

Summary
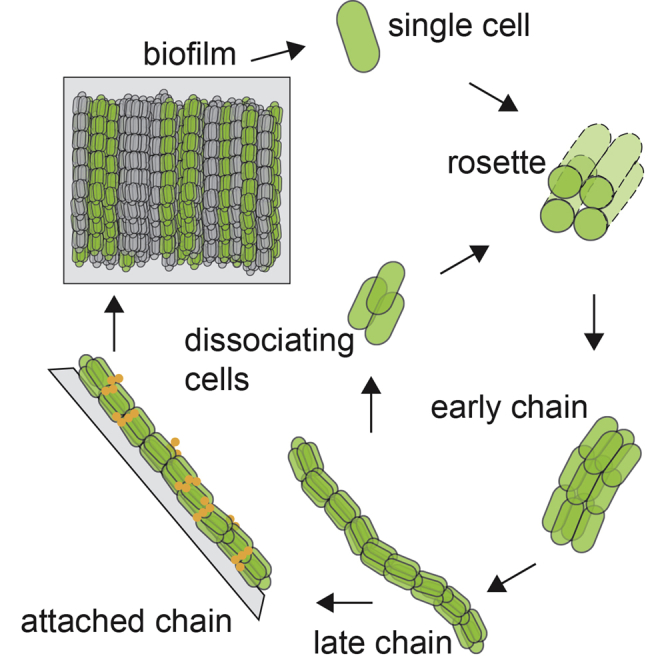
Biofilms are surface-attached multicellular microbial communities. Their genetics have been extensively studied, but the cell-scale morphogenetic events of their formation are largely unknown. Here, we recorded the entirety of morphogenesis in Escherichia coli, and discovered a previously unknown multicellular self-assembly process. Unattached, single-cells formed 4-cell rosettes which grew into constant-width chains. After ∼10 cell generations, these multicellular chains attached to surfaces and stopped growing. Chains remained clonal throughout morphogenesis. We showed that this process generates biofilms, which we found are composed of attached clonal chains, aligned in parallel. We investigated genetics of chain morphogenesis: Ag43 facilitates rosette formation and clonality; type-1 fimbriae and curli promote stability and configuration; and extracellular polysaccharide production facilitates attachment. Our study establishes that E. coli, a unicellular organism, can follow a multistage, clonal, genetically-regulated, rosette-initiated multicellular life cycle. These findings have implications for synthetic biology, multicellular development, and the treatment and prevention of bacterial diseases.
Subject areas: Bacteriology, Microbiofilms
Graphical abstract
Highlights
-
•
A possible multicellular life cycle in bacterium Escherichia coli is reported
-
•
Single-cells assemble 4-cell rosettes that extend into constant-width clonal chains
-
•
Chains attach as clonal units and their parallel-aligned accumulation generates biofilms
-
•
The uncovered life cycle has multiple genetically-regulated stages
BacteriologyMicrobiofilms
Introduction
Bacteria form surface-attached multicellular communities known as biofilms.1 Biofilm cells resist toxic environments, escape immune responses, tolerate antibiotics, and contribute to chronic infections.2,3,4,5 Their structure, cellular differentiation, and coordinated behavior suggest they could represent a bacterial equivalent of development in higher organisms.6 However, the cell-scale morphogenetic events generating biofilms are not fully understood and are under ongoing investigation.7,8,9,10,11,12,13
In Escherichia coli, genes required for biofilm formation have been identified, many of which encode adhesins that are understood in molecular detail (Ag43, type-1 fimbriae, curli, polysaccharides, and pili).14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29 Type-1 fimbriae are rigid protein polymers that extend 2 μm from the cell surface.30,31 Their adhesive tips bind to mannose groups and play a role in host-cell interactions, though their essentiality to biofilm formation on mannose-lacking surfaces suggests they perform cell-cell interactions as well.32 Curli are extracellular fibers that facilitate cell-cell and cell-surface interactions and are a major component of the biofilm extracellular matrix.16,33 Their function is thought to overlap with fimbriae, though their rigidity and adhesion differ.34 Poly-β-1,6-N-acetyl-D-glucosamine (polyglucosamine) is a sugar polymer and the primary component of the extracellular matrix.18,19 It is required for surface attachment during biofilm formation in E. coli, Staphylococcus epidermis, Staphylococcus aureus, and Pseudomonas fluorescens18,19 and its production has a significant metabolic cost,18,20 suggesting growth and attachment might be mutually exclusive behaviors. Antigen-43 (Ag43), encoded by flu, is a self-recognizing adhesion protein that coordinates cell-cell aggregation and has been implicated in E. coli biofilms.22,23,24,25,26 It acts over short distances (∼10 nm from the cell membrane) and can cause auto aggregation when overexpressed.22,23,24,25,26Ag43 has also been shown to localize to the poles and allow sister-cells to remain adhered after cell division.35 However, its expression and precise role in biofilm formation are debated.22,23,24,25,26 Though all these adhesins are understood at the molecular-scale, their precise contributions to morphogenesis at the cell-scale are unclear.
Deciphering the specific roles of biofilm genes will require tracking of the entirety of morphogenesis at the cell-scale, beginning from individual planktonic cells. Hydrodynamic conditions (fluid flow) are often used to study biofilms at the cellular scale as they allow continuous supply of nutrients and enhance biofilm formation in many species.1 However, biofilm formation in commensal and lab K-12 E coli strains occurs in hydrostatic conditions, and is inhibited by fluid flow.36 Moreover, genetic studies of biofilm formation have relied on standardized bulk-scale hydrostatic experiments.15,18,19,21,24,37,38,39 Although robust, these methods cannot track cell-scale behavior.
Results
Morphogenesis of clonal, multicellular chains in E. coli
We developed devices combining microfluidics and agarose pad methods40 to track E. coli multicellular morphogenesis from single cells (Figure S1; STAR Methods). These devices allowed nutrient delivery by diffusion rather than fluid flow, creating a static liquid growth environment between agarose and microscope cover glass. Planktonic cells were loaded into these devices then imaged at 100x magnification. Automated microscopy simultaneously recorded multicellular morphogenesis beginning from separate individual cells. The resulting videos (captured at 10-min intervals) revealed that single cells grew and assembled chain-like communities hundreds-of-μm long (Figures1A and S2–S4; Videos S1, S2, and S3). Equivalent chains were observed in experiments with cells containing green fluorescent protein (GFP, STAR Methods), imaged every 6 min. These experiments showed that chains were initially unattached, moved flexibly, and had constant width (2–3 cells wide) during longitudinal growth for 4–5 h (growing to over 200 μm in length) before ceasing motion by evident surface attachment (Figures 1B, 1C, and S5–S7; Videos S4, S5, and S6). All chains were initiated by the formation of square, 4-cell “rosettes”, where two cells were stacked atop two more cells such that the long axes of all cells were parallel (Figures 1D and 1E). The initial 4-cell configuration and width were maintained as chains grew, with new cells remaining attached to their sister cells at their poles (Videos S4 and S5). Individual cells or small cell clusters occasionally (approximately once per chain life span) separated from an elongating chain (Videos S7 and S8). This breaking away produced new rounds of multicellular chain morphogenesis indicating that chains could effectively self-propagate. Growth and motion simultaneously stopped in each chain, though separate chains could stop growing at different times. Long chains merged with one another and attached to the surface, creating a multicellular community with porous channels (Figures S8–S10). Similar morphogenesis of multicellular chains was observed in multiple strains (MG1655, BW25113) and nutrient environments (Figures S11–S13).
Figure 1.

Multicellular chain formation in E. coli
(A) Micrographs from a representative example of multicellular chain formation in E. coli (MG1655) starting from single-cells (Figures S1–S7; Videos S1, S2, S3, S4, S5, and S6). Exponential phase cells were loaded into microfluidic devices and grown at 37°C while imaging at 100x magnification on 10-min intervals for ∼10 h. Time and scale are indicated on all micrographs; see Figure S1 & STAR Methods for complete experimental details.
(B) Representative example of late-stage chain morphogenesis in E. coli cells containing constitutively-expressed green fluorescent protein (GFP) (pUA66-pompC:gfp), imaged at 6-min intervals.
(C) Micrographs from a representative example of early chain morphogenesis in E. coli cells expressing GFP.
(D) Representative micrographs of a single 4-cell rosette.
(E) Illustrative diagram of rosette geometry with calculated dimensions represented as mean ± standard deviation (SD) (width: n = 30; diagonals: n = 3).
(F) Growth of rosette-configured chains and monolayer colonies calculated as area versus time (see STAR Methods). Data represent mean ± SD (shaded area; n = 3). Inset: representative micrographs of chain and monolayer communities at different growth phases.
