

Abstract
Astrocytes are the most abundant cells in the brain. They have many important functions in the central nervous system (CNS), including the maintenance of glutamate and ion homeostasis, the elimination of oxidative stress, energy storage in glycogen, tissue repair, regulating synaptic activity by releasing neurotransmitters, and participating in synaptic formation. Astrocytes have special highly ramified structure. Their branches contact with synapses of neurons inwardly, with fine structure and wrapping synapses; their feet contact with blood vessels of brain parenchyma outward, almost wrapping the whole brain. The adjacent astrocytes rarely overlap and communicate with each other through gap junction channels. The ideal location of astrocytes enables them to sense the weak changes of their surroundings and provide the structural basis for the energy supply of neurons. Neurons and astrocytes are closely coupled units of energy metabolism in the brain. Neurons consume a lot of ATPs in the process of neurotransmission. Astrocytes provide metabolic substrates for neurons, maintain high activity of neuron, and facilitate information transmission of neurons. This article reviews the characteristics of glucose metabolism, lipid metabolism, and amino acid metabolism of astrocytes. The metabolic interactions between astrocytes and neurons, astrocytes and microglia were also detailed discussed. Finally, we classified analyzed the role of metabolic disorder of astrocytes in the occurrence and development of neurodegenerative diseases.
Keywords: Alzheimer's diseases, astrocytes, metabolism, microglia, neuron, Parkinson disease
Metabolic changes of astrocytes in neurodegenerative diseases. In neurodegenerative diseases, the exchange of metabolites among astrocytes, neurons, and microglia has changed greatly, which aggravates the occurrence and development of neurodegenerative diseases. Astrocytes lose their homeostasis function. On the one hand, they are reactive to inflammatory factors of microglia. On the other hand, they fail to support the health of neurons. In addition, they also secrete many toxic factors that affect neurons.
1. INTRODUCTION
Astrocytes are the greatest number and special ramified cell‐type in CNS. They have several functions: their processes and cerebral vessels form the blood–brain barrier (BBB); their synapses represent the third element of the tripartite synapse; they provide a certain degree of metabolic supplement for neurons.
Astrocytes provide essential metabolic support for neighboring neurons and other cell types, and protect their neighboring cells by ingesting excessive glutamate and K+ and releasing growth factors, lactic acid, glutamine, mitogen, and other necessary chemical messengers. With the development of aging, injury, and disease, astrocytes will undergo significant morphological and molecular phenotypic changes, among which the most extensive features are cell hypertrophy and the upregulation of intermediate filament protein GFAP. Morphological hypertrophy of astrocytes is one of the main pathologies identified by Alois Alzheimer in 1910. 1 It is now considered as a marker of AD and most other forms of brain injury and chronic neurodegeneration. Although the reactive astrocytes of AD have a long history and significant performance, they are not as deeply studied as other major cell types in the brain, neuron, and microglia. Therefore, the effect of reactive astrocytes on the pathophysiology of AD is still unclear and speculated. In recent years, the research on energy metabolism of brain has changed from single center of neuron to three‐way metabolic coupling of neuron, astrocyte, and microglia. More attention has been paid to the energy metabolism cooperation among neurons, astrocytes, and microglia. Large number of studies have found that the mRNA expression of various metabolic enzymes in astrocytes changes in neurodegenerative diseases. 2 , 3 , 4 , 5 The material transfer and interaction among the three are of great significance to maintain the energy metabolism in the brain under various physiological and pathological conditions.
In this review, we discussed the characteristics of glucose metabolism, lipid metabolism, and amino acid metabolism of astrocytes in detail. The metabolic interactions between astrocytes and neurons, astrocytes, and microglia were also discussed. Finally, we analyzed the role of metabolic disorders of astrocytes in the occurrence and development of neurodegenerative diseases.
2. INTRODUCTION OF ASTROCYTES
Glial cells in the central nervous system (CNS) mainly contain microglia, oligodendrocytes, and the most diverse astrocytes. The concept of astrocyte was first put forward by Rudolf Virchow in the 19th century. 6 Later, Camillo Golgi used silver chromate staining to visualize the morphology of astrocytes. 7 Astrocytes were quickly divided into two basic morphological subtypes: protoplast type and fibrous type. 8 Protoplast astrocytes are usually found in gray matter, while fibrous astrocytes are commonly found in white matter of CNS. Due to the deepening of neuroanatomy and morphological analysis, astrocytes are divided into four types: interlayer type, varicose type, protoplast type, and fibrous type. 9
With the progress of transcriptome analysis tools, two types of reactive astrocytes were described in 2012. Compelling evidence suggests that two injury mouse models, neuroinflammation induced by lipopolysaccharide (LPS) and ischemic stroke induced by MCAO, identified two stimulus specific reactive astrocyte subtypes with unique transcriptome characteristics, called A1 astrocytes (detrimental) and A2 astrocytes (beneficial), respectively. 10 A1 astrocytes can secrete a harmful neurotoxin, which can induce apoptosis of neurons and oligodendrocytes. 11 However, transcriptomic analysis of A2 showed upregulated the expression of anti‐inflammatory genes and increased phagocytosis of microglia to protect neurons from Aβ toxicity. 12 The latest development of single cell transcriptomics has led to the identification of five astrocyte subsets (AST) in the mouse nervous system, showing various forms and functions. AST1 is defined as the high expression of Gfap and Agt, and AST2 is defined as the deletion of Agt and the high expression of Unc13c and Slc1a3. The distribution of AST3 is predicted by theexpression of Agt and low expression of Unc13c and Gfap. While AST4 is identified by high expression levels of Frzb, Ascl1, and Slc1a3. Finally, AST5 is identified by low expression of Ogt and high expression of Fam107a and Slc1a3. AST1 and AST4 are mainly distributed in hippocampus, AST2 is mainly distributed in cortex, AST3 and AST5 are evenly distributed between brain regions. 13 Among them, AST2 and AST3 differentially expressed genes related to neurotransmission in the cerebral cortex. AST2 is rich in transcripts related to glutamate neurotransmission. 14 AST3, in contrast, is rich in transcripts associated with GABAergic neurotransmission. 15 , 16 In addition, AST2 is rich in chordin‐like 1 (Chrd1), which is an important factor in stabilizing synapses. 17
3. METABOLISM OF ASTROCYTES
3.1. Carbohydrate metabolism of astrocytes
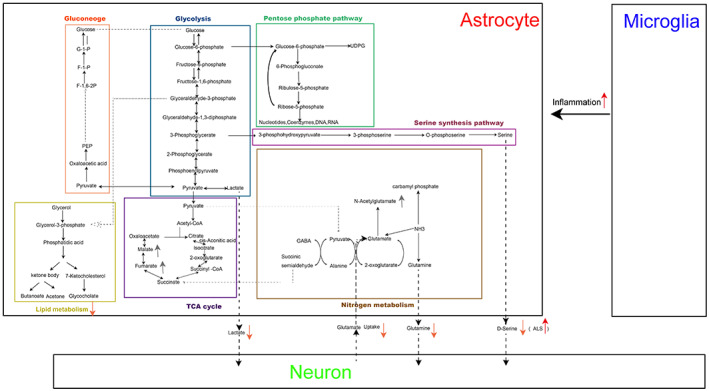
While the brain consumes a lot of energy, it is highly dependent upon the uninterrupted supply of energy substrates from the circulation. Glucose is the main energy source of the brain, which can be directly metabolized or stored in the form of glycogen. The loss of glucose of brain can lead to neurological disorders, unconsciousness, even coma within minutes. 18 However, under specific conditions, brain cells can effectively utilize various energy substrates besides glucose, including lactic acid, pyruvate, glutamate, and glutamine. 19 Most of these metabolites are formed endogenously by using glucose as carbon source. As brain metabolism is a process of compartmentalization, these metabolites undergo complex intercellular transport in the brain.
Through glucose transporter (GLUT) subtypes with different kinetic properties, glucose shuttles back and forth in brain cells reversibly from arterial blood through endothelial cell membrane. 20 GLUT1 is mainly located in endothelial cells and astrocytes, while GLUT3 and GLUT4 are mainly located in neurons. After glucose enters the cell, it is phosphorylated by hexokinase (HK) to produce glucose‐6‐phosphate. Like other organs, glucose‐6‐phosphate can be processed through different metabolic pathways, mainly in three ways 21 : the first is glycolysis, which leads to lactic acid production or mitochondrial metabolism, and the second is pentose phosphate pathway (PPP). The third is oxidative phosphorylation (OXPHOS). The fourth, there is glycogenesis, which is only in astrocytes.
