

Abstract
Cerebral small vessel diseases (CSVDs) are prominent contributors to vascular cognitive impairment and dementia and can arise from a range of etiologies. Cerebral amyloid angiopathy (CAA) and hypertension (HTN), both prevalent in the elderly population, lead to cerebral microhemorrhages, macrohemorrhages, and white matter damage. However, their respective underlying mechanisms and molecular events are poorly understood. Here, we show that the transgenic rat model of CAA type 1 (rTg-DI) exhibits perivascular inflammation that is lacking in the spontaneously hypertensive stroke-prone (SHR-SP) rat model of HTN. Alternatively, SHR-SP rats display notable dilation of arteriolar perivascular spaces. Comparative proteomics analysis revealed few shared altered proteins, with key proteins such as ANXA3, H2A, and HTRA1 unique to rTg-DI rats, and Nt5e, Flot-1 and Flot-2 unique to SHR-SP rats. Immunolabeling confirmed that upregulation of ANXA3, HTRA1, and neutrophil extracellular trap proteins were distinctly associated with rTg-DI rats. Pathway analysis predicted activation of TGF-β1 and TNFα in rTg-DI rat brain, while insulin signaling was reduced in the SHR-SP rat brain. Thus, we report divergent protein signatures associated with distinct cerebral vessel pathologies in the SHR-SP and rTg-DI rat models and provide new mechanistic insight into these different forms of CSVD.
Keywords: Cerebral amyloid angiopathy, Experimental rat models, Hypertension, Proteomics
INTRODUCTION
Cerebral small vessel diseases (CSVDs) are prominent contributors to vascular cognitive impairment and dementia (VCID). They are common among patients in the growing elderly population and are therefore of increasing clinical concern (1–3). CSVDs describe a range of cerebral vessel-related pathologies typically involving thrombotic events in capillaries, arterioles, and small arteries that lead to parenchymal injuries and white matter (WM) damage (4, 5). CSVDs arise from various etiopathogenic origins and can be classified into 2 major categories: amyloidal and nonamyloidal (2). Cerebral amyloid angiopathy (CAA), a common form of CSVD characterized by the deposition of amyloid β-protein (Aβ) fibrils in the cerebral vasculature, occurs sporadically in >50% of individuals over the age of 80 years and is a strong comorbidity in the majority of Alzheimer disease (AD) patients (6–10). Cerebral vessel deposition of Aβ and resultant vessel wall dysfunction commonly leads to cerebral microhemorrhages, intracerebral macrohemorrhages (ICH), WM damage, and VCID (6, 8, 11, 12). CAA can present as 2 forms: CAA type-1 where Aβ deposition occurs primarily in brain microvessels and capillaries, and CAA type-2 where amyloid deposition resides within intracortical small arteries and arterioles and surface meningeal/pial vessels (13). CAA type-1 is characterized by capillary “dyshoric” amyloid, where fibrillar amyloid protrudes into the adjacent parenchyma causing considerable perivascular neuroinflammation (8, 13–15). Ultimately, these severe vascular and perivascular pathologies correlate with cognitive decline and dementia (16, 17).
On the other hand, nonamyloidal forms of CSVD include lifestyle-induced disorders such as hypertension (HTN) (18), diabetes (19), or arteriosclerosis (20). Hypertensive arteriopathy (HA) causes loss of blood-brain barrier (BBB) integrity and endothelial dysfunction, and is another prominent cause of cerebral hemorrhages, WM damage, and VCID (20, 21). Diagnosed in ∼70% of individuals over the age of 65, and present in 80% of CSVD patients, HTN is a prevalent risk factor for VCID, and is the most common cause of ICH in young adults (20, 22–24).
The preclinical transgenic rat model of CAA type 1 (rTg-DI) produces human Aβ containing the Dutch (E22Q) and Iowa (D23N) familial CAA mutations, resulting in extensive cerebral microvascular amyloid deposition and severe perivascular inflammation (25, 26). Recently, we reported regional proteomic analysis, using Sequential Acquisition of all Theoretical Mass Spectra (SWATH-MS), of several brain regions isolated with laser capture microdissection from 12-month rTg-DI rat brains that revealed many proteins commonly elevated in all regions presenting significant vascular amyloid deposition and perivascular inflammation, as well as proteins specific to the thalamus where more severe vasculopathies such as small vessel occlusions and microhemorrhages occur (27). On the other hand, the spontaneously hypertensive stroke-prone rat (SHR-SP) is a well-documented preclinical model for HTN- and HA-related nonamyloidal CSVD pathology (21, 28–30).
While amyloidal and nonamyloidal CSVDs have distinct origins, interplay between the 2 has been suggested (21, 31). Furthermore, studies performed in the SHR-SP rat model reported that HTN and HA can result in failed Aβ clearance, and that CVSD arising from HTN may lead to the development of CAA (21, 31). Despite the prevalence and significant clinical burden of these 2 common CVSDs, the underlying molecular mechanisms responsible for CAA- or HTN-related vasculopathies and neuropathic presentations are not fully understood.
Regardless of their different etiologies, HTN and CAA share a convergence of clinical pathologies, including cerebral microhemorrhages, cerebral infarction, and WM damage. The goal of the present study was to compare brain proteomic analysis in a model of amyloidal CSVD (rTg-DI) and nonamyloidal CSVD (SHR-SP) to identify distinct, and potentially common, proteins and pathways responsible for the respective pathologies observed in each model. Here, we conduct brain SWATH-MS proteomic analysis of the rTg-DI and SHR-SP rat models and reveal comparatively largely different proteomes in each. Additionally, we perform pathway analysis to define regulators and causal networks specific to each model. Thus, we report differential proteomic and pathway signatures for each model of CSVD and describe novel findings to better understand the mechanisms underlying their pathogenesis.
MATERIALS AND METHODS
Animals
All work with animals was approved by the University of Rhode Island Institutional Animal Care and Use Committee and in accordance with the United States Public Health Service’s Policy on Humane Care and Use of Laboratory Animals was and in compliance with the ARRIVE guidelines. The rTg-DI rat model was generated as previously described (25). Briefly, rTg-DI rats, generated on a Sprague-Dawley (SD) background, express low levels of human Swedish/Dutch/Iowa mutant AβPP under the control of the neuronal-specific Thy1.2 promoter and results in production of chimeric Dutch/Iowa CAA mutant Aβ peptides in the brain (25). Microvascular Aβ accumulation and perivascular inflammation begin at ≈3 months of age and increase in a time dependent manner (25). SHR-SP rats were obtained from Charles River Laboratories, Kingston, NY. For the present study, rTg-DI rats (5 female + 5 male), SHR-SP rats (3 female + 3 male), and SD wild-type (WT) rats (5 female + 5 male) were all aged to 12 months. The study was not randomized or blinded. All rats were housed in a controlled room (22 ± 2°C and 40%–60% humidity) on a standard 12-hour light cycle. Rat chow and water were available ad libitum.
Brain Tissue Collection and Preparation
Anesthetized rats were transcardially perfused with PBS and the brains surgically removed. Rat brains were then bisected in the midsagittal plane, with 1 hemisphere subsequently fixed in 4% paraformaldehyde for immunohistochemical analysis and the other hemisphere directly snap-frozen and pulverized on dry ice for protein MS analysis.
Immunolabeling and Histological Analyses
Tissue sections were cut from frozen paraformaldehydefixed brain hemispheres in the sagittal plane at 25-µm thickness using a cryostat (Leica, Buffalo Grove, IL) and placed on slides. Antigen retrieval was conducted via 5-minute incubation with proteinase K (0.2 mg/mL) at 22°C. Tissue sections were then blocked in Superblock blocking buffer (37518, Thermo Fisher, Waltham, MA) containing 0.3% Triton X-100 at room temperature for 30 minutes and incubated with individual primary antibodies at the following dilutions overnight: rabbit polyclonal antibody to collagen IV to identify cerebral blood vessels (1:250, SD2365885, Invitrogen, Waltham, MA); goat polyclonal antibodies to glial fibrillary acidic protein ([GFAP], 1:250, ab53554, Abcam, Cambridge, UK) or ionized calcium-binding adapter molecule 1 ([Iba-1], 1:250, NB100-1028, Novus, St. Louis, MO) used to identify astrocytes and microglia, respectively, and rabbit polyclonal antibodies to Annexin A3 (ANXA3, 1:250, PA5082483, Invitrogen), HTRA1 (1:200, MAB2916, R&D Systems, Minneapolis, MN), neutrophil elastase (NE) (1:250, RRID# AB_2746314, Invitrogen), and histone 2A (1:250, RRID# AB2735313, Invitrogen). Alexa Fluorescent 594- or 488-conjugated secondary antibodies (1:1000) were used for primary antibody detection. Deposits of fibrillar vascular amyloid were detected by staining with either thioflavin S (123H0598, Sigma-Aldrich, St. Louis, MO) or Amylo-Glo (TR-300-AG, Biosensis, Inc., Thebarton, Australia) as described by the manufacturer. Prussian blue iron staining was performed to detect hemosiderin deposits reflecting signs of previous microbleeds (25, 32). Von Kossa calcium staining was used to detect calcified small vessel occlusions in the brain (25, 33). Immunolabeled and histological images were collected with the Keyence BZ-X710 Microscope (RRID: SCR_017202) and analyzed with the Keyence BZ-X Analyzer Software Version 1.3.1.1 (Keyence Corp., Osaka, Japan).
