

Abstract
Purpose
Genotype‐phenotypic correlation of KCNH1 variant remains elusive. This study aimed to expand the phenotypic spectrum of KCNH1 and explore the correlations between epilepsy and molecular sub‐regional locations.
Methods
We performed whole‐exome sequencing in a cohort of 98 patients with familiar febrile seizure (FS) or epilepsy with unexplained etiologies. The damaging effects of variants were predicted by protein modeling and multiple in silico tools. All reported patients with KCNH1 pathogenic variants with detailed neurological phenotypes were analyzed to evaluate the genotype‐phenotype correlation.
Results
Two novel KCNH1 variants were identified in three cases, including two patients with FS with inherited variant (p.Ile113Thr) and one boy with epilepsy with de novo variant (p.Arg357Trp). Variant Ile113Thr was located within the eag domain, and variant p.Arg357Trp was located in transmembrane domain 4 of KCNH1, respectively. Two patients experienced refractory status epilepticus (SE), of which one patient died of acute encephalopathy induced by SE. Further analysis of 30 variants in 51 patients demonstrated that de novo variants were associated with epileptic encephalopathy, while mosaic/somatic or germline variants cause isolated epilepsy/FS. All hotspot variants associated with epileptic encephalopathy clustered in transmembrane domain (S4 and S6), while those with isolated epilepsy/seizures or TBS/ZLS without epilepsy were scattered in the KCNH1.
Conclusions
We found two novel missense variants of KCNH1 in three individuals with isolated FS/epilepsy. Variants in the KCNH1 cause a spectrum of epileptic disorders ranging from a benign form of genetic isolated epilepsy/FS to intractable form of epileptic encephalopathy. The genotypes and variant locations help explaining the phenotypic variation of patients with KCNH1 variant.
Keywords: epilepsy, KCNH1 gene, status epilepticus, Temple‐Baraitser syndrome, Zimmermann‐Laband syndrome
Mutations in the KCNH1 cause a spectrum of epileptic disorders ranging from a benign form of genetic isolated epilepsy/febrile seizure to intractable form of epileptic encephalopathy. The genotypes and variant locations help explaining the phenotypic variation of patients with KCNH1 variant.
1. INTRODUCTION
KCNH1 gene (OMIM* 603305), mapping to 1q32.2, encodes potassium voltage‐gated channel subfamily H member 1 (KCNH1). KCNH1 is a protein with 989 amino acids, containing an eag domain in N‐terminal, which is composed of a Per‐Arnt‐Sim (PAS) domain and a PAS‐cap domain, whereas the C‐terminal region contains a cyclic nucleotide‐binding homology domain (CNBHD), which is connected to the pore through a C‐linker domain. Besides, KCNH1 exhibits a typical Kv membrane topology with six transmembrane domains (S1–S6). 1 , 2 KCNH1 is highly expressed in human brain, being essential for brain development (www.proteinatlas.org/ENSG00000143473‐KCNH1). 3 , 4 Clinically, variants in KCNH1 have been associated with Temple‐Baraitser syndrome (TBS, OMIM# 611816) and Zimmermann‐Laband syndrome (ZLS, OMIM# 135500), two forms of neurodevelopmental disorder charactered by intellectual disability (ID), developmental disorder (DD), coarse face, gingival overgrowth, hypertrichosis, digital/toe anomalies, and seizures. 5 , 6 , 7 , 8 , 9 Although epileptic seizures were usually observed in these syndromes, the genotype‐phenotypic associations of KCNH1 are not fully understood, as pathogenic KCNH1 variants have been identified in uncharacterized patients exhibited a part of the above phenotypes, including isolated epilepsy could be ascribed neither to a TBS nor to a ZLS. Here, we reported three cases harboring novel variants in the KCNH1 gene suffering from epilepsy/febrile seizure (FS) and refractory status epilepticus (SE), but otherwise presenting distinct clinical features of TBS or ZLS, broadening the phenotypic spectrum of KCNH1. We also analyzed all previously reported patients with KCNH1 variant, focusing on the correlations between epilepsy and molecular sub‐regional locations.
2. MATERIALS AND METHODS
2.1. Subjects
Patients with unexplained epilepsy or familial FS were recruited from Department of Pediatrics, Affiliated Hospital of Zunyi Medical University between July 2017 and March 2021. The studies adhered to the guidelines of the International Committee of Medical Journal Editors with regard to patient's consent for research or participation. This study was approved by the ethics committee of the Affiliated Hospital of Zunyi Medical University. We obtained written consents for genetic testing and publication of data for all patients.
Detailed clinical information of the patients was collected, including age, gender, epileptic types and frequencies, general and neurological examination results, family history, response to anti‐seizure medicines (ASMs), results of brain magnetic resonance imaging (MRI), and video‐electroencephalography (EEG). Epileptic seizures or epilepsies were diagnosed according to the criteria of the Commission on Classification and Terminology of the ILAE. 10 , 11 , 12 FS is defined as seizures triggered by fever during aged 6 months to 5 years without a history of an unprovoked seizure or concurrent central nervous system infection. 13 , 14 SE is defined as convulsions persisting for >5 min. Refractory SE is defined as clinical or electroencephalographic seizures lasting >60 min despite treated with at least one first‐line ASMs (e.g., benzodiazepine) and one second‐line ASMs (e.g., phenytoin, phenobarbital, or valproate). Super‐refractory SE is defined as SE that has persisted or recurred for 24 h after the onset of general anesthesia treatment. 15 , 16 , 17 Epilepsies with acquired causes were excluded.
2.2. Trios‐based WES
Blood samples were obtained from the probands and their parents to determine the origin of the identified genetic variants. Genomic DNA was extracted from peripheral blood using a QuickGene DNA whole blood kit (Fujifilm). Exome captures were performed using the IDT xGen Exome Research Panel with paired‐end read sequences generated on NovaSeq 6000 sequencing. Sequences were aligned to Human reference genome GRCh38/hg38. The variants were then annotated through AnnoVar 18 and evaluated according to allele frequencies, pathogenicity prediction, and protein function. Pathogenic variants related to clinical phenotypes will further be verified by Sanger sequencing.
2.3. Mutation analysis
Aiming to evaluate the genotype‐phenotype correlation, we exhaustively searched KCNH1 pathogenic variants on the PubMed up until Mar 2022 to identify studies published in English using the following terms: KCNH1, epilepsy, seizure, TBS, ZLS, Temple‐Baraitser, Zimmermann‐Laband. All pathogenic variants in patients with detailed neurological phenotypes were analyzed.
Molecular modeling analysis was performed to show the variations in protein structure. The human_KCNH1 model was downloaded in the AlphaFold dataset. 19 UCSF Chimera software was used for three‐dimensional protein structure visualization and analysis. DUTE server (http://biosig.unimelb.edu.au/duet/) and Grantham scores 20 were used for prediction of protein stability changes. The changes of the protein stability were assessed using the free energy stability change (DDG, kcal/mol) value.
3. RESULTS
3.1. Identification of KCNH1 variants
A total of 98 patients were recruited; among them, two missense KCNH1 variants were identified in three cases (Table 1; Figure 1A,D), including one inherited variant (c.338T>C; p.Ile113Thr) and one de novo variant (c.1069C>T; p.Arg357Trp). The three cases had no other pathogenic or likely pathogenic variants. Two variants of KCNH1 were annotated based on the transcript NM_172362 and confirmed by Sanger sequencing (Figure 1B,E).
TABLE 1.
Clinical features of the individuals with KCNH1 variants
| Variants (NM_172362) | Gender | Age at report | Diagnosis | Age of onset | Seizure & frequency | ASM | ID/DD | EEG | Brain MRI | Outcomes | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.338T>C; p.Ile113Thr | Male | 4 yrs | FS, supper refractory SE | 1 yrs | 3–4 simple FS, febrile supper refractory SE at age of 4, GTCS | – | No | Severe abnormal. See Figure 2A | Abnormal at age of 4 (see Figure 2B) | Died due supper refractory SE and brain damage |
| 2 | c.338T>C; p.Ile113Thr | Female | 32 yrs | FS, | 1.5 yrs | 4–5 times FS, GTCS | – | No | NA | NA | Seizure‐free after age of 6 yrs |
| 3 | c.1069C>T; p.Arg357Trp | Male | 2 yrs | Dravet syndrome, refractory SE | 8 months | PE or generalized, 10+ times triggered by fever /hot water bath, SE | VPA | Mild DD | Abnormal. See Figure 2C | Normal | Seizure‐free for 10 months |
Abbreviations: ASM, anti‐seizure medicine; DD, developmental disorder; EEG, electroencephalogram; FS, febrile seizure; GTCS, generalized tonic‐clonic seizure; ID, intellectual disability; MRI, magnetic resonance imaging; NA, not available; SE, status epilepticus; VPA, valproate; yr, year.
FIGURE 1.
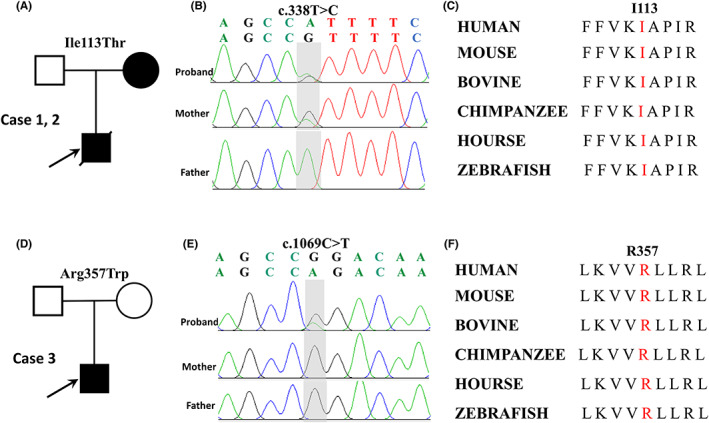
Genetic data on the patients with KCNH1 variants. (A, D) Pedigree of the two families. The filled symbol with arrows identifies the probands. (B, E) The variants c.338T>C and c.1069C>T were identified through whole‐exome sequencing and confirmed by Sanger sequencing. The SNVs were shown in gray shadows, c.338T>C was inherited from his mother, and c.1069C>T was de novo. (C, F) The variant amino acids in our patient were conserved from multiple species.
The amino acid residues of the two missense variants are highly conserved in various species (Figure 1C,F). The two missense variants were suggested to be damaging/disease‐causing/conserved by at least three silico tools (Table 2). The two variants are not present in gnomAD database (Table 2). Variant p.Arg357Trp was predicted to have more severe effects than variant p.Ile113Thr (101 vs. 89) according to Grantham scores.
TABLE 2.
Genetic features of the individuals with KCNH1 variants
| Cases | Position | cDNA change (NM_172362) | Protein change | MAF | MAF‐EAS | Mutation taster | SIFT | CADD | Polyphen2_HDIV | GERP++ | PhyloP | Grantham scores |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1, 2 | Chr1:211090663 | 338T>C | p.Ile113Thr | – | – | DC (1.00) | D (0.09) | D (26.30) | PD (0.99) | C (5.22) | C (8.93) | 89 |
| 3 | Chr1:210920033 | 1069C>T | p.Arg357Trp | – | – | DC (1.00) | D (0.00) | D (34.00) | PD (1.00) | C (5.48) | C (4.26) | 101 |
Abbreviations: B, benign; C, conserved; CADD, combined annotation dependent depletion; D, damaging; DC, disease‐causing; Chr, chromosome; MAF, minor allele frequency from Genome Aggregation Database; MAF‐EAS, minor allele frequency from East Asia population in Genome Aggregation Database; NA: not applicable; PD, probably damaging.
3.2. Clinical information
The three patients showed infancy or childhood‐onset seizures (8 months–1.5 years). The main clinical features of the cases are summarized in Table 1. Three patients were all born to non‐consanguineous parents after an uneventful pregnancy.