(G) Representative example of the dynamic changes in the centerline for an individual chain. Centerlines were calculated through segmented images of morphogenetic chains formed by E. coli cells containing GFP (see also Figures S14, S15, and S19; STAR Methods).
(H) Chain width during morphogenesis represented as mean ± SD (shaded area; n = 6). Rosette width is maintained throughout chain elongation and attachment (Figures S21 and S22; STAR Methods).
(I) Percent growth rate (blue) and area of motion (AOM) (red) of chains during morphogenesis (STAR Methods). AOM of 1 indicates no motion. Data represent mean ± SD (shaded area; n = 3). The decrease in motion (AOM) coincides with the decrease in growth rate.
We next quantified chain growth by tracking morphogenesis (6-min resolution) in cells expressing GFP, which aided image processing (Figures S14 and S15; STAR Methods). Chain area and length grew exponentially from 1 to >1,000 cells per chain (Figures 1F and S16). The rate of this growth (2.3 ± 0.2 doublings per hour) was comparable to the maximum growth rate of planktonic cells (2.9 ± 0.2 doublings per hour) in this medium41 (Figures S17 and S18; STAR Methods). Chain movement stopped when exponential growth stopped (Figure 1F), suggesting attachment coincides with growth cessation (further quantified below). As a control for community shape, we grew cells on standard agarose pads which force cells to grow in monolayers (Figure 1F; STAR Methods). Monolayers were exposed to the same nutrient environment as chains and only differed in their three-dimensional cellular configurations. Monolayer communities grew slower and for longer times than chains, suggesting growth physiology may be influenced by chain structure. Calculated chain centerlines demonstrated repeated bending during elongation (Figures 1G and S19) and centroids had random-like motion, demonstrating a lack of stable surface attachment (Figure S20). We confirmed that chain width was constant (∼3 μm; equivalent to rosette width (Figures 1E and S21A)) and maintained during elongation (Figures 1H, S21, and S22). To quantify chain attachment, we calculated the “area of motion” (AOM; STAR Methods). For most of morphogenesis, AOM was >1 indicating motion (Figure 1I). However, its value ultimately decreased to nearly 1 at the end of morphogenesis, indicating a complete lack of motion (Figure 1I). The decrease in AOM immediately followed the decrease in percent growth rate of chains, demonstrating growth and movement stopped simultaneously (Figure 1I).
We next quantified the clonality of chains by recording videos (10-min resolution) of morphogenesis in a mixed population of cells containing either GFP (green) or mScarlet (red) (Figure S1; STAR Methods). In these experiments, clonal communities would be single-color and polyclonal communities resulting from dissociation and aggregation would be two-color. All chains were observed to be single-color (Figures 2A, 2B, and S23). Even when separate chains came into contact, cellular mixing or exchange was not observed (Figures 2A, 2B, and S24), indicating that chain clonality is stable long-term. Together, these findings demonstrate a previously unreported bacterial morphogenetic process that is clonal, has multiple stages (rosette formation, chain growth, and chain attachment), and spans a range of phenotypes (from single planktonic cells to non-growing surface-attached communities).
Figure 2.

E. coli biofilms are comprised of clonal multicellular chains
(A and B) Micrographs from a representative example of multicellular chain formation in E. coli (MG1655) starting from single-cells containing either GFP (green) or mScarlet (red) (1:1 ratio) (see also Figures S23 and S24). Exponential phase cells were loaded into microfluidic devices and grown at 37°C while imaging at 100x magnification on 10-min intervals for ∼10 h (see STAR Methods). Time and scale are indicated on all micrographs. The highlighted region in (B) is magnified in the inset.
(C) Illustrative diagram of method used to image biofilms (see STAR Methods).
(D) Representative micrograph of an E. coli biofilm grown on a cover glass at 37°C and imaged at 63x magnification. All biofilm micrographs represent X-Y imaging plane (X direction is parallel to the air-liquid interface, Y direction is perpendicular to the air-liquid interface and represents the depth of the biofilm, and Z direction points toward the camera and represents thickness of the biofilm). The inoculum for biofilm growth was a 1:10 mixture of cells containing GFP (green) and mScarlet (red). Air, liquid, and biofilm regions are indicated on the micrograph. Contrast adjustment: red channel is 11x reduced and green channel is 3x reduced in the right section of the image, to provide alternate view. Biofilm edges are indicated by dashed lines (verified by bright-field imaging).
(E) A zoomed-out view of a representative biofilm grown and imaged at 100x magnification as described above (only green fluorescence shown).
(F) Calculated widths of rosettes (illustrated in Figures 1D and 1E), chains during morphogenesis (illustrated in Figure 1H), and the clonal chains within biofilms. Data represent mean ± SD (n = 20).
Chain morphogenesis generates E. coli biofilms
We reasoned that the chain morphogenesis we had observed in microfluidic devices (Figure 1) might contribute to biofilm formation and, moreover, that the stable clonality of chains might make them visible within biofilms (Figures 2A and 2B). Adapting standard methods where biofilms form at the surface-liquid-air interface in hydrostatic cultures,42 we grew biofilms on microscope cover glass from a mixture of cells containing either GFP (green) or mScarlet (red) (Figure 2C; STAR Methods). After 16 h of growth, cover glass with attached biofilms was removed from culture and imaged at 63x or 100x magnification. The resulting biofilms were composed of single-color clonal chains, arranged in parallel to one another (Figures 2D, 2E, and S25). Chains extended perpendicular to the air-liquid interface, over the entire depth of biofilms (i.e. chain length corresponded to biofilm depth), and had equivalent lengths and widths (Figure 2F) to the multicellular chains we observed in microfluidic devices (Figure 1). Randomly aggregated cells were rare and only occurred at the bottom (liquid facing) edge of biofilms, possibly resulting from planktonic single cell adherence to surface-attached chain communities. These findings demonstrate that E. coli biofilms have a clonally organized internal structure which is not evident solely by bright-field imaging43 (Figure S26).
We considered the possibility that the clonal chains in E. coli biofilms might result from steric effects exerted by attached growing communities, as similar patterns have been noted when two microcolonies merge.44,45,46 If this were true, clonal communities would grow longer as biofilms aged, and surface-attached, isolated clonal regions would be observable at early time points before they merged with other colonies. Hence, we grew biofilms on cover glass for different times (Figures 3A–3D and S27; STAR Methods; see diagram in Figure 2C). No biofilms or adhered communities were observed after 4 (Figure S27A) or 8 h (Figure 3A), and only randomly-scattered cells were visible. At 16, 20, and 24 h, biofilms spanned the complete surface-liquid-air interface of the cover glass and were composed of clonal chains (Figures 3C, 3D, and S27B). Regardless of growth time, the depth of biofilms did not substantially vary (∼400 μm) (Figure 3E). These experiments rule out the possibility that clonal chains result from steric effects on growing communities. Firstly, isolated attached clonal communities were never observed. Secondly, we did not observe that clonal chains in biofilms could elongate, suggesting that growth stops at surface attachment, equivalent to our observations of chain morphogenesis in microfluidic devices (Figures 1 and S1; STAR Methods). These implications are further supported by 12-h experiments which capture intermediate biofilm propagation at the surface-liquid-air interface, with regions where biofilm formation had yet to occur (Figure 3B). Biofilm regions in these samples were continuous, had constant depth (∼400 μm) (Figure 3B), and were composed of clonal chains equivalent to those in mature biofilms (Figures 3A–3D) and microfluidic devices (Figure 1). Beyond the propagating edge of biofilm regions, attached communities were not observed in intermediate biofilms (Figure 3B). These findings show that E. coli biofilms are composed of non-growing, attached clonal chains, aligned in parallel.
Figure 3.

Clonal chains assemble prior to attachment in biofilms
(A–D) Representative micrographs of E. coli biofilms grown on cover glass at 37°C for 8 (A), 12 (B), 16 (C) and 20 (D) hours and imaged at 63x magnification (see also Figure S27 for biofilms grown for 4 and 24 h; and see STAR Methods and Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7C). The inoculum for biofilm growth was a 1:10 mixture of cells containing GFP (green) and mScarlet (red) and dashed lines represent edges of biofilm (verified by bright-field imaging). Time and scale are indicated on all micrographs. Calculated biofilm depth is indicated by arrowed line and represents mean ± SD. Highlighted regions in each micrograph are magnified in panels on the right.
(E) Calculated biofilm depth from images after different growth times, represented as mean ± SD (n = 3). Two-sample t test showed that the depth (in the Y-direction) of formed biofilms did not significantly differ at any time points (p values: 12h&16h- 0.930, 12h&20h- 0.435, 12h&24h- 0.264, 16h&20h- 0.365, 16h&24h- 0.230, 20h&24h- 0.563; α = 0.05). † indicates that this depth was calculated from the formed biofilm portion of these images, though 12-h biofilms were intermediate and had regions where biofilm had not yet formed.
