the SAP isomer of Lu-6 also agree better with the experimental ones. While valuable for the characterization of Lu-5 and Lu-6 in solution, these proton NMR spectra and the calculated proton chemical shifts still do not provide direct evidence as to whether Lu-5 and Lu-6 exist as TSAP or SAP in solution. To resolve this, 2D-NOESY studies were performed.
Figure 3.
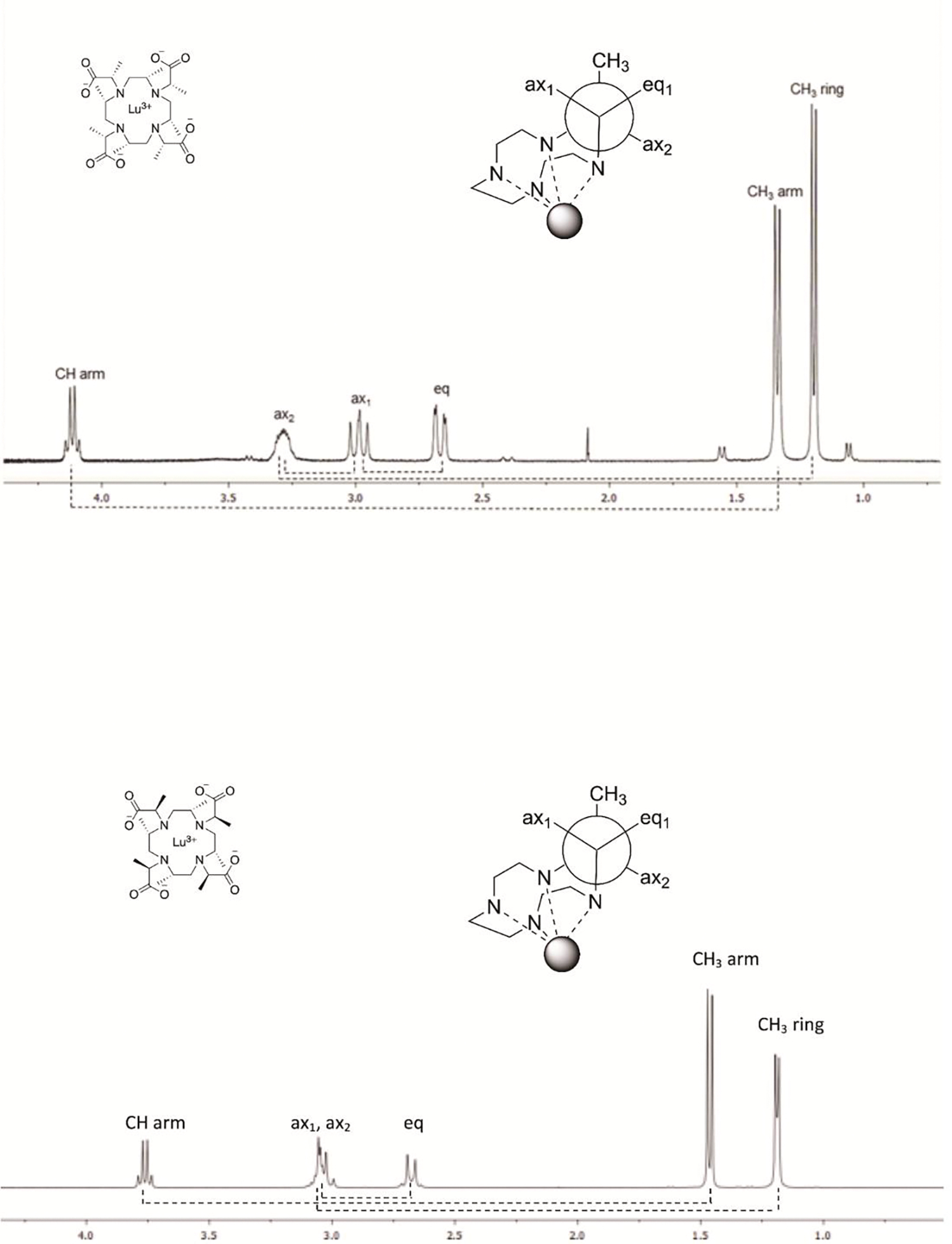
Proton NMR spectra of Lu-5 (top) and of Lu-6 (bottom) at 298 K and 400 MHz. The dashed lines underneath the spectrum indicate the couplings determined from 2D COSY.
Table 1.
Assignments of 1H NMR chemical shifts (δ) for Lu-5 and Lu-6.
| Lu-5 | Lu-6 | |||||
|---|---|---|---|---|---|---|
| Theoreticala | Exp.c | Theoreticala | Exp.c | |||
| TSAP | SAP | TSAP | SAP | |||
| CH3 (ring) | 1.17b | 1.04b | 1.19 | 0.94b | 1.14b | 1.19 |
| CH3 (arm) | 1.39b | 1.68b | 1.33 | 1.38b | 1.54b | 1.46 |
| Heq | 2.53 | 2.33 | 2.66 | 2.62 | 2.61 | 2.67 |
| Hax1 | 2.83 | 3.10 | 2.99 | 2.77 | 2.72 | 3.04 |
| Hax2 | 3.24 | 3.30 | 3.28 | 3.06 | 3.16 | 3.04 |
| CH (arm) | 4.30 | 3.34 | 4.12 | 3.73 | 3.66 | 3.76 |
Calculated at the level of B3LYP/6-311+G(2d,p)/SDD//B3LYP/6-31+G(d)/SDD.
Average value of three protons.
Assignment for the major peaks.
The 2D-NOESY spectrum of Lu-5 at 298 K (Figure 4) shows the cross peaks between the arm CH3 and the ring CH3 ring protons as well as between the eq-H and the arm CH3 protons that can be present only in the TSAP conformation. For example, the distance between the eq-H and the arm CH3 protons in TSAP is 2.18 Å as compared to 4.56 Å in SAP. In contrast, the NOE study of Lu-6 (Figure 5) shows the cross peak between the arm CH3 and the ring CH3 protons that can only arise from the SAP conformation; the average distance between the two methyl protons of SAP is 2.1 Å as compared to 4.07 Å in TSAP. These NOE studies, together with the quantum chemical calculations, thus provide the unequivocal evidence that Lu-5 and Lu-6 exist as TSAP and SAP in solution, respectively.
Figure 4.