Case 1 and case 2 harbored variant p.Ile113Thr. Case 1 was a 4‐year‐old boy. He developed simple FS at frequency of 1–2 times yearly since age of 1 year. Psychomotor development was normal. EEG and brain MRI were unremarkable. He experienced febrile‐induced SE at age of 4, his seizures, which lasted for 1 h, ceased after the administration of intravenous continuous infusion of valproate (1.5 mg/kg/h). He was comatose, despite the disappearance of the seizures. His Glasgow Coma Scale score was 6/15 (E2 + V2 + M2). He developed refractory SE 2 days after admission, as his seizures were unresponsive to standard use of diazepam, intravenous valproate, and phenobarbital. Super‐refractory SE was diagnosed because seizures remained uncontrolled 24 h after initiating continuous intravenous use of midazolam and propofol. No dysmorphic features were observed. Except for obvious intracranial hypertension (310 mmH2O), no abnormality was found in cerebrospinal fluid tests. Routine blood testing results were unremarkable. EEG revealed diffuse slow waves, and subclinical seizures lasted for 10 min originated from bilateral temporal were present (Figure 2A). Brain MRI (5th day of seizure onset) showed bilateral hemisphere cerebral edema characterized by diffuse subcortical white matter lesions. Bright tree appearance (subcortical white matter hyper‐intense signal) in diffuse weighted images and low‐intense signal in apparent diffusion coefficient were observed on MRI (Figure 2B). Subsequently, diagnosis of acute encephalopathy after SE was made based on his clinical and neuroimaging features. Despite aggressive therapeutic strategies were given, the patient eventually died 20 days after this seizure onset due to uncontrollable seizures and severe brain damage.
FIGURE 2.
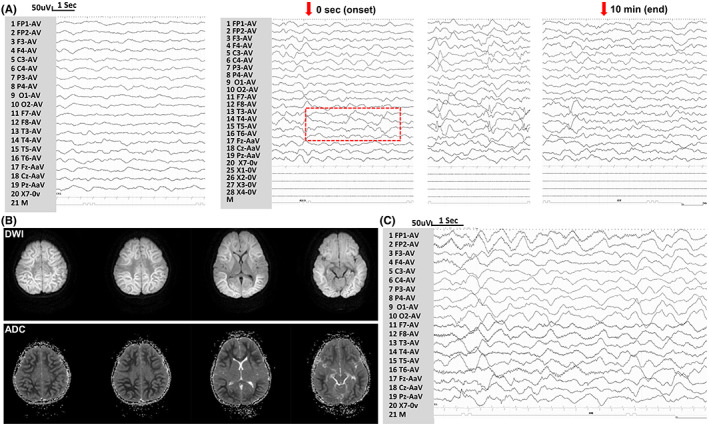
EEG and neuroimaging results in the cases with KCNH1 variants. (A) EEG of case 1 detected diffuse slow waves, and subclinical seizures originated from bilateral temporal (red box indexed the seizure onset). (B) Brain MRI showed bright tree appearance in DWI, and low‐intense signal in ADC. (C) EEG of case 3 indicated diffuse slow waves and drug‐related fast waves. (DWI, diffuse weighted images; ADC, apparent diffusion coefficient)
Case 2, the mother of case 1, is currently 32 years old. She had 4–5 times of FS since the age of 1.5, and she did not have seizure after the age of 5. Neither neuroimaging nor EEG were performed. Her neuropsychology is normal at present.
Variant p.Arg357Trp was identified in case 3. This boy experienced FS (generalized or focal) since age of 8 months. Subsequently, he experienced frequently seizures triggered by low‐grade fever or hot‐water bath, which led to a diagnosis of Dravet syndrome. Brain MRI and EEG were normal at age of 1 year. At age of 1 year and 2 months, he had short‐duration but frequent (>10 times/h) seizures triggered by fever, which were resistant to multiple ASMs, including diazepam, valproate, phenobarbital. Subsequently, his seizures stopped until continuous intravenous infusion of midazolam (0.24 mg/kg/h). EEG showed diffuse slow waves and drug‐related fast waves (Figure 2C). He became seizure‐free with treatment of valproate (22 mg/kg/day) till the last follow‐up at age of 2 years even when the body temperature was as high as 40°C. Mild developmental delay was observed. He can walk without support at age of 1 year and 7 months and can speak 3–4 words at age of 2 years. Gesell Developmental Observation‐Revised screening was performed, and the results showed a mild delay in gross motor development, language, and social‐emotional responses. No any dysmorphic feature or malformations was observed, including hypoplasia of the nails, coarse face (Figure S1).
3.3. Structural alteration of KCNH1 protein
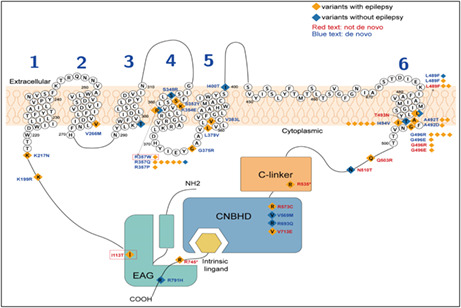
As shown schematically in Figure 3A, KCNH1 contains eag domain and CNBHD located in cytoplasmic and six transmembrane domains (S1–S6). Structural model of KCNH1 indicated variant p.Ile113Thr was located within the eag domain, and variant p.Arg357Trp was located in S4.
FIGURE 3.
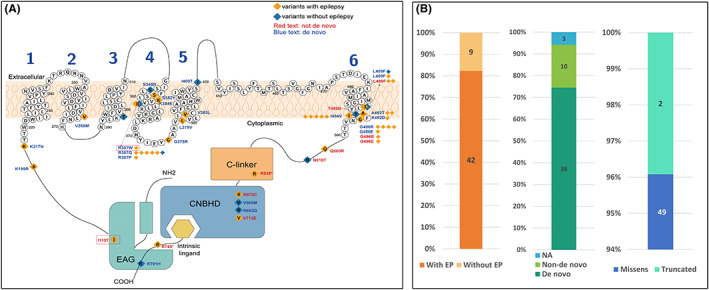
Schematic diagram of variant sites and phenotypes/genotypes characteristics of the patients reported to date. (A) Schematic diagram of the transmembrane structure. The CNBHD, C‐linker, and EAG domains were shown in the picture. The S1–S4 segments act as voltage‐sensor domains. The KCNH1 channel shows the location of the residues affected in individuals with (orange square) or without (blue square) epilepsy. Most of them were de novo variants (blue text), and few were inherited or not known (red text). Variants in our patients were highlighted with red boxes. (B) Showed the clinical phenotypes, and genetic characteristics of the patients reported to date
DUET server was used to analyze the effects of missense variants on protein stability. Results showed destabilizing for the residues' changes. Variant p.Ile113Thr and p.Arg357Trp were predicted to be least stable with DDG value of −3.159 and −0.448 kcal/mol, respectively. Both variants changed the hydrogen bonds (Figure 4A,B). Residue Ile113 originally formed one hydrogen bond with residue Asn93. The missense variant p.Ile113Thr results in an additional hydrogen bond with residue Asn93. Residue Arg357 originally formed hydrogen bonds with residue Lys354 and residue Arg 360, respectively. When arginine was replaced by tryptophan at residue Arg357, a new hydrogen bond with residue Leu304 was formed. We also analyzed the previous reported five reported hotspot variants (p.Arg357, p.Leu489, p.Ala492, p.Leu494, and p.Gly496) and found that all variants changed numbers or distances of hydrogen bonds (Figure 4B–F).
FIGURE 4.
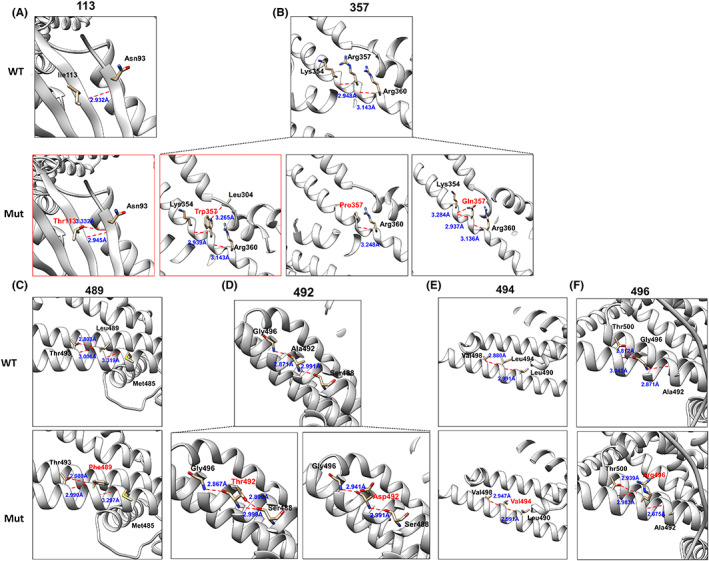
Changes in KCNH1 protein structure. (A–F) The protein changes including our variants and five hotspots (p.Arg357, p.Leu489, p.Ala492, p.Leu494, and p.Gly496). The wild‐type protein structures were shown in the top row, and the mutant structures were in the row below. Hydrogen bonds are represented by red dashed lines and the distances are highlighted with blue text. Structures in our patients were highlighted with red boxes.
3.4. Genotype‐phenotype correlation of KCNH1 variants
We analyzed genotype‐phenotypic associations in all reported KCNH1 pathogenic variants with detailed neurological phenotypes. Previously, 28 KCNH1 variants in 48 patients have been reported, 4 , 5 , 9 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 including the present data, a total of 30 variants in 51 patients. Forty‐two out of 51 patients had epilepsy. Thirty‐eight patients harbored de novo variants, 13 patients harbored non‐de novo variant (inherited, mosaic or unknown origin; Figure 3B). Forty‐nine out of 51 patients harbored missense variants. Among the only two nonsense variants, 34 , 37 variant p.R535* was also found in clinically unaffected father and sister; the patient carrying variant p.R745* also harbored another epileptic encephalopathy‐related, pathogenic variant, CACNA1A (c.2134G>A; p.Ala712Thr), this variant has been reported as a pathogenic variant in epileptic encephalopathy patients. 38 Clinical and molecular details of patients with epilepsy are listed in Table 3, and patients without epilepsy were listed in Table S1.
TABLE 3.
Patients with KCNH1 variant with epilepsy
| References | Age/sex | Variants (NM_172362) | Inheritance | Onset of seizures | Epilepsy/seizure types | TBS/ZLS | Intellectual disability (ID)/developmental delay (DD) | Effective ASM and outcomes of epilepsy |
|---|---|---|---|---|---|---|---|---|
| De novo | ||||||||
| vonWrede 37 | 6 yrs 4 mons/M | c.596A>G; p.Lys199Arg | De novo | 2.5 yrs | FS, DEE, GTCS, myoclonic, SE | No | Psychomotor development was severely impaired | Cortisone, perampanel, and topiramate were effective |
| Simons 33 | 6 yrs/F | c.651G>C; p.Lys217Asn | De novo | 6 yrs | Generalized tonic‐clonic seizure | TBS | Mild psychomotor development delay | NA |
| Rochtus 29 | 11 yrs/M | c.796G>A; p.Val266Met | De novo | 2.5 yrs | Myoclonic, GTCS, absence, and drop seizures | No | Mild delay | NA, but she got seizure‐free and off medication |
| Kortüm 25 | 21 yrs/M | c.1055C>A; p.Ser352Tyr | De novo | Childhood | Frequent seizures, SE | ZLS | Profound ID/DD | NA |
| Gripp 24 | 2.9 yrs/F | c.1060A>G; p.Lys354Glu | De novo | 4 weeks | NA | TBS | Motor and speech delay; skills at 4 m level at age 2.8 | NA |
| Present study | 1 yrs 10 mons/M | c.1069C>T; p.Arg357Trp | De novo | 8 mons | Fever sensitivity, GTCS, focal, SE. | No | Mild delay | Valproic acid, seizure‐free |
| Fukai 23 | 8 yrs/M | c.1070G>A; p.Arg357Gln | De novo | 6 mons | generalized tonic‐clonic | TBS | Severe ID and DD | Epilepsy was controlled by valproic acid and zonisamide |
| Bramswig 5 | 14 yrs/F | c.1070G>A; p.Arg357Gln | De novo | 1.5 yrs | Focal epilepsy with secondary generalization | TBS | Severe ID and DD | NA |
| Bramswig 5 | 4 yrs 4 mons/F | c.1070G>A; p.Arg357Gln | De novo | 2 yrs 9mons | Frequent temporal epilepsy, SE | TBS | Severe ID and DD, ASD | NA |