(F) Representative 3D-view of biofilms grown for 24 h and imaged by confocal microscopy (STAR Methods; see also Figure S29). The Z-direction of biofilms is captured in addition to the X-Y imaging plane captured by the previous micrographs.
(G) Thickness (in the Z-direction) of biofilms grown for 16 and 24 h and imaged by confocal microscopy, represented as mean ± SD (n = 3). Two-sample t test showed that thickness was significantly different between the two time points (p value: 0.018; α = 0.05).
(H and I) Representative micrograph of communities washed off of a growing biofilm. Biofilms grown from a 1:10 mixture of cells containing GFP (green) and mScarlet (red) on cover glass (depicted in Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7C) were washed with phosphate-buffered saline (PBS) (STAR Methods). The run-off was collected on agarose pads and imaged at 100x magnification (see also Figures S30 and S31). The highlighted region is magnified in (I).
(J) Cells per chain versus chain length for clonal communities washed from biofilms (see also Figure S30). Diagram illustrates rosette-configured chain geometry and approximate cell density, ∼2 cells/μm which is also indicated by the red fit line (see also Figures S30, S31, and 1). Inset: Cell density of clonal communities washed from biofilms represented as mean ± SD (n = 15).
Though the depth of biofilms remained constant as they aged, their fluorescence intensity increased (Figure S28A). Dispersing biofilms revealed that this could not be explained by increases in single-cell fluorescence (Figure S28B), therefore suggesting biofilms grow thicker as they age, i.e. away from the glass surface as a pellicle biofilm (toward the camera, or z-direction, in the images). We confirmed this by confocal microscopy (Figures 3F and S29; Video S9), showing that biofilm thickness in the z-direction increased with age (Figure 3G). This finding is consistent with biofilm growth resulting from successive rounds of local chain morphogenesis and attachment. This model for biofilm formation would further suggest that they contain clonal chains that are not yet attached. To test this, we washed red-green biofilms and imaged the resulting liquid run-off at 100x magnification (STAR Methods). This run-off contained single-color (clonal) communities of ∼50–300 cells (Figures 3H and 3I), demonstrating the presence of unattached clonal chain-like communities in growing biofilms. Though clonally organized, most cells in these communities were not directly touching and hence were not arranged in rosette configuration. However, we reasoned that the sample preparation for this experiment, which compresses communities between glass and agarose, might disrupt rosette configuration by forcibly separating cells. If true, there would be 2–4 cells/μm in these communities, equivalent to the cell density in rosettes and rosette-configured chains. We calculated the cell density of washed chains to be 2.6 ± 0.6 cells/μm (Figure 3J inset, STAR Methods) and found that this value was conserved for chains of different lengths (Figures 3J and S30), indicating that the clonal communities washed from biofilms were chains whose organization was statistically equivalent to rosettes. Occasionally, two chains, adhered in parallel, were washed off of biofilms together, suggesting chains might preferentially attach to one another rather than the surface (Figure S31). Together, these findings indicate that E. coli biofilms formed in hydrostatic cultures are composed of clonal multicellular chains that assemble before surface attachment. They further suggest that biofilms are generated by the clonal multicellular morphogenesis we observed in microfluidic devices (Figure 1). We therefore reasoned the separate stages of chain morphogenesis that we had observed in microfluidic devices might be individually regulated by known biofilm genes and, in turn, the role of these genes in chain morphogenesis might explain their role in biofilm formation.
Ag43 is required for rosette formation and chain clonality
The tight binding of cells in chains led us to consider that Ag43 might play a critical role in morphogenesis. Ag43 is expressed at intermediate levels in wild type in most conditions and can be locked into a high expression state by epigenetic regulation when induced.47,48 We verified that our strains were not rare phase on variants (STAR Methods). We recorded videos (5-min resolution) of morphogenesis in Δflu cells, lacking Ag43, and found that they commonly dissociated after division, demonstrating Ag43 facilitated the initial adhesion of sister cells in rosette formation (Figures 4A and S32; Video S10; STAR Methods; see diagram in Figure S1). Δflu cells that dissociated could later aggregate, as cell density increased, to form communities similar to wild type. However, these communities were not clonal, lacked stability, and did not robustly attach to surfaces (Figure 4A; Video S11). We further demonstrated the lack of clonality and stability in Δflu communities through red-green experiments (Figure S1; STAR Methods). These communities were polyclonal, resulting from constant dissociation and aggregation (Figure 4B). To determine if these effects were observable in biofilms, we grew Δflu biofilms on cover glass (STAR Methods; see diagram in Figure 2C). Δflu biofilms were less deep (∼184 μm) and thinner (approximately single-cell thick) compared to wild-type biofilms (Figure 4C). Moreover, the clonal chain structure of wild-type biofilms was abolished by Δflu mutation, leading to random cell arrangement (Figures 4C and S37). Together, these results show that Ag43 is essential for sister-cell adhesion, clonality, and surface attachment in both chain morphogenesis (Figure 4A) and biofilm formation (Figure 4C). The occurrence of some attachment and chain-like aggregates in dense Δflu populations suggests other adhesins also contribute to morphogenesis.
Figure 4.

Ag43 and type-1 fimbriae maintain chain clonality and stability
(A) (left panel) Illustration of the role of antigen 43 (encoded by flu). (right panel) Micrographs from a representative example of Δflu morphogenesis (see also Videos S10 and S11 & Figure S32). Exponential phase Δflu cells were loaded into microfluidic devices and grown at 37°C while imaging at 100x magnification on 5-min intervals (see Figure S1 & STAR Methods). Time, scale, and phenotype description are indicated on all micrographs.
(B) Representative micrograph of Δflu morphogenesis from cells containing either GFP (green) or mScarlet (red) (1:1 ratio; highlighted region is magnified in inset).
(C) Representative micrograph of Δflu biofilms grown for 16 h from 1:10 mixture of cells containing GFP (green) and mScarlet (red) and imaged at 63x magnification (see Figures S37 and Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7C & STAR Methods). Scale and biofilm depth are indicated on the micrograph (highlighted region is magnified in inset).
(D) (left panel) Illustration of type-1 fimbriae. (right panel) Micrographs from a representative example of ΔfimH morphogenesis (see also Videos S12, S13, and S14 & Figures S33–S36). Exponential phase ΔfimH cells were loaded into microfluidic devices and grown at 37°C while imaging at 100x magnification on 6-min intervals (see Figure S1 & STAR Methods). Time, scale, and phenotype description are indicated on all micrographs.
(E) Representative micrograph of ΔfimH morphogenesis from cells containing either GFP (green) or mScarlet (red) (1:1 ratio; highlighted region is magnified in inset).
(F) Representative micrograph of ΔfimH biofilms grown for 16 h from 1:10 mixture of cells containing GFP (green) and mScarlet (red) and imaged at 63x magnification (see also Figure S37). Scale and biofilm depth are indicated on the micrograph (highlighted region is magnified in inset).
(G) Representative micrograph of wild-type biofilms grown for 16 h with 10 mM mannoside from a 1:10 mixture of cells containing GFP (green) and mScarlet (red) (STAR Methods). Scale and biofilm depth are indicated on the micrograph (highlighted region is magnified in inset).
Fimbriae are required for the stability of chains
Type-1 fimbriae are phase-variable, i.e. their genes are either phase on or phase off, ensuring cells have hundreds of fimbriae or none at all. Regulation of fimbriae is not fully understood and is sometimes considered stochastic as wild-type populations can contain rare phase-on cells (∼10−3),47 which were ruled out in our strains (STAR Methods). There is a need to re-evaluate stochastic regulation49 given the induction of fimbriation on hydrated surfaces of solid media.50 We recorded videos (6-min resolution) of morphogenesis in ΔfimH cells, which lack the adhesive, mannose-binding tip of fimbriae (STAR Methods; see diagram in Figure S1). These cells formed rosettes indistinguishable from wild type, indicating that fimbriae are not critical to the initial step of morphogenesis (Figure 4D). However, by the 16-cell stage, ΔfimH chains broke apart or folded over, demonstrating fimbriae are required for chain stability (Figures 4D and S33–S36; Videos S12 and S13). In addition, ΔfimH mutation impaired surface attachment and ΔfimH communities dissociated into single-cell-wide strands adhered only at the cell poles, suggesting fimbriae mediate lateral cell-cell interactions in chains (Figure 4D; Video S14). Red-green experiments demonstrated ΔfimH communities were not clonal and instead resulted from dissociation-aggregation events (Figure 4E, STAR Methods). In these experiments, single-cell-wide clonal strands were observed within polyclonal communities again indicating fimbriae contribute to lateral cell-cell interactions in chains. To determine if these effects were observable in biofilms, we grew ΔfimH biofilms on cover glass for 16 h (STAR Methods; see diagram in Figure 2C). ΔfimH biofilms were less deep (∼127 μm), thinner (approximately single-cell thick) and lacked the clonal chains observed in wild type (Figures 4F andS37). We noted examples of short single-cell-wide strands, indicating fimbriae played equivalent roles in biofilms (Figures 4F and S37) and chain morphogenesis (Figure 4D). Both fimbriae adhesion and biofilm formation are inhibited by mannose analogues.15,51 We found that wild-type biofilms formed on cover glass in the presence of mannoside (10 mM) were indistinguishable in size and internal structure from ΔfimH biofilms (Figure 4G). These findings demonstrate that fimbriae provide chain stability through lateral cell-cell interactions after rosettes are formed, and that this function is critical to E. coli biofilm formation in hydrostatic environments. Similar to those in Δflu, these findings suggest that random aggregation can mask the role of fimbriae when cell density is high.