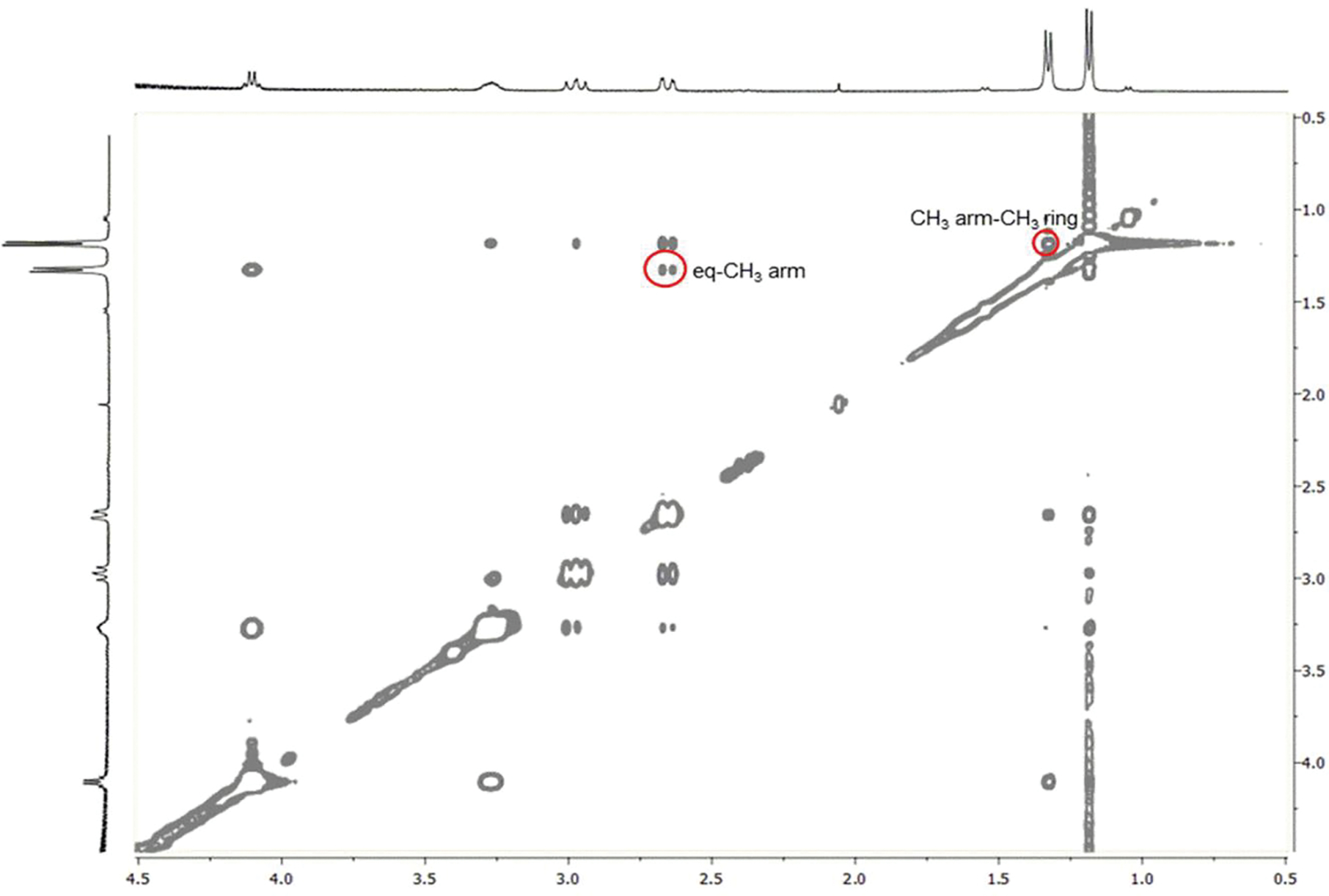
2D-NOESY spectrum of Lu-5 at 298 K and 400 MHz. The red circles show the NOE cross peaks between the CH3 arm (1.33 ppm) and CH3 ring (1.19 ppm) protons and between the eq (2.66 ppm) and CH3 arm (1.33 ppm) protons, which can only be present in the TSAP isomer.
Figure 5.
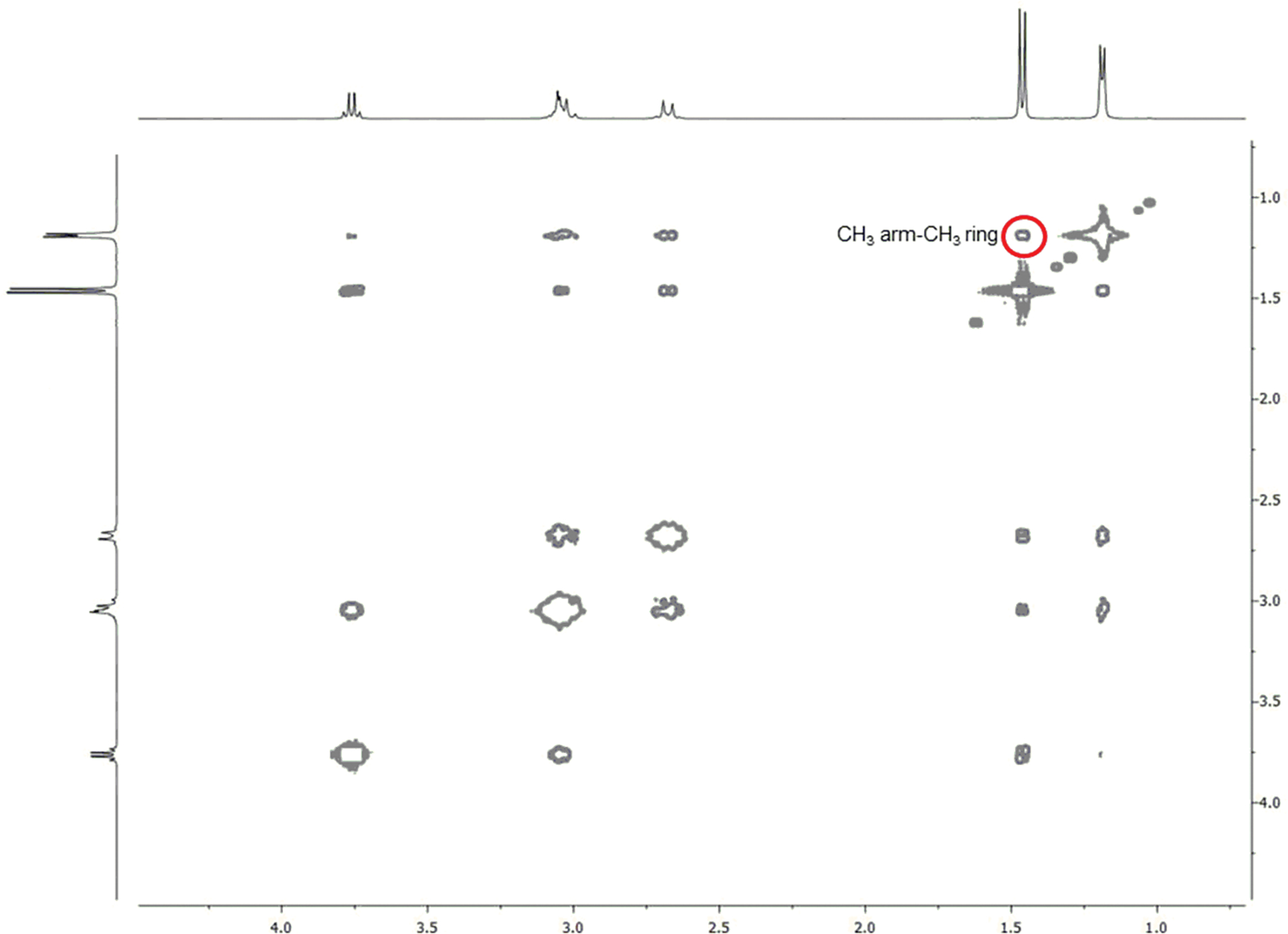
2D-NOESY spectrum of Lu-6 at 298 K and 400 MHz. The red circles show the NOE cross peaks between the CH3 arm (1.46 ppm) and CH3 ring (1.19 ppm) protons that can only be present in the SAP isomer.
Origin of the stability for the conformers of Lu-5 and Lu-6
Having characterized the solution structures for both Lu-5 and Lu-6, we proceeded to pinpoint the structural motifs that modulate their stability. To this end, we introduced the methyl groups in the ring and/or arms of DOTA systematically (Scheme 1); the calculated energetics of these chelates complexed with Lu3+ are listed in Table 2.
Table 2.