Firstly, glucose‐6‐phosphate can be metabolized by glycolysis to produce two pyruvate molecules, ATP and NADH. Pyruvate can enter mitochondria and produce ATP and CO2 while consuming oxygen through the tricarboxylic acid cycle (TCA) and oxidative phosphorylation metabolism. Pyruvate can be reduced to lactic acid by lactate dehydrogenase (LDH). Lactic acid can be released in vitro through monocarboxylate transporter (MCT). Compared with glycolysis (2 ATP), Pyruvate produced by glycolysis can enter the tricarboxylic acid cycle (TCA) and be completely oxidized within the mitochondria to produce more energy in the form of ATP (30–34 ATP). Alternatively, glucose‐6‐phosphate can be treated by PPP to produce NADPH equivalent. It is noteworthy that PPP and glycolysis are associated at glyceraldehyde‐3‐phosphate and fructose‐6‐phosphate levels. Finally, in astrocytes, glucose‐6‐phosphate can also be used as a glycogen storage unit. 22
Importantly, both astrocytes and neurons have the ability to completely oxidize glucose and/or lactate, which is consistent with the observation that both cell types have the same amounts of mitochondria. 23 Although astrocytes show lower oxidative metabolic rates than neurons, they greedily absorb glucose and typically exhibit high glycolysis rates. 24 Most glucose that entering the astrocyte glycolysis pathway is released into lactate in extracellular space. 25 Lactate can be exported from astrocytes through monocarboxylate transporter 1 or 4 (MCT1/4) and transported to neurons through MCT2. 20 In neurons, astrocyte‐derived lactic acid is converted back to pyruvate and transported to mitochondria to produce ATP through the TCA cycle. 21 LDH, of which isoenzyme LDH‐5 is found to be specific to astrocytes, is an important metabolic enzyme in the process of lactate shuttle. 22 The lactate shuttle from astrocytes to neurons plays an important role in the energy supply of neurons. This is the well‐known astrocyte lactate shuttle hypothesis published by Pellerin and Magistretti in 1994, in which lactic acid is transferred from astrocytes to neurons in conjunction with the neurotransmitter glutamine. 23 According to this metabolic mechanism, astrocytes are in the perfect position of balanced metabolism. The glycolytic properties of astrocytes and their preference for the production of lactic acid rather than the entry of pyruvate in the TCA cycle are the result of a specific gene expression profile, which involve a variety of enzyme and transporter phenotypes in the brain. 26 Studies have shown that lactic acid produced by glycolysis of astrocytes is essential for the potential regulation of long‐term memory formation and the molecular changes required for long‐term memory formation. 27 Therefore, glucose metabolized by glycolysis in astrocyte can promote and meet high energy requirements related to cellular changes in learning, memory formation, and memory storage. But neurons are not entirely dependent on the lactic acid supplied by astrocytes. Lundgaard et al. and Diaz‐Garcia et al. 28 , 29 found that glucose is metabolized in situ by neurons in an activity‐dependent manner in awake mice. To be precise, neurons do metabolize lactic acid, but lactic acid is more like an “opportunistic” substrate that, if present, will indeed help support energy metabolism. 30
Other studies have suggested that 31 the high energy requirements of stimulated neurons are supported by astrocytes, which provide neurons with lactic acid produced by aerobic glycolysis, thus providing the energy required for the activity to induce neuron function. A lot of evidences agree that neurons have high oxidative activity, indicating that glycolysis mainly occurs in astrocytes. 23 , 32 That is to say that astrocytes are mainly glycolytic cells, not neurons.
Although the level of glycogen in the CNS is relatively low compared with the surrounding tissues, it is the largest energy reserve of the brain. Interestingly, at the cellular level, glycogen has been found to be almost confined to astrocytes in the adult brain. 33 It represents an advantageous form of glucose storage because it can be metabolized rapidly without the need for ATP, and unlike fatty acids, it can produce ATP under anaerobic conditions.
In addition to its role as an emergency energy reserve, glycogen in brain has many important physiological functions. Glycogen content is dynamically controlled by neurotransmitters, neurohumoral factors, and local energy state. 34 It was also found that decreased neuronal activity observed during sleep and anesthesia was associated with increased brain glycogen levels, consistent with the concept of awake brain utilization of glycogen. 35 In addition, the increase of neuronal activity induced by sensory stimulation is related to the decrease of glycogen level in the activated area, indicating that there is a close coupling between neuronal activity and glycogen mobilization. 36 Furthermore, astrocyte glycogen mobilization is possibly related to the transfer of lactic acid from astrocytes to neurons, as the absolutely necessary part of maintaining neuronal activity 37 and glutamatergic synaptic transmission. 38 These strong evidences indicate that glycogen plays a key and indispensable role in normal brain function and activity.
3.2. Lipid metabolism of astrocytes
Lipids are important components of nerve cell membranes, which participates in brain function in different ways. It is indispensable for astrocytes to participate in functions, such as energy generation, membrane fluidity, and intercellular signal conduction. It has been found that the lipid droplets stored by astrocytes play an essential physiological and protective role in the CNS. 39
Interestingly, the brain is considered autonomous in lipid metabolism as the BBB prevents lipid from entering the brain. 40 Neurons have a limited capacity for lipid metabolism, so they must take enough lipids from the external environment. 41 Astrocytes have strong lipid metabolism enzyme activity, and manage the nutritional support needed by brain for neuron consumption. 42 Therefore, it is generally believed that the lipids produced by astrocytes are absorbed by neurons to support the formation and function of synapses. In recent years, the synthesis of cholesterol and fatty acids in astrocyte has been studied, which depends on the sterol regulatory element binding protein (SREBPs). 43 SREBPs regulate the transcription activation of fatty acid and cholesterol metabolism‐related genes, 44 and the transcription factors mainly depend on the post‐translation activation of SREBP cleavage activator (SCAP) involved in sterol sensor. It is proved that astrocytes are the main SREBP expression cell types in hippocampus, and the decrease of SREBP activity in astrocytes leads to the damage of pre synaptic terminal function and synaptic plasticity. 45 In summary, these studies show that lipids produced by astrocyte is significant in synaptic development and brain function.
Cholesterol and unsaturated fatty acids are enriched in synaptic membrane, which specially affect a series of biochemical processes, including membrane fluidity, vesicle formation and fusion, ion channel function, and contribute to the formation of special micro areas of cell communication. 46 Cholesterol is the main form of cholesterol lipid in the brain. Among all the lipids in astrocytes, cholesterol may play the first part in astrocyte structure. Cholesterol helps regulate the elasticity of cell membrane by interacting with nearby phospholipids, 47 and also includes lipid raft formation, glucose transport, and inflammatory signals. 48 It is necessary to form synaptic vesicles before synapses and to form neurotransmitter receptors after synapses. Because of the existence of BBB, astrocytes' cholesterol metabolism is independent of other cholesterol metabolism in vivo. In addition, astrocytes had higher cholesterol level and lower expression of cholesterol synthetase in neurons. Therefore, astrocytes were considered to be the main place for cholesterol synthesis, mainly occurred in ATP‐dependent process in endoplasmic reticulum. 49 Cholesterol is produced through a complex process, which can be roughly divided into two biochemical pathways: Bloch pathway and the Kandutsch–Russell pathway. And the research has found that astrocytes synthesized cholesterol mainly through Bloch pathway, which is dominant for cholesterol biosynthesis in mouse models. 50
Astrocytes respond to ischemia by AMP‐activated protein kinase (AMPK) in vitro to promote the production of ketone body. The ketone body produced by astrocytes are derived from fatty acids, which may be used as the energy substrate of TCA cycle, rather than l‐lactate, because pyruvate dehydrogenase is susceptible to ischemia. 51 Therefore, fatty acids can be used to initially produce ketone body from astrocytes and then exchange between astrocytes and neurons through monocarboxylate transporter to generate energy through TCA cycle.
Sphingomyelin is a kind of lipid characterized by sphingosine skeleton. This includes a range of lipids, including ceramide, sphingosine, and sphingomyelin. The lipids produced by astrocytes are supplied to neurons and oligodendrocytes as part of synaptic and myelin membranes. 38 Compared with the oxidative metabolism of glucose in resting brain, lipid oxidation metabolism was inhibited, 52 but it was also regulated by the interaction between astrocytes and neurons. Sphingolipids have some functions in astrocytes, including inflammatory regulation. 53 At present, it has been found that normal production of ganglioside by astrocytes can also promote the growth of neurites, regulate the inflammation of neurons, and stabilize the interaction between neurons and glial cells. 54
Apolipoprotein E (ApoE) is a lipid carrier in the CNS. ApoE‐positive vesicles are swallowed by adjacent astrocytes, and are degraded into free fatty acids (FAS) by lysosome. 55 Then the free FAS adhere to lipid drops (LDS), which will mediate metabolism through mitochondria β‐Oxidation and phosphorylation. Astrocytes absorb these ApoE positive LDS through endocytosis, and provide energy for mitochondrial‐mediated apoptosis β‐Oxidation, as well as this coupling of lipid metabolism between neurons and astrocytes, protects neurons from fatty acid toxicity. 56 Astrocytes are not only the key cells to ingest and export FAS and apolipoprotein, but also the main cell group that mediates apoptosis β‐Oxidation of FAS in the brain. 57 Astrocytes transport lipids from BBB to neurons by binding and internalizing the permeable FAS of BBB from endothelial cells, and by the action of ATP binding box transporters (such as ABCA‐1), and loading ApoE into the cargo. 58 Brain is one of the largest sites for ApoE synthesis, second only to liver. 59 Most of the ApoE is derived from astrocytes, which are bound to receptors in the low‐density lipoprotein (LDL) receptor family, which are expressed in astrocytes and neurons. 60 Therefore, astrocytes are the vital parts of the lipid uptake‐ApoE‐lipidation axis, and are essential to maintain the lipid homeostasis and normal neuronal function in the brain. The role of ApoE from astrocyte and microglia in tau lesions is unclear. A recent study found that astrocyte derived ApoE particles were much larger than microglia, and thus contained more lipids. It suggesting that the two cell‐derived ApoE have different roles and functions. 61 Holtzman DM et al. 60 found that astrocytic ApoE4 removal decreasing disease‐associated gene signatures in neurons, oligodendrocytes, astrocytes, and microglia. Removal of astrocytic ApoE4 decreased tau‐induced synaptic loss and microglial phagocytosis of synaptic elements, suggesting a key role for astrocytic ApoE in synaptic degeneration. 62
3.3. Amino acid metabolism of astrocytes
Amino acids have various functions in astrocytes. In addition to their roles in protein biosynthesis, they also represent components of several other biosynthetic pathways. Astrocytes are obviously involved in the metabolism of all amino acids, but the main research direction is glutamate and GABA, two main neuroactive amino acids, which play a role in astrocytes. 63 Therefore, we focus on the glutamate/GABA‐glutamine cycle in astrocytes. Glutamate/GABA‐glutamine cycle is a process in which the two neurotransmitters are removed from synaptic cleft by astrocyte absorption, then transformed into glutamine. The newly‐synthesized glutamine is transferred into neurons to re‐synthesize neurotransmitters. 64 , 65 Glutamine is abundant in the CNS. It is the precursor of many neurotransmitter amino acids, including the excitatory amino acids, glutamic and aspartic acid, and the inhibitory amino acids, γ‐amino butyric acid (GABA). 66
Studies have shown that different responses in this cycle are clearly divided between astrocytes and neurons. 63 For example, Glutamine synthetase (Gs) is an enzyme that transforms glutamate into glutamine, which is expressed only in astrocytes. 65 Glutamine is converted into glutamate by phosphate activated glutaminase (PAG), and its expression in neurons is much higher than that in astrocytes. 67 In addition, GABA is synthesized by glutamic acid decarboxylases (GADs), which are widely present in GABAergic neurons. 68
In conclusion, glutamates are released into synaptic cleft in glutamate neurons, absorbed by astrocytes, and then transformed into glutamines by GS. The newly‐synthesized glutamines are transported to glutamate neurons, and then transformed into glutamates by PAG to participate in the next cycle. In GABAergic neurons, GABA is removed from synaptic cleft by astrocytes. In astrocytes, GABA and α‐ketoglutarate are converted to ammonia, then produced succinic acid. Glutamine is synthesized from succinic acid through tricarboxylic acid cycle (TCA), including citric acid formed by oxaloacetic acid and acetyl‐CoA and synthesized of ketoglutarate and glutamate. The newly‐synthesized Glutamine is transferred to GABAergic neurons and hydrolyzed to produce Glu, some of them can be decarboxylate to GABA. 69
Although the glutamate/GABA glutamine cycle is widely accepted, it is the result of deliberate simplification. For example, the cycle does not take exogenous glutamate into account. The studies have proved that glutamate not only comes from neurotransmitters, but also from glucose, lactic acid and so on. 70 Even some branched‐chain amino acids can be used as the source of glutamic acid. 63 Moreover, astrocytes can not only synthesize glutamine, but also oxidize glutamine. 71
4. METABOLIC INTERACTION OF ASTROCYTES
4.1. Metabolic interaction between astrocytes and neurons
Lactate shuttle between astrocytes and neurons is essential for maintaining normal brain function. Lactate not only provides energy to neurons, but also mediates long‐term memory and increases neuronal excitability. 72
Glutamate is an excitatory neurotransmitter in the brain, and its excessive aggregation in the brain can easily stimulate glutamatergic receptors (GluRs) on neurons and cause neurotoxic damage. 21 Glutamatergic neurons excitedly release glutamate into the extracellular space and absorb it into the cells through astrocyte specific Na+‐dependent GLT‐1(glutamate transporter 1) and GLAST (glutamate/aspartate transporter), with three Na+ ions entering the cells. Because the above reaction can disturb the ion balance inside and outside the cell, it can activate Na+/K+ ATP pump and increase ATP consumption. 73 Glutamic acid in astrocytes is converted to glutamine under GS catalysis, which is also a process of energy consumption. The glutamine produced shuttles back and forth into neurons and produces glutamate under the action of glutaminase (GLSs). Glutamate shuttling triggers energy consumption, which can increase the glucose absorption of astrocytes, and then lead to the increase of glycolysis and glycogen decomposition, the increase of lactic acid production, shuttling into neurons, as an energy substrate to provide ATP for neuron activation. 73 Astrocytes are also efficient at consuming glutamate to offset the energy expend in glutamine synthesis. 74 The pyruvate carboxylase of astrocytes converts glucose into glutamate to supplement the amount of glutamate in the brain. 75 The latter can also be converted into α‐ketoglutarate to enter the TCA cycle for oxidative metabolism.
Astrocytes convert glucose to l‐serine through a series of enzymatic reactions such as 3‐phosphoglycerate dehydrogenase (3PGDH). Selective knockout of astrocytes 3PGDH can reduce the levels of l‐serine and d‐serine in neurons. 76 n‐methyl‐d‐aspartate (NMDA) is a glutamate receptor. The agonist is d‐serine, which binds with glutamate to open its channel and activate glutamatergic neurons. In astrocytes, l‐serine reaches the extracellular matrix through ASCT1 (Slc1a4) transporter, enters neurons through Asc‐1 (Slc7a10) transporter, and transforms into d‐serine, or serine shuttle. Astrocyte ASCT1 was knocked out, l‐serine transport was impaired, and l‐serine and d‐serine were expressed at low levels in neurons. 77 However, glycine in astrocytes is not sensitive to ASCT1. When ASCT1 is injured, a large number of astrocytes can penetrate into neurons and generate l‐serine under the catalysis of serine hydroxymethyl transferase. 77 ASCT1 knockout mice cause serine deficiency and extensive amino acid metabolic disorders (alanine, threonine and glycine), which lead to exercise impairment and learning and emotional behavior defects in mice. 77 Astrocytes can also use d‐serine synthase serine racemase (SRR) to transfer l‐serine.
However, the synthesis of d‐serine is less than that of neurons. This is mainly due to the fact that glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) produced by astrocyte glycolysis can bind to SRR and inhibit its activation, and the product NADH can also allosterically destroy the stability of SRR. ATP produced by glycolysis can also amplify the interaction between GAPDH and SRR, and finally make SRR stable. In the inactive state, the production of d‐serine decreased. 78
When neurons were activated, they released norepinephrine (NA), vasoactive intestinal peptide (VIP), and adenylate cyclase receptor on the surface of astrocytes. cAMP concentration increased and astrocytes glycogen metabolism and glycolysis were stimulated. cAMP‐dependent protein kinase (AMPK) phosphorylates acetyl coenzyme A carboxylase (ACC), which reduces the activity of ACC, resulting in the reduction of malonyl coenzyme a synthesis and the inhibition of carnitine palmitoyl transferase (CPT‐I). CPT‐I carries fatty acids into mitochondria for β oxidation and produces ketone bodies, which are used as substrates for neuronal oxidation and biosynthesis. 79 During ischemia/hypoxia, the ratio of AMP/ATP in astrocytes increased, AMPK activity increased, and ACC activity was inhibited. As mentioned above, ketone body production increased, and neurons preferred to use ketone body as energy substrate. 79 Hypoxia can also increase the non‐oxidative metabolic pathway of non‐esterified fatty acids in astrocytes, increase the production of ceramide, activate extracellular signal regulated kinase (ERK), and lead to neuronal apoptosis, while AMPK activation can increase the β oxidation of fatty acids and the production of ketone bodies, reduce the de novo synthesis of ceramide, which is conducive to the survival of neurons. 79 Therefore, ketogenesis can be used as the main metabolic regulation mechanism of brain protection.
Cholesterol is abundant in the brain and is an important part of cell membrane, synapse and myelin sheath. It is synthesized in astrocytes and regulated by 3‐hydroxy‐3‐methyl‐glutaryl coenzyme A reductase (HMGCR) and sterol regulatory element binding protein (SREBP2). 41 , 80 When brain cholesterol content is high, SREBP cleavage activating protein (SCAP) isolates SREBP2 precursor in endoplasmic reticulum; when cholesterol is needed, it can induce SREBP2 to shuttle to Golgi body, shear into transcriptional active form, and then translocate into nucleus, bind to sterol regulatory element DNA, activate cholesterol synthase transcription, and synthesize cholesterol. 80 Cholesterol binds to LXR‐RXR, regulates the expression of efflux genes (ApoE, ABCA1, and ABCG1), transports lipoprotein granules and internalizes them into neurons. Cholesterol 24 hydroxylase (CYP46A1) is mainly expressed in neurons, catalyzing the degradation of cholesterol to 24 s hydroxycholesterol (24OHC), which reaches the blood through the BBB and enters the liver for further degradation. 41 Defective cholesterol synthesis can lead to impaired neurite growth and changes in memory and motor behavior. 80 Therefore, cholesterol synthesis and metabolism are very important for brain physiological function.
Maintenance and restoration of ion gradients dissipated by signaling processes, as well as uptake and recycling of neurotransmitters after neuronal firing, are the main brain energy needs. 81 Unlike other mesenchymal cells from other organs, astrocytes are involved in the formation of the BBB, so the process of blood supplying nutrients to neurons requires the participation of astrocytes. And astrocytes are responsible for sensing changes in the energy demand of neurons, and are the essential contributors to vasomotor responses, participating in both vasoconstriction and vasodilation. 82 Neurons do not metabolize lipid and could hardly perform gluconeogenesis, making them highly dependent on other supportive cells and external energy material supply, and highly susceptible for injury arising from energy supply failure, which results in two characteristics of astrocytes. One is that astrocytes are highly glycolytic. 21 The other is its ability to regulate vasodilation and blood supply by sensing the energy demand of surrounding neurons. Increased astrocytic activity—reflected by increased astrocytic Ca2+ concentrations—led to the activation of glycolysis and an elevation of extracellular lactate and adenosine concentrations, particularly during lower‐oxygen conditions. High external lactate hinders PGE2 clearance, thus increasing extracellular PGE2, which induces arteriolar dilatation. In addition, adenosine released under low oxygen inhibits astrocyte‐mediated vasoconstrictions at the level of smooth muscle cells by blocking the effect of arachidonic acid. 83 Recent study 84 showed a line of the transgenic mice which express a step function type of light‐gated cation channel (channelrhodopsine‐2; ChR2) in astrocytes (Mlc1‐positive). Photo‐activation of ChR2‐expressing astrocytes resulted in a widespread increase in cerebral blood flow (CBF), extending to the non‐stimulated periphery. Such optogenetic manipulation may make it possible to regulate CBF supply by regulating astrocytes.
4.2. Metabolic interaction between astrocytes and microglia
Microglia are resident myeloid cells of the mammalian CNS, and colonized in CNS during early embryonic development. They are highly motile cells that interact with all cells of the CNS to regulate normal development, homeostasis, and general brain physiology. In the process of neurodegenerative diseases, the polarization and function of microglia are closely related to the disease process.
Molecular dialogue between microglia and astrocytes begins shortly after they begin to fill the brain parenchyma. Astrocytes and microglia actively regulate neuronal activity and brain function. These glial cells also play an important role in the development and progression of various neuropathology. Their activation and malfunction occur in unique spatiotemporal patterns, and during these activations, close molecular dialogue is always present between these cells. 85 , 86 , 87 , 88 Molecular dialogue between microglia and astrocytes in brain development, function, and homeostasis.
The metabolism of microglia and astrocytes is studied relative independently, and there are few studies on the energy interaction between them. It has been found that the growth medium of dense astrocytes culture for three days (ACM) containing CSF‐1/IL‐34, TGF‐β2, and cholesterol can promote the survival of microglia, and microglia recognize and bind to liposomes through TREM2 and transfer cholesterol from astrocytes to microglia is essential for normal physiological function of microglia. 89 It has also been shown that astrocytes can regulate NO production by microglia by secreting serine and glycine. 90
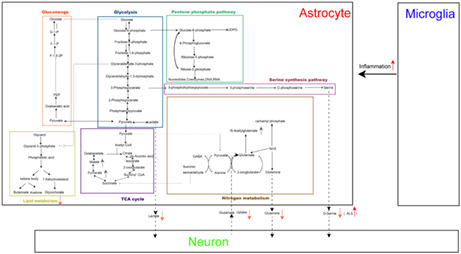
In neurodegenerative diseases, the exchange of metabolites among astrocytes, neurons, and microglia has changed greatly, which aggravates the occurrence and development of neurodegenerative diseases. On the one hand, astrocytes are reactive to inflammatory factors of microglia. Upon activation by Aβ, astrocytes participate in the secretion of inflammatory cytokines, thereby contributing to the neurodegenerative changes observed. On the other hand, the lactic acid, glutamine, and d‐serine provided by astrocytes to neurons are reduced, and the glutamate uptake is insufficient, resulting in the inability to support the health of neurons. However, d‐serine supply was enhanced in ALS. d‐serine induces glutamate excitotoxicity in motor neurons and increases glutamate excitotoxicity in both familial and sporadic ALS patients. In addition, they also secrete many toxic factors that affect neurons (Figure 1).
FIGURE 1.
Metabolic changes of astrocytes in neurodegenerative diseases. In neurodegenerative diseases, the exchange of metabolites among astrocytes, neurons, and microglia has changed greatly, which aggravates the occurrence and development of neurodegenerative diseases. Astrocytes lose their homeostasis function. On the one hand, they are reactive to inflammatory factors of microglia. On the other hand, they fail to support the health of neurons. In addition, they also secrete many toxic factors that affect neurons.
5. DYSMETABOLISM OF ASTROCYTES IN NEURODEGENERATIVE DISEASES
5.1. Dysmetabolism of astrocytes and aging
Aging means that many physiological changes of body function, including the decline of cell and body function over time, gradually reducing the biological function of cells, the reduction of basic substances of tissues and cells, and the decline of body organ structure and function. 91 Aging and metabolism are intricately linked. There is strong evidence that aging and aging‐related secretory phenotypes are sensitive to cell metabolic states, which in turn can drive phenotypes related to metabolic dysfunction. 92 That is, senescent cells are the driving factor of metabolic disorder, which will destroy the metabolic balance of multiple tissues. In fact, metabolic disorders in turn promote aging, thus forming a vicious circle.
Under normal circumstances, the interaction between cell homeostasis and system metabolism is dynamic. With the increase of age, aerobic glycolysis and glucose consumption in the brain are seriously reduced, especially in the temporal, parietal and frontal lobes, and motor cortex. The decrease of brain energy will lead to aging, which is related to the occurrence and development of neurodegeneration. Brain hypometabolism is the first symptom of aging‐related homeostasis disorder.
Astrocyte‐mediated glycolysis is closely related to aging and aging‐related neurodegenerative diseases. Aging is characterized by reduced energy consumption in specific areas of the brain. And astrocytes are the key to regulate neuronal metabolism and neurovascular coupling, so as to consume brain energy for neuronal activity. 93 Especially, in response to neuronal activity, astrocytes absorb glutamate at synapses, triggering astrocytic aerobic glycolysis, and leading to glucose uptake and lactate release. 21 Aging survey report shows that, reduced aerobic glycolysis in astrocytes and decreased mitochondrial oxidative phosphorylation in neurons are the main characteristics of aging. 94 , 95 On the one hand, neurons mainly rely on mitochondrial oxidative phosphorylation, and their ability to regulate glycolysis and inhibit the accumulation of oxidative stress is limited. Therefore, they are more prone to mitochondrial dysfunction related to aging. On the other hand, astrocyte dysfunction has harmful consequences for neurons, making neurons prone to degeneration, resulting in normal aging to neurodegeneration. 96 Aging limits the availability of metabolic substrates and the expression of metabolic mechanisms, which is closely related to abnormal energy metabolism, dysfunctional glutamate circulation of astrocytes, and neurovascular coupling changes. 97 In addition, the decreasing number of astrocytes and the weakened support of neurons through the lactate shuttle are also an important cause of the aging process.
In summary, further dysfunction of astrocytes leads to the progression of aging. These evidences suggest that astrocytes are deeply involved in the vulnerability of the nervous system to the pathological state of aging.
5.2. Dysmetabolism of astrocytes in Alzheimer's disease
Alzheimer's disease (AD) is a complex neurodegenerative disorder characterized. It shows a special cognitive and functional decline associated with age together with a neuropathology, including memory loss and language impairment. The pathogenesis of AD is complex and unclear. 98
The effect of astrocytes on Aβ in AD is still a controversy with no consistent answer. Numerous studies have indicated that astrocytes participate in the clearance of Aβ both in vivo and in vitro, suggesting an important role for these astrocytes in the attenuation of the neurodegenerative processes in AD (Figure 2a). For example, astrocytes plated on Aβ‐laden brain sections from an AD mouse model associate with the Aβ deposits and reduce overall Aβ levels in these sections. 99 Therefore, some articles suggest that the deficits in astroglial clearance of Aβ leads to AD. However, astrocytes can also produce Aβ in some inflammatory states. For instance, TGF‐β1 drives the production of Aβ40/42 by astrocytes leading to Aβ production in TGF‐β1 transgenic mice. 100 In addition, astrocytes can also rapidly engulf large amounts of Aβ, but then store, rather than completely degrade. The N‐terminal degraded Aβ was produced and partially release to the outside of the cell, causing cortical neuron apoptosis. 101 The function of dimorphism is closely related to different metabolic patterns.
FIGURE 2.
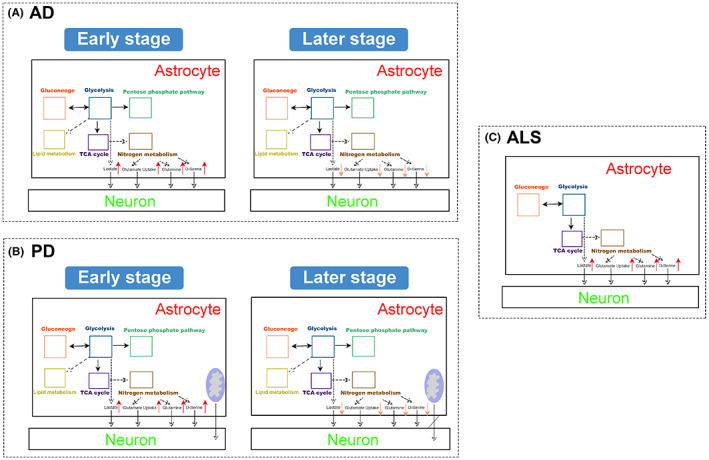
Metabolic changes of astrocytes in AD, PD, and ALS. In the early stage of AD and PD, astrocytes reprogram the metabolic mode to actively response the adverse effects of the disease on neurons, trying to make up for the adverse effects of the pathological process. In the late stage of AD and PD, astrocytes are also damaged by pathological features and cannot compensate for metabolic function.
Jens V. Andersen et al. 102 found that before Aβ formation, APPswe/PSEN1dE9 mice had altered glucose metabolism, and hampered glutamine processing and mitochondrial. Jens V. Andersen et al. 103 found that before Aβ formation in 5XFAD mice, reduced astrocyte TCA cycle activity and decreased glutamine synthesis led to hampered neuronal GABA synthesis in the 5xFAD hippocampus. Claudia Salcedo et al. 104 found that human‐induced pluripotent stem cells (hiPSCs) with APP and PSEN‐1 mutations show reduced leucine‐derived synthesis of glutamate, glutamine, and aspartic acid. The impaired metabolism of branched amino acids may lead to the imbalance of neurotransmitters and energy metabolism in AD. Jens V. Andersen et al. 105 also found that de novo glutamate and glutamine synthesis were reduced in astrocytes in 8 M 5XFAD. Staurenghi Erica et al. 106 indicated that in the AD brain, astrocyte cholesterol metabolism is abnormal and increased oxysterols promote increased the release of Lcn2, cytokines, and chemokines, ultimately affecting neuronal toxicity. Thus, these all imply that astrocytes are an important component of pathogenesis of AD, and also become an important therapeutic targets of AD. Libin Liu et al. 107 found that GLP‐1 increased astrocyte support for neurons by activating the PI3K/Akt signaling pathway and alleviating the reduced level of Aβ‐induced astrocyte glycolysis and the production of ROS. However, the development of glia‐aiming drugs remains in a nascent state, and the pathogenesis needs further research.
5.3. Dysmetabolism of astrocytes in Parkinson's disease
Parkinson's disease (PD) is the second most common neurodegenerative disorder. Its main pathological features are the loss of dopaminergic (DA) neurons in the dense substantia nigra and the aggregation of intracellular a‐synuclein. 108 The exact cause for PD is not yet clear, but the underlying molecular mechanisms have been identified in PD pathology. These include a‐synuclein protease stabilization, mitochondrial function, oxidative stress, calcium homeostasis, axonal transport, and neuroinflammation. 109 There is no cure for PD and currently the treatments are targeted to alleviate motor symptoms by dopamine replacement therapy and surgery.
While PD has been considered as a disease of DA neurons, it is becoming more and more evident that other nonneuronal cell types, including astrocytes, play an important role in the pathogenesis of PD (Figure 2b). Several genes known to have a causative role in the development of PD, such as PARK2, PARK7, PINK1, LRRK2, SNCA, ATP13A2, PLA2G6, and GBA are highly expressed in astrocytes and affect astrocyte metabolism:
PINK1, Parkin, GBA, and DJ‐1 have all been shown to have a role in the maintaining of healthy mitochondria. 110 , 111 , 112 , 113 IPLA2 regulate the release of fatty acid arachidonic acid (AA) from phospholipids, and ATP‐mediated calcium response. 114 , 115 A‐synuclein (a‐SYN) regulates the incorporation and distribution of AA and palmitic acid, 116 and glutamate transporter GLST1 and GLT1 expression and BBB function, and water transport of aquaporin 4(AQP4) localization. 117 DJ‐1 regulate the degradation of lipid rafts protein flotillin‐1 and caveolin‐1 and affect the assembly of lipid rafts (lipid rafts), thereby maintaining the glutamate transporters GLAST on the cell membrane. 118 Parkin regulate the secretion of glutathione in astrocytes and affect neurotrophic ability. 119 , 120 DJ‐1 terminate TLR4 signaling pathway through receptor endocytosis and inhibit IFN‐g inflammatory response. 121 GBA regulates the expression of cathepsin lysosomal proteases, 122 LRRK2 and ATP13A2 regulate the PH of lysosomal, 123 , 124 LRPK2 affects LC3 lipidation, 124 ATP13A2 maintains lysosome stability, 125 , 126 all above jointly regulate protein degradation. Parkin and PINK1 regulate astrocyte proliferation, which, in the case of PINK, function by regulating EGFR protein levels through AKT/p38‐dependent pathways. 119 , 120 , 123
Previous research 127 showed that vitamin d‐activating enzyme CYP27B1 positive astrocytes could display neuroprotective features as they sequester α‐Synuclein oligomers and are associated with Lewy body negative neurons.
Although there are few studies on the role of astrocytes in the pathogenesis of PD compared with the study of neuronal function, indications of astrocytes' involvement in PD have begun to emerge. Samanta Mazzetti et al. revealed that CYP27B1 is increased in astrocyte subpopulations with neuroprotective characteristics in PD patients, and that CYP27B1 positive astrocytes are involved in autophagy‐mediated ‐α‐synuclein uptake. Vitamin D can treat PD by preventing astrocyte changes. 127 Yuqi Zhang et al. 128 suggest that astrocytes regulate neuronal synaptic plasticity by releasing adenosine triphosphate, glutamate, and d‐serine, which may provide a new direction for the treatment of PD.
5.4. Dysmetabolism of astrocytes in ALS
Astrocytes play a crucial role in the pathogenesis of both sporadic and hereditary familial ALS which is associated with the mutation of the human superoxide dismutase 1 (hSOD1) gene. In hSOD1/G93A ALS model mice, astrocytes undergo morphological and pathological remodeling, loss of function, and cell death. The changes of astrocytes further lead to the aggravation of neuronal abnormalities and the appearance of clinical symptoms. 129 Selective silencing of hSOD1 gene in astrocytes delays the progression of ALS. 129
Oliver J Ziff et al. 130 showed that astrocytes in ALS are characterized by upregulation of genes related to extracellular matrix, endoplasmic reticulum stress, and immune response, and downregulation of genes related to synaptic integrity, glutamate uptake, and neural support (Figure 2c). These results suggest that astrocytes in ALS enhance inflammatory processes and inhibit neuronal support mechanisms. The key pathogenic factor associated with neurotoxicity is insufficient glutamate uptake by astrocytes. A fundamental function of astrocytes is to control and reduce the concentration of extracellular glutamate. Excessive glutamate in synaptic space, which is toxic to neurons, can lead to excessive discharge of neurons and increase calcium influx. In ALS mice, glutamate is also released by microglia. 131 , 132 Therefore, the neurotoxicity of glutamate in ALS is closely related to neuroinflammation. Another neurotoxicity molecule closely related to astrocytes in ALS is d‐serine, which is an endogenous comonomer of NMDA receptor. d‐serine induces glutamate excitotoxicity in motor neurons and increases glutamate excitotoxicity in both familial and sporadic ALS patients. 133 In addition, mutations in the gene encoding d‐amino acid oxidase (which controls the level of d‐serine) are associated with many cases of ALS families. 134
Mitochondrial dysfunction is a key mechanism of motor neuron degeneration in ALS. In addition to motoneurons, mitochondrial abnormalities in astrocytes lead to elevated reactive oxygen species (ROS) levels, which is associated with neurodegeneration in SOD1‐ALS mice. 135 Similarly, increased levels of NADPH oxidase (NOX2), inducible nitric oxide synthase (iNOS), and ROS were observed in human astrocytes expressing mutant SOD1, which induced non‐cellular autonomic toxicity in human ES cell‐derived motor neurons. 136 Consistent with these observations, in the sod1‐als model, an experimental treatment for overexpression of Nrf2 (nuclear factor erythroid‐2 related factor 2), a transcription factor, upregulates genes encoding antioxidants, especially in astrocytes.
In addition, Stella Robertoh et al. found changes in lipid metabolism in primary astrocytes in SOD1 (G93A), characterized by an increase in FAS and a reduction in the two intermediates of glycerol metabolism (i.e., galactosyl glycerol and phospholipic acid). These results indicate the changes of lipid metabolism/signal transduction in astrocytes of ALS. 137 Madji Hounoum Blandine et al. showed that compared with those cultured alone, astrocytes co‐cultured with motor neurons upregulated the glutamate and induced the expression of glutamate transporter (GLT/EAAT2), increased the capacity of glutamate/aspartic acid transporter (GLAST/EAAT1) and most metabolites involved in purine and pyrimidine pathways. However, compared with wild‐type motor neuron co‐cultured astrocytes,the accumulation of glutamate in SOD1G93A astrocytes indicated that dysregulation of the glutamate‐glutamine cycle, and SOD1G93A astrocytes treated with glutamate showed decreased levels of lactic acid, creatine, creatinine, deoxycarnitine, and l‐acetylcarnitine. 138 Therefore, astrocytes in ALS have metabolic dysfunction, and these metabolic pathways can be used to open up new therapeutic targets for ALS.
AUTHOR CONTRIBUTIONS
L.LG., and J.Z. conceptualized the study and wrote the manuscript. C.ZL., Y.ZQ., Y.SC. and X.MQ. discussed and edited the manuscript. L.LG. supervised the project. All authors reviewed and gave final approval to the manuscript.
FUNDING INFORMATION
National Nature Science Foundation of China Grant 81,801,337 (L.L.). National Nature Science Foundation of China Grant 82,071,520 (L.L.). Fujian Province Nature Science Foundation (Grant: 2019 J05006 to L.L.). Xiamen Youth Innovation Fund Grant 3502Z20206031 (L.L.).
CONFLICT OF INTEREST
The authors declare no competing interests.
Chen Z, Yuan Z, Yang S, et al. Brain Energy Metabolism: Astrocytes in Neurodegenerative Diseases. CNS Neurosci Ther. 2023;29:24‐36. doi: 10.1111/cns.13982
Zhenlei Chen, Ziqi Yuan, Shangchen Yang, and Yufei Zhu contributed equally to this work.
Contributor Information
Jie Zhang, Email: jiezhang@xmu.edu.cn.
Lige Leng, Email: lenglige@xmu.edu.cn.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available.
REFERENCES
- 1. Verkhratsky A, Parpura V, Rodriguez‐Arellano JJ, Zorec R. Astroglia in Alzheimer's disease. Adv Exp Med Biol. 2019;1175:273‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee M, Lee HJ, Park IS, et al. Aβ pathology downregulates brain mGluR5 density in a mouse model of Alzheimer. Neuropharmacology. 2018;133:512‐517. [DOI] [PubMed] [Google Scholar]
- 3. Paula GE, Daniela DL, Marina A, et al. Glutamate transporter GLT1 expression in Alzheimer disease and dementia with lewy bodies. Front Aging Neurosci. 2018;10:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ruminot I, Schmälzle J, Leyton B, Barros LF, Deitmer JW. Tight coupling of astrocyte energy metabolism to synaptic activity revealed by genetically encoded FRET nanosensors in hippocampal tissue. J Cereb Blood Flow Metab. 2017;39(3):513‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y, Chen K, Sloan SA, et al. An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34(36):11929‐11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deckert‐Schlüter M. Rudolf‐Virchow prize 1998. Award lecture. Toxoplasmosis: a model infection for studying systemic and intracerebral immune reactions. Verh Dtsch Ges Pathol. 1998;82(82):9‐22. [PubMed] [Google Scholar]
- 7. Kenez J. Camillo Golgi and the histology of the brain. Orv Hetil. 1963;104:1806‐1808. [PubMed] [Google Scholar]
- 8. Andriezen WL. The neuroglia elements in the human brain. Br Med J. 1893;2(1700):227‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oberheim NA, Takano T, Han X, et al. Uniquely hominid features of adult human astrocytes. J Neurosci. 2009;29(10):3276‐3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zamanian JL, Xu L, Foo LC, Nouri N, Barres BA. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32(18):6391‐6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liddelow SA, Barres BA. Reactive Astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957‐967. [DOI] [PubMed] [Google Scholar]
- 12. Jeong LH, Ok LJ, Woo LY, et al. LIF, a novel myokine, protects against amyloid‐beta‐induced neurotoxicity via Akt‐mediated autophagy signaling in hippocampal cells. Int J Neuropsychopharmacol. 2019;22(6):402‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Batiuk MY, Martirosyan A, Wahis J, Vin FD, Holt MG. Identification of region‐specific astrocyte subtypes at single cell resolution. Nat Commun. 2020;11(1):1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papouin T, Henneberger C, Rusakov DA, Oliet S. Astroglial versus Neuronal d‐serine: fact checking. Trends Neurosci. 2017;40:517‐520. [DOI] [PubMed] [Google Scholar]
- 15. Yoshimura Y, Dantzker J, Callaway E. Excitatory cortical neurons form fine‐scale functional networks. Nature. 2005;433(7028):868‐873. [DOI] [PubMed] [Google Scholar]
- 16. Lee I, Yoganarasimha D, Rao G, Knierim JJ. Comparison of population coherence of place cells in hippocampal subfields CA1 and CA3. Nature. 2004;430:456‐459. [DOI] [PubMed] [Google Scholar]
- 17. Blanco‐Suarez E, Liu TF, Kopelevich A, Allen NJ. Astrocyte‐secreted chordin‐like 1 drives synapse maturation and limits plasticity by increasing synaptic GluA2 AMPA receptors. Neuron. 2018;100:1116‐1132.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Verberne AJM, Sabetghadam A, Korim WS. Neural pathways that control the glucose counterregulatory response. Front Neurosci. 2014;8(8):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zielke HR, Zielke CL, Baab PJ. Direct measurement of oxidative metabolism in the living brain by microdialysis: a review. J Neurochem. 2010;109(s1):24‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vannucci SJ, Simpson IA. Developmental switch in brain nutrient transport expression in the rat. Am J Physiol Endocrinol Metab. 2003;285(5):E1127‐E1134. [DOI] [PubMed] [Google Scholar]
- 21. Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte‐neuron metabolic cooperation. Cell Metab. 2011;14:724‐738. [DOI] [PubMed] [Google Scholar]
- 22. Dienel GA. Brain glucose metabolism: integration of energetics with function. Physiol Rev. 2019;99(1):949‐1045. [DOI] [PubMed] [Google Scholar]
- 23. Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, Nedergaard M. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci. 2007;27(45):12255‐12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Itoh Y, Esaki T, Shimoji K, et al. Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc Natl Acad Sci. 2003;100(8):4879‐4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bouzier‐Sore AK, Voisin P, Bouchaud V, Bezancon E, Pellerin L. Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: a comparative NMR study. Eur J Neurosci. 2006;24(6):1687‐1694. [DOI] [PubMed] [Google Scholar]
- 26. Mason S. Lactate shuttles in neuroenergetics—homeostasis, allostasis and beyond. Front Neurosci. 2017;11:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suzuki A, Stern S, Bozdagi O, et al. Astrocyte‐neuron lactate transport is required for long‐term memory formation. Cell. 2011;144(5):810‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lundgaard I, Li B, Xie L, et al. Direct neuronal glucose uptake heralds activity‐dependent increases in cerebral metabolism. Nat Commun. 2015;6:6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Díazgarcía C, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G. Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab. 2017;26(2):361‐374.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dienel GA. Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab. 2012;32(7):1107‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci. 1994;91(22):10625‐10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bolaños J, Almeida A, Moncada S. Glycolysis: a bioenergetic or a survival pathway? Trends Biochem Sci. 2010;35(3):145‐149. [DOI] [PubMed] [Google Scholar]
- 33. Brown AM. Brain glycogen re‐awakened. J Neurochem. 2004;89(3):537‐552. [DOI] [PubMed] [Google Scholar]
- 34. Magistretti PJ, Morrison JH, Shoemaker WJ, Bloom S. Vasoactive intestinal polypeptide induces glycogenolysis in mouse cortical slices: a possible regulatory mechanism for the local control of energy metabolism. Proc Natl Acad Sci U S A. 1981;78(10):6535‐6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Magistretti PJ, Sorg O, Yu N, Martin JL, Pellerin L. Neurotransmitters regulate energy metabolism in astrocytes: implications for the metabolic trafficking between neural cells. Dev Neurosci. 1994;15(3–5):306‐312. [DOI] [PubMed] [Google Scholar]
- 36. Dienel GA, Ball KK, Cruz NF. A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: implications for glycogen turnover. J Neurochem. 2007;102(2):466‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tekkök SB, Brown AM, Westenbroek R, Pellerin L, Ransom BR. Transfer of glycogen‐derived lactate from astrocytes to axons via specific monocarboxylate transporters supports mouse optic nerve activity. J Neurosci Res. 2010;81(5):644‐652. [DOI] [PubMed] [Google Scholar]
- 38. Mozrzymas J, Szczęsny T, Rakus D. The effect of glycogen phosphorolysis on basal glutaminergic transmission. Biochem Biophys Res Commun. 2011;404(2):652‐655. [DOI] [PubMed] [Google Scholar]
- 39. Verheijen M, Camargo N, Verdier V, et al. SCAP is required for timely and proper myelin membrane synthesis. Proc Natl Acad Sci U S A. 2009;106(50):21383‐21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saeed AA, Genove G, Li T, et al. Effects of a disrupted blood‐brain barrier on cholesterol homeostasis in the brain. J Biol Chem. 2014;289(34):23712‐23722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pfrieger FW, Ungerer N. Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res. 2011;50(4):357‐371. [DOI] [PubMed] [Google Scholar]
- 42. Lee JA, Hall B, Allsop J, Alqarni R, Allen SP. Lipid metabolism in astrocytic structure and function. Semin Cell Dev Biol. 2021;112:123‐136. [DOI] [PubMed] [Google Scholar]
- 43. Camargo N, Brouwers JF, Loos M, Gutmann DH, Smit AB, Verheijen MH. High‐fat diet ameliorates neurological deficits caused by defective astrocyte lipid metabolism. FASEB J. 2012;26(10):4302‐4315. [DOI] [PubMed] [Google Scholar]
- 44. Eberlé D, Hegarty B, Bossard P, Ferré P, Foufelle F. SREBP transcription factors: master regulators of lipid homeostasis. Biochimie. 2004;86(11):839‐848. [DOI] [PubMed] [Google Scholar]
- 45. Camargo N, Smit AB, Verheijen M. SREBPs: SREBP function in glia‐neuron interactions. FEBS J. 2009;276(3):628‐636. [DOI] [PubMed] [Google Scholar]
- 46. Van Deijk ALF, Camargo N, Timmerman J, et al. Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia. 2017;65(4):670‐682. [DOI] [PubMed] [Google Scholar]
- 47. Engberg O, Hautala V, Yasuda T, et al. The affinity of cholesterol for different phospholipids affects lateral segregation in bilayers. Biophys J. 2016;111(3):546‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sonnino S, Prinetti A. Membrane domains and the "lipid raft" concept. Curr Med Chem. 2013;20:4‐21. [PubMed] [Google Scholar]
- 49. Reinhart MP, Billheimer JT, Faust JR, Gaylor JL. Subcellular localization of the enzymes of cholesterol biosynthesis and metabolism in rat liver. J Biol Chem. 1987;262(20):9649‐9655. [PubMed] [Google Scholar]
- 50. Nieweg K, Schaller H, Pfrieger FW. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J Neurochem. 2010;109(1):125‐134. [DOI] [PubMed] [Google Scholar]
- 51. Takahashi S, Iizumi T, Mashima K, Abe T, Suzuki N. Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. ASN Neuro. 2014;6(5):175909141455099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Camargo N, Goudriaan A, Deijk ALFV, Otte WM, Verheijen MHG. Oligodendroglial myelination requires astrocyte‐derived lipids. PLoS Biol. 2017;15(5):e1002605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Novgorodov SA, Voltin JR, Wang W, Tomlinson S, Riley CL, Gudz TI. Acid sphingomyelinase deficiency protects mitochondria and improves function recovery after brain injury. J Lipid Res. 2019;60:609‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tobias M, Shaw PJ, Johnathan CK. Disrupted glycosylation of lipids and proteins is a cause of neurodegeneration. Brain.2020;143(5):1332‐1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mukherjee S, Suresh SN. Neuron–astrocyte liaison to maintain lipid metabolism of brain. Trends Endocrinol Metab. 2019;30(9):573‐575. [DOI] [PubMed] [Google Scholar]
- 56. Farmer BC, Kluemper J, Johnson LA. Apolipoprotein E4 alters astrocyte fatty acid metabolism and lipid droplet formation. Cell. 2019;8:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ebert D, Haller RG, Walton ME. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci. 2003;23(13):5928‐5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yassine HN, Qingru F, Jiarong C, et al. ABCA1‐mediated cholesterol efflux capacity to cerebrospinal fluid is reduced in patients with mild cognitive impairment and Alzheimer's disease. J Am Heart Assoc. 2016;5(2):e002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Elshourbagy NA, Liao WS, Mahley RW, Taylor JM. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc Natl Acad Sci U S A. 1985;82:203‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E Receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(3):a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huynh T, Wang C, Tran A, et al. Lack of hepatic apoE does not influence early Aβ deposition: observations from a new APOE knock‐in model. Mol Neurodegener. 2019;14(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang C, Xiong M, Gratuze M, et al. Selective removal of astrocytic APOE4 strongly protects against tau‐mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron. 2021;109(10):1657‐74.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yudkoff M. Interactions in the metabolism of glutamate and the branched‐chain amino acids and ketoacids in the CNS. Neurochem Res. 2017;42(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Albrecht J, Sidoryk‐Wegrzynowicz M, Zielinska M, Aschner M. Roles of glutamine in neurotransmission. Neuron Glia Biol. 2010;6(4):263‐276. [DOI] [PubMed] [Google Scholar]
- 65. Kreft M, Bak LK, Waagepetersen HS, Schousboe A. Aspects of astrocyte energy metabolism, amino acid neurotransmitter homoeostasis and metabolic compartmentation. ASN Neuro. 2012;4(3):e00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Segovia G, Del Arco A, Mora F. Role of glutamate receptors and glutamate transporters in the regulation of the glutamate‐glutamine cycle in the awake rat. Neurochem Res. 1999;24(6):779‐783. [DOI] [PubMed] [Google Scholar]
- 67. Kvamme E, Torgner IA, Roberg B. Kinetics and localization of brain phosphate activated glutaminase. J Neurosci Res. 2001;66(5):951‐958. [DOI] [PubMed] [Google Scholar]
- 68. Lee SE, Lee Y, Lee GH. The regulation of glutamic acid decarboxylases in GABA neurotransmission in the brain. Arch Pharm Res. 2019;42(12):1031‐1039. [DOI] [PubMed] [Google Scholar]
- 69. Bak LK, Schousboe A, Waagepetersen HS. The glutamate/GABA‐glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem. 2006;98(3):641‐653. [DOI] [PubMed] [Google Scholar]
- 70. Berent‐Spillson A, Russell JW. Metabotropic glutamate receptor 3 protects neurons from glucose‐induced oxidative injury by increasing intracellular glutathione concentration. J Neurochem. 2007;101(2):342‐354. [DOI] [PubMed] [Google Scholar]
- 71. Cardona C, Sanchez‐Mejias E, Davila JC, et al. Expression of GLS and GLS2 glutaminase isoforms in astrocytes. Glia. 2015;63(3):365‐382. [DOI] [PubMed] [Google Scholar]
- 72. Magistretti PJ, Allaman I. Lactate in the brain: from metabolic end‐product to signalling molecule. Nat Rev Neurosci. 2018;19:235‐249. [DOI] [PubMed] [Google Scholar]
- 73. Magistretti PJ, Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86(4):883‐901. [DOI] [PubMed] [Google Scholar]
- 74. Sonnewald U, Westergaard N, Petersen SB, Unsgrd G, Schousboe A. Metabolism of [U‐13C]glutamate in astrocytes studied by 13C NMR spectroscopy: incorporation of more label into lactate than into glutamine demonstrates the importance of the tricarboxylic acid cycle. J Neurochem. 1993;61(3):1179‐1182. [DOI] [PubMed] [Google Scholar]
- 75. Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 2007;27(2):219‐249. [DOI] [PubMed] [Google Scholar]
- 76. Ehmsen JT, Ma TM, Sason H, et al. d‐serine in glia and neurons derives from 3‐phosphoglycerate dehydrogenase. J Neurosci. 2013;33(30):12464‐12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Eitan K, Salman Z, Inna R, et al. ASCT1 (Slc1a4) transporter is a physiologic regulator of brain D‐serine and neurodevelopment. Proc Natl Acad Sci U S A. 2018;115:9628‐9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Suzuki M, Sasabe J, Miyoshi Y, et al. Glycolytic flux controls d‐serine synthesis through glyceraldehyde‐3‐phosphate dehydrogenase in astrocytes. Proc Natl Acad Sci. 2015;112:E2217‐E2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Guzmán M, Blázquez C. Is there an astrocyte–neuron ketone body shuttle? TRENDS in Endocrinology & Metabolism. Elsevier; 2001. [DOI] [PubMed] [Google Scholar]
- 80. Ferris HA, Perry RJ, Moreira GV, Shulman GI, Horton JD, Kahn CR. Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole‐body metabolism. Proc Natl Acad Sci U S A. 2017;114(5):1189‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Alle H, Roth A, Geiger JR. Energy‐efficient action potentials in hippocampal mossy fibers. Science. 2009;325(5946):1405‐1408. [DOI] [PubMed] [Google Scholar]
- 82. Carmignoto G, Gomez‐Gonzalo M. The contribution of astrocyte signalling to neurovascular coupling. Brain Res Rev. 2010;63(1–2):138‐148. [DOI] [PubMed] [Google Scholar]
- 83. Gordon GR, Choi HB, Rungta RL, Ellis‐Davies GC, MacVicar BA. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature. 2008;456(7223):745‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hatakeyama N, Unekawa M, Murata J, et al. Differential pial and penetrating arterial responses examined by optogenetic activation of astrocytes and neurons. J Cereb Blood Flow Metab. 2021;41(10):2676‐2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Barres BA. Normal aging induces A1‐like astrocyte reactivity. Proc Natl Acad Sci. 2018;115(8):E1896‐E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jha MK, Lee WH, Suk K. Functional polarization of neuroglia: Implications in neuroinflammation and neurological disorders. Biochem Pharmacol. 2016;1‐16:1‐16. [DOI] [PubMed] [Google Scholar]
- 87. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vainchtein ID, Chin G, Cho FS, et al. Astrocyte‐derived interleukin‐33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359:1269‐1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA. Diverse requirements for microglial survival, specification, and function revealed by defined‐medium cultures. Neuron. 2017;94(4):759‐73.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yang L, Tanaka J, Zhang B, Sakanaka M, Maeda N. Astrocytes modulate nitric oxide production by microglial cells through secretion of serine and glycine. Biochem Biophys Res Commun. 1998;251(1):277‐282. [DOI] [PubMed] [Google Scholar]
- 91. Dodig S, Epelak I, Pavi I. Hallmarks of senescence and aging. Biochem Med. 2019;29(3):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Coppé J‐P, Patil CK, Rodier F, et al. Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853‐2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Manninen T, Saudargiene A, Linne ML. Astrocyte‐mediated spike‐timing‐dependent long‐term depression modulates synaptic properties in the developing cortex. PLoS Comput Biol. 2020;16(11):e1008360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Goyal MS, Vlassenko AG, Blazey TM, et al. Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 2017;26(2):353‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jiang T, Cadenas E. Astrocytic metabolic and inflammatory changes as a function of age. Aging Cell. 2014;13:1059‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Grimm A, Eckert A. Brain aging and neurodegeneration: from a mitochondrial point of view. J Neurochem. 2017;143:418‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tarantini S, Tran C, Gordon GR, Ungvari Z, Csiszar A. Impaired neurovascular coupling in aging and Alzheimer's disease: contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp Gerontol. 2016;94:52‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Fakhoury M. Microglia and Astrocytes in Alzheimer's Disease: Implications for Therapy. Curr Neuropharmacol. 2018;16(5):508‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wyss‐Coray T, Loike JD, Brionne TC, Lu E, Husemann J. Adult mouse astrocytes degrade amyloid‐beta in vitro and in situ. Nat Med. 2003;9(4):453‐457. [DOI] [PubMed] [Google Scholar]
- 100. Lesné S, Docagne F, Gabriel C, Liot G, Vivien D. Transforming growth factor‐beta 1 potentiates amyloid‐beta generation in astrocytes and in transgenic mice. J Biol Chem. 2003;278(20):18408‐18418. [DOI] [PubMed] [Google Scholar]
- 101. Söllvander S, Nikitidou E, Brolin R, et al. Accumulation of amyloid‐β by astrocytes result in enlarged endosomes and microvesicle‐induced apoptosis of neurons. Mol Neurodegener. 2016;11(1):1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Andersen JV, Christensen SK, Aldana BI, Nissen JD, Tanila H, Waagepetersen HS. Alterations in cerebral cortical glucose and glutamine metabolism precedes amyloid plaques in the APPswe/PSEN1dE9 mouse model of Alzheimer's disease. Neurochem Res. 2016;42(6):1589‐1598. [DOI] [PubMed] [Google Scholar]
- 103. Andersen JV, Skotte N, Christensen S, Polli F, Waagepetersen HS. Hippocampal disruptions of synaptic and astrocyte metabolism are primary events of early amyloid pathology in the 5xFAD mouse model of Alzheimer's disease. SSRN Electron J. 2021;12(11):954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Salcedo C, Andersen JV, Vinten KT, et al. Functional metabolic mapping reveals highly active branched‐chain amino acid metabolism in human astrocytes, which is impaired in iPSC‐derived astrocytes in Alzheimer's disease. Front Aging Neurosci. 2021;13:736580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Andersen JV, Christensen S, Westi EW, Diaz‐Delcastillo M, Waagepetersen H. Deficient astrocyte metabolism impairs glutamine synthesis and neurotransmitter homeostasis in a mouse model of Alzheimer's disease. Neurobiol Dis. 2021;148:105198. [DOI] [PubMed] [Google Scholar]
- 106. Staurenghi E, Cerrato V, Gamba P, Testa G, Leonarduzzi G. Oxysterols present in Alzheimer's disease brain induce synaptotoxicity by activating astrocytes: a major role for lipocalin‐2. Redox Biol. 2020;39(1):101837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Zheng J, Xie Y, Ren L, Qi L, Liu L. GLP‐1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in Alzheimer's disease. Mol Metab. 2021;47:101180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Samii A, Nutt JG, Ransom BR. Parkinson's disease. Lancet. 2004;363(9423):1783‐1793. [DOI] [PubMed] [Google Scholar]
- 109. Rocha EM, De Miranda B, Sanders LH. Alpha‐synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson's disease. Neurobiol Dis. 2017;109:249‐257. [DOI] [PubMed] [Google Scholar]
- 110. Choi I, Kim J, Jeong HK, Kim B, Joe EH. PINK1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced AKT and increased p38 MAPK activation, and downregulation of EGFR. Glia. 2013;61(5):800‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Saskia S, Bettina L, Sonja M, et al. Genetic mouse models for Parkinson's disease display severe pathology in glial cell mitochondria. Hum Mol Genet. 2011;6:1197‐1211. [DOI] [PubMed] [Google Scholar]
- 112. Vitner EB, Dekel H, Zigdon H, et al. Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum Mol Genet. 2010;19(18):3583‐3590. [DOI] [PubMed] [Google Scholar]
- 113. Larsen NJ, Ambrosi G, Mullett SJ, Berman SB, Hinkle DA. DJ‐1 knock‐down impairs astrocyte mitochondrial function. Neuroscience. 2011;196(33):251‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gui YX, Xu ZP, Wen‐Lv LHM, Zhao JJ, Hu XY. Four novel rare mutations of PLA2G6 in Chinese population with Parkinson's disease. Parkinsonism Relat Disord. 2013;19(1):21‐26. [DOI] [PubMed] [Google Scholar]
- 115. Mikhail S, Seburn KL, Cox GA, Martens KA, Georg R. Severe disturbance in the Ca2+ signaling in astrocytes from mouse models of human infantile neuroaxonal dystrophy with mutated Pla2g6. Hum Mol Genet. 2012;12:2807‐2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Castagnet PI, Golovko MY, Barceló‐Coblijn G, Nussbaum RL, Murphy EJ. Fatty acid incorporation is decreased in astrocytes cultured from alpha‐synuclein gene‐ablated mice. J Neurochem. 2010;94(3):839‐849. [DOI] [PubMed] [Google Scholar]
- 117. Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H. Astrocytic expression of Parkinson's disease‐related A53T α‐synuclein causes neurodegeneration in mice. Mol Brain. 2010;3(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kim JM, Cha SH, Choi YR, Jou I, Joe EH, Park SM. DJ‐1 deficiency impairs glutamate uptake into astrocytes via the regulation of flotillin‐1 and caveolin‐1 expression. Sci Rep. 2016;6:28823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Solano RM, Casarejos MJ, Menéndez‐Cuervo J, Rodriguez‐Navarro JA, García de Yébenes J, Mena MA. Glial dysfunction in parkin null mice: effects of aging. J Neurosci. 2008;28(3):598‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Solano RM, Menéndez J, Casarejos MJ, Rodríguez‐Navarro JA, Mena MA. Midbrain neuronal cultures from parkin mutant mice are resistant to nitric oxide‐induced toxicity. Neuropharmacology. 2006;51(2):327‐340. [DOI] [PubMed] [Google Scholar]
- 121. Kim KS, Kim JS, Park JY, et al. DJ‐1 Associates with lipid rafts by palmitoylation and regulates lipid rafts‐dependent endocytosis in astrocytes. Hum Mol Genet. 2013;22(23):4805‐4817. [DOI] [PubMed] [Google Scholar]
- 122. Pontikis CC, Cella CV, Parihar N, et al. Late onset neurodegeneration in the Cln3−/− mouse model of juvenile neuronal ceroid lipofuscinosis is preceded by low level glial activation. Brain Res. 2004;1023(2):231‐242. [DOI] [PubMed] [Google Scholar]
- 123. Manzoni C, Mamais A, Dihanich S, et al. Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim Biophys Acta. 2013;1833(12):2900‐2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Henry AG, Soheil A, Harry S, et al. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet. 2015;21:6013‐6028. [DOI] [PubMed] [Google Scholar]
- 125. Rinaldi DE, Corradi GR, Cuesta LM, Adamo HP, Felicitas D. The Parkinson‐associated human P5B‐ATPase ATP13A2 protects against the iron‐induced cytotoxicity. Biochim Biophys Acta. 2015;1848(8):1646‐1655. [DOI] [PubMed] [Google Scholar]
- 126. Ramirez A, Heimbach A, Gründemann J, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P‐type ATPase. Nat Genet. 2006;38(10):1184‐1191. [DOI] [PubMed] [Google Scholar]
- 127. Mazzetti S, Barichella M, Giampietro F, et al. Astrocytes expressing Vitamin d‐activating enzyme identify Parkinson's disease. CNS Neurosci Ther. 2022;28(5):703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zhang Y, Lu K. Potential protective role of astrocytes in the pathogenesis of astrocyte‐mediated synaptic plasticity of Parkinson's disease. J Integr Neurosci. 2021;20(2):515‐525. [DOI] [PubMed] [Google Scholar]
- 129. Yamanaka K, Komine O. The multi‐dimensional roles of astrocytes in ALS. Neurosci Res. 2017;126:31‐38. [DOI] [PubMed] [Google Scholar]
- 130. Ziff O, Clarke B, Taha D, Crerar H, Luscombe N, Patani R. Meta‐analysis of human and mouse ALS astrocytes reveals multi‐omic signatures of inflammatory reactive states. Genome Res. 2022;32(1):71‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Takeuchi H, Mizoguchi H, Doi Y, et al. Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer's disease. PLoS One. 2011;6:e21108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Pinar M, Sakina Z, Lobsiger CS, Stéphanie M, Carole E, Danielle S, et al. System xC‐ is a mediator of microglial function and its deletion slows symptoms in amyotrophic lateral sclerosis mice. Brain A Journal of Neurology. 2012;138(Pt 1):53 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Sasabe J, Miyoshi Y, Suzuki M, et al. d‐Amino acid oxidase controls motoneuron degeneration through d‐serine. Proc Natl Acad Sci. 2011;109:627‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Mitchell J, Paul P, Chen HJ, et al. Familial amyotrophic lateral sclerosis is associated with a mutation in d‐amino acid oxidase. Proc Natl Acad Sci U S A. 2010;107(16):7556‐7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Cassina P, Cassina A, Pehar M, Castellanos R, Radi R. Mitochondrial dysfunction in SOD1G93A‐bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial‐targeted antioxidants. J Neurosci. 2008;28(16):4115‐4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Marchetto M, Muotri AR, Mu Y, Smith AM, Cezar GG, Gage FH. Non‐cell‐autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell. 2008;3(6):649‐657. [DOI] [PubMed] [Google Scholar]
- 137. Stella R, Bonadio RS, Cagnin S, Massimino ML, Peggion C. Perturbations of the proteome and of secreted metabolites in primary astrocytes from the hSOD1(G93A) ALS mouse model. Int J Mol Sci. 2021;22(13):7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Madji Hounoum B, Mavel S, Coque E, et al. Wildtype motoneurons, ALS‐Linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling. Glia. 2017;65(4):592‐605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are openly available.