| Fukai 23 | 6 yrs/M | c.1070G>A; p.Arg357Gln | De novo | 1 mon | Focal seizures | No | Severe ID and DD | His seizures were controlled by frisium, trileptin, and risperdal |
| Gripp 24 | 9.7 yrs/F | c.1070G>A; p.Arg357Gln | De novo | 2.75 yrs | Focal, GTCS, and absences | TBS | Severe DD/ID | NA |
| Fukai 23 | 7 yrs/M | c.1070G>C; p.Arg357Pro | De novo | 4 mons | Generalized tonic‐clonic | No | Severe ID and DD | Carbamazepine, and clobazam, relative effectiveness |
| Kortüm 25 | 19 yrs/F | c.1123G>A; p.Gly375Arg | De novo | Adolescence | NA | ZLS | Severe/profound ID | NA |
| Kortüm 25 | 7 yrs/M | c.1135C>G; p.Leu379Val | De novo | 8 mons | Focal clonic secondary generalized seizures, SE | ZLS | Severe intellectual and motor disability | Generalized drug‐resistant seizures |
| Kortüm 25 | 21 yrs/M | c.1147G>C; p.Val383Leu | De novo | Childhood | Frequent seizures, | ZLS | Profound DD/ID | NA |
| Bramswig 5 | 14 yrs/F | c.1465C>T; p.Leu489Phe | De novo | 9 mons | GTCS, absences | TBS | Severe ID and DD | Seizures were responding well to lamotrigine and ethymal |
| Rochtus 29 | 3 yrs/M | c.1474G>A; p.Ala492Thr | De novo | Neonatal | DEE | No | Severe ID and DD | NA |
| Froukh 22 | 5 mons/M | c.1474G>A; p.Ala492Thr | De novo | NA | DEE | TBS | Mild DD/ID | NA |
| Mastrangelo 28 | NA | c.1475C>A; p.Ala492As | De novo | Infancy | GTCS | No | Severe ID/DD | Responsive to levetiracetam |
| Simons 33 | 3 yrs 7 mons/M | c.1480A>G; p.Ile494Val | De novo | 3 yrs | FS, GTCS | TBS | Moderate ID/DD | NA |
| Simons 33 | 4 yrs/M | c.1480A>G; p.Ile494Val | De novo | 4 yrs | NA | TBS | Severe ID/DD | NA |
| Simons 33 | 9 yrs/F | c.1480A>G; p.Ile494Val | De novo | 8 yrs | NA | TBS | Severe ID/DD | NA |
| Kortüm 25 | 12 yrs 8 mons/F | c.1480A>G; p.Ile494Val | De novo | 10 yrs 6 mons | Generalized tonic‐clonic seizures | TBS | Severe DD/ID, autism | NA |
| Kortüm 25 | 4 yrs 6 mons/F | c.1480A>G, p.Ile494Val | De novo | Neonatal | Focal motor seizure, SE | TBS | Severe DD/ID | Seizures were responsive to phenytoin, valproic acid |
| Kortüm 25 | 12 yrs/F | c.1486G>A; p.Gly496Arg | De novo | 6 mons | Focal epilepsy | TBS | Moderate DD/ID | Controlled by tegretol |
| Mastrangelo 28 | 15 yrs/F | c.1486G>A; p.Gly496Arg | De novo | Infancy | Tonic, tonic‐clonic, 2 SE | No | Severe ID | Responsive to lamotrigine, carbamazepine and clobazam |
| Mastrangelo 28 | 9 yrs/F | c.1486G>A; p.Gly496Arg | De novo | Infancy | Focal clonic, generalized tonic‐clonic, myoclonic | No | Mild/moderate ID/DD | Responsive to carbamazepine |
| Mastrangelo 28 | 1.7 yrs/F | c.1486G>A; p.Gly496Arg | De novo | Infancy | Generalized tonic‐clonic, focal clonic, myoclonic | No | Mild ID | Responsive to levetiracetam |
| Fukai 23 | 3 yrs/M | c.1487G>A; p.Gly496Glu | De novo | Neonatal | Generalized convulsions | No | Severe DD and ID | Valproate, clobazam, and levetiracetam were effective |
| Inherited or mosaic | ||||||||
| Present study | 32 yrs/F | c.338T>C; p.Ile113Thr | Parents not tested | Childhood | GTCS | No | No | Medicine was not given, seizure‐free |
| Present study | 4 yrs/M | c.338T>C; p.Ile113Thr | Maternal | 1.5 yrs | GTCS, super SE | No | No | Medication was not given, dead due to SE |
| Simons 33 | NA/F a | c. 1465C>T; p.Leu489Phe | Mosaic (blood 13.6%) | NA | NA | No | No | NA |
| Simons 33 | 14 yrs/F b | c. 1465C>T; p.Leu489Phe | Maternal? | 4 yrs | NA | TBS | Mild/moderate DD/ID | Controlled by topiramate and carbamazepine |
| Simons 33 | NA/F c | c.1508A>G; p.Gln503Arg | Mosaic (blood 4.7%; saliva 4.5%; fibroblast 3.4%) | Childhood | Generalized seizure | No | No | Epilepsy was well controlled with carbamazepine. |
| Simons 33 | 4 yrs/M d | c.1508A>G; p.Gln503Arg | Maternal? | 2 yrs | Generalized seizure | TBS | Mild ID/DD | Well controlled on phenytoin |
| vonWrede 37 | 36 yrs/F | c.1603C>T; p.Arg535* | Paternal | 2 yrs | GTCS, atonic, focal seizures | No | Mild/moderate ID/DD | Controlled by sultiame, phenytoin, and clonazepam |
| Rochtus 29 | 6 yrs/M | c.1717C>T; p.Arg573Cys | Paternal | 2 mons | Focal seizures | No | Global developmental delay | NA |
| vonWrede 37 | 40 yrs/F | c.2138T>A; p.Val713Glu | Maternal | 13 yrs | Absence, tonic, GTCS) | No | No | Drug‐resistant, cannabidiol was partial effective |
| vonWrede 37 | 40 yrs/M | c.2138T>A; p.Val713Glu | Mosaic (brain tissue) | 5 yrs | Multiple seizures, FCD IIb | No | No | Drug‐resistant epilepsy |
| Unknown origin | ||||||||
| Gripp 24 | 34 yrs/F | c.1486G>A; p.Gly496Arg | Father not tested | Neonatal | GTCS, several times SE | ZLS | Mild DD, moderate ID | NA |
| Gripp 24 | 39 yrs/F | c.1487G>A; p.Gly496Glu | Parents not tested | Prenatal | Myoclonic jerks, mixed seizure type | TBS | Severe DD and ID | NA, improved but not stopped by medication |
| Takata 34 | NA | c.2233C>T; p.Arg745* | NA | NA | DEE | NA | Developmental encephalopathy | NA |
Abbreviations: DEE, developmental epileptic encephalopathy; F, female; FS, febrile seizure; GTCS, generalized tonic‐clonic seizure; ID/DD, intellectual disability/developmental disorder; M, male; mon, month; NA, not available; SE, status epilepticus; TBS, Temple‐Baraitser syndrome; ZLS, Zimmermann‐Laband syndrome.
We further analyzed the sub‐regional locations of all variants demonstrating that 83% (35/42) variants in patients with epilepsy/seizures were located in the transmembrane domains. De novo variants associated with epilepsy have obvious spatial clustering properties, five hotspot/recurrent variants including p.Arg357 (eight patients), p.Leu489 (four patients), p.Ala492 (three patients), p.Leu494 (five patients), and p.Gly496 (seven patients) were observed and all located in the transmembrane domains. Except for p.Arg357 located in S4, all other hotspot variants are located in S6 (Figure 3A).
Among the 42 patients with epilepsy, more than half of patients (58%, 20/34) were pharmaco‐responsive, of which 10 patients became seizure‐free. 21% (9/42) of patients had SE. We also found that patients with inherited or mosaic/somatic variants have a more mild phenotypes than patients with de novo variants, specifically: later age of seizure onset (2.1 vs. 2.7 years of age), fewer incidences of ID/DD (40%, 4/10 vs. 100%, 41/41), and higher rate of seizure‐free (63%, 5/8 vs. 19%, 5/26).
4. DISCUSSION
Present study provided a clinical description of three individuals with two novel missense variants of KCNH1 with FS/epilepsy and refractory SE without features of TBS/ZLS. One patient had mild ID with drug‐responsive Dravet syndrome and finally got seizure‐free. In the familial cases of FS, one patient died of super refractory SE. Both variants had no allele frequency in the gnomAD. The two variants affected residues conserved through evolution and invariantly observed among vertebrates. The two variants were predicted to be damaging by multiple in silico tools and altered the protein conformation. Taking together the evidence that KCNH1 gene is predominantly expressed in brain and associated with neurodevelopment and neural excitability, 3 , 4 the two variants of KCNH1 were suggested to be the pathogenic gene of the current cases.
We analyzed the largest cohort of 51 patients with KCNH1 variants to date and found that epilepsy/seizures were present in 82% individuals suggesting a direct role of KCNH1 in epileptogenesis. However, the seizure types, severity, and response to ASMs varied widely. Patients with inherited or mosaic/somatic variants have milder phenotypes than patients with de novo variants, including later age of seizure onset, fewer incidences of ID/DD, and higher rate of seizure‐free. Of the 10 patients with hereditary or mosaic/somatic variant, most of them presented with isolated epilepsy without TBS/ZLS. These findings provided possible evidence that a low level of mosaic/somatic variant or a weaker effect on KCNH1 function of inherited variant may contribute to isolated epilepsy phenotype.
In addition to inheritance and variant patterns that determine phenotypic differences, recent studies have showed that molecular sub‐regional location of variants was also a critical factor to determine the pathogenicity of variants and associated with phenotypic variations. 39 , 40 , 41 In this study, all patients with hotspot variants (p.Arg357, p.Leu489, p.Ala492, p.Ile494, and p.Gly496) associated with epilepsy with moderate to severe ID/DD clustered in S4 and S6, while those with isolated epilepsy/seizures or TBS/ZLS without epilepsy were scattered, suggesting a molecular sub‐regional effect of KCNH1 variants. One of our newly reported patients presented severe phenotype of Dravet syndrome had a variant (p.Arg357Trp) in S4. Another patient had variant (p.Ile113Thr) in the eag domain near N‐terminal; this patient showed a mild phenotype‐FS. These results demonstrate the important role of voltage‐sensing transmembrane helix S4 and S6 of the KCNH1 channel in maintaining neuronal excitability and development.
However, factors influencing clinical phenotypic heterogeneity of patients with KCNH1 variants remain not fully elucidated, because we found that even patients harbored variants located in S6, their clinical presentations varied widely. Fourteen patients with variants at p.Leu489, p.Ala492, and p.Gly496; all showed early onset of epilepsy within 2 years of age, while five patients with p.Ile494Val variant had late‐onset of epilepsy, with an average age of 5. We found variant Ile494Val appears to have the least effect on hydrogen bonds or distances of inter‐amino acid, which may help explain the mild phenotype of patients with p.Ile494Val variant. However, the specific mechanism is currently unknown; further accumulation of cases is needed for detailed research on effect of KCNH1 variant on spatial structure of KCNH1.
When comparing de novo variants in patients with neurodevelopmental disorders, individuals with missense variants have been reported to be generally more likely to develop epilepsy than individuals with truncating variants, and enrichment was observed in genes associated with ion channel‐encoding genes (KCNQ2, SCN1A, and KCNH1), 42 , 43 , 44 suggesting a dominant‐negative or gain‐of‐function effect of KCNH1 variant in the pathophysiology of epilepsy. In KCNH1, studies in both Xenopus laevis oocytes and human HEK293T cells have revealed that missense variants lead to deleterious gain‐of‐function effect. 2 , 25 , 33 Present study further supports this view, as 96% of patients harbored missense variants. There are evidences of non‐pathogenicity for only two truncated variants: variant p.Arg535* had very low penetrance, as two healthy individuals in the family also carried this variant; patient with variant p.Arg745* also carried another pathogenic epileptic encephalopathy‐related gene. These findings indicated that gain‐of‐function effect is the pathogenic mechanism of KCNH1 variant, because variants lead to haploinsufficiency in this gene appear to be better tolerated.
In this study, more than half of patients with epilepsy (20/34) responded well to the ASMs. However, these patients are prone to develop SE, because two of our newly reported patients developed super refractory SE, and 21% of the reported patients experienced SE. SE may induce acute encephalopathy which is characterized by non‐inflammatory encephalopathy, followed by prolonged consciousness disturbance, and often followed by severe neurological sequelae. 45 , 46 As occurred in our case 1, patients with KCNH1 variant may also develop acute encephalopathy after SE like patients with other ion channel‐encoding genes variant, such as KCNQ2 and SCN1A. 46
This study has several limitations. The direct functional effects of the variants were not examined. More cases are needed to elucidate genotype‐phenotype correlation of KCNH1 gene in future studies.
5. CONCLUSIONS
In conclusion, we found two novel missense variants of KCNH1 in three individuals with FS/epilepsy. Variants in the KCNH1 cause a spectrum of epileptic disorders ranging from benign isolated epilepsy/FS to severe epileptic encephalopathy. Low doses of mosaic/somatic variants and less deleterious germline variants may cause isolated epilepsy/FS, while de novo gain‐of‐function variants always cause developmental syndromic disorders with or without epilepsy. The genotypes and variant locations help explain the phenotypic heterogeneity of patients with KCNH1 variant.
AUTHOR CONTRIBUTIONS
Mao‐Qiang Tian, Ren‐Ke Li, and Xiao‐Mei Shu collected the data from patients, reviewed the literatures, and wrote the paper; Juan Li analyzed genetic pathogenicity; Jing Chen and Fan Yang performed whole exome sequencing data analysis; Long‐Ying Peng, Xiao‐Hua Yu, and Chang‐Jian Yang analyzed EEG recordings and neuroimaging data.
FUNDING INFORMATION
This work was supported by grants from Basic Research Program of Guizhou Province: Guizhou Science and Technology Foundation (Grant No. ZK [2021] General 418).
CONFLICT OF INTEREST
All authors claim that there are no conflicts of interest.
INFORMED CONSENT
The patients gave their informed consents for this report.
Supporting information
Figure S1
Appendix S1
Table S1
ACKNOWLEDGMENTS
The authors are deeply grateful to the patients and clinicians who participated in this work.
Tian M‐Q, Li R‐K, Yang F, et al. Phenotypic expansion of KCNH1 ‐associated disorders to include isolated epilepsy and its associations with genotypes and molecular sub‐regional locations. CNS Neurosci Ther. 2023;29:270‐281. doi: 10.1111/cns.14001
Mao‐Qiang Tian and Ren‐Ke Li contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study will be available from the corresponding author upon reasonable request.
REFERENCES
- 1. Haitin Y, Carlson AE, Zagotta WN. The structural mechanism of KCNH‐channel regulation by the eag domain. Nature. 2013;501(7467):444‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hamilton MJ, Suri M. "Electrifying dysmorphology": potassium channelopathies causing dysmorphic syndromes. Adv Genet. 2020;105:137‐174. [DOI] [PubMed] [Google Scholar]
- 3. Cazares‐Ordonez V, Pardo LA. Kv10.1 potassium channel: from the brain to the tumors. Biochem Cell Biol. 2017;95(5):531‐536. [DOI] [PubMed] [Google Scholar]
- 4. Niday Z, Tzingounis AV. Potassium channel gain of function in epilepsy: an unresolved paradox. Neuroscientist. 2018;24(4):368‐380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bramswig NC, Ockeloen CW, Czeschik JC, et al. 'Splitting versus lumping': temple‐Baraitser and Zimmermann‐Laband syndromes. Hum Genet. 2015;134(10):1089‐1097. [DOI] [PubMed] [Google Scholar]
- 6. Chacon‐Camacho OF, Vazquez J, Zenteno JC. Expanding the phenotype of gingival fibromatosis‐mental retardation‐hypertrichosis (Zimmermann‐Laband) syndrome. Am J Med Genet A. 2011;155A(7):1716‐1720. [DOI] [PubMed] [Google Scholar]
- 7. Gabbett MT, Clark RC, McGaughran JM. A second case of severe mental retardation and absent nails of hallux and pollex (Temple‐Baraitser syndrome). Am J Med Genet A. 2008;146A(4):450‐452. [DOI] [PubMed] [Google Scholar]
- 8. Temple IK, Baraitser M. Severe mental retardation and absent nails of hallux and pollex. Am J Med Genet. 1991;41(2):173‐175. [DOI] [PubMed] [Google Scholar]
- 9. Megarbane A, Al‐Ali R, Choucair N, et al. Temple‐Baraitser syndrome and Zimmermann‐Laband syndrome: one clinical entity? BMC Med Genet. 2016;17(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1989;30(4):389‐399. [DOI] [PubMed] [Google Scholar]
- 11. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Younus I, Reddy DS. Epigenetic interventions for epileptogenesis: a new frontier for curing epilepsy. Pharmacol Ther. 2017;177:108‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sawires R, Buttery J, Fahey M. A review of febrile seizures: recent advances in understanding of febrile seizure pathophysiology and commonly implicated viral triggers. Front Pediatr. 2021;9:801321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Subcommittee on Febrile Seizures , American Academy of Pediatrics . Neurodiagnostic evaluation of the child with a simple febrile seizure. Pediatrics. 2011;127(2):389‐394. [DOI] [PubMed] [Google Scholar]
- 15. Abend NS, Loddenkemper T. Pediatric status epilepticus management. Curr Opin Pediatr. 2014;26(6):668‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson EL, Kaplan PW. Status epilepticus: definition, classification, pathophysiology, and epidemiology. Semin Neurol. 2020;40(6):647‐651. [DOI] [PubMed] [Google Scholar]
- 17. Trinka E, Cock H, Hesdorffer D, et al. A definition and classification of status epilepticus ‐ Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia. 2015;56(10):1515‐1523. [DOI] [PubMed] [Google Scholar]
- 18. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20(9):1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185(4154):862‐864. [DOI] [PubMed] [Google Scholar]
- 21. Cappi C, Oliphant ME, Peter Z, et al. De Novo damaging DNA coding mutations are associated with obsessive‐compulsive disorder and overlap with Tourette's disorder and autism. Biol Psychiatry. 2020;87(12):1035‐1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Froukh T, Nafie O, Al Hait SAS, et al. Genetic basis of neurodevelopmental disorders in 103 Jordanian families. Clin Genet. 2020;97(4):621‐627. [DOI] [PubMed] [Google Scholar]
- 23. Fukai R, Saitsu H, Tsurusaki Y, et al. De novo KCNH1 mutations in four patients with syndromic developmental delay, hypotonia and seizures. J Hum Genet. 2016;61(5):381‐387. [DOI] [PubMed] [Google Scholar]
- 24. Gripp KW, Smithson SF, Scurr IJ, et al. Syndromic disorders caused by gain‐of‐function variants in KCNH1, KCNK4, and KCNN3‐a subgroup of K(+) channelopathies. Eur J Hum Genet. 2021;29(9):1384‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kortum F, Caputo V, Bauer CK, et al. Mutations in KCNH1 and ATP6V1B2 cause Zimmermann‐Laband syndrome. Nat Genet. 2015;47(6):661‐667. [DOI] [PubMed] [Google Scholar]
- 26. Kortum F, Niceta M, Magliozzi M, et al. Cantu syndrome versus Zimmermann‐Laband syndrome: report of nine individuals with ABCC9 variants. Eur J Med Genet. 2020;63(9):103996. [DOI] [PubMed] [Google Scholar]
- 27. Lelieveld SH, Wiel L, Venselaar H, et al. Spatial clustering of de novo missense mutations identifies candidate neurodevelopmental disorder‐associated genes. Am J Hum Genet. 2017;101(3):478‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mastrangelo M, Scheffer IE, Bramswig NC, et al. Epilepsy in KCNH1‐related syndromes. Epileptic Disord. 2016;18(2):123‐136. [DOI] [PubMed] [Google Scholar]
- 29. Rochtus A, Olson HE, Smith L, et al. Genetic diagnoses in epilepsy: the impact of dynamic exome analysis in a pediatric cohort. Epilepsia. 2020;61(2):249‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rossi M, El‐Khechen D, Black MH, Farwell Hagman KD, Tang S, Powis Z. Outcomes of diagnostic exome sequencing in patients with diagnosed or suspected autism spectrum disorders. Pediatr Neurol. 2017;70:34‐43 e2. [DOI] [PubMed] [Google Scholar]
- 31. Sangkhathat S, Laochareonsuk W, Maneechay W, Kayasut K, Chiengkriwate P. Variants associated with infantile cholestatic syndromes detected in extrahepatic biliary atresia by whole exome studies: a 20‐case series from Thailand. J Pediatr Genet. 2018;7(2):67‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Satterstrom FK, Kosmicki JA, Wang J, et al. Large‐scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180(3):568‐84 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Simons C, Rash LD, Crawford J, et al. Mutations in the voltage‐gated potassium channel gene KCNH1 cause Temple‐Baraitser syndrome and epilepsy. Nat Genet. 2015;47(1):73‐77. [DOI] [PubMed] [Google Scholar]
- 34. Takata A, Nakashima M, Saitsu H, et al. Comprehensive analysis of coding variants highlights genetic complexity in developmental and epileptic encephalopathy. Nat Commun. 2019;10(1):2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Turner TN, Wilfert AB, Bakken TE, et al. Sex‐based analysis of de novo variants in neurodevelopmental disorders. Am J Hum Genet. 2019;105(6):1274‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang H, Zhang X, Ding H. Temple‐Baraitser syndrome with KCNH1 Asn510Thr: a new case report. Clin Dysmorphol. 2021;30(1):27‐31. [DOI] [PubMed] [Google Scholar]
- 37. von Wrede R, Jeub M, Arioz I, et al. Novel KCNH1 mutations associated with epilepsy: broadening the phenotypic spectrum of KCNH1‐associated diseases. Genes. 2021;12(2):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Epi KC. De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet. 2016;99(2):287‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tang B, Li B, Gao LD, et al. Optimization of in silico tools for predicting genetic variants: individualizing for genes with molecular sub‐regional stratification. Brief Bioinform. 2020;21(5):1776‐1786. [DOI] [PubMed] [Google Scholar]
- 40. Ye XG, Liu ZG, Wang J, et al. YWHAG mutations cause childhood myoclonic epilepsy and febrile seizures: molecular sub‐regional effect and mechanism. Front Genet. 2021;12:632466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zou D, Qin B, Wang J, et al. AFF2 is associated with X‐linked partial (focal) epilepsy with antecedent febrile seizures. Front Mol Neurosci. 2022;15:795840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heyne HO, Singh T, Stamberger H, et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet. 2018;50(7):1048‐1053. [DOI] [PubMed] [Google Scholar]
- 43. Wagnon JL, Barker BS, Hounshell JA, et al. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol. 2016;3(2):114‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Weckhuysen S, Mandelstam S, Suls A, et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. 2012;71(1):15‐25. [DOI] [PubMed] [Google Scholar]
- 45. Tian X, Ye J, Zeng Q, et al. The clinical outcome and neuroimaging of acute encephalopathy after status epilepticus in Dravet syndrome. Dev Med Child Neurol. 2018;60(6):566‐573. [DOI] [PubMed] [Google Scholar]
- 46. Shibata A, Kasai M, Terashima H, et al. Case‐control association study of rare nonsynonymous variants of SCN1A and KCNQ2 in acute encephalopathy with biphasic seizures and late reduced diffusion. J Neurol Sci. 2020;414:116808. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Appendix S1
Table S1
Data Availability Statement
The data that support the findings of this study will be available from the corresponding author upon reasonable request.