Relative stability of SAP and TSAP of Lu3+ complexed with DOTA and its methylated chelates
| Chelate | ΔE (SAP − TSAP) (kcal/mol) | ΔG (SAP − TSAP) (kcal/mol) |
|---|---|---|
| 1 (δδδδ) | 0.0 | −0.3 |
| 2 (δδδδ) | −1.2 | −2.0 |
| 3 (δδδδ) | −17.9 | −20.3 |
| 3 (λλλλ) | 12.0 | 14.7 |
| 4 (δδδδ) | 12.0 | 14.7 |
| 5 (δδδδ) | 1.3 | 2.1 |
| 6 (δδδδ) | −11.0 | −12.5 |
Conformers of Lu-1 and Lu-2
The QM calculations indicate that the TSAP isomer of Lu-1 with the δδδδ conformation (ϕ1 = 56.8°; ϕ2 = 18.5°) is slightly less stable than SAP (ϕ1 = 58.7°; ϕ2 = −22.4°). The ring flip of these two conformers gives rise to two additional conformational enantiomers, namely, SAP [Δ(λλλλ)] (ϕ1 = −58.7° ) and TSAP [Λ(λλλλ)] (ϕ1 = −56.8°), as shown in Figure 1. The X-ray structure of the racemic mixture of homo-chiral Lu-1 in SAP had provided direct evidence of inversion in solution.[11]
The ring S-methylation on DOTA makes the SAP isomer of Lu-2 more stable than TSAP by ΔE of 1.2 kcal/mol. However, unlike Lu-1, the ring S-methylation renders the ring-flip from (δδδδ) to (λλλλ) untenable since it introduces the S-methyl to the axial position, resulting in a huge steric repulsion with the neighboring axial hydrogens. For example, the QM calculations with the single S-methyl substitution on the ring of Lu-1 indicate that SAP [Λ(δδδδ)] is the most stable conformer (Figure S1). Furthermore, the ring flip of both SAP [Λ(δδδδ)] and TSAP [Δ(δδδδ)] results in less stable TSAP [Λ(λλλλ)] (ΔE = 7.6 kcal/mol) and SAP [Δ(λλλλ)] (ΔE = 9.1 kcal/mol), respectively, due to the 1,3-diaxial-like steric strain seen in methylcyclohexane but much stronger in magnitude. Our calculated geometries and energetics are consistent with the previous NMR study, reporting that Yb-MDOTA with a single ring methyl substituent prevents the ring inversion.[13] These results indicate that ring methylated Ln3+ tags can substantially simplify the analysis of a protein of interest since they prevent the ring inversion completely.
Conformer stability of Lu-3 and Lu-4
The arm R-methyl substitution also has a large effect on the isomer stability of Lu-3. Figure 6A depicts that each arm methyl of TSAP [Δ(δδδδ)] is in close proximity to the neighboring carboxyl, as shown by the distance of 3.68 Å between the two carbon atoms. As a consequence, while absent in SAP [Λ(δδδδ)], the TSAP isomer incurs a sizable electrostatic (ES) repulsion as discussed below, and this largely explains the stability of SAP (ΔE = −17.9 kcal/mol) over the TSAP isomer. Both isomers, however, can undergo a ring inversion to have the λλλλ conformation with the enhanced stability. The ring inversion of the TSAP [Δ(δδδδ)] isomer results in a more stable SAP [Δ(λλλλ)] by ΔE of 7.1 kcal while the inversion of the SAP [Λ(δδδδ)] isomer imparts 1.2 kcal/mol stability to TSAP [Λ(λλλλ)]. This finding is in line with the previous work, which reported that the TSAP [Λ(λλλλ)] isomer of Lu-3 is the most stable one in solution.[7a] Moreover, our calculated TSAP [Λ(λλλλ)] isomer of Lu-3 (Figure. 6A) has the same ring conformation of the X-ray structures of the TSAP isomer of Ln (Ce3+-Yb3+)-DOTMA [7a, 7b], which further the notion that the stability is gained by a ring inversion. Without the arm methyl groups, as in the case of Lu-1, the ring inversion resulted in no stabilization in ΔE. It is noted that previous theoretical work[7g] attributed the stability of the TSAP [Λ(λλλλ)] isomer of Lu-3 over SAP [Λ(δδδδ)] to the hydration energy difference.
Figure 6.
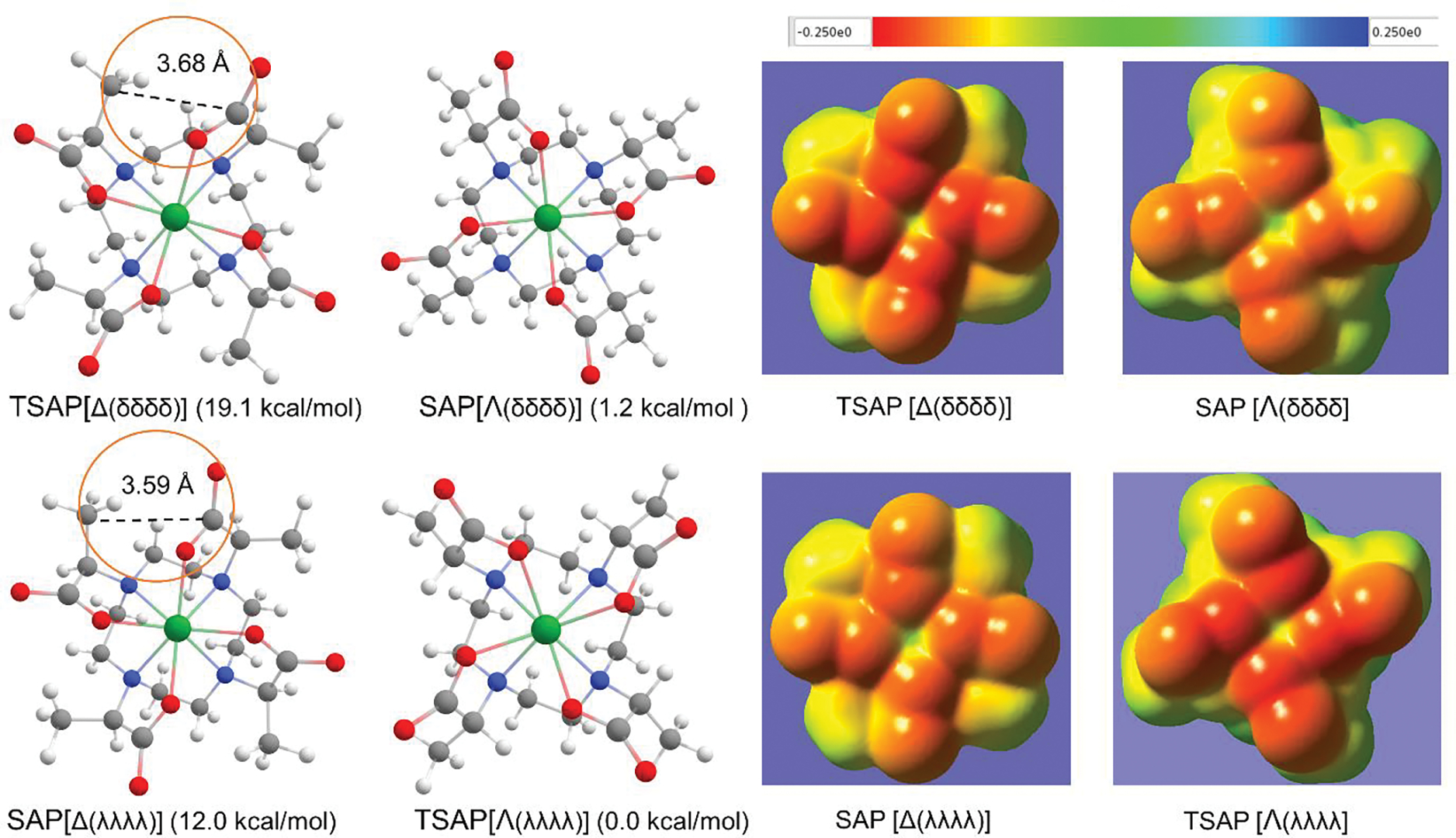
A: Geometry optimized conformers of Lu-3. The isomers on the top and at the bottom have the respective δδδδ and λλλλ conformation. The value in parentheses represents the electronic energy (ΔE) with respect to that of the TSAP [Λ(λλλλ)] conformer. The circles indicate the proximity between the arm methyl and the neighboring carboxyl group. B: Electrostatic potential maps based on the isodensity contour at 0.001e/bohr3 for the conformers. The potentials in au are colored according to the inset.
To better illustrate the ES repulsion and its impact on the isomer stability, the electrostatic potential (EP) maps for these isomers projected onto their iso-electron density surface of 0.001 e/bohr3 were constructed (Figure 6B). The arm methyl groups in TSAP [Δ(δδδδ)] are positioned between the negatively charged carboxylate groups and thus face the strong ES repulsion. In contrast, the arm methyl groups of SAP [Λ(δδδδ)] are pointing into the page, and thus, do not encounter the ES repulsion which, in turn, accounts for the ΔE of 17.9 kcal/mol difference between the two. The ring inversion of TSAP [Δ(δδδδ)] resulted in SAP [Δ(λλλλ)], which has less electron density on the carboxylate oxygens; a weakened ES repulsion gives the ΔE of 7.1 kcal/mol stability over TSAP [Δ(δδδδ)]. These four isomers can be ordered in decreasing stability: namely, TSAP [Λ(λλλλ)] > SAP [Λ(δδδδ)] > SAP [Δ(λλλλ)] > TSAP [Δ(δδδδ)]. While the arm R-methyl groups thus hinder the formation of rotamers such as TSAP [Δ(δδδδ)] and SAP [Δ(λλλλ)] due to the ES repulsion, the ring inversion can still take place rendering the highest stability to TSAP with the λλλλ conformation. By symmetry, the arm S-methyl substitution makes TSAP [Δ(δδδδ)] of Lu-4, which is enantiomeric to TSAP [Λ(λλλλ)] of Lu-3, the most stable conformer. The structures of Lu-4 are shown in Figure S2 to further analyze the conformer stability of Lu-5 having both arm and ring S-methyl groups.
Conformers of Lu-5 and Lu-6: interaction between the ring methyl and the arm methyl
How does the ring S-methyl interact with the arm methyl of Lu-5 and Lu-6? As pointed out for Lu-2 and Lu-MDOTA (Figure S1), the steric repulsion between the axial ring S-methyl and the neighboring axial protons prevents both Lu-5 and Lu-6 from having the λλλλ conformation. It is worth pointing out that our recent X-ray structure of the TSAP isomer of Pr-5 also has the δδδδ conformation.[10] Whereas both Lu-5 and Lu-6 thus have the locked-in δδδδ conformation, they still can undergo arm rotation. As listed in Table 2, the TSAP isomer of Lu-5 is more stable than SAP by ΔE of only 1.3 kcal/mol. The SAP isomer of both Lu-4 (Figure S2) and Lu-5 (Figure 2) faces a comparable ES repulsion between the arm S-methyl and the neighboring carboxyl. However, the ring methyl of TSAP of Lu-5 is also positioned to have a steric repulsion with the arm S-methyl; the NOE spectrum (Figure 4) confirmed the proximity of these two methyl groups. In addition, the arm methyl of the TSAP conformer is in eclipse to the ring methyl while that of SAP is in gauche, and thus faces a stronger repulsion (Figure 2). This methyl-methyl repulsion in TSAP was calculated to be ~2.7 kcal/mol higher than in SAP (see Table S1). Since the TSAP isomer of Lu-5 has four such methyl-methyl repulsions, it becomes destabilized by the electronic energy in 11 kcal/mol, thereby decreasing ΔE just to 1.3 kcal/mol with respect to the SAP isomer.
The SAP isomer of Lu-6 with the arm R-methyl was calculated to be significantly more stable (ΔE = −11.0 kcal/mol) than TSAP. This sizable ΔE, as well as the switch in conformer stability to SAP relative to Lu-5, can be explained as follows: Since the ring S-methyl groups force Lu- 6 to adopt only the δδδδ conformation, the more stable λλλλ conformation as shown for Lu-3 (Figure 6A) is untenable. The ES repulsion between the arm R-methyl and the neighboring carboxyl makes the TSAP isomer of Lu-3 [Λ(δδδδ)] 17.9 kcal/mol unstable relative to SAP [Λ(δδδδ)]. The TSAP isomer of Lu-6 encounters such an ES repulsion (Figure 2). However, the ring methyl groups of the SAP isomer of Lu-6 are also positioned to face a steric repulsion with the arm-methyl; the NOE spectrum of SAP of Lu-6 (Figure 5) has indeed shown the cross peak between the two methyl groups. The steric repulsion between the two methyl groups in SAP was calculated to be ~1.6 kcal/mol higher than in TSAP (Table S1), and thus imparts the SAP isomer of Lu-6 the stability of ΔE = −11.0 kcal/mol.
The EP map for the SAP isomer of Lu-5 (Figure 7) displays a weakened ES repulsion between the arm methyl and the neighboring carboxylate, as indicated by the low electron density on the carboxylate oxygen atoms. This ES repulsion, albeit stronger, is also seen for TSAP of Lu-6. The methyl-methyl repulsions are also observed in both TSAP of Lu-5 and SAP of Lu-6.
Figure 7.
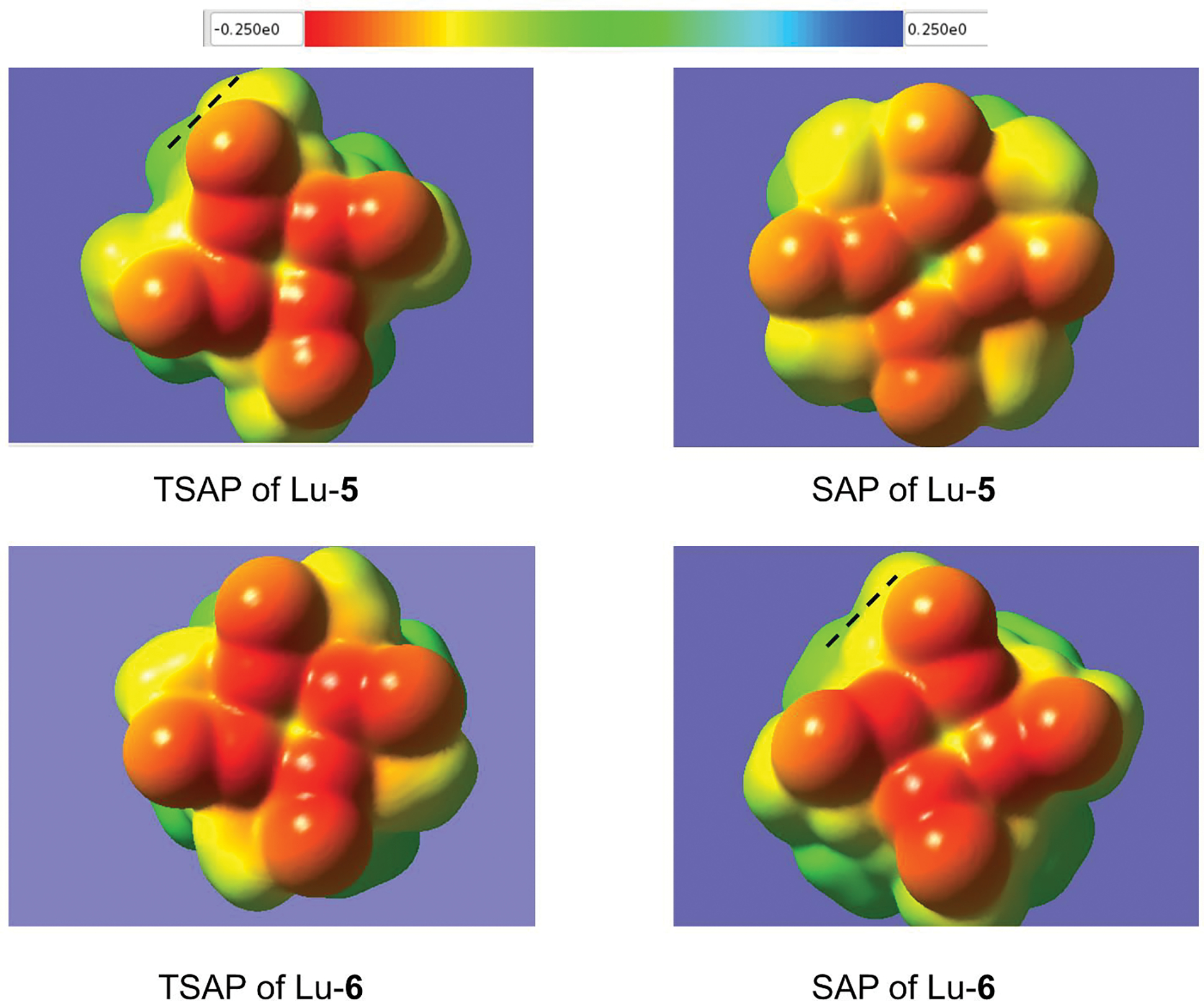
Electrostatic potential maps based on the isodensity contour at 0.001 e/bohr3 for the conformers of Lu-5 and Lu-6 all with the δδδδ ring. The broken lines indicate the methyl-methyl repulsion. The potentials in au are colored according to the inset.
Conformers of 5 and 6 complexed with La, Pr, and Yb
The strong ES repulsion between the arm methyl and the neighboring carboxyl as shown in the TSAP isomer of Ln-6 (Figure 7), likely makes SAP more stable in solution across the Ln series.[10] The present QM calculations, however, indicate that the SAP stabilization of Ln-6 becomes smaller with increasing radius of the Ln3+ ion. The ΔE decreases to −5.1 kcal/mol for Yb-6, to −3.4 kcal/mol for Pr-6, and then to −2.1 kcal/mol for La-6 (Table 3). A progressive weakening of the ES repulsion in the TSAP isomer with the increase of the Ln3+ ion size might account for this trend. Our calculations further indicate that the apical water bound to the Ln complexes attenuates the ES repulsion. For example, the ΔE for La-6 without bound water was estimated as −3.1 kcal/mole rather than −2.1 kcal/mol with the bound water. While less pronounced, a similar trend was observed for the Ln-5 concomitant with the stability transition from TSAP to SAP as the ion size increases.
Table 3.
Stability of the conformers of Ln-5 and Ln-6
| Ln-5 ΔE (SAP − TSAP) (kcal/mol) |
Ln-6 ΔE (SAP − TSAP) (kcal/mol) |
||
|---|---|---|---|
| La | −2.1 | La | −2.1 |
| Pra | −1.8 | Pra | −3.4 |
| Yba | 0.5 | Yba | −5.1 |
| Lu | 1.3 | Lu | −11.0 |
Ref. [10]. For the complexes of La3+ and Pr3+, energies were calculated with the bound water.
Dynamics of interconversion in the Lu complexes
The solution structures of Ln complexes and its dynamics had been investigated at both experimental [9a,11,12a] and theoretical[12, 14] levels. Herein, the reaction paths for the ring flip and the arm-rotation of Lu-1 were constructed first to visualize the interconversion processes depicted in Figure 1, and also to compare the rotational energy barrier for Lu-1 with those for Lu-4, Lu-5, and Lu-6 (Table 4).
Table 4.
Calculated bond distances and energy barriers via arm rotation for Lu3+ complexed with DOTA and its methylated derivatives
| Chelate | State | Lu−N (Å) | Lu−O (Å) | ΔE (kcal/mol) | ΔG≠ (kcal/mol) |
|---|---|---|---|---|---|
| 1 | TSAP | 2.55 | 2.26 | 0 | 0 |
| 1 | TS | 2.68 | 2.23 | 20.9 | 18.7 |
| 4 | TSAP | 2.56 | 2.25 | 0 | 0 |
| 4 | TS | 2.68 | 2.25 | 24.2 | 23.5 |
| 5 | TSAP | 2.57 | 2.20 | 0 | 0 |
| 5 | TS | 2.70 | 2.22 | 32.0 | 32.2 |
| 6 | SAP | 2.56 | 2.25 | 0 | 0 |
| 6 | TS | 2.71 | 2.22 | 26.9 | 27.5 |
The path for the ring inversion for TSAP [Δ(δδδδ)] of Lu-1 to SAP [Δ(λλλλ)] was constructed by successively varying the dihedral angle of ϕ1 clockwise. As shown in Figure 8, this inversion involves the three intermediate states that are higher in ΔG of 6.2 to 8.7 kcal/mol with respect to the TSAP isomer, as well as the three TSs with the free energy of activation (ΔG≠) ranging from 15.6 to 17.9 kcal/mol. The energy barriers mainly arise from the constrained dihedral angle of ϕ1, as shown for the TS3 having the dihedral angle of ϕ1 = 7.2°. While the TS3 has the highest energy barrier, it sits only 9.2 kcal/mol above the second intermediate state (Int2) having the ring conformation of (λλδδ). Accordingly, the rate-determining step for the ring flip is the formation of the Int1 via TS1 with ΔG≠ of 15.6 kcal/mol. Likewise, the inversion of SAP to TSAP has a ΔG≠ of 15.9 kcal/mol, which is in good agreement with the experimental ΔG≠ of 15.8 kcal/mol[11]. The attempt to calculate the barrier for the synchronized TS was not successful due to the high ring strain ( > 40 kcal/mol), strongly suggesting that the ring inversion occurs in a stepwise manner. A previously reported high-energy barrier (~61.9 kcal/mol) for the ring inversion of La-DOTA via a symmetrical TS[12e] is in line with our finding.
Figure 8.
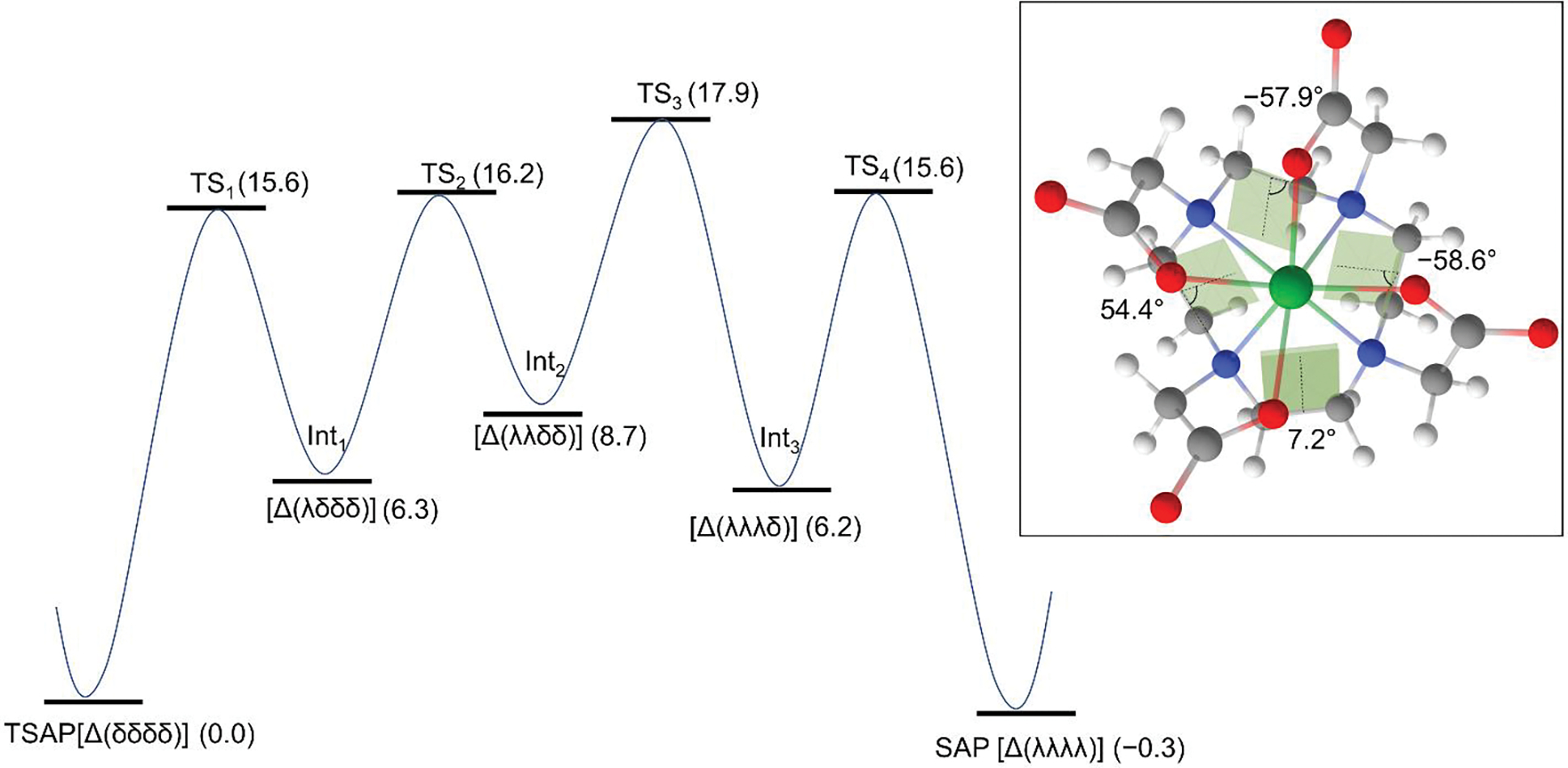
Reaction path for the ring inversion of TSAP [Δ(δδδδ)] to SAP [Δ(λλλλ)] for Lu-DOTA(1). The path for the ring inversion was constructed by successively varying the dihedral angle of ϕ1 clockwise. The values in parentheses represent the relative Gibbs free energy (ΔG) in kcal/mol at 298.15 K with respect to the TSAP isomer. The inset depicts the structure of the TS3; the shaded areas represent the dihedral angles of ϕ1.
Unlike the ring inversion, the variation of the dihedral angle (Lu−N−C−C) of a single arm of TSAP [Δ(δδδδ)] from −35° to 40° gave rise to a continuous increase in energy due to the ES repulsion with the neighboring carboxyl. The absence of intermediate state indicates that the conversion of TSAP [Δ(δδδδ)] to SAP [Λ(δδδδ)] via arm rotation occurs in a synchronized manner to minimize the ES repulsion, as indicated by the symmetrical TS geometry (Figure 9A). The Lu−N bond distance in the TS is weakened to 2.68 Å, as compared to 2.55 Å in TSAP, while the Lu−O distance (2.23 Å) of the TS remains comparable to that of the TSAP isomer (2.26 Å). The weakening of the Lu−N bond in the TS is likely responsible for the ΔG≠ of 18.7 kcal/mol for the TSAP to SAP conversion and the ΔG≠ of 19.0 kcal for the reverse process. A previous study[12a] also reported the comparable ΔG≠ of 19.8 kcal/mol for the conversion of SAP to TSAP. The experimental ΔG≠ of 15.6 kcal/mol for the SAP to TSAP conversion[11] is 3.4 kcal/mol lower than our calculated ΔG≠, and this discrepancy likely arises from the lack of explicit solvation in QM calculations that may attenuate the ES repulsion, particularly in the TS. This notion is further supported by the better agreement between experiment and theory of the energy barrier (ΔΔG≠ = ~1 kcal/mol) for the arm rotation of Eu-DOTA[14a] when two water molecules were included in the calculations.
Figure 9.
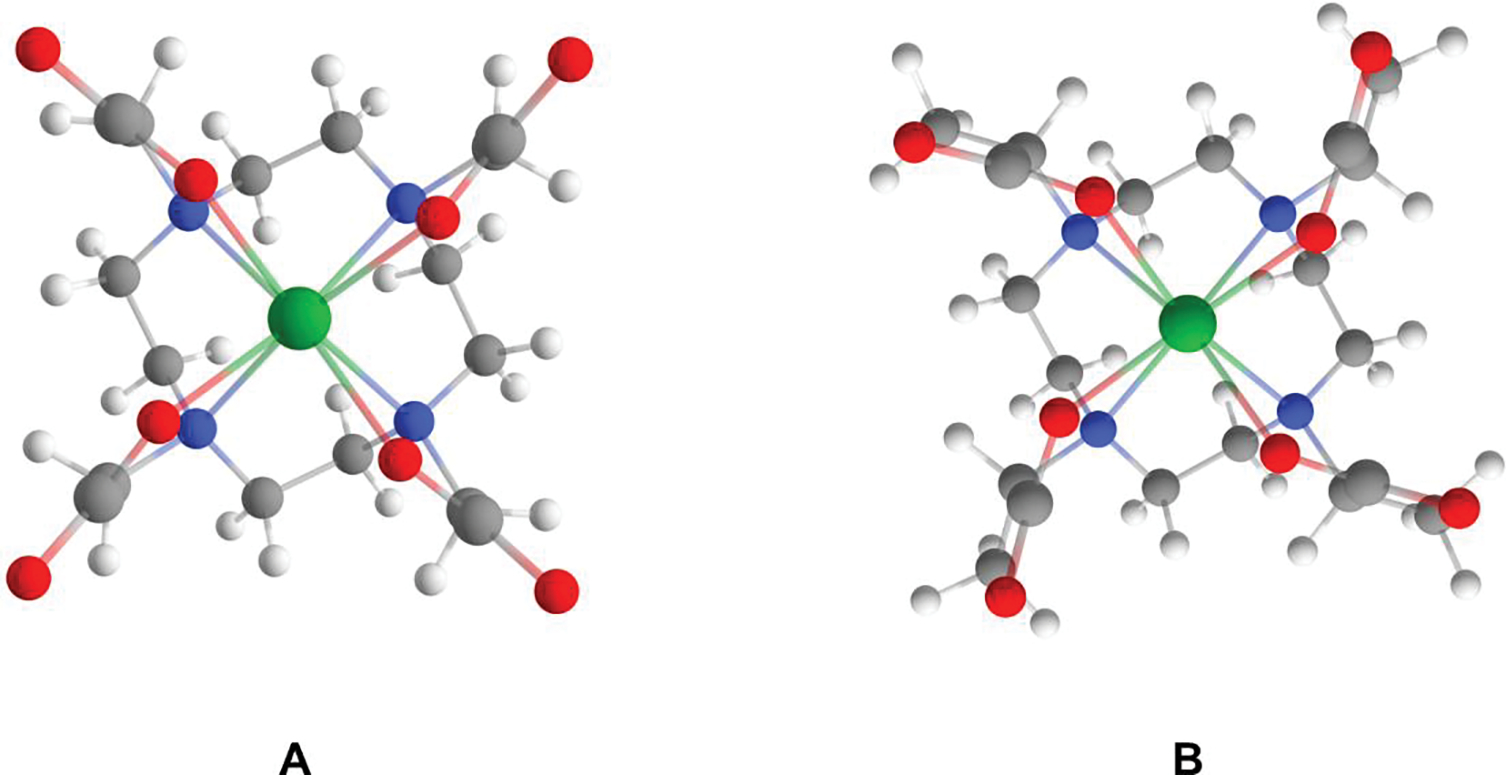
Geometry optimized structures of TSs for the conversion of TSAP[Δ(δδδδ)] to SAP[Λ(δδδδ)] via arm rotation in Lu-1 (A) and Lu-4 (B).
Due to the ring strain, the enantiomerization of Lu-1 (Figure 1), i.e., Δ(δδδδ) ↔ Λ(λλλλ) or Λ(δδδδ) ↔ Δ(λλλλ) is unlikely to occur in a concerted manner. Instead, a discrete path either via ring flipping followed by synchronized arm rotation or vice versa is kinetically much more feasible for the enantiomerization.
The Lu−N and Lu−O distances of Lu-4 in both ground state (GS) and TS differ no more than 0.01 Å with respect to those of Lu-1. The ΔG≠ for the arm rotation in Lu-4, however, was calculated to be 4.8 kcal/mol higher than the ΔG≠ of 18.7 kcal/mol for Lu-1, which can be attributed to the steric repulsion between the arm methyl and the ethylenic ring hydrogens in the TS (Figure 9B).
Interconversion in Lu-5 and Lu-6 and Variable Temperature 1H NMR Study
The TS geometry of Lu-5 (Figure S3, inset) is characterized by the longer distance of Ln−N (2.70 Å) as compared to that (2.57 Å) of TSAP; the average dihedral angle ϕ2 is 8.5°, intermediate between TSAP (−47.2°) and SAP (27.0°). At the TS, both the ring and the arm methyl groups are close to one another and thus manifest a stronger steric repulsion than in TSAP, resulting in the high energy barrier of 32.2 kcal/mol. Variable temperature dependance of 1H NMR spectra of Lu-5 (Figure S3) indicated no observation of the increase in the intensity of the minor peaks and the coalescence of any peaks as the temperature increased from 25 °C to 85 °C. These observations are consistent with the calculated ΔG≠ of 32.2 kcal/mol, which significantly hinders the conversion of the TSAP isomer of Lu-5 to a less stable SAP, even at 85 °C. By contrast, our previous NMR study on both Pr-5 and Eu-5 had shown a marked increase in the intensity of the minor TSAP peaks (e.g., from 3 % to 18 % for Eu-5) relative to the SAP peaks as the temperature increased from 25 °C to 80 °C[8], indicating that the interconversion energy barrier for both Pr-5 and Eu-5 is substantially lower as compared with Lu-5.
The rotational energy barrier for Lu-6 was calculated to be 27.5 kcal/mol, 4.7 kcal/mol lower than for Lu-5. This energy lowering is due to the arm R-methyl pointing away from the ring methyl in the TS (Figure S4, inset). As in the case of Lu-5, no new peaks appeared for Lu-6 (Figure S4) at high temperatures. The calculated ΔG≠ of 27.5 kcal/mol, however, is not high enough to prevent the conversion of the more stable SAP to TSAP, indicating that thermodynamics, not kinetics, is responsible for the absence of any new peaks for Lu-6 at high temperatures.
Future Study
Conjugating lanthanide DOTA derivatives to a macromolecule is of value to extract structural information of proteins in solution. Rigidification of the lanthanide tag minimizes motional averaging in solution, which generates large PCS. The rigidified polymethylated Ln-5 and Ln-6 are therefore excellent Ln-tag candidates for this purpose. To be utilized as Ln-tag for protein characterization, one of the arms of both Ln-5 and Ln-6 has to be modified with a thiol reactive moiety.[7c,e–f,8,10] Both Ln-5 and Ln-6 were modified with a pyridyl disulfide. However, despite the fact that Ln-(4S,4R)-M8Spy complexes have only SAP isomer in solution, the Dy-(4S,4R)-M8 tag upon conjugation to the protein had shown paramagnetic susceptibility tensor with smaller values for Δχax and Δχrh compared to Dy-(4S,4S)-M8.[7e] Previous studies had shown that magnetic anisotropy and in consequence, the pseudocontact shifts of lanthanide complexes can be significantly affected by small changes in the lanthanide ion coordination environment.[19] Recently, Ln-6 analogs with a rigid non-flexible aromatic moiety (thiazole linker) yielded larger PCS when compared to the pyridyl disulfide Ln-6 analogs were reported.[7f] This finding emphasizes the important role of the rigid and short linkers in obtaining large PCS. In this endeavor, the synthesis of DOTA-like chelates bearing various alkyl substituents with electron donating or withdrawing group and rigid linker is ongoing with the aim of relating the isomer stability and/or rotational energy barriers to the magnitude of PCS.
Conclusions
Via NOE and quantum chemical studies, we have demonstrated that the newly synthesized Lu-5 and Lu-6 in solution exist as TSAP and SAP, respectively. Our QM calculations have quantified that even a single S-methyl substitution in the ring of DOTA makes the SAP [Λ(δδδδ)] isomer of the Lu complex more stable (ΔE = −7.6 kcal/mol) than TSAP with the inverted ring [Λ(λλλλ)], and thus locks the ring in the δδδδ conformation. By contrast, due to the ES repulsion with the neighboring acetate, the arm methyl freezes out the high energy rotational conformer TSAP or SAP, depending upon the chirality of the arm methyl as shown for Lu-3 and Lu-4. Of the two methyl groups, the ring methyl introduces a larger thermochemical and structural perturbation. When both the ring and the arm are methylated, the interplay of the steric and ES repulsions modulates the conformer stability, as seen in Lu-5 and Lu-6. The ES repulsion between the arm methyl and the neighboring carboxylate is a driving force for destabilizing both SAP of Lu-5 and TSAP of Lu-6. However, the steric repulsion between the ring and arm methyl groups attenuates the stability of TSAP of Lu-5 and SAP of Lu-6 over their corresponding conformers.
We have also shown that Lu-5 has a high free energy of activation for the arm rotation ( ΔG≠ = 32.2 kcal/mol) consistent with the absence of the increase in the intensity of the minor SAP peaks for Lu-5 at high temperatures. The calculated ΔG≠ of 27.5 kcal/mol for Lu-6 is not high enough to prevent the conversion of the more stable SAP to TSAP at all temperatures. The significantly higher stability of SAP (ΔE = −11.0 kcal/mol) over TSAP, however, indicates that thermodynamics is responsible for the absence of new peaks for Lu-6 at all temperatures.
The present study has provided a structural basis for the development of DOTA-like chelates that give rise to a single isomer in solution when complexed with Ln ions, and this may lead to design of better NMR shift probes for investigating protein dynamics and interactions with the paramagnetic Ln ions such as Pr3+, Gd3+, Tm3+, and Yb3+. Our in-depth investigation of the Lu-DOTA-based complexes will also provide insight for the development of DOTA based chelators with the enhanced thermochemical stability for 177Lu in nuclear medicine.
Experimental
NMR Spectroscopy
Lu-4S-4S-M4DOTMA and Lu-4S-4R-M4DOTMA complexes were synthesized according to the published procedure. [7d, 8, 13] The 1H, COSY, NOESY/ROESY NMRs were measured on a Varian 400 MHz NMR at 25°C. [7d, 8, 13] The 1H, COSY, NOESY/ROESY NMRs were measured on a Varian 400 MHz NMR at 25°C. Variable temperature 1H NMR spectra were recorded on a Varian 400 MHz NMR equipped with a standard temperature control unit at 25, 45, 65 and 85°C. The samples were pre-equilibrated for 15 minutes prior to measurements.
Quantum Chemistry
All quantum chemical calculations were done with Gaussian 09[15] on the anionic Ln-complexes. We employed M06-L[16] for geometry optimization and energy calculations. B3LYP was utilized for calculating the proton shielding tensors with the gauge-independent atomic orbital (GIAO) method.[17] For the Ln atom, the pseudopotential SDD[18] for 28 core electrons and the SDD basis set was used for the valence electrons; for all other atoms, 6-31+G(d) basis set was utilized for geometry minimization, and 6-311+G(2d,p) was used for NMR calculations on the geometry optimized at the level of B3LYP/6-31+G(d). The calculated proton chemical shift for TMS is 31.94 ppm. Water solvent effect was adopted for all calculations using the Polarizable Continuum Model (PCM) as implemented in Gaussian 09 software. TSs were obtained with the keyword of opt=qst3, which allows us to ascertain a TS via the interpolation of the two GS conformers and an approximate TS. Further refinements of the TS were also carried out with the keywords opt=(ts, calcfc, noeigentest). Frequency calculations were performed to verify the minima and TS. All real frequencies were observed for minima and only one imaginary frequency for TS. Gibbs free energy were calculated at 298.15 K. The calculated structures of Pr-4S,4S-M4DOTMA from the previous study[8] were utilized to obtain the Lu and La complexes. The hydration state of all Lu complexes was assumed to be zero.
Supplementary Material
Acknowledgments.
This study was supported by the Intramural Research Program of the National Institutes of Heart, Lung, and Blood and the Center for Information Technology.
The quantum chemical study utilized PC/LINUX clusters at the Center for Molecular Modeling of the NIH (http://cit.nih.gov) and the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). YS Lee thanks Sergio Hassan of the BCBB/NIAID for valuable discussion and comments.
Footnotes
Supporting Information: Structures of Lu-MDOTA and Lu-4, variable temperature dependance of 1H NMR spectra of Lu-5 and Lu-6 as well as the coordinates for the Lu complexes are available.
References
- [1].(a) Aime S, Barge A, Botta M, De Sousa AS, Parker D, Angew. Chem. Int. Ed. Engl. 1998, 37, 2673–2675; [DOI] [PubMed] [Google Scholar]; (b) Zhang S, Winter P, Wu K, Sherry AD, J. Am. Chem. Soc. 2001, 123, 1517–1518. [DOI] [PubMed] [Google Scholar]
- [2].(a) Bertini I, Luchinat C, Curr. Opin. Chem. Biol. 1999, 3, 145–151; [DOI] [PubMed] [Google Scholar]; (b) Mittag T, Forman-Kay JD, Curr. Opin. Struct. Biol. 2007, 17, 3–14; [DOI] [PubMed] [Google Scholar]; (c) Pintacuda G, John M, Su XC, Otting G, Acc. Chem. Res. 2007, 40, 206–212; [DOI] [PubMed] [Google Scholar]; (d) Rodriguez-Castaneda F, Haberz P, Leonov A, Griesinger C, Magn. Reson. Chem. 2006, 44, S10–S16. [DOI] [PubMed] [Google Scholar]
- [3].Brittain HG, Desreux JF, Inorg. Chem. 1984, 23, 4459–4466. [Google Scholar]
- [4].(a) Gourni E, Henriksen G, Molecules 2017, 22; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, Mittra E, Kunz PL, Kulke MH, Jacene H, Bushnell D, O’Dorisio TM, Baum RP, Kulkarni HR, Caplin M, Lebtahi R, Hobday T, Delpassand E, Van Cutsem E, Benson A, Srirajaskanthan R, Pavel M, Mora J, Berlin J, Grande E, Reed N, Seregni E, Oberg K, Lopera Sierra M, Santoro P, Thevenet T, Erion JL, Ruszniewski P, Kwekkeboom D, Krenning E, Investigators N-T, Engl N. J. Med. 2017, 376, 125–135; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Czerwinska M, Bilewicz A, Kruszewski M, Wegierek-Ciuk A, Lankoff A, Molecules 2020, 25; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dai L, Jones CM, Chan WTK, Pham TA, Ling X, Gale EM, Rotile NJ, Tai WC, Anderson CJ, Caravan P, Law GL, Nat. Commun. 2018, 9, 857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Helm L, Merbach AE, Tóth É, ProQuest (Firm), The chemistry of contrast agents in medical magnetic resonance imaging, 2nd ed., John Wiley & Sons Inc., Hoboken, N.J., 2013. [Google Scholar]
- [6].Regueiro-Figueroa M, Platas-Iglesias C, J. Phys. Chem. A 2015, 119, 6436–6445. [DOI] [PubMed] [Google Scholar]
- [7].(a) Aime S, Botta M, Garda Z, Kucera BE, Tircso G, Young VG, Woods M, Inorg. Chem. 2011, 50, 7955–7965; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Woods M, Katherine MP, Valente EJ, Kucera BE, Young VG Jr., Chem. Eur. J. 2019, 25, 9997–10005; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Haussinger MD, Huang JR, Grzesiek S, J. Am. Chem. Soc. 2009, 131, 14761–14767; [DOI] [PubMed] [Google Scholar]; (d) Ranganathan RS, Pillai RK, Raju N, Fan H, Nguyen H, Tweedle MF, Desreux JF, Jacques V, Inorg. Chem. 2002, 41, 6846–6855; [DOI] [PubMed] [Google Scholar]; (e) Joss D, Walliser RM, Zimmerman K, Haussinger D, J. Biomol. NMR 2018, 72, 29–38; [DOI] [PubMed] [Google Scholar]; (f) Muntener T, Kottelat J, Huber A, Haussinger D, Bioconjugate Chem. 2018, 29, 3344–3351; [DOI] [PubMed] [Google Scholar]; (g) Kumas C, Fernando WS, Zhao P, Reguiero-Figueroa M, Kiefer GE, Martins AF, Platas-Igelsias C, Sherry AD, Inorg. Chem. 2016, 55, 9297–9305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Opina ACL, Strickland M, Lee YS, Tjandra N, Byrd RA, Swenson RE, Vasalatiy O, Dalton Trans. 2016, 45, 4673–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].(a) Aime S, Botta M, Ermondi G, Inorg. Chem. 1992, 31, 4291–4299; [Google Scholar]; (b) Aime S, Botta M, Fasano M, Marques MPM, Geraldes CFGC, Pubanz D, Merbach AE, Inorg. Chem. 1997, 36, 2059–2068. [DOI] [PubMed] [Google Scholar]
- [10].Opina ACL, Strickland M, Lee YS, Tjandra N, Swenson RE, Vasalatiy O, Inorg. Chem. 2019, 58, 15788–15800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Aime S, Barge A, Botta M, Fasano M, Ayala JD, Bombieri G, Inorg. Chim. Acta. 1996, 246, 423–429. [Google Scholar]
- [12].(a) Purgel M, Baranyai Z, de Blas A, Rodriguez-Blas T, Banyai I, Platas-Iglesias C, Toth I, Inorg. Chem. 2010, 49, 4370–4382 ; [DOI] [PubMed] [Google Scholar]; (b) Regueiro-Figueroa M, Ruscsák E, Esteban-Gómez D, Charbonnière LJ, Tircsó G, Tóth I, de Blas A, Rodríguez-Blas T, Platas-Iglesias C, Inorg. Chem. 2011, 50, 4125–4141; [DOI] [PubMed] [Google Scholar]; (c) Regueiro-Figueroa M, Esteban-Gómez D, de Blas A, Rodríguez-Blas T, Platas-Iglesias C, Eur. J. Inorg. Chem. 2010, 3586–3595; [DOI] [PubMed] [Google Scholar]; (d) Natrajan L, Khoabane NM, Dadds BL, Muryn CA, Pritchard RG, Heath SL, Kenwright AM, Kuprov I, Faulkne S, Inorg. Chem. 2010, 49, 7700–7709; [DOI] [PubMed] [Google Scholar]; (e) Di Vaira M, Stoppioni P, New J Chem. 2002, 26, 136–144. [Google Scholar]
- [13].Ranganathan RS, Raju N, Fan H, Zhang X, Tweedle MF, Desreux JF, Jacques V, Inorg. Chem. 2002, 41, 6856–6866. [DOI] [PubMed] [Google Scholar]
- [14].(a) Blahut J, Hermann P, Tosner Z, Platas-Iglesias C, Phys. Chem. Chem. Phys. 2017, 19, 26662–26671; [DOI] [PubMed] [Google Scholar]; (b) Jacques V, Desreux JF, Inorg. Chem. 1994, 33, 4048–4053; [Google Scholar]; (c) Cosentino U, Villa A, Pitea D, Moro G, Barone V, Maiocchi A, J. Am. Chem. Soc. 2002, 124, 4901–4909 [DOI] [PubMed] [Google Scholar]
- [15].Frisch MJT, G. W.; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ, Gaussian 09, (Revision D.01), Gaussian Inc., 2013. [Google Scholar]
- [16].Zhao Y, Truhlar DG, J. Chem. Phys. 2006, 125 194101. [DOI] [PubMed] [Google Scholar]
- [17].(a) Wolinski K, Hinton JF, Pulay P, J. Am. Chem. Soc. 1990, 112, 8251–8260; [Google Scholar]; (b) Cheeseman JR, Trucks GW, Keith TA, Frisch MJ, J. Chem. Phys.1996, 104, 5497–5509. [Google Scholar]
- [18].Cao XY, Dolg M, J Chem Phys 2001, 115, 7348–7355. [Google Scholar]
- [19].(a) Parker D, Suturina EA, Kuprov I, Chilton NF, Acc. Chem. Res. 2020, 53, 1520; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Harnden AC, Suturina EA, Batsanov AS, Senanayake PK, Fox MA, Mason K, Vonci M, McInnes EJL, Chilton NF, Parker D, Angew. Chem. 2019, 131, 10396 –10400; [DOI] [PubMed] [Google Scholar]; (c) Kreidt E, Bischof C, Platas-Iglesias C, Seitz M, Inorg. Chem. 2016, 55, 5549–5557; [DOI] [PubMed] [Google Scholar]; (d) Blackburn OA, Chilton NF, Keller K, Tait CE, Myers WK, McInnes EJL, Kenwright AM, Beer PD, Timmel CR, Faulkner S, Angew. Chem. Int. Ed. 2015, 54, 10783; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu T, Nonat A, Beyler M, Regueiro-Figueroa M, Nchimi Nono K, Jeannin O, Camerel F, Debaene F, Cianfeŕani - Sanglier S, Tripier R, Platas-Iglesias C, Charbonnier̀e LJ, Angew.Chem.,Int. Ed. 2014, 53, 7259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.