The density of microglia and astrocytes was quantified in the regions of the cortex, thalamus, and hippocampus of rTg-DI, SHR-SP, and age-matched WT rats using stereological principles (26, 27) The total numbers of microglia and astrocytes were estimated using the Stereologer software system (Systems Planning and Analysis, Alexandria, VA). Every tenth section cut at 25 µm was selected and generated ≈20 sections per reference space in a systematic-random manner. Immunopositive cells were counted using the optical fractionator method with the dissector principle and unbiased counting rules (26, 27). A minimum of 8–10 reference spaces within each brain region from each slide were randomly selected for counting. Criteria for counting cells required that cell bodies exhibited positive GFAP or Iba-1 immunostaining, for astrocytes or microglia, respectively. Statistical significance was determined by 2-way ANOVA.
For perivascular space (PVS) area measurements, multiple brain tissue sections (4–6 sections per animal) were stained with hematoxylin and eosin. A total of 30 random cortical arterioles were imaged per animal. The total area occupied by the PVS and vessel together and area of vessel alone was measured utilizing the area measurement tool on the Keyence BZ-X Analyzer Software. The vessel area was then subtracted from the total area to reveal the PVS area. A ratio of PVS area to vessel area was then calculated for each animal. Animals from each cohort (n = 3) were pooled for a total of 90 PVS determinations for comparison. Statistical significance was determined by 1-way ANOVA.
Protein Digest of Collected Brain Tissue Regions
Aliquots from pulverized whole brain tissue from SHR-SP (n = 6), rTg-DI (n = 10), and WT (n = 10) rats were lysed in RIPA buffer via sonication, and proteins were denatured with 25 µL DTT (100 mM) and 15-minute shaking (300 rpm) incubation at 95°C. Twenty-five microliters IAA (200 mM) was added for alkylation, and samples were incubated at 20°C in the dark for 30 minutes. Proteins were then precipitated and concentrated via chloroform-methanol-water (1:2:1) precipitation and resuspended in sodium deoxycholate ([DOC], 3% w/v in 50 mM ammonium bicarbonate). Pressure cycling technology was used for proteins digestion with TPCK-treated trypsin at a concentration of 1:20 (trypsin:protein) (Sciex, Framingham, MA), in a barocycler (Pressure Biosciences Inc, Easton MA) as previously described (27, 34). Formic acid (5% v/v, in 50% v/v acetonitrile in water) addition for final concentration of 0.5% v/v, followed by centrifugation (15 300g, 5 minutes, at RT) was performed to precipitate the DOC. Sample supernatants were then collected and subjected to LC-MS/MS analysis.
Analysis by LC-QTOF/MS
Proteomic experiments using a SCIEX 5600 TripleTOF mass spectrometer, in positive ion mode using a DuoSpray ion source (AB Sciex, Concord, Canada), following chromatographic separation using an Acquity UPLC HClass system (Waters Corp., Milford, MA) were done as previously described (27, 34). An Acquity UPLC Peptide BEH C18 (2.1 mm × 150 mm, 300 Å, 1.7 µm) column preceded by an Acquity VanGuard precolumn (2.1 mm × 5 mm, 300 Å, 1.7 µm) were used for peptide separation following the previously described solvent method (27). TOF mass calibration was monitored with trypsin-digested β-galactosidase injected every 5 samples. Analyst TF 1.7.1 software (AB Sciex, Concord) in data-independent acquisition mode was used for data acquisition, and mass spec settings were exactly as previously described (27, 34).
Data Processing
Spectronaut (Biognosys, Schlieren, Switzerland) software, referencing our previously formed in-house rat brain spectral library (27) was used for protein identification and quantification. Spectronaut factory defaults were used for all settings, except “used Biognosys’ iRT kit” and “PTM localization” were deselected. Using the total protein approach (35, 36), raw protein intensities were converted to molar concentrations (pmol/mg total protein). We imputed a baseline concentration (0.013 pmol/mg tissue) for protein concentrations of zero in individual samples as previously described (27). Effect threshold cutoffs for differentially expressed proteins (DEPs) were set at ≥33% increase or ≥33% decrease, and statistical significance of p < 0.05 determined via Student t-test. Primary component analysis (PCA) considering the DEPs in each disease model was performed using the statistical computing software “R,” and custom scripts in RStudio.
RESULTS
Accumulation of Vascular Amyloid in the Brains of rTg-DI Rats but Not Hypertensive SHR-SP Rats
We previously reported that rTg-DI rats are a compelling model of CAA type-1 with robust accumulation of microvascular amyloid in the cortex, hippocampus, and thalamus (25–27). Despite being a nonamyloidal model of CSVD hypertensive (HA), there have been mixed reports of cerebral vascular amyloid accumulation and Aβ deposition in the SHR-SP model (21, 31, 37, 38). Therefore, we first sought to determine if vascular amyloid is present in our cohort of 12-month SHR-SP rats. Robust microvascular amyloid was confirmed in the cortex, hippocampus, and thalamus of rTg-DI rats (Fig. 1B, E, H), consistent with our previous studies (25–27). In contrast, we found no evidence of vascular amyloid deposition in any brain region of the SHR-SP rats (Fig. 1C, F, I), similar to the WT rats (Fig. 1A, D, G). The presence of extensive microvascular amyloid in rTg-DI rats had no appreciable effect on systolic blood pressure and were comparable to WT rats (109 ± 23 vs 108 ± 18, respectively) in contrast to SHR-SP rats (163 ± 28) (Supplementary DataFig. S1). Thus, we found that in contrast to the normotensive rTg-DI rats, the present cohort of hypertensive SHR-SP rats does not show evidence of microvascular amyloid deposition.
FIGURE 1.

Prominent cerebral microvascular amyloid in rTg-DI rats but not in SHR-SP rat brain. Brain sections from 12-month WT (A, D, G), rTg-DI (B, E, H), and SHR-SP (C, F, I) rats were immunolabeled with rabbit polyclonal antibody to collagen IV to specifically detect cerebral microvessels (red) and stained with thioflavin S to identify fibrillar amyloid (green). Cerebral microvascular fibrillar amyloid deposits are only observed in rTg-DI rats. Scale bars = 50 µm.
Increased Perivascular Glial Cells in rTg-DI Rats but Not Hypertensive SHR-SP Rats
Cerebral microvascular amyloid deposition results in a robust neuroinflammatory response in the rTg-DI rat brain marked by activation and dramatic increases in the number of perivascular astrocytes and microglia in the cortex, hippocampus, and thalamus (26, 27). Given the disparity in vascular amyloid deposition between the rTg-DI and SHR-SP rats, we compared the neuroinflammatory cell responses in each model compared to WT rats. Consistent with previous findings, rTg-DI rats displayed elevated numbers of astrocytes in the cortex, hippocampus, and thalamus, representing a 75%, 55%, and 182% increase compared to WT rat astrocyte densities, respectively (Fig. 2B, D). On the other hand, astrocyte numbers in the SHR-SP rats showed no significant changes compared to WT rats (Fig. 2C, D). Similarly, microglia numbers were dramatically increased in the cortex, hippocampus, and thalamus the rTg-DI rats, representing 420%, 446%, and 746% increases over WT rat microglia densities, respectively, and adopt an activated phenotype (Fig. 3B, D). The SHR-SP rats however, showed no significant change in the microglia densities compared to WT rats in any of the brain regions and retained their resting state with extended processes (Fig. 3C, F). Thus, unlike the amyloidal-based rTg-DI rats the nonamyloidal hypertensive phenotype of the SHR-SP model does not promote appreciable glial cell responses.
FIGURE 2.

Increased astrocytes in rTg-DI rats but not in SHR-SP rats. Brain sections from 12-month WT rats (A), rTg-DI rats (B), and SHR-SP rats (C) were labeled with Amylo-Glo to detect microvascular fibrillar amyloid (blue), goat polyclonal antibody to collagen IV to detect cerebral microvessels (red), and rabbit polyclonal antibody to GFAP to detect astrocytes (green). Images from the cortex are shown. Scale bars = 50 µm. (D) Quantitation of astrocyte numbers in different brain regions from WT rats (gray circles), rTg-DI rats (red circles), and SHR-SP rats (blue circles) at 12 months. Data points show the results from each rat and the group mean±SD of n = 6 rats of each type. ***p < 0.001 determined by 2-way ANOVA.
FIGURE 3.

Increased microglia in rTg-DI rats but not in SHR-SP rats. Brain sections from 12-month WT rats (A), rTg-DI rats (B), and SHR-SP rats (C) were labeled with Amylo-Glo to detect microvascular fibrillar amyloid (blue), goat polyclonal antibody to collagen IV to detect cerebral microvessels (red), and rabbit polyclonal antibody to Iba-1 to detect microglia (green). Images from the cortex are shown. Scale bars = 50 µm. (D) Quantitation of microglia numbers in different brain regions from WT rats (gray circles), rTg-DI rats (red circles), and SHR-SP rats (blue circles) at 12 M. Data points show the results from each rat and the group mean±SD of n = 6 rats of each type. ***p < 0.001 determined by 2-way ANOVA.
Dilated PVSs in Hypertensive SHR-SP Rats
Enlargement of PVS is a common presentation in CSVD (39, 40). Recently, it has been reported that SHR-SP rats display enlarged PVS (41). Therefore, we investigated the presence of dilated PVS in the rTg-DI rat model of CAA and in hypertensive SHR-SP rats. While there was no PVS enlargement in the rTg-DI rats compared to WT rats, the hypertensive SHR-SP rats displayed significant dilation of arteriolar PVS (Fig. 4).
FIGURE 4.