Robust biofilm formation requires both Ag43 and fimbriae
Our data indicate that chain morphogenesis can be initiated by a single cell and is self-propagating (Figure 1). This suggests biofilm formation might also be self-propagating and would be robust to reductions in the initial cell density. Our findings suggest the opposite would be true for ΔfimH and Δflu strains, both of which require density-dependent random aggregation for chain morphogenesis and biofilm formation. To test this, we grew biofilms on cover glass from wild-type, Δflu, and ΔfimH strains at different initial cell densities (Figures 5A–5F and S38; STAR Methods; see diagram in Figure 2C). At 109 cfu/mL initial concentration, Δflu and ΔfimH biofilms were less deep and thinner than wild-type biofilms, and lacked their characteristic clonal structure (Figures 5A–5C). Lowering the inoculum concentration reduced the depth of Δflu and ΔfimH biofilms, completely abolishing them at 100x dilution (107 cfu/mL initial concentration), whereas wild-type cells generated biofilms with indistinguishable structures (Figures 5D–5F and S38). This confirms wild-type biofilm formation is robust and demonstrates that the stability and clonality of chains are essential to this robustness. Furthermore, these findings show that high cell density can mask the roles of biofilm genes in some conditions. These findings also suggest a possible explanation for why some E. coli isolate and mutant strains form biofilms under fluid flow whereas commensal and K-12 biofilms are prevented. Many of these mutants cause auto-aggregation (increased cell-cell binding) in planktonic cultures which would normally separate cells (e.g., Ag43 overexpression and specific fimH mutations).23,27 Our data in Δflu and ΔfimH show that defects in chain morphogenesis can be partly overcome by cell-cell aggregation which facilitates aspects of wild-type morphogenesis and attachment, possibly by activating components of the extracellular matrix (e.g., curli and extracellular polyglucosamine).
Figure 5.

flu and fimH are required for robust biofilm formation
(A–C) (A) Wild-type, (B)Δflu, and (C)ΔfimH biofilms grown for 16 h from a 1:10 mixture of cells containing GFP (green) and mScarlet (red) at an initial dilution of 109 cfu/mL, using method illustrated in Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7C (STAR Methods).
(D–F) (D) Wild-type, (E)Δflu, and (F)ΔfimH biofilms grown for 16 h from a 1:10 mixture of cells containing GFP (green) and mScarlet (red) at an initial dilution of 107 cfu/mL, using method illustrated in Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7C (STAR Methods) (see also Figure S38). Scale and biofilm depth are indicated on micrographs.
Curli are required for the structure but not stability of chains
Curli production is activated in a switch-like manner and is repressed in planktonic cells under most conditions.16,33 We recorded videos (6-min resolution) of morphogenesis in ΔcsgA cells, which lack the major polymer component of curli (STAR Methods; see diagram in Figure S1). These cells formed rosettes indistinguishable from wild type, indicating that curli are not critical to initiating morphogenesis (Figure 6A; Video S15). However, by the ∼16-cell stage, ΔcsgA chains lost rosette configuration and flattened into short thick shapes (Figures 6A and S39–S42; Video S15). These communities folded over on themselves, but unlike ΔfimH, they did not break apart, demonstrating curli maintain the unique structure of chains but are not as critical as fimbriae or Ag43 to stability. Further differing from wild type, Δflu, and ΔfimH, ΔcsgA mutation abolished surface attachment and led to full dissociation of communities in the long term (Figure 6A; Video S16). Red-green experiments demonstrated that most ΔcsgA communities were clonal, though some single-cell-wide strands resembling ΔfimH were noted (Figure 6B). We next attempted to grow ΔcsgA biofilms on cover glass, but noted that this mutation effectively abolished biofilm formation (Figure 6C; STAR Methods; see diagram in Figure 2C). These results show that curli production is essential to maintaining chain structure after rosette formation, but is less critical to stability than Ag43 or fimbriae. These findings also show that curli are critical to the later attachment stage, either by adhering to surfaces or by facilitating other adhesins, such as extracellular polyglucosamine.
Figure 6.

Curli and extracellular polysaccharide mediate chain structure and attachment
(A) (left panel) Illustration of the role of curli. (right panel) Micrographs from a representative example of ΔcsgA morphogenesis (see also Videos S15 and S16; Figures S39–S42). Exponential phase ΔcsgA cells were loaded into microfluidic devices and grown at 37°C while imaging at 100x magnification on 6-min intervals (see Figure S1 & STAR Methods). Time, scale, and phenotype description are indicated on all micrographs.
(B) Representative micrograph of ΔcsgA morphogenesis from cells containing either GFP (green) or mScarlet (red) (1:1 ratio; highlighted region is magnified in inset).
(C) Representative micrograph of ΔcsgA biofilms grown for 16 h from 1:10 mixture of cells containing GFP (green) and mScarlet (red) and imaged at 100x magnification (see Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7C & STAR Methods). Scale is indicated on the micrograph (highlighted region is magnified in inset).
(D) (left panel) Illustration of extracellular polysaccharide polyglucoseamine. (right panel) Micrographs from a representative example of ΔpgaB morphogenesis (see also Video S17; Figures S43–S46). Exponential phase ΔpgaB cells were loaded into microfluidic devices and grown at 37°C while imaging at 100x magnification on 6-min intervals (see Figure S1 & STAR Methods). Time, scale, and phenotype description are indicated on all micrographs.
(E) AOM of wild-type and ΔpgaB chains (STAR Methods). AOM of 1 indicates no motion. Data represent mean ± SD (shaded area; n = 3).
(F) Representative micrograph of ΔpgaB biofilms grown for 16 h from 1:10 mixture of cells containing GFP (green) and mScarlet (red) and imaged at 100x magnification (see Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7C & STAR Methods). Scale is indicated on the micrograph (highlighted region is magnified in inset).
Polyglucosamine is required for chain attachment
Extracellular polyglucoasmine production is necessary for surface attachment during biofilm formation18,19 and could be sufficient given that most biofilm genes are dispensable if polyglucosamine is supplied exogenously.52 We recorded videos (6-min resolution) of morphogenesis in ΔpgaB cells, which are deficient in polyglucosamine export20 (STAR Methods; Figure S1). These cells proceeded through rosette formation and chain elongation equivalently to wild type, however chains never attached to surfaces (Figures 6D and S43–S46; Video S17). Instead, they continued to grow and move before eventually breaking apart into small clusters. The AOM further demonstrated the lack of attachment in ΔpgaB chains relative to wild type (Figure 6E; STAR Methods). We next attempted to grow ΔpgaB biofilms on cover glass, but found that this mutation abolished biofilm formation (Figure 6F; STAR Methods; see diagram in Figure 2C), consistent with previous reports.18,19 These results indicate that polyglucosamine is responsible for the attachment of chains to surfaces at the end of multicellular morphogenesis.
Chain morphogenesis in E. coli UTI-isolate biofilms
We studied additional E. coli strains to explore the generality of chain morphogenesis in biofilm formation. Similar to MG1655, BW25113 (another K-12 derivative) biofilms were constant depth and consisted of parallel-aligned clonal chains (Figure S47; STAR Methods; see diagram in Figure 2C). The internal structure of these biofilms was also interrupted by blocking fimbriae adhesion with mannoside (10 mM) (Figure S48). We considered that chain morphogenesis might partly result from domestication of strains by long-term laboratory cultivation and therefore might not be a native behavior of fully wild-type E. coli. Hence, we sought to test an E. coli strain that had never been propagated in a lab and acquired a strain directly isolated from a urinary tract infection (UTI). We transformed this strain separately with plasmids carrying mScarlet and GFP and grew biofilms on cover glass (STAR Methods; see diagram in Figure 2C). These biofilms (Figure 7A) were composed of clonal chains similar to those observed in the lab strains (Figures S47 and S49). However, the structure of chains in these biofilms was better defined (Figures 7A and S49) and individual chains clearly spanned the biofilm from the air to the liquid edges. We reasoned this might be due to increased lateral adhesion between cells, either through increased fimbriation or increased fimbriae binding targets. Consistent with this hypothesis, higher concentrations of mannoside (100 mM) were required to disrupt the internal structure of UTI-isolate biofilms (Figures 7B and S50). These findings suggest that the uncovered multicellular morphogenesis in E. coli is not limited to lab strains. Occasionally, clonal aggregates were adhered to the bottom of the chain biofilm in the UTI strain (Figure S51), which had not occurred in other strains or mutants. This indicates that different forms of multicellular organization (aggregates versus chains) may not be mutually exclusive within some strains.
Figure 7.

UTI-isolate biofilms and proposed model for E. coli biofilm formation
(A and B) Representative micrograph of an E. coli UTI-isolate biofilm grown on a cover glass at 37°C without (A) or with (B) 100 mM mannoside and imaged at 63x and 100x magnification respectively. The inoculum for biofilm growth was a 1:10 mixture of cells containing GFP (green) and mScarlet (red) (see Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7C & STAR Methods). Scale is indicated on micrographs (highlighted region is magnified in inset) (see also Figures S49–S51).
(C and D) Representative micrograph of an E. coli UTI-isolate intermediate biofilm grown on cover glass before (C) and after (D) washing with PBS (STAR Methods) imaged at 100x magnification. Scale is indicated on the micrograph (highlighted region is magnified in inset; only green fluorescence shown).
(E) (top) Illustration of a proposed model for E. coli biofilm formation through chain morphogenesis and attachment. Chain clonality is indicated by green- or no-fill, and polyglucosamine is indicated in orange. (Bottom) Illustrations of the genes, and their interaction distance and selectivity, regulating each stage of this process.
We reasoned that the increased fimbriae-dependent stability of the UTI-isolate might enable detection of unattached chains at the propagating edge of biofilms. We suspected that such local unattached chains in MG1655 biofilms had been disrupted by the imaging sample preparation. Hence, we grew intermediate biofilms on cover glass from the UTI-isolate (STAR Methods; see diagram in Figure 2C). Unlike lab strains of E. coli, the intermediate UTI-isolate biofilms contained regions of shorter chains with increased bending (Figure 7C), suggesting that these chains were still growing and were unattached. We reasoned that washing these samples would remove these short chains, without disturbing the attached longer chains (STAR Methods). Washing biofilms confirmed this hypothesis, entirely removing short chain regions without displacing attached long-chain regions (Figure 7D). We noted the length of unattached chains increased as their distance from the propagating edge decreased, indicating they were closer to completing morphogenesis and attaching. Combined, with the self-propagation of chains (Figure 1) and the robustness of chain morphogenesis for biofilm formation (Figure 5), these findings support the idea that the uncovered multicellular morphogenesis in E. coli generates self-propagating biofilms.
Discussion
E. coli is one of the best studied unicellular organisms, though our understanding of its natural existence, in hosts and the environment, is incomplete. Here, we have uncovered a previously unknown multicellular morphogenesis in E. coli and demonstrated that it has reproducible, well-regulated growth and dynamic community shape properties (Figure 1); is initiated by the formation of rosettes and is clonal (Figures 1 and 2) similar to multicellular organisms; can self-propagate and reproduce new chains by the dissociation of cell clusters (Videos S7 and S8); ends with surface attachment and growth arrest (Figure 1); and has multiple stages (Figure 1), each of which is temporally and dependently organized and is regulated by a specific genetic mechanism (Figures 4, 5, and 6). Together, these findings suggest that E. coli can follow a multicellular life cycle that occurs at the microscopic scale. We show that this life cycle can generate the macroscopic communities known as biofilms (Figures 2, 3, 4, 5, 6, and 7) in E. coli K-12 (a model commensal strain), and in a “wild” strain isolated from a urinary tract infection (Figure 7). The similar morphogenesis uncovered in these two strains, which were isolated 100 years apart, could suggest the intriguing possibility of a general multicellular life cycle in E. coli.
Based on our findings, we propose a preliminary model for a multicellular life cycle in E. coli (Figure 7E). Single cells divide and remain adhered to create 4-cell rosettes. Communities grow longitudinally into chains hundreds-of-μm long while maintaining rosette configuration for ∼8 additional cell generations. Chains then attach to surfaces or to each other and stop elongating. Though attached chains do not elongate, they are not completely dormant, given the metabolic potentiation of antibiotics against biofilms.53,54,55 Each morphogenetic stage relies on specific adhesins (Figure 7E): Ag43 facilitates sister-cell adhesion, rosette formation, and clonality; type-1-fimbriae and curli organize chain stability and structure after rosette formation, and polyglucosamine production attaches chains to surfaces. The temporal organization of these adhesins has a potential logic (Figure 7E): as morphogenesis progress, adhesin interaction distance increases and specificity decreases. Cells initially only adhere to sister cells (Ag43), then they generate clonal communities (fimbriae and curli), and ultimately they attach to surfaces or other cellular communities (polyglucosamine). The insights we have uncovered into E. coli’s morphogenesis could serve as a blueprint to engineer biofilms, design “living matter,” and create “synthetic morphogenesis”.56,57,58,59,60,61,62,63,64,65
Rosettes are not generally observed in bacteria, with a few exceptions66,67,68 including E. coli invasion of the host epithelial cells in UTI pathogenesis. Invasion of host cells is critical to UTIs, which are the most common type of infection and are caused by E. coli in 80% of cases.69 The host cell cytoplasm is a hydrostatic environment and the reported rosette geometry67 is equivalent to the 4-bacterial cell rosettes we have described. Within host cells, E. coli creates biofilm-like intracellular bacterial communities (IBCs) which express Ag43 and type-1 fimbriae, and produce polyglucosamine (and possibly curli)69 despite lacking surface attachment and containing only hundreds of bacterial cells. IBCs are clonal70 and their requirement for type-1 fimbriae is independent of host-cell surface attachment32,71 indicating fimbriae organize the bacterial communities themselves. These parallels may indicate that E. coli IBC formation, chain morphogenesis, and biofilm formation result from the same underlying processes. Rosette formation is often considered a distinguishing feature between multicellular organisms and their unicellular relatives.72 Hence, rosette-initiated morphogenesis in E. coli may represent an example of convergent evolution and an alternative model system for studying the principles of multicellular development.
The significance of E. coli’s multicellular life cycle is not fully clear, and many questions remain unanswered. For example, does E. coli follow this life cycle in its natural habitats: the mucus layer coating the large intestine (where it lives as a commensal organism) or on plants and soil (where it lives as a saprophyte)? Does this life cycle have advantages in such environments, which can be exposed to antibiotic treatment, phage attack, interspecies competition,73 bacterial predators like amoeba and nematodes,74 and dramatic fluctuations in hydration, temperature, and ultraviolet light?75 There are also more immediate questions to resolve, such as: how do rosettes form, how is stage-specific gene expression regulated, and how does this life cycle relate to strains that form biofilms under fluid flow?23,36,76,77 Addressing these questions will enhance our understanding of E. coli which, in addition to being the best-studied unicellular organism, is critical to human health and disease.
Limitations of the study
This study does not address biofilm formation in hydrodynamic conditions by adherent E. coli strains; does not directly measure the adherent force among cells in clonal chains; does not quantify the “breaking-away” phenomena during morphogenesis which could indicate the “reproductive rate” of multicellular chains; and does not directly observe the behavior of unattached chains within a growing biofilm or host environment itself.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus strains | ||
| E. coli MG1655 (E. coli K-12 F-, λ-, ilvG, rfb-50, rph-1) | American Type Culture Collection (ATCC) | ATCC 700926 |
| E. coli BW25113 (E. coli K-12 F-, Δ(araD-araB)567, ΔlacZ4787(::rrnB-3), λ-, rph-1, Δ(rhaD-rhaB)568, hsdR514) | Yale stock collection78 | CGSC#: 7636 |
| E. coli MG1655 (Δflu::kan(kanR); Δflu (cured)) | Tranduced from KEIO Collection78 | N/A |
| E. coli MG1655 (ΔfimH::kan(kanR); ΔfimH (cured)) | Tranduced from KEIO Collection78 | N/A |
| E. coli MG1655 (ΔcsgA::kan(kanR); ΔcsgA (cured)) | Tranduced from KEIO Collection78 | N/A |
| E. coli MG1655 (ΔpgaB::kan(kanR); ΔpgaB (cured)) | Tranduced from KEIO Collection78 | N/A |
| E. coli Urinary Tract Infection isolate | This study (Clinical isolate received from Emory Investigational Clinical Microbiology Core) | N/A |
| P1 vir | Yale stock collection | CGSC#: 12133 |
| pUA66 pompC::gfp (pUA66 derivative containing gfpmut2 gene under control of ompC promoter) | (Zaslaver et al.)79 | N/A |
| pEB2-mScarlet-I (mScarlet) | Addgene80 | Plasmid # 104007 |
| pCP20 (thermosensitive helper plasmid encoding FLP recombinase) | (Datsenko and Wanner)81 | N/A |
| Software and algorithms | ||
| LAS X software | Leica Microsystems | N/A |
| MATLAB (R2021b) | MathWorks | N/A |
| Imaris | Bitplane | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kyle R. Allison (kyle.r.allison@emory.edu).
Materials availability
This study does not contain newly generated materials.
Experimental model and subject details
E. coli stains used in this study are listed in the key resources table.
Method details
Strains, plasmids, and media
Escherichia coli K-12 derivative MG1655 was the primary strain used in all experiments. E. coli BW25113 and an E. coli UTI clinical isolate were also used where specified. To facilitate imaging and analysis, strains were transformed with pUA66-pompC::gfp (green)79 or pEB2-mScarlet-I (red).80 In our hands, both plasmids demonstrated high fluorescence and low variability making them useful for fluorescent tracking experiments. Additionally, neither plasmid affected cell growth or morphogenesis, as equivalent phenotypes were observed with and without them. Δflu, ΔfimH, ΔcsgA, and ΔpgaB strains were created by transducing Δflu::kan(kanR), ΔfimH::kan(kanR), ΔcsgA::kan(kanR), and ΔpgaB::kan(kanR) respectively from the KEIO Collection78 using P1vir into E. coli MG1655 following standard methods,82 and were cured with pCP20.81 Colony morphology and flocculation were assessed and demonstrated that strains were not rare, phase-on variants for either ag43 or type-1-fimbriae.47 Prior to microfluidic experiments, strains were cultured in light-insulated shakers at 37°C and 300 RPM in 14 mL Falcon Round-Bottom Test Tubes with aerating caps (VWR). Cells were cultured in Luria Borth (LB, Miller; Difco™) for all experiments unless noted or Neidhardt Supplemented MOPS Defined Medium (EZ-rich; Teknova) (Figure S13). When plasmids were used, media were supplemented with 50 μg/mL of kanamycin (kanamycin monosulfate; Goldbio).
Microfluidic growth devices
Microfluidic devices were designed to enable imaging of multicellular morphogenesis in a hydrostatic liquid environment. Device layout is illustrated in Figure S1. Following standard approaches for bacterial microscopy,40 LB 2% agarose (Fisher Bioreagents) pads were created, but by adding the molten agarose within a silicone layer (Bio-Rad) (∼1 mm thick) with a 0.5–1 cm square cut out. Exponential phase cultures were diluted 1:100 in fresh LB and 3 μL was added to agarose pads. Agarose pads were not allowed to dry and were instead immediately covered with cover glass (18 × 18 mm; VWR) sealing a thin (∼0.5 mm) cell-containing liquid layer between the agarose and cover glass. On the opposite side from the cover glass, a μ-Slide I Flow Channel (ibidi®) was adhered to the silicone-agarose layer, completely sealing the agarose pad between a liquid growth chamber on one side and a fluidic channel on the other. μ-Slide I Flow Channels were then connected to a pressure and vacuum microfluidic flow controller system (OB1 MK3, Elveflow Instruments) using silicone tubing (ibidi®) pre-loaded with media and controlled through programmable software (Elveflow). Media (supplemented with antibiotics when appropriate) were perfused over the agarose pad through the flow channels at 5 μL/minute throughout experiments using Microfluidic Flow Sensor (MFS; Elveflow). Devices for monolayer (flat colony) growth were made similarly, with the modification that agarose pads were allowed to fully dry after cells were added and hence no liquid space was created between agarose and cover glass (Figure 1F). Imaging and temperature control were performed as described below.
Microscope imaging
Images tracking morphogenesis and of biofilms were acquired using a Leica DMi8 microscope equipped with a DIC HCPL APO 63X oil immersion objective (1.6x mag changer) Hamamatsu ORCA-Flash 4.0 camera, and Lumencor Spectra-X light engine. Microfluidic devices were secured in a microscope-stage-top incubator (Tokai-Hit, STX Stage Top Incubator Temp and Flow; STXF-WSKMX-SET), and the incubator and the microscope objective were maintained at 37°C prior to and throughout all experiments. Leica LAS X software was programmed to automatedly acquire tiled images (25–80 fields of view per image) at regular and specific time intervals at a 100X magnification to track morphogenesis. Images were acquired in the differential interference contrast (DIC) configuration or in the red (excitation 510 nm; emission 592–668 nm) or green (excitation 470 nm; emission 500–550 nm) fluorescent channels. Images were uniformly contrast adjusted, unless otherwise noted, in LAS X software. Biofilms were imaged using the same settings used for morphogenesis tracking, at 63X and 100X magnification. Biofilms were additionally imaged by confocal microscopy at the Emory Integrated Cellular Imaging Core. Confocal microscopy of the biofilm depicted in Video S9 was captured using Olympus FV1000 using 488 nm and 599 nm excitation lasers for green and red fluorescence, respectively. Z-stack images were collected at a step height of 1 μm. The confocal image depicted in Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7F was acquired using the Nikon Crestoptics X-Light V2 FOV Spinning Disc Microscope, paired with Nikon Ti2-E inverted microscope (APO 100x oil objective) using 477 nm and 546 nm excitation lasers for green and red fluorescence, respectively. Z-stack images were collected at a step height of 0.2 μm and captured a total height of 40 μm. Confocal images were rendered using NIS-Element Viewer and Imaris Viewer.
Image analysis
Raw images were segmented and analyzed using custom MATLAB scripts to calculate dynamic geometric properties of multicellular communities (Figures 1G, 1H, S14–S16, S19, S20, S22, S33–S36, S39–S42, and S46). Calculations of geometric properties were made using multicellular chains that had not yet branched or merged with neighboring communities. Rosette width was calculated using the LAS X software from 30 width measurements, 3 widths each from 10 images (Figures 1E and S21). Value for mean ± standard deviation is reported. Rosette diagonals were calculated using LAS X software from 3 images (Figure 1E). Value for mean ± standard deviation is reported. Width of elongating chains was calculated by area/length at each time point (Figures 1H, S21, and S22). For z-stack analysis of assembled chains in microfluidic devices, micrographs were captured at different focal planes, with a step height of 1.644 μm, capturing a total height of 14.8 μm (Figure S8). All cross-sectional images were overlaid using Fiji ImageJ software. 3D rendering of assembled chains represented in Figure S8 was performed using the LAS X software (Figures S9 and S10). Percent growth rate was calculated from the derivative for area, calculated over a moving 30-minute window and normalized by the initial area at each time point, during morphogenesis (Figure 1I). Area of motion (AOM), a heuristic we developed for quantifying community motion, was calculated using custom MATLAB scripts as the moving average (over 5 times points) of the normalized span in multicellular community area between consecutive time points (Figures 1I and 6E). AOM is 1 for no motion and is otherwise >1. For microfluidic chain clonality experiments, overlapping areas from red and green fluorescence images were calculated (Figure S24). Percent overlap (overlap area/total area:red + green channel) as a function of time, and overlap area as a function of total area were plotted (Figure S24). Biofilm width was calculated using the LAS X software from 9 width measurements taken for each biofilm condition (Figures 3, 4, 5, 6, 7, S27, S47, and S49). Values for mean ± standard deviation are reported. Biofilm chain widths were calculated using LAS X software by measuring the width of individual chains viewed in green fluorescence channel from 20 different chains (Figure 2F). For biofilm intensity and dispersed-biofilm single-cell intensity, raw images were analyzed using custom MATLAB scripts to segment and extract fluorescence intensity (Figure S28). Biofilm height was measured from confocal microscopy images using NIS software (Figure 3G).
Growth rate for planktonic cells
Overnight cultures of E. coli MG1655 were diluted (1:1000) in LB and cultured in light-insulated shakers at 37°C and 300 RPM for 8 hours. Every hour, optical density at 600 nm (OD600) was measured using a spectrophotometer (Genesys 20 Thermo Spectronic) (Figures S17 and S18).
Sample preparation for biofilm imaging
Standard microtitre plate biofilm assays42 were adapted for biofilm imaging (illustrated in Figure 2C). Wells in 96-well plates were loaded with 200 μL of fresh LB with overnight cultured cells (∼109 CFU/mL) unless otherwise noted. For red-green imaging, cells were mixed at a ratio of 1:10 green (GFP) and red (mScarlet) fluorescence. Cover glass pieces were placed in each well, perpendicular to the air-liquid interface in order to allow for biofilm formation. Plates were then incubated without shaking at 37°C for 16 hours, or otherwise specified times, to allow biofilm formation to occur. After incubation, cover glass pieces were removed from the wells and placed between a glass slide and a fresh piece of cover glass in order to image by microscopy (as described above) (Figures 2, 3, 4, 5, 6, 7, S25–S27, S29, S37, S38, and S47–S51). This ensures the optical light path is perpendicular to the plane of the biofilm surface and allows for high magnification imaging of a formed biofilm directly. For mannoside experiments (Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7G, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7B, S48, and S51), biofilms were grown using the procedure described above with the specified amount of Methly α-D-mannopyranoside (Sigma-Aldrich) added in LB. For fluorescence measurements of single biofilm cells (Figure S28), grown biofilms were washed with PBS then dispersed by sonication for 15 minutes in PBS and loaded onto PBS agarose pads for imaging. To image unattached biofilm cells (Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7H–3J, S30, and S31), cover glass with growing biofilms was washed by pipetting PBS over the surface and allowing the run-off to land on fresh PBS agarose pads. Cover glass was then placed over these agarose pads and samples were imaged (Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7H–3J, S30, and S31). For imaging washed UTI isolate biofilms (Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7D), intermediate biofilms grown on cover glass for 12 hours were washed by pipetting PBS over the surface. Cover glass with biofilm was imaged before and after washing.
Quantification and statistical analysis
Details of statistical analysis are mentioned in the corresponding figure legends and in the STAR Methods. Data represent mean ± standard deviation.
Acknowledgments
We thank Sarah Satola and the Emory Investigational Clinical Microbiology Core for supplying the UTI clinical isolate strain; April Reedy and the Emory Integrated Cellular Imaging Core for assistance with the confocal microscopy; and the manuscript’s peer reviewers for their insightful comments. This work was supported by funding from the NIH Director’s Early Independence Award to Kyle R. Allison (NIH DP5OD019792).
Author contributions
D.P. performed all experiments and analyses with assistance from X.F. and K.R.A. D.P. and K.R.A. designed experiments, discussed results, and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: January 20, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105795.
Supplemental information
Data and code availability
-
•
All reported data in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Donlan R.M., Costerton J.W. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002;15:167–193. doi: 10.1128/CMR.15.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall-Stoodley L., Costerton J.W., Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat. Rev. Microbiol. 2004;2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 3.Costerton J.W., Stewart P.S., Greenberg E.P. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 4.Mah T.F., O'Toole G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001;9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 5.Flemming H.C., Wingender J., Szewzyk U., Steinberg P., Rice S.A., Kjelleberg S. Biofilms: an emergent form of bacterial life. Nat. Rev. Microbiol. 2016;14:563–575. doi: 10.1038/nrmicro.2016.94. [DOI] [PubMed] [Google Scholar]
- 6.O'Toole G., Kaplan H.B., Kolter R. Biofilm formation as microbial development. Annu. Rev. Microbiol. 2000;54:49–79. doi: 10.1146/annurev.micro.54.1.49. [DOI] [PubMed] [Google Scholar]
- 7.Yan J., Sharo A.G., Stone H.A., Wingreen N.S., Bassler B.L. Vibrio cholerae biofilm growth program and architecture revealed by single-cell live imaging. Proc. Natl. Acad. Sci. USA. 2016;113:E5337–E5343. doi: 10.1073/pnas.1611494113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drescher K., Dunkel J., Nadell C.D., van Teeffelen S., Grnja I., Wingreen N.S., Stone H.A., Bassler B.L. Architectural transitions in Vibrio cholerae biofilms at single-cell resolution. Proc. Natl. Acad. Sci. USA. 2016;113:E2066–E2072. doi: 10.1073/pnas.1601702113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin B., Fei C., Bridges A.A., Mashruwala A.A., Stone H.A., Wingreen N.S., Bassler B.L. Cell position fates and collective fountain flow in bacterial biofilms revealed by light-sheet microscopy. Science. 2020;369:71–77. doi: 10.1126/science.abb8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berk V., Fong J.C.N., Dempsey G.T., Develioglu O.N., Zhuang X., Liphardt J., Yildiz F.H., Chu S. Molecular architecture and assembly principles of Vibrio cholerae biofilms. Science. 2012;337:236–239. doi: 10.1126/science.1222981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pamp S.J., Sternberg C., Tolker-Nielsen T. Insight into the microbial multicellular lifestyle via flow-cell technology and confocal microscopy. Cytometry A. 2009;75:90–103. doi: 10.1002/cyto.a.20685. [DOI] [PubMed] [Google Scholar]
- 12.Hartmann R., Singh P.K., Pearce P., Mok R., Song B., Díaz-Pascual F., Dunkel J., Drescher K. Emergence of three-dimensional order and structure in growing biofilms. Nat. Phys. 2019;15:251–256. doi: 10.1038/s41567-018-0356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Q., Li J., Nijjer J., Lu H., Kothari M., Alert R., Cohen T., Yan J. Morphogenesis and cell ordering in confined bacterial biofilms. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2107107118. e2107107118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merritt J.H., Kadouri D.E., O'Toole G.A. Growing and analyzing static biofilms. Curr. Protoc. Microbiol. 2011 doi: 10.1002/9780471729259.mc01b01s22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pratt L.A., Kolter R. Genetic analysis of Escherichia coli biofilm formation: roles of flagella, motility, chemotaxis and type I pili. Mol. Microbiol. 1998;30:285–293. doi: 10.1046/j.1365-2958.1998.01061.x. [DOI] [PubMed] [Google Scholar]
- 16.Barnhart M.M., Chapman M.R. Curli biogenesis and function. Annu. Rev. Microbiol. 2006;60:131–147. doi: 10.1146/annurev.micro.60.080805.142106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cegelski L., Pinkner J.S., Hammer N.D., Cusumano C.K., Hung C.S., Chorell E., Åberg V., Walker J.N., Seed P.C., Almqvist F., et al. Small-molecule inhibitors target Escherichia coli amyloid biogenesis and biofilm formation. Nat. Chem. Biol. 2009;5:913–919. doi: 10.1038/nchembio.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X., Preston J.F., Romeo T. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J. Bacteriol. 2004;186:2724–2734. doi: 10.1128/JB.186.9.2724-2734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itoh Y., Wang X., Hinnebusch B.J., Preston J.F., Romeo T. Depolymerization of β-1, 6-N-acetyl-D-glucosamine disrupts the integrity of diverse bacterial biofilms. J. Bacteriol. 2005;187:382–387. doi: 10.1128/JB.187.1.382-387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Itoh Y., Rice J.D., Goller C., Pannuri A., Taylor J., Meisner J., Beveridge T.J., Preston J.F., Romeo T. Roles of pgaABCD genes in synthesis, modification, and export of the Escherichia coli biofilm adhesin poly-β-1, 6-N-acetyl-D-glucosamine. J. Bacteriol. 2008;190:3670–3680. doi: 10.1128/JB.01920-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niba E.T.E., Naka Y., Nagase M., Mori H., Kitakawa M. A genome-wide approach to identify the genes involved in biofilm formation in E. coli. DNA Res. 2007;14:237–246. doi: 10.1093/dnares/dsm024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diderichsen B. flu, a metastable gene controlling surface properties of Escherichia coli. J. Bacteriol. 1980;141:858–867. doi: 10.1128/jb.141.2.858-867.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kjærgaard K., Schembri M.A., Ramos C., Molin S., Klemm P. Antigen 43 facilitates formation of multispecies biofilms. Environ. Microbiol. 2000;2:695–702. doi: 10.1046/j.1462-2920.2000.00152.x. [DOI] [PubMed] [Google Scholar]
- 24.Danese P.N., Pratt L.A., Dove S.L., Kolter R. The outer membrane protein, antigen 43, mediates cell-to-cell interactions within Escherichia coli biofilms. Mol. Microbiol. 2000;37:424–432. doi: 10.1046/j.1365-2958.2000.02008.x. [DOI] [PubMed] [Google Scholar]
- 25.Heras B., Totsika M., Peters K.M., Paxman J.J., Gee C.L., Jarrott R.J., Perugini M.A., Whitten A.E., Schembri M.A. The antigen 43 structure reveals a molecular Velcrolike mechanism of autotransporter-mediated bacterial clumping. Proc. Natl. Acad. Sci. USA. 2014;111:457–462. doi: 10.1073/pnas.1311592111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasman H., Chakraborty T., Klemm P. Antigen-43-mediated autoaggregation of Escherichia coli is blocked by fimbriation. J. Bacteriol. 1999;181:4834–4841. doi: 10.1128/jb.181.16.4834-4841.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schembri M.A., Christiansen G., Klemm P. FimH-mediated autoaggregation of Escherichia coli. Mol. Microbiol. 2001;41:1419–1430. doi: 10.1046/j.1365-2958.2001.02613.x. [DOI] [PubMed] [Google Scholar]
- 28.Schembri M.A., Sokurenko E.V., Klemm P. Functional flexibility of the FimH adhesin: insights from a random mutant library. Infect. Immun. 2000;68:2638–2646. doi: 10.1128/IAI.68.5.2638-2646.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prigent-Combaret C., Prensier G., Le Thi T.T., Vidal O., Lejeune P., Dorel C. Developmental pathway for biofilm formation in curli-producing Escherichia coli strains: role of flagella, curli and colanic acid. Environ. Microbiol. 2000;2:450–464. doi: 10.1046/j.1462-2920.2000.00128.x. [DOI] [PubMed] [Google Scholar]
- 30.Sauer F.G., Mulvey M.A., Schilling J.D., Martinez J.J., Hultgren S.J. Bacterial pili: molecular mechanisms of pathogenesis. Curr. Opin. Microbiol. 2000;3:65–72. doi: 10.1016/s1369-5274(99)00053-3. [DOI] [PubMed] [Google Scholar]
- 31.Hahn E., Wild P., Hermanns U., Sebbel P., Glockshuber R., Häner M., Taschner N., Burkhard P., Aebi U., Müller S.A. Exploring the 3D molecular architecture of Escherichia coli type 1 pili. J. Mol. Biol. 2002;323:845–857. doi: 10.1016/S0022-2836(02)01005-7. [DOI] [PubMed] [Google Scholar]
- 32.Wright K.J., Seed P.C., Hultgren S.J. Development of intracellular bacterial communities of uropathogenic Escherichia coli depends on type 1 pili. Cell Microbiol. 2007;9:2230–2241. doi: 10.1111/j.1462-5822.2007.00952.x. [DOI] [PubMed] [Google Scholar]
- 33.Mika F., Hengge R. Small RNAs in the control of RpoS, CsgD, and biofilm architecture of Escherichia coli. RNA Biol. 2014;11:494–507. doi: 10.4161/rna.28867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klemm P., Schembri M. Type 1 fimbriae, curli, and antigen 43: adhesion, colonization, and biofilm formation. EcoSal Plus. 2004;1 doi: 10.1128/ecosalplus.8.3.2.6. [DOI] [PubMed] [Google Scholar]
- 35.Vejborg R.M., Klemm P. Cellular chain formation in Escherichia coli biofilms. Microbiology. 2009;155:1407–1417. doi: 10.1099/mic.0.026419-0. [DOI] [PubMed] [Google Scholar]
- 36.Schembri M.A., Klemm P. Biofilm formation in a hydrodynamic environment by novel FimH variants and ramifications for virulence. Infect. Immun. 2001;69:1322–1328. doi: 10.1128/IAI.69.3.1322-1328.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pratt L.A., Kolter R. Genetic analyses of bacterial biofilm formation. Curr. Opin. Microbiol. 1999;2:598–603. doi: 10.1016/S1369-5274(99)00028-4. [DOI] [PubMed] [Google Scholar]
- 38.Danese P.N., Pratt L.A., Kolter R. Exopolysaccharide production is required for development of Escherichia coli K-12 biofilm architecture. J. Bacteriol. 2000;182:3593–3596. doi: 10.1128/JB.182.12.3593-3596.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Genevaux P., Muller S., Bauda P. A rapid screening procedure to identify mini-Tn10 insertion mutants of Escherichia coli K-12 with altered adhesion properties. FEMS Microbiol. Lett. 1996;142:27–30. doi: 10.1016/0378-1097. [DOI] [PubMed] [Google Scholar]
- 40.Young J.W., Locke J.C.W., Altinok A., Rosenfeld N., Bacarian T., Swain P.S., Mjolsness E., Elowitz M.B. Measuring single-cell gene expression dynamics in bacteria using fluorescence time-lapse microscopy. Nat. Protoc. 2011;7:80–88. doi: 10.1038/nprot.2011.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sezonov G., Joseleau-Petit D., D'Ari R. Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 2007;189:8746–8749. doi: 10.1128/JB.01368-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Toole G.A. Microtiter dish Biofilm formation assay. JoVE. 2010:2437. doi: 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monds R.D., O'Toole G.A. The developmental model of microbial biofilms: ten years of a paradigm up for review. Trends Microbiol. 2009;17:73–87. doi: 10.1016/j.tim.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 44.Nuñez I.N., Matute T.F., Del Valle I.D., Kan A., Choksi A., Endy D., Haseloff J., Rudge T.J., Federici F. Artificial symmetry-breaking for morphogenetic engineering bacterial colonies. ACS Synth. Biol. 2017;6:256–265. doi: 10.1021/acssynbio.6b00149. [DOI] [PubMed] [Google Scholar]
- 45.Rooney L.M., Amos W.B., Hoskisson P.A., McConnell G. Intra-colony channels in E. coli function as a nutrient uptake system. ISME J. 2020;14:2461–2473. doi: 10.1038/s41396-020-0700-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rudge T.J., Federici F., Steiner P.J., Kan A., Haseloff J. Cell polarity-driven instability generates self-organized, fractal patterning of cell layers. ACS Synth. Biol. 2013;2:705–714. doi: 10.1021/sb400030p. [DOI] [PubMed] [Google Scholar]
- 47.Hasman H., Schembri M.A., Klemm P. Antigen 43 and type 1 fimbriae determine colony morphology of Escherichia coli K-12. J. Bacteriol. 2000;182:1089–1095. doi: 10.1128/JB.182.4.1089-1095.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chauhan A., Sakamoto C., Ghigo J.M., Beloin C. Did I pick the right colony? Pitfalls in the study of regulation of the phase variable antigen 43 adhesin. PLoS One. 2013;8:e73568. doi: 10.1371/journal.pone.0073568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Der Woude M.W. Re-examining the role and random nature of phase variation. FEMS Microbiol. Lett. 2006;254:190–197. doi: 10.1111/j.1574-6968.2005.00038.x. [DOI] [PubMed] [Google Scholar]
- 50.Lim J.K., Gunther N.W., 4th, Zhao H., Johnson D.E., Keay S.K., Mobley H.L. In vivo phase variation of Escherichia coli type 1 fimbrial genes in women with urinary tract infection. Infect. Immun. 1998;66:3303–3310. doi: 10.1128/iai.66.7.3303-3310.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nagahori N., Lee R.T., Nishimura S.I., Pagé D., Roy R., Lee Y.C. Inhibition of adhesion of type 1 fimbriated Escherichia coli to highly mannosylated ligands. Chembiochem. 2002;3:836–844. doi: 10.1002/1439-7633(20020902)3:9<836::Aid-cbic836>3.0.Co;2-2. [DOI] [PubMed] [Google Scholar]
- 52.Amini S., Goodarzi H., Tavazoie S. Genetic dissection of an exogenously induced biofilm in laboratory and clinical isolates of E. coli. PLoS Pathog. 2009;5:e1000432. doi: 10.1371/journal.ppat.1000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allison K.R., Brynildsen M.P., Collins J.J. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature. 2011;473:216–220. doi: 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosenberg C.R., Fang X., Allison K.R. Potentiating aminoglycoside antibiotics to reduce their toxic side effects. PLoS One. 2020;15:e0237948. doi: 10.1371/journal.pone.0237948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lebeaux D., Chauhan A., Létoffé S., Fischer F., de Reuse H., Beloin C., Ghigo J.M. pH-mediated potentiation of aminoglycosides kills bacterial persisters and eradicates in vivo biofilms. J. Infect. Dis. 2014;210:1357–1366. doi: 10.1093/infdis/jiu286. [DOI] [PubMed] [Google Scholar]
- 56.Teague B.P., Guye P., Weiss R. Synthetic morphogenesis. Cold Spring Harb. Perspect. Biol. 2016;8:a023929. doi: 10.1101/cshperspect.a023929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brenner K., Karig D.K., Weiss R., Arnold F.H. Engineered bidirectional communication mediates a consensus in a microbial biofilm consortium. Proc. Natl. Acad. Sci. USA. 2007;104:17300–17304. doi: 10.1073/pnas.0704256104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo S., Dubuc E., Rave Y., Verhagen M., Twisk S.A.E., van der Hek T., Oerlemans G.J.M., van den Oetelaar M.C.M., van Hazendonk L.S., Brüls M., et al. Engineered living materials based on adhesin-mediated trapping of programmable cells. ACS Synth. Biol. 2020;9:475–485. doi: 10.1021/acssynbio.9b00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karig D., Martini K.M., Lu T., DeLateur N.A., Goldenfeld N., Weiss R. Stochastic Turing patterns in a synthetic bacterial population. Proc. Natl. Acad. