

Abstract
The direct binding of bacteria to platelets is a postulated major interaction in the pathogenesis of infective endocarditis. To identify bacterial components that mediate platelet binding by Streptococcus mitis, we screened a Tn916ΔE-derived mutant library of S. mitis strain SF100 for reduced binding to human platelets in vitro. Two distinct loci were found to affect platelet binding. The first contains a gene (pblT) encoding a highly hydrophobic, 43-kDa protein with 12 potential membrane-spanning segments. This protein resembles members of the major facilitator superfamily of small-molecule transporters. The second platelet binding locus consists of an apparent polycistronic operon. This region includes genes that are highly similar to those of Lactococcus lactis phage r1t and Streptococcus thermophilus phage 01205. Two genes (pblA and pblB) encoding large surface proteins are also present. The former encodes a 107-kDa protein containing tryptophan-rich repeats, which may serve to anchor the protein within the cell wall. The latter encodes a 121-kDa protein most similar to a tail fiber protein from phage 01205. Functional mapping by insertion-duplication mutagenesis and gene complementation indicates that PblB may be a platelet adhesin and that expression of PblB may be linked to that of PblA. The combined data indicate that at least two genomic regions contribute to platelet binding by S. mitis. One encodes a probable transmembrane transporter, while the second encodes two large surface proteins resembling structural components of lysogenic phages.
The pathogenesis of infective endocarditis is a complex phenomenon, involving numerous host-pathogen interactions. Infection of the endocardium is initiated by the attachment of blood-borne organisms to platelets, fibrin, and extracellular matrix proteins on the damaged valve surface (10, 19). The relative importance of these host binding factors to colonization is uncertain. However, since a variety of endocarditis-associated organisms, including streptococci and staphylococci, can bind platelets directly in vitro (16, 36, 42, 43), it is likely that direct binding to platelets in vivo contributes to the initiation of endocardial infection.
The subsequent development of mature, macroscopic vegetations may also be mediated in part by the direct binding of platelets to bacteria. Binding of circulating platelets to organisms on the valve surface may result in the further accumulation of platelets at the site of endocardial infection. In addition, such binding may be a mechanism for the reattachment of bacteria shed into the circulation back onto the valve surface (33). Evidence for these processes in vivo came initially from histologic studies of animals with experimental endocarditis, in which the progressive accumulation of platelets and bacteria was observed at the outer margins of maturing vegetations (9). More recently, the induction of selective thrombocytopenia in rabbits with early endocarditis has resulted in vegetations of significantly reduced mass, indicating that platelets continue to be deposited on the infected endocardium and are a major structural component of vegetations (35). In addition, diminished platelet binding in vitro by Staphylococcus aureus has been associated with reduced virulence in an animal model of endocarditis, as manifested by decreased concentrations of bacteria within vegetations and a reduced incidence of peripheral embolization and hematogenous dissemination of infection (34).
Among the viridans group streptococci, Streptococcus mitis is a leading cause of infective endocarditis (8, 27). In addition to its long-recognized association with endocardial infection, this organism has recently emerged as a major cause of bacteremia in the immunocompromised host (4, 15, 26, 38). Therapy of S. mitis infections has become problematic, due to a high prevalence of multidrug resistance reported in this organism (6, 25). Despite the clinical importance of S. mitis, few studies have addressed its virulence determinants, particularly with regard to endocarditis and the role of platelets in pathogenesis (23).
In view of the role of platelet binding by endocarditis-associated organisms and the importance of S. mitis as an endocardial pathogen, we sought to identify bacterial components that contribute to platelet binding by strain SF100. These studies indicate that platelet binding by this organism is a complex interaction that involves at least two distinct loci. As shown below, one is likely to encode a small molecule transmembrane transporter and the other encodes cell surface proteins resembling structural components of streptococcal phages.
MATERIALS AND METHODS
Bacterial strains, plasmids, and reagents.
The bacterial strains and plasmids used in this study are listed in Table 1. Strain SF100 is a streptomycin-resistant variant of S. mitis 12021, which was isolated from the blood of a patient with infective endocarditis. The species of this strain was confirmed by biochemical testing at the Centers for Disease Control (Atlanta, Ga.) and by 16S rRNA sequencing (MIDI Labs, Newark, Del.). Enterococcus faecalis RH110 carries Tn916ΔE, a derivative of Tn916 in which the tet gene has been replaced by erm (28). These strains and their variants were grown in Todd-Hewitt broth (THB; Difco Laboratories, Detroit, Mich.) or on sheep blood agar (Remel, Lenexa, Kans.) at 37°C in a 5% CO2 environment. When indicated, antibiotics were added to the media at the following concentrations: 15 μg of erythromycin per ml, 750 μg of streptomycin per ml, and 5 μg of chloramphenicol per ml. Escherichia coli strains were grown in Luria-Bertani broth containing 100 μg of ampicillin per ml or 15 μg of chloramphenicol per ml when appropriate. Tyrode's salts, trypsin, sodium lauroyl sarcosine (SLS), and Dulbecco's phosphate-buffered saline (DPBS) were purchased from Sigma (St. Louis, Mo.).
TABLE 1.
Strains and plasmids used in this work
| Strain or plasmid | Relevant characteristicsa | Reference or source |
|---|---|---|
| E. coli | ||
| DH5α | F− r−m+ ø80dlacZΔM15 | Gibco BRL |
| MC1061 | F−araD139 Δ(ara-leu)7696 galE15 galK16 Δ(lac)X74 rpsL (Strr) hsdR2 (rk− mK+) mcrA mcrBl | 41 |
| BL21 (DE3) | Expression host, inducible T7 RNA polymerase | Novagen |
| Streptococcus | ||
| RH110 | E. faecalis OG1RF::Tn916ΔE rif fus erm | 28 |
| 12021 | S. mitis | This study |
| SF100 | Smr derivative of S. mitis strain 12021 | This study |
| PS101 | SF100 pblT::Tn916ΔE | This study |
| PS116 | SF100 pblA::Tn916ΔE | This study |
| PS163 | SF100 pblT::pVA891 | This study |
| PS301 | SF100 pblA::pVA891 | This study |
| PS344 | SF100Δ[ORF1-pblB] | This study |
| PS345 | SF100 pblB::pVA891 | This study |
| PS361 | SF100 pblR::pVA891 | This study |
| PS385 | PS301(pDC123) | This study |
| PS386 | PS301(pSF116A) | This study |
| PS389 | PS345(pDC123) | This study |
| PS390 | PS345(pSF116B) | This study |
| PS416 | PS301(pSF116B) | This study |
| Plasmids | ||
| pWE15 | Cos, Apr | 39 |
| pBluescript | Apr | Stratagene |
| pVA891 | Tcr, cat, ori (E. coli), erm (gram positive) | 18 |
| pDC123 | cat phoZ, streptococcal expression vector with blue-white screening | 7 |
| pSF116A | pblA cloned in pDC123 | This study |
| PSF116B | pblB cloned in pDC123 | This study |
Smr, streptomycin resistance; Apr, ampicillin resistance; Tcr, tetracycline resistance. Cos, recognition site for packaging by phage lambda.
Quantitative assay for binding to immobilized platelets.
The binding of streptococci to human platelets was assessed quantitatively as described previously (34). In brief, washed, fixed human platelets were immobilized in poly-l-lysine-coated 22-mm-diameter tissue culture wells, producing monolayers of 75 to 90% confluence. To reduce nonspecific adherence, the wells were then treated with a casein solution (1× blocking reagent [Roche, Indianapolis, Ind.] in DPBS) for 1 h at room temperature. After the blocking solution was removed by aspiration, the wells were inoculated with approximately 5 × 106 CFU of streptococci suspended in 0.5 ml of DPBS and incubated at 37°C for 2 h, with gentle rocking to enhance mixing. Unbound bacteria were then removed by washing, and the platelet-bound organisms were recovered by treating the platelet monolayers with 1 mg of trypsin per ml in DPBS. The number of organisms bound was determined by plating serial dilutions of the suspension onto blood agar, and binding was expressed as a percentage of the inoculum. In control studies, trypsinization of SF100 had no effect on viability and was found to allow complete recovery of bound organisms. For experiments in which SF100 was treated with trypsin before being tested in the binding assay, bacteria were incubated for 60 min at room temperature in DPBS containing 1 mg of trypsin per ml and then washed repeatedly. All studies were done in triplicate, using platelets from multiple human donors.
Transposon mutagenesis and selection of low-binding variants.
Transposon mutagenesis of SF100 was by done by filter mating, as described previously (29). In brief, 1 ml of an exponential-phase culture of SF100 (∼5 × 108 CFU) was combined with 10 ml of an exponential-phase culture of E. faecalis strain RH110. Cells were collected on a sterile 0.2-μm-pore-size filter (Millipore, Bedford, Mass.), placed on a blood agar plate, and incubated overnight at 37°C. The filter was then transferred to 10 ml of THB containing streptomycin and erythomycin and incubated for 3 h at 37°C to select for SF100 transconjugants.
To enrich for low-binding mutants, the bacterial suspension was washed twice with TEN buffer (50 mM Tris-HCl, 20 mM EDTA, 100 mM NaCl [pH 7.25]), suspended in 1 ml of Tyrode's solution, and centrifuged (100 × g for 10 min) onto platelets immobilized in a 35-mm tissue culture well. The plate was vortexed for 10 s to suspend nonadherent organisms, which were then collected and passaged again over immobilized platelets. After a total of 12 passages, the enriched suspension was plated on blood agar and incubated at 37°C for 18 h. The resultant colonies were picked and individually tested for reduction in platelet binding, using a previously described turbidimetric screening assay (22, 34). Clones thought to represent low-binding variants were then tested individually by the above quantitative binding assay.
Southern blot analysis.
Chromosomal DNA was isolated from streptococci by adding 6.5 ml of fresh THB and 0.1 g of solid glycine to 3.5 ml of an overnight culture and incubating it for 90 min at 37°C. The bacteria were centrifuged, resuspended in 1 ml of distilled H2O, and transferred to a microcentrifuge tube. Cell pellets were suspended in 100 μl of TE 50:5 (50 mM Tris, 5 mM EDTA [pH 8.0]), 50 μl of a lysis solution (50 mg of lysozyme per ml and 200 U of mutanolysin per ml in TE 50:5) was added, and the mixture was incubated for 1 h at 37°C. After addition of 340 μl of TE 50:5, 7.5 μl of 20% sodium dodecyl sulfate (SDS), and 2 μl of proteinase K (25 mg/ml), the suspensions were gently mixed and then incubated for 1 h at 37°C. An additional 200 μl of TE 50:5 was added, and the mixtures were extracted with 700 μl of phenol. After centrifugation, the aqueous phase was transferred to a Phase Lock Gel tube (Eppendorf, Westbury, N.Y.) and extracted twice with 2.5 ml of phenol-chloroform (1:1). The aqueous phase was then extracted with an equal volume of chloroform-isoamyl alcohol (24:1), and nucleic acid was precipitated from 400 μl of the aqueous phase by adding 40 μl of 3 M sodium acetate (pH 5.2) and 1 ml of ethanol. Pellets were rinsed with 70% ethanol in dH2O, resuspended in 100 μl of 10 mM Tris–1 mM EDTA (pH 8) containing 0.5 μg of DNase-free RNase (Roche), and incubated at 37°C until completely dissolved. Following treatment with restriction enzymes and electrophoresis, digested chromosomal DNA was transferred to positively charged nylon membranes (Roche) using a Trans-Blot semidry transfer apparatus (Bio-Rad, Hercules, Calif.). The membranes were hybridized with digoxigenin-labeled probes and developed with the CDP-Star chemiluminescent substrate as recommended by the supplier (Roche).
DNA sequence analysis.
To identify the sites of Tn916ΔE insertion in the low-binding mutants, chromosomal regions flanking the transposon were isolated by cloning EcoRI fragments in the cosmid vector pWE15 as described previously (3). Following Tn916ΔE excision from the cosmid clones, the EcoRI fragments (or portions thereof) were subcloned in pBluescript KS(−) or pBluescript SK(−) (Stratagene, La Jolla, Calif.) for sequence analysis by primer walking. Sequences upstream and downstream from the EcoRI fragment of PS101 (Fig. 1) were obtained by marker rescue of pVA891 from SacI- or ClaI-digested chromosomal DNA from strain PS163, which has pVA891 integrated in pblT. For PS116, a full-length EcoRI fragment (later estimated to be 20 kb by Southern blot analysis of SF100 chromosomal DNA) was not maintained in DH5α but, rather, tended to undergo deletion or rearrangement. However, a 2.3-kb HindIII fragment (Fig. 2, segment d) was maintained in one spontaneously deleted cosmid construct and was subcloned for sequence analysis. A chromosomal segment flanking the left end of Tn916ΔE, which includes the erythromycin resistance marker (Fig. 2, segment b), was obtained by direct cloning of HindIII-digested PS116 chromosomal DNA in pBluescript. Since these constructs were also unstable, the cloned fragment was amplified by PCR, using the M13 −40 universal primer and a primer complementary to bases 85 to 104 of the left end of the transposon (5′-CGAAAGCACATAGAATAAGG-3′), and the 2.0-kb PCR product was sequenced directly. The location of this fragment relative to segment d was confirmed by PCR amplification of SF100 chromosomal DNA, using a forward primer upstream from the PstI site in segment b and a reverse primer downstream from the XbaI site in segment d. The PstI-XbaI fragment (segment c) was then cloned in pBluescript and sequenced. Additional sequence was obtained after cloning products derived by inverse PCR of XbaI- or SacI-digested chromosomal DNA, using primers reading outward from the known sequence (segment e or segments a, f, and g, respectively).
FIG. 1.

Map of the PS101 locus, and site of insertion of Tn916ΔE. The broken arrow indicates an incomplete ORF. Bold letters correspond to the ribosome binding site of pblR. The underlined segment corresponds to the probable −10 region of the promoter element. The −35 region is within an inverted repeat, underscored with arrows. The site of insertion of Tn916ΔE was determined by cloning the left flank of the transposon (containing the erythromycin resistance gene) and then sequencing with a primer complementary to the left end of the transposon. E, EcoRI; H, HincII; C, ClaI.
FIG. 2.

Map of the PS116 locus. (A) Location of selected restriction sites, and fragments used for DNA sequence analysis. Segment d corresponds to the initially sequenced HindIII fragment. The sequence of segment b was obtained from a PCR product that was amplified from a metastable clone of the left flank of Tn916ΔE. Segment c was cloned after PCR amplification of the region from SF100 chromosomal DNA. Segment e was cloned from a PCR product derived by inverse PCR of XbaI-digested chromosomal DNA. Segments a, f, and g were cloned from products derived by inverse PCR of SacI-digested chromosomal DNA. E, EcoRI; H, HindIII; Hp, HpaI; N, NheI; P, PstI; RV, EcoRV; S, SacI; X, XbaI. (B) Genes and gene products. The speckled region of pblB encodes a protein sequence showing 47% similarity to PspA of S. pneumoniae and 42% similarity to M protein of S. pyogenes. cc, coiled-coil domain.
DNA sequencing was performed by the Biomolecular Resource Center at the University of California, San Francisco, Calif., using the ABI-Prism system (Applied Biosystems, Foster City, Calif.). The sequences were assembled, formatted, and translated using the Gene Construction Kit 2 software (Textco, Inc., West Lebanon, N.H.). Protein similarity searches were conducted using BLAST programs to search sequences in the nonredundant databases available at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov). Analysis of protein characteristics and secondary structure predictions were carried out using the Genetics Computer Group programs available from the Computer Graphics Laboratory at the University of California, San Francisco (http://www.sacs.ucsf.edu/Resources/webgcg) or using programs available from the Swiss Institute of Bioinformatics (http://www.expasy.ch/) indicated below.
Directed mutations.
Selected genes were mutated by insertion-duplication or by gene replacement, using pVA891 (18). For insertion-duplication mutagenesis, internal portions of the target genes were cloned in pVA891 and the resulting plasmids were propagated in Escherichia coli strain DH5α prior to introduction to SF100 by natural transformation. To create strain PS344, in which open reading frame 3 (ORF3), pblA, ORF4, and pblB were deleted by gene replacement, the HindIII-SacI fragment (Fig. 2, segment g) was cloned adjacent to the SacI-BglII fragment (from segment a [Fig. 2]) in the BamHI site of pVA891. This plasmid was linearized with SacI and then used to transform SF100. Recombination at the expected site was confirmed by Southern blot analysis of chromosomal DNA isolated from the transformants.
Complementation.
For trans-complementation of wild-type or mutated chromosomal genes, pblA and pblB were each cloned in the streptococcal expression vector pDC123 (7). The ribosome binding site and entire coding sequence of pblA or pblB was amplified by PCR, using the following BamHI-linked primers: 5′-AAGGATCCAATAGGAGGTGAGGATTAATGGCTACAG-3′ and 5′-AAGGATCCATTAGATTCCCTCCCTTGC-3′ for pblA or 5′-AAGGATCCTTGGAGGTATAAAATATGATTTACTT-3′ and 5′-AAGGATCCTTTGTTTGTCCTGTTCGTTCATGC-3′ for pblB. PCR products were then cloned in the BamHI site of pDC123 downstream from the constitutive cat/tet promoter. Plasmids were propagated in E. coli strain MC1061, and those with pblA or pblB in the proper orientation were used to transform S. mitis by electroporation.
Transformation of S. mitis.
Introduction of pVA891 derivatives into SF100 was accomplished by natural transformation. In pilot studies, the frequency of transformation of SF100 was found to be very low (<10−8 transformant per μg of DNA). To enhance the efficiency of transformation, a competence-stimulating peptide (CSP) specific for this S. mitis strain was identified, using the strategy of Håvarstein et al. (13). The comCDE locus of SF100 was amplified by PCR and sequenced. Analysis of comC indicated that the amino acid sequence of the CSP was DWRISETIRNLIFPRRK. This peptide was synthesized (Biomolecular Resource Center, University of California, San Francisco, Calif.) and was subsequently found to increase the transformation frequencies to approximately 10−5 transformant per μg of DNA. To transform SF100 with pVA891 derivatives, overnight cultures were diluted 100-fold in fresh THB supplemented with 20% heat-inactivated horse serum, 200 ng of CSP per ml, and 1 μg of DNA per ml. Transformation mixtures were incubated 8 h at 37°C and then plated on blood agar containing erythromycin.
Transformation with autonomously replicating plasmids was accomplished by electroporation. To generate electrocompetent bacteria, cells growing exponentially in THB were washed three times with ice-cold dH2O and then twice with ice-cold 0.3 M sucrose–10% glycerol in dH2O. The bacteria were then suspended in 0.3 M sucrose–10% glycerol at 1/50 of the original culture volume. A 40-μl aliquot of the cell suspension was combined with 300 ng of DNA, transferred to a 0.1-cm electroporation cuvette, and pulsed with 1.5 kV at a capacitance of 25 μF and resistance of 200 Ω, using a Gene Pulser apparatus (Bio-Rad). Then 500 μl of sterile THB–0.3 M sucrose was added immediately to the cuvette, and the cell suspension was incubated for 2 h at 37°C before being plated on blood agar containing the appropriate antibiotic.
Production of polyclonal antisera.
Since overexpression of full-length PblA or PblB was found to be toxic to E. coli host strains, two subdomains of each protein were used to generate antisera. For PblA, one domain was generated by in-frame fusion of the HindIII-BglII fragment of segment b (Fig. 2) to the glutathione S-transferase (GST) moiety of pGEX-3X (Amersham Pharmacia Biotech, Piscataway, N.J.). The second domain was created by fusing the entire pblA coding sequence with that of GST and then deleting the internal HindIII fragment of pblA, producing an in-frame fusion of the N- and C-terminal regions of PblA to GST. For PblB, the EcoRV-EcoRV or HpaI-EcoRV fragment of pblB (Fig. 2) was fused in frame with GST in pGEX-3X. These plasmids were introduced into E. coli strain BL21(DE3), and protein expression was induced as recommended (Amersham Pharmacia Biotech). Expressed proteins were separated by SDS-polyacrylamide gel electrophoresis and then stained with zinc (Pierce, Rockford, Ill.). Regions of the gel containing the overexpressed proteins were excised and used to immunize goats (Caltag Laboratories, South San Francisco, Calif.). To enhance the specificity of the antisera for PblA or PblB, each antiserum was adsorbed repeatedly with S. mitis strain PS344, a deletion mutant (described above) that does not express PblA or PblB.
Western blot analysis.
Cell surface proteins were extracted from S. mitis strains using SLS or mutanolysin as described by Jenkinson (14). The extracted proteins were separated by electrophoresis through SDS–6.8% polyacrylamide gels under reducing conditions, transferred to Biotrace NT nitrocellulose membranes (Pall Corp., Ann Arbor, Mich.), and incubated with antisera. Antibody binding was detected with horseradish peroxidase-conjugated anti-goat immunoglobulin G IgG (Sigma) and developed with the Super Signal chemiluminescent detection system (Pierce).
RNA isolation and blotting.
Total RNA was extracted from S. mitis strains using the RNeasy kit as recommended by the manufacturer (Qiagen, Valencia, Calif.), except that 500 U of mutanolysin per ml was included in the cell resuspension buffer. For dot blot analysis of pblR transcripts, 2 μg of RNA was spotted onto nylon membranes, hybridized with a digoxigenin-labeled DNA fragment of pblR, and developed with the CDP-Star chemiluminescent substrate as specified in the Genius System protocol (Roche). For Northern blot analysis of pblB transcripts, approximately 5 μg of RNA was denatured, loaded into wells of a 1% agarose gel, electrophoresed as described previously (2), and then transferred to a nylon membrane using the TurboBlotter system (Schleicher & Schuell, Keene, N.H.). The membrane was probed with a digoxigenin-labeled DNA fragment spanning the pblB coding region and developed as described above.
Statistical methods.
Differences in platelet binding were compared by the unpaired t test, using the Welch modification when appropriate.
Nucleotide sequence accession numbers.
The sequences generated from strains PS101 and PS116 have been deposited in GenBank under accession numbers AY007504 and AY007505, respectively. The sequence of the SF100 comC has been deposited under accession number AY007503.
RESULTS
Platelet binding by SF100 and isolation of low-binding mutants.
Strain SF100 readily bound to human platelets immobilized in tissue culture wells. Initial characterization of platelet binding by this strain indicated that after 2 h of incubation with platelet monolayers, binding ranged from 1.4 to 3.7% of the applied inoculum, with a mean ± standard deviation of 3.3% ± 1.9%. In contrast, only 0.33% ± 0.24% of the inoculum bound to polylysine-coated plastic wells (P = 0.0002 compared with platelet binding; n = 12). Trypsinization of bacteria prior to testing reduced platelet binding by 88.7% ± 8.8% compared with that of untreated organisms (P < 0.0001; n = 4). These results indicated that the binding of SF100 to platelets was relatively selective and was mediated predominantly by one or more surface proteins.
To define further the molecular basis of binding, a pool of approximately 2,000 Tn916ΔE mutants of SF100 were screened for reduced binding to platelets. Twenty-three potential low-binding variants were identified by the initial turbidimetric screening assay. Two of these mutants, PS101 and PS116, were subsequently found to be consistent, low-binding mutants when tested repeatedly in the quantitative binding assay. Compared with the parental strain, binding of PS101 to platelet monolayers was reduced by 65.2% ± 32.1% (P < 0.05; n = 4). Binding of PS116 was also reduced significantly (52.5% ± 23.7% [P < 0.05; n = 4). PS101 and PS116 were otherwise phenotypically normal, as measured by growth rate in THB, hemolysin production, and 30 additional biochemical characteristics included in the Vitek Gram Positive Identification Card (bioMérieux, Marcy L'Etoile, France). Southern blot analysis of chromosomal DNA from the two mutants indicated that each mutant carried a single copy of Tn916ΔE and that the sites of insertion in PS101 and PS116 were different (data not shown).
DNA sequence analysis of the PS101 locus.
To characterize the sites of Tn916ΔE insertion in the low-binding mutants, chromosomal regions flanking the transposon were cloned and sequenced as described in Materials and Methods. The initial sequence of the PS101 locus was obtained from a 1.2-kb EcoRI fragment of chromosomal DNA. A 0.6-kb EcoRI-HincII fragment upstream from the site of the Tn916ΔE insertion (Fig. 1) was then cloned in the suicide vector pVA891 and used to generate strain PS163 by insertion-duplication mutagenesis of strain SF100. Additional sequences downstream and upstream of the 1.2-kb EcoRI fragment were obtained by recovery of pVA891 and flanking DNA from PS163.
Analysis of the combined sequences indicated that Tn916ΔE was inserted at the extreme 3′ end of a 1.2-kb ORF encoding a protein of 399 amino acids. The predicted protein has a molecular mass of 43 kDa and a pI of 10.6. The first 31 amino acids are predicted to comprise a signal peptide (21). The remainder of the protein is extremely hydrophobic, with 12 potential transmembrane-spanning regions, indicating that it is likely to be an integral membrane protein. Searches for similarity of this protein to others listed in the current databases indicated that it is likely to be a member of the major facilitator superfamily of small-molecule transporters (24) and is most similar (48% similarity and 29% identity) to the Oxalobacter formigenes oxalate:formate antiporter (1). The gene therefore has been designated pblT (for “platelet binding locus transporter”).
Immediately downstream from pblT is a 0.9-kb ORF encoding a 293-amino-acid protein. This gene has been designated pblR, since the encoded protein is predicted to be a member of the AraC/XylS family of transcriptional regulators, as defined by profile PS01124 from the PROSITE database (12). PblR is most similar to MsmR of Streptococcus mutans (36% similarity and 24% identity). An apparent promoter element is located just upstream from pblR. This sequence is similar to the pneumococcal extended promoter consensus (30) at 13 of 15 positions, with a spacing of 15 bp between the −35 and −10 elements.
PblT contributes to platelet binding.
To confirm that the reduced platelet binding observed with PS101 was due to transposon insertion within pblT, strain PS163 was tested for platelet binding. Compared with the parental strain, platelet binding by PS163 was reduced by 42.0% ± 10.1% (P < 0.0001) (Fig. 3). To assess whether the low-binding phenotype was due to a polar effect on downstream gene expression, strain PS361 was generated by insertion-duplication mutagenesis of pblR. Platelet binding by PS361 was not significantly different from that by SF100 (P = 0.0794) (Fig. 3). In addition, dot blot analysis of RNA from PS101 showed no difference in pblR transcript levels compared with RNA from SF100 (data not shown). These results indicated that platelet binding by SF100 is in part mediated by pblT and that the loss of binding seen with pblT disruption was not due to polar effects on the expression of pblR.
FIG. 3.

Platelet binding by insertion-duplication and deletion mutants. Values presented are percent of wild-type binding (mean ± standard deviation). Values that are significantly different from those of SF100 (P < 0.05) are indicated by asterisks. Strain genotypes: SF100, wild type; PS163, pblT::pVA891; PS361, pblR::pVA891; PS344, ΔORF1-pblB; PS301, pblA::pVA891; PS345, pblB::pVA891.
DNA sequence analysis of the PS116 locus.
The chromosomal region of PS116 flanking Tn916ΔE was sequenced in several stages, as diagrammed in Fig. 2. A total of 8.5 kb of sequence was compiled for this locus, including 3.9 kb upstream and 4.6 kb downstream from the point of the Tn916ΔE insertion. Analysis of the combined sequences indicated that the entire region was likely to be part of a polycistronic operon, since there are just a few nucleotides between adjacent ORFs.
The upstream gene products (ORF1 to ORF3) are highly similar to those of the lactococcal phage r1t (Table 2). The protein encoded by pblA is large (107 kDa) and acidic (pI = 4.1) and is predicted to have a 71-amino-acid signal peptide. PblA is most similar to proteins encoded by ORF40 and ORF42 of phage r1t. In contrast to the r1t ORFs, which are split by an intron (37), pblA consists of a single large reading frame. In addition, PblA has an expanded region with regular repeats of a semiconserved, tryptophan-rich pentapeptide motif (Fig. 2) that is not present in phage r1t ORF42. The presence of tryptophan-rich repeats suggests that PblA might be a cell wall-associated protein, similar to choline binding proteins of pneumococci and listeriae (5, 44).
TABLE 2.
Characteristics of gene products encoded by the PS116 locus
| Gene | Size (nt) | Predicted mol mass (kDa) | Homologs (% similarity/% identity) |
|---|---|---|---|
| ORF1 | 549 | 19 | L. lactis phage r1t ORF37a (76/61) |
| ORF2 | 309 | 12 | L. lactis phage r1t ORF38a (77/47) |
| ORF3 | 378 | 14 | L. lactis phage r1t ORF39a (69/51) |
| pblA | 3,021 | 107 | L. lactis phage r1t ORF40a (62/52) and ORF42a (42/27) |
| ORF4 | 693 | 26 | No significant similarity |
| pblB | 3,186 | 121 | S. thermophilus phage 01205 ORF45 (48/33)b |
ORF37 through ORF42 of phage r1t are within a region encoding structural proteins.
The similarity to ORF45 is limited to the carboxy-terminal half of PblB.
Analysis of the sequence downstream from the site of the Tn916ΔE insertion indicated the presence of two additional ORFs. A 231-amino-acid protein encoded by ORF4 shows no significant similarity to any reported sequences. The ORF4 protein has no membrane-spanning segments and is predicted to be cytoplasmic. The downstream gene, pblB, is predicted to encode a 121-kDa protein with a pI of 8.1. The N-terminal half of PblB is predicted to have a coiled-coil domain that lies within a region similar to the pneumococcal surface protein A (PspA) and the Streptococcus pyogenes M proteins (Fig. 2). The C-terminal half of PblB is most similar to a tail fiber protein from the Streptococcus thermophilus phage 01205 (32) and to receptor recognition or host specificity proteins of various coliphages.
Role of PblA and PblB in platelet binding.
To confirm the role of this second locus in platelet binding, the platelet binding phenotype of a mutant (PS344) carrying a deletion of ORF1 through pblB was assessed. Compared with the parental strain, platelet binding by PS344 was reduced by 36.1% ± 16.4% (P < 0.0001) (Fig. 3), confirming that disruption of the PS116 locus was linked to loss of binding.
To determine subsequently whether either of the two large genes in this locus affected platelet binding, pblA and pblB were each disrupted by insertion-duplication mutagenesis. The resultant mutants, PS301 (PblA−) and PS345 (PblB−), were then tested for binding to platelet monolayers. Both mutants showed a decrease in platelet binding similar to that of the PS344 deletion mutant. Compared with the parental strain, platelet binding by PS301 was reduced by 22.9% ± 13.5% (P < 0.0001) (Fig. 3) and binding by PS345 was reduced by 29.2% ± 25.2% (P = 0.0014) (Fig. 3). Complementation of PS301 with a copy of pblA carried in trans (strain PS386) led to only a slight increase in platelet binding (13.2% ± 15.9% increase compared with PS301 carrying the vector alone [strain PS385]). However, complementation of PS345 with a copy of pblB carried in trans (strain PS390) led to a 47.8% ± 24.0% increase in binding compared with PS345 carrying the vector only (strain PS389) (P = 0.0002). These results indicated that PblB was primarily responsible for platelet binding. The findings also suggested that the reduction in platelet binding caused by interruption of pblA might be due to polar effects on pblB transcription.
Disruption of pblA is not transcriptionally polar.
To determine whether disruption of pblA caused a polar mutation, RNA from strain PS301 was compared with RNA from strain SF100 by using a pblB probe. Northern blot analysis indicated that the pblB mRNA is part of a very large (much greater than 6.9 kb) polycistronic transcript in SF100 (Fig. 4, lane 1). Although the level of transcription of pblB in PS301 (lane 2) was somewhat reduced compared with the level in SF100, it was not abolished. Thus, integration of pVA891 into pblA in strain PS301 was not transcriptionally polar. However, the pblB transcript in PS301 was smaller. The difference in transcript size was probably due to either differential processing within pVA891 sequences or initiation of transcription from a promoter within pVA891.
FIG. 4.

Northern blot of RNA isolated from S. mitis strains and hybridized with a pblB probe. Lanes: 1, RNA from the parental strain SF100; 2, RNA from the pblA mutant strain PS301. Arrows indicate the predominant prgB transcripts. The bands seen at 2.5 kb correspond to nonspecific hybridization to the 23S rRNA.
Expression of PblB may be linked to that of PblA.
To compare the expression of PblA and PblB in the wild-type and mutant strains, polyclonal antisera were generated to each of these proteins and expression was assessed by Western blotting. Preliminary experiments showed that both PblA and PblB could be extracted with SLS but not with a combination of mutanolysin and lysozyme (data not shown), indicating that they were associated with the cell wall but not covalently linked to the peptidoglycan (20). The anti-PblA serum reacted with proteins of 120, 110, and 80 kDa that were absent from the PS344 deletion mutant (Fig. 5A, lanes 1 and 2). The anti-PblB serum reacted with two protein bands: the predominant form had an apparent molecular mass of 110 kDa, and a minor form was apparent near the predicted molecular mass of 121 kDa (Fig. 5B, lane 1).
FIG. 5.

Western blot of SLS-extracted proteins from strain SF100 and from pblA and pblB mutant strains. Lanes: 1, SF100; 2, PS344 (PblA− PblB−); 3, PS301 (PblA−); 4, PS345 (PblB−). (A) The blot was reacted with anti-PblA serum. (B) The blot was reacted with anti-PblB serum.
To determine whether PblB expression was altered in the pblA mutant strain PS301 and whether PblA expression was normal in the pblB mutant strain PS345, SLS extracts of surface proteins from these strains were also analyzed by Western blotting. Surface expression of PblB was markedly reduced in the pblA mutant compared with that in the parental strain (Fig. 5B, lane 3), whereas disruption of pblB had no effect on PblA expression (Fig. 5A, lane 4). Since PS345, which was defective in platelet binding, shows normal levels of PblA expression, these results support the finding from complementation of the mutant strains (described above) that PblB, rather than PblA, was primarily responsible for the platelet binding phenotype. That is, reduction in platelet binding is correlated with a loss of PblB expression.
To determine if PblB was sufficient for platelet binding, pblB was added in trans to the pblA mutant strain PS301. Surprisingly, platelet binding by this strain (PS416) was even lower than that of PS301 carrying the vector only (PS385). Compared with PS385, binding by strain PS416 was reduced by 39.7% ± 14.3% (P = 0.0005). The combined results indicate that although PblB is required, it is not sufficient for platelet binding, since PblA is also needed.
DISCUSSION
These results demonstrate that at least two distinct loci contribute to platelet binding by S. mitis strain SF100. DNA sequence analysis of the first region indicated that it contains an assortment of divergent and convergent ORFs. The gene responsible for platelet binding, pblT, is likely to encode a transmembrane transporter that resembles members of the major facilitator superfamily of small-molecule transporters. Solute binding proteins of the ATP binding cassette transporter family have been shown to facilitate adhesion by other microbes, such as Streptococcus parasanguis (11). Thus, it is possible that PblT affects platelet binding directly. Alternatively, it could play a secondary role, such as transport of a signal molecule or transport of a substrate required for adhesin assembly that cannot be produced de novo.
Binding is also mediated by one or more surface proteins encoded by the PS116 locus. Our data indicate that PblB is a likely adhesin, since the surface expression of this protein correlated with platelet binding, and PblB has several features typical of binding proteins. Of note, the N-terminal half of PblB shows similarity to two well-characterized streptococcal adhesins, PspA of S. pneumoniae and M protein of S. pyogenes. PblB is also predicted to form a coiled coil, which is characteristic of fibrillar proteins (17). Moreover, PblB is similar to receptor recognition proteins of various phages. Alternatively, it may serve as a scaffolding protein for another adhesin or may be part of a larger binding complex that includes PblA.
PblA has features that are more characteristic of a surface structure. It is predicted to have a signal peptide and has five regular repeats of a tryptophan-rich pentapeptide motif. Although the significance of these repeats for PblA is unknown, tryptophan-rich repeats have been shown to allow the association of proteins with choline residues in the cell wall of pneumococci and listeriae (5, 44) and the cell wall polysaccharides of S. mitis strains do contain choline (31). PblA may be associated with PblB on the cell surface, which could either stabilize PblB or affect its conformation. The possibility of such a multicomponent adhesin is suggested by the relatedness of the gene products to components of phage capsids.
The similarity of PblA and PblB to phage proteins also raises the question whether the locus can transfer between strains. A search for sequences similar to PblA and PblB within the unfinished genome sequences available from the University of Oklahoma Advanced Center for Genome Technology (http://www.genome.ou.edu) and The Institute for Genomic Research (http://www.tigr.org) indicated that homologs are present in S. pyogenes and E. faecalis but not in the sequences reported to date for S. mutans or S. pneumoniae. Interestingly, if present, the homologs again reside in a phage or cryptic phage locus (Fig. 6). That is, S. pyogenes has an arrangement of genes identical to that of pblA-ORF4-pblB, which are flanked by phage r1t-like sequences. E. faecalis has two sets of homologs: one set lies within a region of sequences similar to phage r1t sequences, and the other lies within a region of sequences similar to phage 01205 sequences. The sequence of the corresponding locus of SF100, by comparison, appears to be a mosaic that may have evolved from reassortment of the two streptococcal phage genomes. It is interesting to consider that, as with other virulence properties, they may be encoded within a mobile genetic element (40). Although it is unknown whether pblA and pblB reside within a functional phage, we have found that expression is greatly induced by mitomycin C and UV light (unpublished results), agents commonly used to induce the lytic cycle of prophages.
FIG. 6.
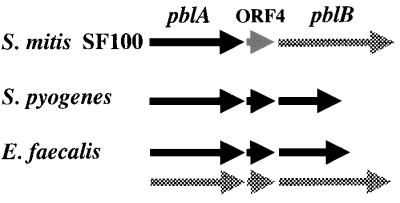
Homologs of PblA and PblB in other streptococcal species. Black arrows indicate similarity to Lactococcus lactis phage r1t sequences, and checkered arrows indicate similarity to S. thermophilus phage 01205 sequences. The gray arrow corresponds to ORF4, which was not similar to any sequences reported in the standard databases. In each of the loci diagrammed here, the pblA and pblB homologs are flanked by at least 6 kb of sequences corresponding to the structural protein region of the indicated phage. S. pyogenes has a cryptic or functional phage similar to r1t. E. faecalis has sequences similar to those of both r1t and 01205. The pblAB locus of S. mitis strain SF100, by comparison, appears to be a mosaic of the two streptococcal phage genomes.
These results indicate that platelet binding by S. mitis is a complex process, involving multiple bacterial factors. In part, binding appears to be mediated by PblA, PblB, and PblT. The precise mechanisms by which these proteins mediate binding are as yet unknown. These proteins may serve as direct adhesins or may be part of more complicated binding structures. It is also possible that other, as yet unidentified streptococcal adhesins contribute to platelet binding by S. mitis. Thus, additional studies are needed to address these issues. Further work is also required to determine whether platelet binding mediated by PblA, PblB, or PblT is important in the pathogenesis of infective endocarditis.
ACKNOWLEDGMENTS
This work was supported by grants AI41513 and AI22152 from the National Institutes of Health and by the Department of Veterans Affairs. B.B. was the recipient of a fellowship from the American Heart Association, Western Branch Affiliates.
We thank Richard Facklam at the Centers for Disease Control (Atlanta, Ga.) for the original typing of strain SF100, John Bartell from MIDI Labs for rRNA typing, and Wendy McKinley for excellent technical assistance.
REFERENCES
- 1.Abe K, Ruan Z S, Maloney P C. Cloning, sequencing, and expression in Escherichia coli of OxlT, the oxalate:formate exchange protein of Oxalobacter formigenes. J Biol Chem. 1996;271:6789–6793. doi: 10.1074/jbc.271.12.6789. [DOI] [PubMed] [Google Scholar]
- 2.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons, Inc.; 1997. pp. 4.9.1–4.9.4. [Google Scholar]
- 3.Bensing B A, Dunny G M. Cloning and molecular analysis of genes affecting expression of binding substance, the recipient-encoded receptor s mediating mating aggregate formation in Enterococcus faecalis. J Bacteriol. 1993;175:7421–7429. doi: 10.1128/jb.175.22.7421-7429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bochud P Y, Calandra T, Francioli P. Bacteremia due to viridans streptococci in neutropenic patients: a review. Am J Med. 1994;97:256–264. doi: 10.1016/0002-9343(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 5.Braun L, Dramsi S, Dehoux P, Bierne H, Lindahl G, Cossart P. InlB: an invasion protein of Listeria monocytogenes with a novel type of surface association. Mol Microbiol. 1997;25:285–294. doi: 10.1046/j.1365-2958.1997.4621825.x. [DOI] [PubMed] [Google Scholar]
- 6.Carratala J, Alcaide F, Fernandez-Sevilla A, Corbella X, Linares J, Gudiol F. Bacteremia due to viridans streptococci that are highly resistant to penicillin: increase among neutropenic patients with cancer. Clin Infect Dis. 1995;20:1169–1173. doi: 10.1093/clinids/20.5.1169. [DOI] [PubMed] [Google Scholar]
- 7.Chaffin D O, Rubens C E. Blue/white screening of recombinant plasmids in Gram-positive bacteria by interruption of alkaline phosphatase gene (phoZ) expression. Gene. 1998;219:91–99. doi: 10.1016/s0378-1119(98)00396-5. [DOI] [PubMed] [Google Scholar]
- 8.Douglas C W, Heath J, Hampton K K, Preston F E. Identity of viridans streptococci isolated from cases of infective endocarditis. J Med Microbiol. 1993;39:179–182. doi: 10.1099/00222615-39-3-179. [DOI] [PubMed] [Google Scholar]
- 9.Durack D T. Experimental bacterial endocarditis. IV. Structure and evolution of very early lesions. J Pathol. 1975;115:81–89. doi: 10.1002/path.1711150204. [DOI] [PubMed] [Google Scholar]
- 10.Durack D T, Beeson P B. Experimental bacterial endocarditis. I. Colonization of a sterile vegetation. Br J Exp Pathol. 1972;53:44–49. [PMC free article] [PubMed] [Google Scholar]
- 11.Fenno J C, Shaikh A, Spatafora G, Fives-Taylor P. The fimA locus of Streptococcus parasanguis encodes an ATP-binding membrane transport system. Mol Microbiol. 1995;15:849–863. doi: 10.1111/j.1365-2958.1995.tb02355.x. [DOI] [PubMed] [Google Scholar]
- 12.Gallegos M T, Schleif R, Bairoch A, Hofmann K, Ramos J L. AraC/XylS family of transcriptional regulators. Microbiol Mol Biol Rev. 1997;61:393–410. doi: 10.1128/mmbr.61.4.393-410.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Håvarstein L S, Gaustad P, Nes I F, Morrison D A. Identification of the streptococcal competence-pheromone receptor. Mol Microbiol. 1996;21:863–869. doi: 10.1046/j.1365-2958.1996.521416.x. [DOI] [PubMed] [Google Scholar]
- 14.Jenkinson H F. Cell-surface proteins of Streptococcus sanguis associated with cell hydrophobicity and coaggregation properties. J Gen Microbiol. 1986;132:1575–1589. doi: 10.1099/00221287-132-6-1575. [DOI] [PubMed] [Google Scholar]
- 15.Kern W, Kurrle E, Schmeiser T. Streptococcal bacteremia in adult patients with leukemia undergoing aggressive chemotherapy. A review of 55 cases. Infection. 1990;18:138–145. doi: 10.1007/BF01642101. [DOI] [PubMed] [Google Scholar]
- 16.Klotz S A, Harrison J L, Misra R P. Aggregated platelets enhance adherence of Candida yeasts to endothelium. J Infect Dis. 1989;160:669–677. doi: 10.1093/infdis/160.4.669. [DOI] [PubMed] [Google Scholar]
- 17.Lupas A. Prediction and analysis of coiled-coil structures. Methods Enzymol. 1996;266:513–525. doi: 10.1016/s0076-6879(96)66032-7. [DOI] [PubMed] [Google Scholar]
- 18.Macrina F L, Evans R P, Tobian J A, Hartley D L, Clewell D B, Jones K R. Novel shuttle plasmid vehicles for Escherichia-Streptococcus transgeneric cloning. Gene. 1983;25:145–150. doi: 10.1016/0378-1119(83)90176-2. [DOI] [PubMed] [Google Scholar]
- 19.McGowan D A, Gillett R. Scanning electron microscopic observations of the surface of the initial lesion in experimental streptococcal endocarditis in the rabbit. Br J Exp Pathol. 1980;61:164–171. [PMC free article] [PubMed] [Google Scholar]
- 20.McNab R, Jenkinson H F. Lipoproteins and other cell-surface associated proteins in streptococci. Methods Cell Sci. 1998;20:209–216. [Google Scholar]
- 21.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 22.Nizet V, Smith A L, Sullam P M, Rubens C E. A simple microtiter plate screening assay for bacterial invasion or adherence. Methods Cell Sci. 1998;20:107–111. [Google Scholar]
- 23.Ohkuni H, Todome Y, Okibayashi F, Watanabe Y, Ohtani N, Ishikawa T, Asano G, Kotani S. Purification and partial characterization of a novel human platelet aggregation factor in the extracellular products of Streptococcus mitis, strain Nm-65. FEMS Immunol Med Microbiol. 1997;17:121–129. doi: 10.1111/j.1574-695X.1997.tb01004.x. [DOI] [PubMed] [Google Scholar]
- 24.Pao S S, Paulsen I T, Saier M H., Jr Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poutanen S M, de Azavedo J, Willey B M, Low D E, MacDonald K S. Molecular characterization of multidrug resistance in Streptococcus mitis. Antimicrob Agents Chemother. 1999;43:1505–1507. doi: 10.1128/aac.43.6.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richard P, Amador Del Valle G, Moreau P, Milpied N, Felice M P, Daeschler T, Harousseau J L, Richet H. Viridans streptococcal bacteraemia in patients with neutropenia. Lancet. 1995;345:1607–1609. doi: 10.1016/s0140-6736(95)90117-5. [DOI] [PubMed] [Google Scholar]
- 27.Roberts R B, Krieger A G, Schiller N L, Gross K C. Viridans streptococcal endocarditis: the role of various species, including pyridoxal-dependent streptococci. Rev Infect Dis. 1979;1:955–966. doi: 10.1093/clinids/1.6.955. [DOI] [PubMed] [Google Scholar]
- 28.Rubens C E, Heggen L M. Tn916ΔE: a Tn916 transposon derivative expressing erythromycin resistance. Plasmid. 1988;20:137–142. doi: 10.1016/0147-619x(88)90016-9. [DOI] [PubMed] [Google Scholar]
- 29.Rubens C E, Wessels M R, Heggen L M, Kasper D L. Transposon mutagenesis of type III group B Streptococcus: correlation of capsule expression with virulence. Proc Natl Acad Sci USA. 1987;84:7208–7212. doi: 10.1073/pnas.84.20.7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabelnikov A G, Greenberg B, Lacks S A. An extended −10 promoter alone directs transcription of the DpnII operon of Streptococcus pneumoniae. J Mol Biol. 1995;250:144–155. doi: 10.1006/jmbi.1995.0366. [DOI] [PubMed] [Google Scholar]
- 31.Schenkein H A, Gunsolley J C, Best A M, Harrison M T, Hahn C L, Wu J, Tew J G. Antiphosphorylcholine antibody levels are elevated in humans with periodontal diseases. Infect Immun. 1999;67:4814–4818. doi: 10.1128/iai.67.9.4814-4818.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanley E, Fitzgerald G F, Le Marrec C, Fayard B, van Sinderen D. Sequence analysis and characterization of phi O1205, a temperate bacteriophage infecting Streptococcus thermophilus CNRZ1205. Microbiology. 1997;143:3417–3429. doi: 10.1099/00221287-143-11-3417. [DOI] [PubMed] [Google Scholar]
- 33.Sullam P M. Host-pathogen interactions in the development of bacterial endocarditis. Curr Opin Infect Dis. 1994;7:304–309. [Google Scholar]
- 34.Sullam P M, Bayer A S, Foss W M, Cheung A L. Diminished platelet binding in vitro by Staphylococcus aureus is associated with reduced virulence in a rabbit model of infective endocarditis. Infect Immun. 1996;64:4915–4921. doi: 10.1128/iai.64.12.4915-4921.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sullam P M, Frank U, Yeaman M R, Tauber M G, Bayer A S, Chambers H F. Effect of thrombocytopenia on the early course of streptococcal endocarditis. J Infect Dis. 1993;168:910–914. doi: 10.1093/infdis/168.4.910. [DOI] [PubMed] [Google Scholar]
- 36.Sullam P M, Payan D G, Dazin P F, Valone F H. Binding of viridans group streptococci to human platelets: a quantitative analysis. Infect Immun. 1990;58:3802–3806. doi: 10.1128/iai.58.11.3802-3806.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Sinderen D, Karsens H, Kok J, Terpstra P, Ruiters M H, Venema G, Nauta A. Sequence analysis and molecular characterization of the temperate lactococcal bacteriophage r1t. Mol Microbiol. 1996;19:1343–1355. doi: 10.1111/j.1365-2958.1996.tb02478.x. [DOI] [PubMed] [Google Scholar]
- 38.Villablanca J G, Steiner M, Kersey J, Ramsay N K, Ferrieri P, Haake R, Weisdorf D. The clinical spectrum of infections with viridans streptococci in bone marrow transplant patients. Bone Marrow Transplant. 1990;5:387–393. [PubMed] [Google Scholar]
- 39.Wahl G M, Lewis K A, Ruiz J C, Rothenberg B, Zhao J, Evans G A. Cosmid vectors for rapid genomic walking, restriction mapping, and gene transfer. Proc Natl Acad Sci USA. 1987;84:2160–2164. doi: 10.1073/pnas.84.8.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waldor M K. Bacteriophage biology and bacterial virulence. Trends Microbiol. 1998;6:295–297. doi: 10.1016/s0966-842x(98)01320-1. [DOI] [PubMed] [Google Scholar]
- 41.Wertman K F, Wyman A R, Botstein D. Host/vector interactions which affect the viability of recombinant phage lambda clones. Gene. 1986;49:253–262. doi: 10.1016/0378-1119(86)90286-6. [DOI] [PubMed] [Google Scholar]
- 42.Yeaman M R, Sullam P M, Dazin P F, Ghannoum M A, Edwards J E, Jr, Bayer A S. Fluconazole and platelet microbicidal protein inhibit Candida adherence to platelets in vitro. Antimicrob Agents Chemother. 1994;38:1460–1465. doi: 10.1128/aac.38.7.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yeaman M R, Sullam P M, Dazin P F, Norman D C, Bayer A S. Characterization of Staphylococcus aureus-platelet binding by quantitative flow cytometric analysis. J Infect Dis. 1992;166:65–73. doi: 10.1093/infdis/166.1.65. [DOI] [PubMed] [Google Scholar]
- 44.Yother J, White J M. Novel surface attachment mechanism of the Streptococcus pneumoniae protein PspA. J Bacteriol. 1994;176:2976–2985. doi: 10.1128/jb.176.10.2976-2985.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]