Dilated perivascular spaces in the SHR-SP rats. Brain sections from 12-month WT rats (A), SHR-SP rats (B), and rTg-DI rats (C) stained with hematoxylin and eosin revealing dilated PVS in SHR-SP brains. (D) Measurement of PVS area was determined by subtracting the area occupied by the vessel from the total combined PVS and vessel area. A ratio for PVS to vessel area was then determined. Data shown are the mean±SD of 90 randomly selected individual cortical arterioles (30 vessels selected from n = 3 rats per group). ****p < 0.0001 determine by 1-way ANOVA.
Cerebral Microhemorrhages Are Present in Both rTg-DI and SHR-SP Rat Brains
We previously reported that 12-month rTg-DI rats exhibit numerous cerebral microhemorrhages and calcified, occluded small vessels that are predominantly found in the thalamus (25–27, 42, 43). Consistent with our previous findings, histological staining revealed perivascular hemosiderin deposits and calcified, occluded small vessels in the thalamus of the rTg-DI rats (Fig. 5A, C, respectively). On the other hand, hemosiderin staining in the SHR-SP rats revealed that cerebral microhemorrhages were more random and present in the cortical regions and olfactory bulb (Fig. 4B). However, in contrast to rTg-DI rats we observed no small vessel occlusions in the WT or SHR-SP rat brains (Supplementary DataFig. S2). Thus, while the amyloidal-based rTg-DI model and nonamyloidal hypertensive SHR-SP model both present with cerebral microhemorrhages, their anatomical distribution is different and small vessel calcified occlusions were unique to the rTg-DI rats.
FIGURE 5.
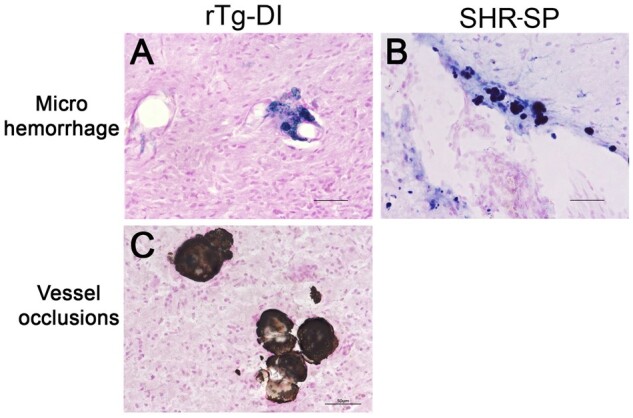
Cerebral microhemorrhages in rTg-DI rats and SHR-SP rats. Brain sections from 12-month rTg-DI rats (A, C) and SHR-SP rats (B) were stained for hemosiderin with Prussian blue to identify microhemorrhages (blue). Scale bars = 50 µm. Representative images show that cerebral microhemorrhages in the thalamus of rTg-DI rats (A) and cortex of SHR-SP rats (B). In addition, rTg-DI rats typically show calcified, occluded small vessels detected by von Kossa calcium stain in the thalamic region (C) that are not observed in SHR-SP rats.
Comparison of Elevated Proteins in the Brains of SHR-SP and rTg-DI Rats
Together, the above studies show that although amyloidal rTg-DI rats and nonamyloidal hypertensive SHR-SP rats share certain features (e.g. cerebral microhemorrhages) they are largely distinct forms of CSVD presenting with different cerebral vascular pathologies. To further understand the unique, and potentially common, molecular impacts of CAA and HTN in these models, we conducted global proteomic analysis of the rTg-DI and SHR-SP rat brains and compared the DEPs (relative to WT). Proteins isolated from pulverized whole brain tissue from rTg-DI rats and SHR-SP rats were subjected to the data-independent acquisition approach, Sequential Window Acquisition of all Theoretical Mass Spectra (SWATH-MS), as previously described (27). Data-independent acquisition data was analyzed with Spectronaut (Biognosys) for protein identification and quantification referencing a recently compiled spectral library (27), and protein quantities were converted to molar concentration (pmol/mg total protein) according to the “total protein approach” (35). In total, 2685 proteins were identified and quantified, and the number of proteins found in each analyzed sample is displayed in Supplementary DataFigure S3. Protein concentrations were then compared to identify DEPs. Because multiple testing corrected false discovery rates (FDR) can be too restrictive in small n proteomic studies (44, 45), we imposed threshold cutoffs to manage FDR and considered uncorrected p values as previously performed (27, 34). Additionally, FDR, set to 0.01, is managed during protein identification and quantification by Spectronaut at the protein, peptide, and protein spectrum match levels. We defined significantly enhanced proteins as ≥33% increase in expression compared to WT, with statistical significance defined as p ≤ 0.05. Comparative analysis of detected protein concentrations revealed 101 and 276 proteins were significantly elevated in the rTg-DI and SHR-SP rats, respectively, of which 28 proteins were commonly enhanced (Fig. 6A). Lists of the elevated proteins in each model can be found in Supplementary DataTables S1 and S2, and heat maps depicting the most enhanced proteins in the rTg-DI, SHR-SP, and those common to both models are depicted in Figure 6B. Anxa3, HTRA1, S100A4, CLU, SDC4, Histone H2A, and Histone H4 were all specifically elevated in the rTg-DI rat brain (Fig. 6B), all of which we previously reported elevated in different regions of rTg-DI rat brains (27). Anxa3 may be an important marker of microglia activation in the rTg-DI rats and CAA (27, 34), while HTRA1 has been reported as a possible marker of CAA in humans (46). H2A and H4 histone proteins are reported markers of neutrophil extracellular traps (NETs), a neutrophil mediated immune response causing breakdown of blood vessels, thrombosis, and severe perivascular tissue damage (47–52). Thus, the unique differential expression of these proteins may contribute to the distinctive pathologies associated with CAA in rTg-DI rats.
FIGURE 6.

Differentially expressed proteins in SHR-SP and rTg-DI brains. (A) A Venn diagram comparing significantly enhanced proteins by ≥33% of the WT concentration in the rTg-DI and SHR-SP brains (n = 10 for rTg-DI and 6 for SHR-SP, p ≤ 0.05 determined by t-test). (B) Heat map comparing relative expression of the enhanced proteins with the greatest increases specific to the rTg-DI, SHR-SP, and those overlapping between the 2 models. Red indicates enhanced expression, green reduced expression, and color intensity relative to the degree of change. (C) A Venn diagram comparing significantly reduced proteins by ≥33% of the WT concentration in the rTg-DI and SHR-SP brains (n = 10 for rTg-DI and 6 for SHR-SP, p ≤ 0.05). (D) Heat map comparing relative expression of the decreased proteins with the greatest relative decreases specific to the rTg-DI, SHR-SP, and those overlapping between the 2 models. Color shading as in panel (B).
Globally the 2 models share few commonly elevated proteins, with the 28 overlapping proteins representing 27.7% and 10% of the enhanced proteins in the rTg-DI and SHR-SP rats, respectively. Among the overlapping enhanced proteins was Metallothionein I (MT1, Mt1m). MT1 can mediate inflammatory responses in the brain, and enhanced expression of MT1 in astrocytes and microglia during neuroinflammation has been reported (53, 54). Though MT1 upregulation has been reported in the liver of spontaneously hypertensive (SHR) rats (55), to our knowledge it has not yet been reported in SHR-SP brains. Additionally, upregulation of MT1 in cells surrounding AD amyloid plaques and in the brains of the Tg-SwDI mouse model of CAA has been reported (53, 56). Heat shock protein B1 (HSPB1, HSP27) was also commonly elevated in both models. We have previously reported enhanced HSPB1 expression in rTg-DI rat brains (27), while enhanced expression in AD and colocalization with senile plaques and CAA in humans have also been reported (57, 58). Also, HSPB1 upregulation in peri-ischemic microglia and astrocytes following cerebral ischemia in rat brain has been reported (59). Aquaporin 4 (AQP4) was also enhanced in both rTg-DI rats and SHR-SP rats. We have previously reported AQP4 elevation in rTg-DI rat brains (27), and elevated expression of AQP4 in brains of AD and CAA patients surrounding senile plaques and vascular amyloid deposits has been established (60–62). Increases in AQP4 expression in astrocyte end feet following the development of HTN in SHR-SP rats has been reported in the mixed vascular dementia (HTN and AD) SHR-SP/FAD model rats (63, 64).
Proteins specifically elevated in the SHR-SP rat brains included milk fat globule EGF factor V/VIII (MFGE8), 5′ ectonucleotidase (Nt5e, CD73), and flotillins 1 and 2 (FLOT1, FLOT2). MFGE8 has been reported to be neuroprotective via anti-inflammatory mechanisms and inhibiting neuronal apoptosis in rodent models of cerebral ischemia (65, 66). CD73 can contribute to chronic HTN by modulating angiotensin-II signaling via the induction of the A2B adenosine receptor (67). Other reports indicate roles for CD73 limiting vascular leakage, leukocyte migration, and cerebral infarct severity during hypoxia/ischemia and stroke (68, 69). Thus, CD73 may contribute to the incidence of HTN or mitigating responses to the cerebral vascular pathologies in the SHR-SP rats. Enhancement of FLOT1 and FLOT2 was previously reported in both skeletal muscle of SHR-SP rats and renal and mesenteric arteries of related SHR rats (70, 71). To our knowledge, this is the first-time FLOT1 and FLOT2 upregulation has been characterized in the SHR-SP brain.
Comparison of Reduced Proteins in the Brains of SHR-SP and rTg-DI Rats
Fifty-four and 157 proteins were significantly reduced (≥33%, p < 0.05) in the rTg-DI and SHR-SP rat brains, respectively, of which 11 overlapped between the 2 models (Fig. 6C). Lists of these proteins can be found in Supplementary DataTables S3 and S4. Among the specifically reduced proteins in the rTg-DI brains was biliverdin (BLVRA). BLVRA deficiency has been reported to trigger a cascade leading to endothelial to mesenchymal transition, and endothelial dysfunction (72). Thus, BLVRA deficiency could be a contributing factor to the vascular pathologies observed in the rTg-DI rats. Sirtuin 1 (SIRT1) was also uniquely reduced in the rTg-DI rats. Inhibition of neuroinflammation and resultant neuroprotective effects of SIRT1 activation have been reported (73–75). SIRT1 activation has also been reported to inhibit Aβ induced inflammation in age-related macular degeneration (76). Therefore, reduced expression of SIRT1 in rTg-DI rats might contribute to an impaired regulation of Aβ induced neuroinflammation. Adam10, also specifically reduced in the rTg-DI brains, has been reported as the α-secretase responsible for APP cleavage, and therefore is vital in lowering Aβ production (77). Furthermore, ADAM10 levels in the CSF of AD patients are reduced (78). Thus, decreases in ADAM10 expression may contribute to the cerebral vascular accumulation of Aβ in rTg-DI rats.
Only a small fraction of the significantly reduced proteins overlapped between the 2 models. It is noteworthy that out of the 11 overlapping proteins, 6 (Krt1, Krt2, Krt14, Krt10, Krt6a, and Krt15) were keratin proteins. To our knowledge, none of these proteins has been previously associated with either CAA, AD, or HTN. Still, the small number of overlapping proteins further highlights the distinct nature of the rTg-DI and SHR-SP proteomes.
Among the uniquely reduced proteins in the SHR-SP rats was ATPase Na+/K+ Transporting Subunit β1 (ATP1B1). Genetic polymorphisms in the ATP1B1 gene have been associated with an increased risk of HTN (79). In mice, reduced ATP1B1 expression is associated with elevated blood pressure, and circadian expression rhythms of AT1B1 are antiphasic to that of blood pressure (80). It is possible that reduced AT1B1 expression in SHR-SP rats contributes to elevated blood pressure. Interestingly, many hemoglobin subunit proteins (Hba-a3, Hbb, Hba1, Hba-a2, Hbb2) showed reduced expression in the SHR-SP rats. To our knowledge, downregulation of hemoglobin has not been reported in SHR-SP rats.
PCA of DEPs
To further highlight the distinct nature of the DEP signatures in the rTg-DI and SHR-SP models, we conducted PCA using custom scripts in the program “R.” Two PCAs of the DEPs were conducted, first considering the DEPs in the rTg-DI rats (Fig. 7A), and second considering the DEPs in the SHR-SP rats (Fig. 7B). In the rTg-DI DEP PCA, there is clear separation of both models from the WT and from each other, despite indication of some similarity between the rTg-DI and SHR-SP. This is expected as ∼25% of the DEPs in the rTg-DI model overlap with the SHR-SP. On the other hand, PCA according to the DEPs observed in the SHR-SP rats, showed large separation between the SHR-SP rats from the rTg-DI rats and WT rats, with PC1 accounting for 42.88% of the variance, while rTg-DI rats and WT rats did not fully separate from each other. This again, is not surprising, as only ∼9% of the SHR-SP DEPs overlap with the rTg-DI. Nevertheless, PCA analysis of the DEPs in each model reveals that the 2 models display largely unique changes in their respective proteomes.
FIGURE 7.
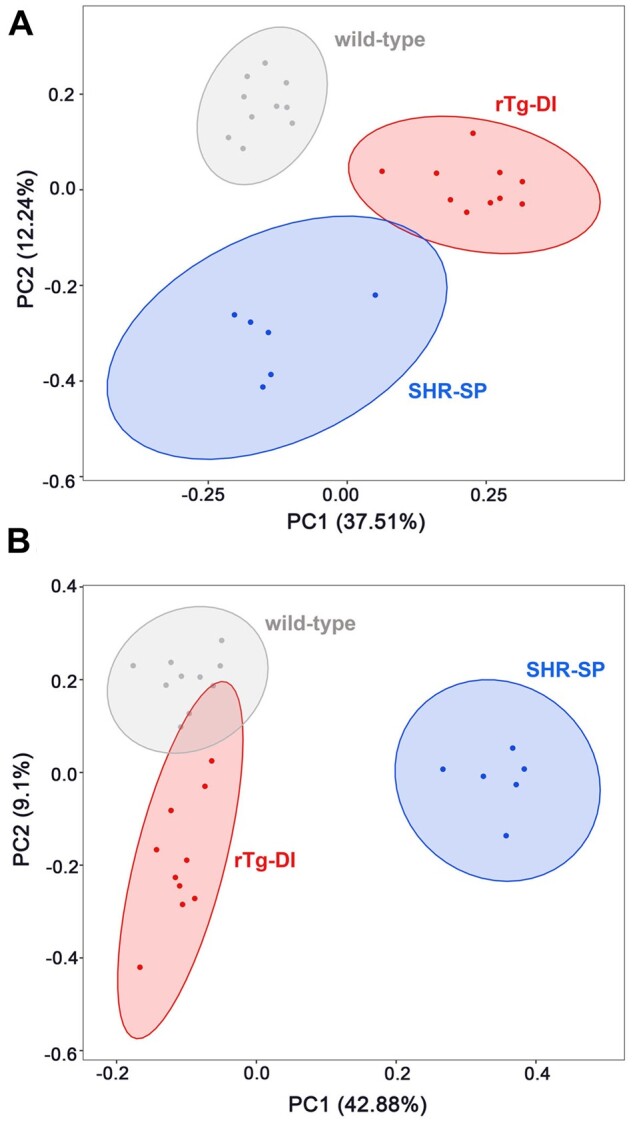
Primary component analysis (PCA) of differentially expressed proteins in the rTg-DI and SHR-SP brains. (A) PCA conducted in RStudio computing software, considering only the DEPs identified in the rTg-DI rats with PC1 accounting for 37.51% and PC2 accounting for 12.24% of the variance. The individual animals and probability ellipses for WT show in gray, rTg-DI red, and SHR-SP blue. (B) PCA conducted in RStudio computing software, considering only the DEPs identified in the SHR-SP rats with PC1 accounting for 42.88% and PC2 accounting for 9.1% of the variance. The individual animals and probability ellipses are indicated as in panel (A).
Ingenuity Pathway Analysis Reveals Distinct Activities in rTg-DI and SHR-SP Rat Brains
To discern pathway/disease-related functional context to the observed proteomic changes, we conducted comparison analysis with ingenuity pathway analysis (IPA) (Qiagen, Redwood City, CA) of the DEPs (≥33% increase or ≥33% decrease, p < 0.05). In our study, IPA analyzes the directional differential global expression of proteins to predict activation or inhibition of pathways, upstream regulators, causal networks, and disease functions, and providing a z score for each prediction (81). We recently reported regional differential regulator, network, and disease function IPA predictions in the cortex, hippocampus, and thalamus of the rTg-DI rats (27). Here, comparative IPA analysis revealed predicted upstream regulator activation of TNFα and TGFβ-1 in the rTg-DI (z = 2.467, 2.606, respectively), but not in the SHR-SP rats. Heat maps displaying relative global expression of DEPs associated with TNFα and TGFβ-1 networks in the rTg-DI rat brains are displayed in Figure 8A, B, respectively, along with their corresponding SHR-SP expression. We have previously reported increased TNFα mRNA expression and predicted activation of TNFα in distinct rTg-DI rat brain regions (26, 27), and TNFα is elevated in the CSF of AD patients (82). Important model differences in this network include Syndecan 4 (SDC4), a protein we have previously reported as elevated in multiple rTg-DI rat brain regions (27). TNFα has been shown to regulate SDC4 expression in multiple cell types (83–85), and SDC4 can contribute to both cellular uptake and fibrillization of Aβ (86). Thus, SDC4 may play an important role in mediating proinflammatory effects of TNFα, contribute to Aβ fibrillization, and be an important marker of CAA as evidenced by the rTg-DI rats.
FIGURE 8.

Ingenuity pathway analysis (IPA) identified upstream regulators, causal networks, and disease functions. (A) Heat map depicting the differentially expressed proteins (DEPs) (≥33% increase or ≥33% decrease) in the rTg-DI and SHR-SP rat brains associated with the upstream regulator TNFα. (B) Heat map depicting the DEPs (≥33% increase or ≥33% decrease) in the rTg-DI and SHR-SP rat brains associated with the upstream regulator TGF-β1. (C) Heat map depicting the DEPs (≥33% increase or ≥33% decrease) in the rTg-DI and SHR-SP rat brains associated with the Insulin causal network. Red indicates increased, green indicates decreased, and white indicates not DEPs; color intensity correlates with degree of change.
TGF-β1 released by pericytes and astrocytes is reported to support BBB function (87), however, hyperactivation of TGF-β1 in astrocytes is associated with loss of BBB integrity (88). Notable differences in expression include CLU and HTRA1, which we recently reported elevated in several brain regions of rTg-DI rats (27). CLU has been previously suggested as a possible marker of CAA in human samples (89). TGF-β1 induces the release of CLU, and CLU has been reported to mediate effects of TGF-β1, including epithelial to mesenchymal transition (90). The differential expression of HTRA1 between these 2 CVSD models is also interesting, as mutations in the Htra1 gene that cause loss of proteolytic function have been reported to cause cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL), another nonamyloidal CSVD (91–94).
Insulin was indicated as inhibited by IPA analysis (z = −1.837) in the SHR-SP but not the rTg-DI rats (Fig. 8C). The SHR-SP rat has been previously described as a model of insulin resistance (70, 93, 95). Therefore, our findings here are consistent with this previous characterization of the SHR-SP model. Insulin resistance and HTN have been linked to increased severity of CSVD in human patients, as well as increased incidence of small vessel stroke in nondiabetic patients (96, 97). Thus, the delineating differential proteomics of the SHR-SP from the rTg-DI rats reported here is consistent with previous findings that suggest insulin resistance as a distinct mechanism contributing to the vascular pathologies observed in the SHR-SP model.
Validation of CAA-Specific Regional Proteins
To further characterize and validate some the distinguishing rTg-DI-specific proteomic results we conducted regional immunolabeling of select proteins of interest in the rTg-DI and SHR-SP models. ANXA3 is commonly elevated in microglia of the rTg-DI rat cortex, hippocampus, and thalamus (27), and ANXA3 has been suggested as a marker of microglial activation (98). We also recently reported that ANXA3 was elevated in microglia of the rTg-DI corpus callosum, a region which displays little CAA burden (34). Based on these findings, ANXA3 may be an important marker of microglial activation in CAA. The proteomic results indicated that ANXA3 was not elevated in the SHR-SP brain. Immunolabeling studies were performed to confirm the differential expression of this potentially important marker for CAA in both CSVD models. Consistent with the proteomic results, immunolabeling for ANXA3 revealed strong elevation in the rTg-DI rats only, with no significant changes in any region of the SHR-SP rats. Figure 9A–C shows representative images of ANXA3 staining in the thalamus of WT, rTg-DI, and SHR-SP rats, respectively, confirming ANXA3 elevation was specific to the rTg-DI rats.
FIGURE 9.

Increased immunolabeling for specific protein markers in rTg-DI rats. Brain sections from 12-month WT rats (A, D, G, J), rTg-DI rats (B, E, H, K), and SHR-SP rats (C, F, I, L) were stained with thioflavin S to detect microvascular fibrillar amyloid (green) and rabbit polyclonal antibody to annexin 3 (A–C), Htra1 (D–F), histone 2A (G–I), and neutrophil elastase (J–L) (red). Scale bars = 50 µm. Representative images show each protein is increased only in the thalamus of rTg-DI rats.
Because of its implication in the TGF-β1 pathway (Fig. 8A), its specific upregulation in the rTg-DI rats (Fig. 6) (27), and its reported role in the CSVD, CARASIL (91–94), we further characterized expression of HTRA1 in the rTg-DI and SHR-SP brain regions. Consistent with our proteomic results, immunolabeling revealed elevation in HTRA1 in the rTg-DI thalamus (Fig. 9E) and other regions, while no significant changes in HTRA1 expression were observed in the SHR-SP thalamus (Fig. 9F), or other regions. Therefore, HTRA1 may be another important distinctive marker of CAA-related pathology.
The observed incidence of microhemorrhages and calcified vessel occlusions, and the DEPs in the rTg-DI thalamus led us to further characterize the presence of NET markers in the rTg-DI and SHR-SP brains. We previously reported the presence of NET markers only in the thalamus of rTg-DI rats, the only region to display microbleeds and thrombotic events (27). In the present study, elevation of NET markers H2A and H4 was specific to the rTg-DI rats (Fig. 6A). Therefore, we investigated the presence of NET markers in the rTg-DI and SHR-SP via immunolabeling for Histone 2A (H2A) and NE (27, 48). Consistent with our previous results, H2A and NE show robust immunolabeling in the rTg-DI thalamus confirming the presence of NETs in this region (Fig. 9H, K). On the other hand, we did not find any enhancement of H2A or NE in the thalamus (Fig. 9I, L) or any other brain region of SHR-SP rats. Thus, the presence of NET markers was specific to the rTg-DI model and may not contribute to the microbleeds observed in the SHR-SP brains.
DISCUSSION
The goal of the present study was to compare proteomic signatures in the nonamyloidal, hypertensive CSVD rat model SHR-SP with that of the CAA type-1 CSVD rat model rTg-DI, to identify DEPs and implicated mechanisms. The rTg-DI rat is a well-documented model of CAA type-1 that faithfully produces microvascular fibrillar amyloid deposition, significant perivascular neuroinflammation and glial cell responses, along with numerous microhemorrhages and occluded small vessels that lead to diffuse WM loss and behavioral deficits (25–27, 42, 99). Investigating the similar and disparate regional pathologies using proteomics and pathway analysis allowed for the identification of DEPs and mechanisms associated with the presence of CAA. On the other hand, the SHR-SP rat is a well-studied model of nonamyloidal HTN CSVD and the investigation of HA (21, 28–30, 37). To our knowledge, the SWATH-MS and subsequent pathway analysis of the brain performed here is the first such study in the SHR-SP rat model. This comparative analysis with the rTg-DI rat model allows for the identification of important proteins and mechanisms common or distinct to both types of CSVD. By uncovering changes that are unique to rTg-DI rats our study is advantageously positioned to report novel potential biomarkers and important proteins related to mechanisms specific to CAA.
SWATH-MS analysis revealed 155 and 433 DEPs in the rTg-DI and SHR-SP rats, respectively. The 39 overlapping proteins represented ∼25% and ∼9% of the total DEPs in the rTg-DI and SHR-SP models, respectively, signifying that the protein changes relative to the WT animals were largely unique between the 2 models. PCA of the DEPs in each model further displayed the unique nature of the proteomic changes in each model of CSVD (Fig. 7A, B).
In general, significantly more DEPs were identified in the SHR-SP rat brains. A possible explanation for this, and limitation of this study, is that the different genetic backgrounds between the SHR-SP rats and SD WT controls can produce numerous “false positives” or DEPs unrelated to the hypertensive state. Indeed, many studies have used Wistar Kyoto (WKY) rats as a WT control for the SHR-SP model (21, 28–30). However, WKY and SHR-SP contain large strain difference in their genome as the WKY rats were developed independently of SHR-SP from a separate ancestral pair in the original closed colony (100, 101), and it has been suggested that WKY rats are not a suitable control for SHR-SP rats (100, 102). Therefore, a more appropriate WT control for the SHR-SP may not be available. In any case, if the used SD WT control has resulted in overestimating the number of DEPs in the SHR-SP rats, then subsequent comparison with the rTg-DI results would lead to underestimating the number of proteins uniquely altered in that model. Thus, background differences and false discoveries in the SHR-SP model do not reduce our confidence in proteins identified as uniquely altered in the rTg-DI rat brains.
FLOT1 and FLOT2 upregulation has been previously reported in SHR and SHR-SP rats (70, 71). Flotillins are lipid raft associated proteins with reported functions in cell adhesion maintenance, endocytosis, and a suggested role in insulin signaling (103–105). Based on their pronounced enhancement in SHR-SP skeletal muscle, and SHR renal and mesenteric arteries, it has been suggested that aberrant expression of FLOT1 and FLOT2 contributes to alterations in the insulin signaling cascade and HTN pathology observed in these animals (70, 71). Previously, we observed significant elevation of FLOT1 and FLOT2 in the hippocampal and thalamic regions of the rTg-DI rats (27). However, in the present global proteomic analysis the flotillins did not meet this threshold in rTg-DI rats and was unique to SHR-SP rats.
Other specifically enhanced proteins in SHR-SP rats included MFGE8 and CD73. MFGE8 harbors neuroprotective and anti-inflammatory functions and could be upregulated in response to HTN stress (65, 66). CD73 can contribute to the incidence of HTN via angiotensin-II signaling and upregulation of the A2B adenosine receptor (67). Thus, CD73 upregulation in the SHR-SP rats, which exhibit HTN, is not surprising. On the other hand, CD73 is also reported to restrict vascular leakage, leukocyte migration, and cerebral infarct severity during cerebral ischemia/stroke (68, 69). Hence, it is also possible that CD73 upregulation in SHR-SP is in response to the HTN pathologies.
Many elevated proteins in rTg-DI rat brain including ANXA3, S100A4, HTRA1, and CLU were not elevated in the SHR-SP rats. We recently reported regional elevation of each of these proteins in the cortex, hippocampus, thalamus, and corpus callosum of 12-month rTg-DI rats (27, 34). The identification of these uniquely elevated proteins in the rTg-DI brains is important, as they harbor potential as biomarkers for differentiating CAA type-1 from other CSVDs. The identification of HTRA1 as a unique protein, both by proteomic analysis and immunolabeling in rTg-DI rats is also quite intriguing. Mutations in the Htra1 gene and resultant HTRA1 dysfunction are reported to cause CARASIL, a hereditary nonamyloidal CSVD (92–94). While loss of HTRA1 function is already implicated in nonamyloidal CSVD, the implications of elevated expression of HTRA1 in the rTg-DI model and its mechanistic ramifications for CAA type-1 pathogenesis have yet to be determined. Similar to our findings in rTg-DI rats, elevated HTRA1 was recently reported in human CAA type-1 further supporting a role for this protein in this disease (46). Thus, HTRA1 may play differential roles in CARASIL (nonamyloidal) and CAA (amyloidal) CSVD from a mechanistic perspective and distinct from HTN. ANXA3 is upregulated in all brain regions of rTg-DI rats and immunolabeling has revealed its expression in microglia (27, 34). In the present study, both proteomic analysis and immunolabeling revealed that ANXA3 upregulation is unique to the rTg-DI rats. Thus, ANXA3 is potentially an important identifying marker of CAA-type 1.
NETs are an important innate immune response mediated by neutrophils that consist of web-like structures of nucleic acids budded with histones and proteinases (48, 106). NETs have been reported to promote thrombosis, blood vessel wall disruption/breakdown, and result in significant inflammation induced tissue damage (47–52). It has also been reported that Aβ fibrils promote NET release (107). We previously reported elevation of NET histone markers (H3, H4, H2A, and H2B) in rTg-DI thalamus, and immunolabeling with NE and H2A confirmed the presence of NETs in the rTg-DI thalamus (27). Here, global elevation of the H2A and H4 NET markers was specific to the rTg-DI rats. Further, immunolabeling with NE and H2A revealed no evidence of these NET markers in any SHR-SP brain region. Therefore, NETs may be a distinguishing mechanism leading to the thrombotic vasculopathies observed in CAA.
IPA revealed further divergence of the SHR-SP and rTg-DI models. In the comparative analysis, predicted TNFα and TGF-β1 activation was specific to the rTg-DI rats. Elevation of TNFα in the CSF of AD patients has been reported, while we have previously shown upregulation of Tnfα mRNA expression in the brains of rTg-DI rats (26). Associated with this network is the rTg-DI specific upregulation of SDC4. We have previously reported regional upregulation of SDC4 in the cortex, hippocampus, and thalamus of rTg-DI rats (27), and SDC4 can contribute to the fibrillar assembly of Aβ (86). Therefore, TNFα activation may contribute to the CAA-specific pathologies, and SDC4 may be an important marker of CAA. On the other hand, TGF-β1 may have diverse implications regarding CAA. For example, TGF-β1 has been reported to increase BBB permeability and endothelial dysfunction or promote BBB integrity, and, thus, may have differing localized effects on cerebral microvascular function stability (87, 108–111). Elevation of CLU and HTRA1, potential identifying markers of CAA (46, 89), was associated with activation of TGF-β1. CLU has been reported to mediate TGF-β1 induced epithelial to mesenchymal transition (90). On the other hand HTRA1 is reported to regulate TGF-β1 signaling through processing/proteolysis of TGF-β1 (93, 112, 113). Thus, both proteins, CLU through mediating and HTRA1 via regulation, likely have significant impacts on the proinflammatory effects of TGF-β1 activity in response to accumulation of cerebral vascular amyloid in the rTg-DI rats, and while already suggested as potential markers, may have important mechanistic impacts in human CAA as well.
Insulin resistance in the SHR-SP rats is another distinguishing feature between the 2 CSVD models. IPA analysis revealed decreased insulin activity in the SHR-SP rats but not in the rTg-DI. SHR and SHR-SP rats have been previously described as a model of insulin resistance, and it has been postulated that FLOT1 and FLOT2, both elevated in the current study, contribute to the insulin resistance observed in these animals (70, 71). Insulin resistance has been linked to an increased incidence of cerebral ischemia and stroke and CSVD severity in nondiabetic HTN patients (96, 97). Thus, insulin resistance is an important distinguishing feature between these 2 models of CSVD and presents a unique mechanism for the development of vasculopathies observed in the SHR-SP rats not shared in the amyloidal rTg-DI rat model.
In conclusion, the global brain proteomic analysis of SHR-SP hypertensive rats and comparison to the global brain proteomic profile of rTg-DI CAA rats reveals largely distinct patterns of differential expression in these 2 models of CSVD. Even though HTN and CAA, and their respective models, display some convergent downstream pathological presentations, the present results suggest they arise from very different underlying mechanisms. Furthermore, uniquely differentiated proteins identified here in rTg-DI rats harbor potential for development as disease-specific protein biomarkers for CAA. Thus, this study presents important distinguishing mechanistic and diagnostic insight from the SHR-SP hypertensive and the rTg-DI CAA models of CSVD.
DATA AVAILABILITY
Raw mass spectrometry data can be found in the MassIVE repository [massive.ucsd.edu/ProteoSAFe/static/massive.jsp], project ID No.: MSV000089526, and password: SHRSPwb2022.
ACKNOWLEDGMENTS
The authors would like to thank Ms. Tyler DeVos for her assistance with the statistical analysis.
Supplementary Material
Contributor Information
Joseph M Schrader, From the George and Anne Ryan Institute for Neuroscience; Department of Biomedical and Pharmaceutical Sciences, University of Rhode Island, Kingston, Rhode Island, USA.
Aleksandra Stanisavljevic, From the George and Anne Ryan Institute for Neuroscience; Department of Biomedical and Pharmaceutical Sciences, University of Rhode Island, Kingston, Rhode Island, USA.
Feng Xu, From the George and Anne Ryan Institute for Neuroscience; Department of Biomedical and Pharmaceutical Sciences, University of Rhode Island, Kingston, Rhode Island, USA.
William E Van Nostrand, From the George and Anne Ryan Institute for Neuroscience; Department of Biomedical and Pharmaceutical Sciences, University of Rhode Island, Kingston, Rhode Island, USA.
The present work was supported by National Institutes of Health Research grant RO1NS094201 (WEVN) and an Advance Clinical and Translational Research Pilot Award (JMS).
The authors have no duality or conflicts of interest to declare.
Supplementary Data can be found at academic.oup.com/jnen.
REFERENCES
- 1. Shi Y, Wardlaw JM.. Update on cerebral small vessel disease: A dynamic whole-brain disease. Stroke Vasc Neurol 2016;1:83–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mustapha M, Nassir C, Aminuddin N, et al. Cerebral small vessel disease (CSVD) – Lessons from the animal models. Front Physiol 2019;10:1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pasi M, Cordonnier C.. Clinical relevance of cerebral small vessel diseases. Stroke 2020;51:47–53 [DOI] [PubMed] [Google Scholar]
- 4. Doubal FN, MacLullich AMJ, Ferguson KJ, et al. Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke 2010;41:450–4 [DOI] [PubMed] [Google Scholar]
- 5. Smith EE, Biessels GJ, De Guio F, et al. Harmonizing brain magnetic resonance imaging methods for vascular contributions to neurodegeneration. Alzheimers Dement (Amst) 2019;11:191–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rensink AAM, de Waal RMW, Kremer B, et al. Pathogenesis of cerebral amyloid angiopathy. Brain Res Brain Res Rev 2003;43:207–23 [DOI] [PubMed] [Google Scholar]
- 7. Arvanitakis Z, Leurgans SE, Wang Z, et al. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 2011;69:320–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Attems J, Jellinger K, Thal DR, et al. Review: Sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 2011;37:75–93 [DOI] [PubMed] [Google Scholar]
- 9. Viswanathan A, Greenberg SM.. Cerebral amyloid angiopathy in the elderly. Ann Neurol 2011;70:871–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jäkel L, De Kort AM, Klijn CJM, et al. Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimers Dement 2022;18:10–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greenberg SM, Vernooij MW, Cordonnier C, et al. ; Microbleed Study Group. Cerebral microbleeds: A guide to detection and interpretation. Lancet Neurol 2009;8:165–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boyle PA, Yu L, Nag S, et al. Cerebral amyloid angiopathy and cognitive outcomes in community-based older persons. Neurology 2015;85:1930–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thal DR, Ghebremedhin E, Rüb U, et al. Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 2002;61:282–93 [DOI] [PubMed] [Google Scholar]
- 14. Eikelenboom P, Veerhuis R, Familian A, et al. Neuroinflammation in plaque and vascular beta-amyloid disorders: Clinical and therapeutic implications. Neurodegener Dis 2008;5:190–3 [DOI] [PubMed] [Google Scholar]
- 15. Richard E, Carrano A, Hoozemans JJ, et al. Characteristics of dyshoric capillary cerebral amyloid angiopathy. J Neuropathol Exp Neurol 2010;69:1158–67 [DOI] [PubMed] [Google Scholar]
- 16. Thal DR, Ghebremedhin E, Orantes M, et al. Vascular pathology in Alzheimer disease: Correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol 2003;62:1287–301 [DOI] [PubMed] [Google Scholar]
- 17. Bailey TL, Rivara CB, Rocher AB, et al. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res 2004;26:573–8 [DOI] [PubMed] [Google Scholar]
- 18. Cuadrado-Godia E, Dwivedi P, Sharma S, et al. Cerebral small vessel disease: A review focusing on pathophysiology, biomarkers, and machine learning strategies. J Stroke 2018;20:302–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu J, Rutten-Jacobs L, Liu M, et al. Causal impact of type 2 diabetes mellitus on cerebral small vessel disease. Stroke 2018;49:1325–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Q, Yang Y, Reis C, et al. Cerebral small vessel disease. Cell Transplant 2018;27:1711–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jandke S, Garz C, Schwanke D, et al. The association between hypertensive arteriopathy and cerebral amyloid angiopathy in spontaneously hypertensive stroke-prone rats. Brain Pathol 2018;28:844–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Welsh TJ, Gladman JR, Gordon AL.. The treatment of hypertension in people with dementia: A systematic review of observational studies. BMC Geriatr 2014;14:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics–2015 update: A report from the American Heart Association. Circulation 2015;131:e29–322 [DOI] [PubMed] [Google Scholar]
- 24. Broderick M, Rosignoli L, Lunagariya A, et al. Hypertension is a leading cause of non-traumatic intracerebral hemorrhage in young adults. J Stroke Cerebrovasc Dis off J Natl Stroke Assoc 2020;29:104719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Davis J, Xu F, Hatfield J, et al. A novel transgenic rat model of robust cerebral microvascular amyloid with prominent vasculopathy. Am J Pathol 2018;188:2877–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu X, Hatfield J, Sullivan JK, et al. Robust neuroinflammation and perivascular pathology in rTg-DI rats, a novel model of microvascular cerebral amyloid angiopathy. J Neuroinflammation 2020;17:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schrader JM, Xu F, Van Nostrand WE.. Distinct brain regional proteome changes in the rTg-DI rat model of cerebral amyloid angiopathy. J Neurochem 2021;159:273–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Braun H, Bueche CZ, Garz C, et al. Stases are associated with blood–brain barrier damage and a restricted activation of coagulation in SHRSP. J Neurol Sci 2012;322:71–6 [DOI] [PubMed] [Google Scholar]
- 29. Bueche CZ, Hawkes C, Garz C, et al. Hypertension drives parenchymal β-amyloid accumulation in the brain parenchyma. Ann Clin Transl Neurol 2014;1:124–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Held F, Morris AWJ, Pirici D, et al. Vascular basement membrane alterations and β-amyloid accumulations in an animal model of cerebral small vessel disease. Clin Sci (Lond) 2017;131:1001–13 [DOI] [PubMed] [Google Scholar]
- 31. Schreiber S, Drukarch B, Garz C, et al. Interplay between age, cerebral small vessel disease, parenchymal amyloid-β, and tau pathology: Longitudinal studies in hypertensive stroke-prone rats. J Alzheimers Dis 2014;42:S205–15 [DOI] [PubMed] [Google Scholar]
- 32. Gomori G. Microtechnical demonstration of iron: A criticism of its methods. Am J Pathol 1936;12:655–64 [PMC free article] [PubMed] [Google Scholar]
- 33. Rungby J, Kassem M, Eriksen EF, et al. The von Kossa reaction for calcium deposits: Silver lactate staining increases sensitivity and reduces background. Histochem J 1993;25:446–51 [DOI] [PubMed] [Google Scholar]
- 34. Schrader JM, Xu F, Lee H, et al. Emergent white matter degeneration in the rTg-DI rat model of cerebral amyloid angiopathy exhibits unique proteomic changes. Am J Pathol 2022;192:426–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wiśniewski JR, Rakus D.. Multi-enzyme digestion FASP and the ‘Total Protein Approach’-based absolute quantification of the Escherichia coli proteome. J Proteomics 2014;109:322–31 [DOI] [PubMed] [Google Scholar]
- 36. Wiśniewski JR, Ostasiewicz P, Duś K, et al. Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol Syst Biol 2012;8:611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schreiber S, Bueche CZ, Garz C, et al. The pathologic cascade of cerebrovascular lesions in SHRSP: Is erythrocyte accumulation an early phase? J Cereb Blood Flow Metab 2012;32:278–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schreiber S, Bueche CZ, Garz C, et al. Blood brain barrier breakdown as the starting point of cerebral small vessel disease? – New insights from a rat model. Exp Transl Stroke Med 2013;5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown R, Benveniste H, Black SE, et al. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc Res 2018;114:1462–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Benveniste H, Nedergaard M.. Cerebral small vessel disease: A glymphopathy? Curr Opin Neurobiol 2022;72:15–21 [DOI] [PubMed] [Google Scholar]
- 41. Monte B, Constantinou S, Koundal S, et al. Characterization of perivascular space pathology in a rat model of cerebral small vessel disease by in vivo magnetic resonance imaging. J Cereb Blood Flow Metab 2022;0271678X221105668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee H, Xu F, Liu X, et al. Diffuse white matter loss in a transgenic rat model of cerebral amyloid angiopathy. J Cereb Blood Flow Metab 2021;41:1103–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhu X, Xu F, Hoos MD, et al. Reduced levels of cerebrospinal fluid/plasma Aβ40 as an early biomarker for cerebral amyloid angiopathy in RTg-DI rats. Int J Mol Sci 2020;21:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pascovici D, Handler DCL, Wu JX, et al. Multiple testing corrections in quantitative proteomics: A useful but blunt tool. Proteomics 2016;16:2448–53 [DOI] [PubMed] [Google Scholar]
- 45. Hondius DC, Eigenhuis KN, Morrema THJ, et al. Proteomics analysis identifies new markers associated with capillary cerebral amyloid angiopathy in Alzheimer’s disease. Acta Neuropathol Commun 2018;6:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zellner A, Müller SA, Lindner B, et al. Proteomic profiling in cerebral amyloid angiopathy reveals an overlap with CADASIL highlighting accumulation of HTRA1 and its substrates. Acta Neuropathol Commun 2022;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martinod K, Wagner DD.. Thrombosis: Tangled up in NETs. Blood 2014;123:2768–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kolaczkowska E, Jenne CN, Surewaard BGJ, et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun 2015;6:6673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pietronigro EC, Della Bianca V, Zenaro E, et al. NETosis in Alzheimer’s. Front Immunol 2017;8:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lim CH, Adav SS, Sze SK, et al. Thrombin and plasmin alter the proteome of neutrophil extracellular traps. Front Immunol 2018;9:1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou P, Li T, Jin J, et al. Interactions between neutrophil extracellular traps and activated platelets enhance procoagulant activity in acute stroke patients with ICA occlusion. EBioMedicine 2020;53:102671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hsieh IN, Deluna X, White MR, et al. Histone H4 directly stimulates neutrophil activation through membrane permeabilization. J Leukoc Biol 2021;109:763–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hidalgo J, Penkowa M, Espejo C, et al. Expression of metallothionein-I, -II, and -III in Alzheimer disease and animal models of neuroinflammation. Exp Biol Med (Maywood) 2006;231:1450–8 [DOI] [PubMed] [Google Scholar]
- 54. Dai H, Wang L, Li L, et al. Metallothionein 1: A new spotlight on inflammatory diseases. Front Immunol 2021;12:739918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Apostolova M, Bontchev PR, Nachev C, et al. Metallothioneins in spontaneously hypertensive rat liver. Jpn J Med Sci Biol 1992;45:185–98 [DOI] [PubMed] [Google Scholar]
- 56. Carrasco J, Adlard P, Cotman C, et al. Metallothionein-I and -III expression in animal models of Alzheimer disease. Neuroscience 2006;143:911–22 [DOI] [PubMed] [Google Scholar]
- 57. Wilhelmus MMM, Otte-Höller I, Wesseling P, et al. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol Appl Neurobiol 2006;32:119–30 [DOI] [PubMed] [Google Scholar]
- 58. Wilhelmus MMM, Boelens WC, Kox M, et al. Small heat shock proteins associated with cerebral amyloid angiopathy of hereditary cerebral hemorrhage with amyloidosis (Dutch type) induce interleukin-6 secretion. Neurobiol Aging 2009;30:229–40 [DOI] [PubMed] [Google Scholar]
- 59. Kato H, Kogure K, Liu XH, et al. Immunohistochemical localization of the low molecular weight stress protein HSP27 following focal cerebral ischemia in the rat. Brain Res 1995;679:1–7 [DOI] [PubMed] [Google Scholar]
- 60. Owasil R, O’Neill R, Keable A, et al. The pattern of AQP4 expression in the ageing human brain and in cerebral amyloid angiopathy. Int J Mol Sci 2020;21:1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hoshi A, Yamamoto T, Shimizu K, et al. Characteristics of aquaporin expression surrounding senile plaques and cerebral amyloid angiopathy in Alzheimer disease. J Neuropathol Exp Neurol 2012;71:750–9 [DOI] [PubMed] [Google Scholar]
- 62. Moftakhar P, Lynch MD, Pomakian JL, et al. Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. J Neuropathol Exp Neurol 2010;69:1201–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ishida H, Takemori K, Dote K, et al. Expression of glucose transporter-1 and Aquaporin-4 in the cerebral cortex of stroke-prone spontaneously hypertensive rats in relation to the blood-brain barrier function. Am J Hypertens 2006;19:33–9 [DOI] [PubMed] [Google Scholar]
- 64. Denver P, D’Adamo H, Hu S, et al. A novel model of mixed vascular dementia incorporating hypertension in a rat model of Alzheimer’s disease. Front Physiol 2019;10:1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cheyuo C, Aziz M, Wang P.. Neurogenesis in neurodegenerative diseases: Role of MFG-E8. Front Neurosci 2019;13:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cheyuo C, Jacob A, Wu R, et al. Recombinant human MFG-E8 attenuates cerebral ischemic injury: Its role in anti-inflammation and anti-apoptosis. Neuropharmacology 2012;62:890–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang W, Zhang Y, Wang W, et al. Elevated Ecto-5’-nucleotidase-mediated increased renal adenosine signaling via A2B adenosine receptor contributes to chronic hypertension. Circ Res 2013;112:1466–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Petrovic-Djergovic D, Hyman MC, Ray JJ, et al. Tissue-resident Ecto-5′ nucleotidase (CD73) regulates leukocyte trafficking in the ischemic brain. J Immunol 2012;188:2387–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Thompson LF, Eltzschig HK, Ibla JC, et al. Crucial role for Ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med 2004;200:1395–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. James DJ, Cairns F, Salt IP, et al. Skeletal muscle of stroke-prone spontaneously hypertensive rats exhibits reduced insulin-stimulated glucose transport and elevated levels of caveolin and flotillin. Diabetes 2001;50:2148–56 [DOI] [PubMed] [Google Scholar]
- 71. Bastrup JA, Aalkjær C, Jepps TA.. Identification of novel proteins and mechanistic pathways associated with early-onset hypertension by deep proteomic mapping of resistance arteries. J Biol Chem 2022;298:101512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Klóska D, Kopacz A, Piechota-Polańczyk A, et al. Biliverdin reductase deficiency triggers an endothelial-to-mesenchymal transition in human endothelial cells. Arch Biochem Biophys 2019;678:108182. [DOI] [PubMed] [Google Scholar]
- 73. Yan J, Luo A, Gao J, et al. The role of SIRT1 in neuroinflammation and cognitive dysfunction in aged rats after anesthesia and surgery. Am J Transl Res 2019;11:1555–68 [PMC free article] [PubMed] [Google Scholar]
- 74. Jiao F, Gong Z.. The beneficial roles of SIRT1 in neuroinflammation-related diseases. Oxid Med Cell Longev 2020;2020:e6782872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Velagapudi R, El-Bakoush A, Lepiarz I, et al. AMPK and SIRT1 activation contribute to inhibition of neuroinflammation by thymoquinone in BV2 microglia. Mol Cell Biochem 2017;435:149–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cao L, Liu C, Wang F, et al. SIRT1 negatively regulates amyloid-beta-induced inflammation via the NF-κB pathway. Braz J Med Biol Res 2013;46:659–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yuan XZ, Sun S, Tan CC, et al. The role of ADAM10 in Alzheimer’s disease. J Alzheimers Dis 2017;58:303–22 [DOI] [PubMed] [Google Scholar]
- 78. Sogorb-Esteve A, García-Ayllón MS, Gobom J, et al. Levels of ADAM10 are reduced in Alzheimer’s disease CSF. J Neuroinflammation 2018;15:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Faruque MU, Chen G, Doumatey A, et al. Association of ATP1B1, RGS5 and SELE polymorphisms with hypertension and blood pressure in African–Americans. J Hypertens 2011;29:1906–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nakashima A, Kawamoto T, Noshiro M, et al. Dec1 and CLOCK regulate Na+/K+-ATPase β1 subunit expression and blood pressure. Hypertension 2018;72:746–54 [DOI] [PubMed] [Google Scholar]
- 81. Krämer A, Green J, Pollard J, et al. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014;30:523–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Culpan D, Palmer J, Miners JS, et al. The influence of tumour necrosis factor-α (TNF-α) on amyloid-β (Aβ)-degrading enzymes in vitro. Int J Mol Epidemiol Genet 2011;2:409–15 [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang B, Ouyang P, Chen Y, et al. [Tumor necrosis factor-alpha regulates the proliferation and syndecan-4 expression of human umbilical vein endothelial-like cells cultured in vitro]. Nan Fang Yi Ke Da Xue Xue Bao 2007;27:496–8 [PubMed] [Google Scholar]
- 84. Yang H, Liu H, Li X, et al. TNF-α and TGF-β1 regulate Syndecan-4 expression in nucleus pulposus cells: Role of the mitogen-activated protein kinase and NF-κB pathways. Connect Tissue Res 2015;56:281–7 [DOI] [PubMed] [Google Scholar]
- 85. Okuyama E, Suzuki A, Murata M, et al. Molecular mechanisms of syndecan-4 upregulation by TNF-α in the endothelium-like EAhy926 cells. J Biochem 2013;154:41–50 [DOI] [PubMed] [Google Scholar]
- 86. Letoha T, Hudák A, Kusz E, et al. Contribution of syndecans to cellular internalization and fibrillation of amyloid-β(1–42). Sci Rep 2019;9:1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Derada Troletti C, de Goede P, Kamermans A, et al. Molecular alterations of the blood–brain barrier under inflammatory conditions: The role of endothelial to mesenchymal transition. Biochim Biophys Acta 2016;1862:452–60 [DOI] [PubMed] [Google Scholar]
- 88. Senatorov VV, Friedman AR, Milikovsky DZ, et al. Blood-brain barrier dysfunction in aging induces hyperactivation of TGFβ signaling and chronic yet reversible neural dysfunction. Sci Transl Med 2019;11:eaaw8283. [DOI] [PubMed] [Google Scholar]
- 89. Manousopoulou A, Gatherer M, Smith C, et al. Systems proteomic analysis reveals that clusterin and tissue inhibitor of metalloproteinases 3 increase in leptomeningeal arteries affected by cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 2017;43:492–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shiota M, Zardan A, Takeuchi A, et al. Clusterin mediates TGF-β-induced epithelial-mesenchymal transition and metastasis via Twist1 in prostate cancer cells. Cancer Res 2012;72:5261–72 [DOI] [PubMed] [Google Scholar]
- 91. Fukutake T. Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL): From discovery to gene identification. J Stroke Cerebrovasc Dis 2011;20:85–93 [DOI] [PubMed] [Google Scholar]
- 92. Chen Y, He Z, Meng S, et al. A novel mutation of the high-temperature requirement A serine peptidase 1 (HTRA1) gene in a Chinese family with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). J Int Med Res 2013;41:1445–55 [DOI] [PubMed] [Google Scholar]
- 93. Beaufort N, Scharrer E, Kremmer E, et al. Cerebral small vessel disease-related protease HtrA1 processes latent TGF-β binding protein 1 and facilitates TGF-β signaling. Proc Natl Acad Sci USA 2014;111:16496–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Uemura M, Nozaki H, Koyama A, et al. HTRA1 mutations identified in symptomatic carriers have the property of interfering the trimer-dependent activation cascade. Front Neurol 2019;10:693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Collison M, Glazier AM, Graham D, et al. Cd36 and molecular mechanisms of insulin resistance in the stroke-prone spontaneously hypertensive rat. Diabetes 2000;49:2222–6 [DOI] [PubMed] [Google Scholar]
- 96. Yang X, Zhang S, Dong Z, et al. Insulin resistance is a risk factor for overall cerebral small vessel disease burden in old nondiabetic healthy adult population. Front Aging Neurosci 2019;11:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhou M, Li H, Wang Y, et al. Causal effect of insulin resistance on small vessel stroke and Alzheimer’s disease: A Mendelian randomization analysis. Eur J Neurol 2022;29:698–706 [DOI] [PubMed] [Google Scholar]
- 98. Junker H, Suofu Y, Venz S, et al. Proteomic identification of an upregulated isoform of annexin A3 in the rat brain following reversible cerebral ischemia. Glia 2007;55:1630–7 [DOI] [PubMed] [Google Scholar]
- 99. Popescu DL, Van Nostrand WE, Robinson JK.. Longitudinal cognitive decline in a novel rodent model of cerebral amyloid angiopathy type-1. Int J Mol Sci 2020;21:2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nabika T, Ohara H, Kato N, Isomura M.. The stroke-prone spontaneously hypertensive rat: Still a useful model for post-GWAS genetic studies? Hypertens Res 2012;35:477–84 [DOI] [PubMed] [Google Scholar]
- 101. Johnson ML, Ely DL, Turner ME.. Genetic divergence between the Wistar-Kyoto rat and the spontaneously hypertensive rat. Hypertension 1992;19:425–7 [DOI] [PubMed] [Google Scholar]
- 102. Rapp JP. Use and misuse of control strains for genetically hypertensive rats. Hypertension 1987;10:7–10 [DOI] [PubMed] [Google Scholar]
- 103. Baumann CA, Ribon V, Kanzaki M, et al. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature 2000;407:202–7 [DOI] [PubMed] [Google Scholar]
- 104. Otto GP, Nichols BJ.. The roles of flotillin microdomains–endocytosis and beyond. J Cell Sci 2011;124:3933–40 [DOI] [PubMed] [Google Scholar]
- 105. Banning A, Babuke T, Kurrle N, et al. Flotillins regulate focal adhesions by interacting with α-actinin and by influencing the activation of focal adhesion kinase. Cells 2018;7:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Selders GS, Fetz AE, Radic MZ, et al. An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regen Biomater 2017;4:55–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Azevedo EPC, Guimarães-Costa AB, Torezani GS, et al. Amyloid fibrils trigger the release of neutrophil extracellular traps (NETs), causing fibril fragmentation by NET-associated elastase. J Biol Chem 2012;287:37206–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Garcia CM, Darland DC, Massingham LJ, et al. Endothelial cell–astrocyte interactions and TGFβ are required for induction of blood–neural barrier properties. Brain Res Dev Brain Res 2004;152:25–38 [DOI] [PubMed] [Google Scholar]
- 109. Ueberham U, Ueberham E, Brückner MK, et al. Inducible neuronal expression of transgenic TGF-beta1 in vivo: Dissection of short-term and long-term effects. Eur J Neurosci 2005;22:50–64 [DOI] [PubMed] [Google Scholar]
- 110. Derada Troletti C, Fontijn RD, Gowing E, et al. Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis 2019;10:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Moursel LG, Munting LP, Graaf LM, et al. TGFβ pathway deregulation and abnormal phospho-SMAD2/3 staining in hereditary cerebral hemorrhage with amyloidosis-Dutch type. Brain Pathol 2018;28:495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Graham JR, Chamberland A, Lin Q, et al. Serine protease HTRA1 antagonizes transforming growth factor-β signaling by cleaving its receptors and loss of HTRA1 in vivo enhances bone formation. PLoS One 2013;8:e74094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Launay S, Maubert E, Lebeurrier N, et al. HtrA1-dependent proteolysis of TGF-β controls both neuronal maturation and developmental survival. Cell Death Differ 2008;15:1408–16 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw mass spectrometry data can be found in the MassIVE repository [massive.ucsd.edu/ProteoSAFe/static/massive.jsp], project ID No.: MSV000089526, and password: SHRSPwb2022.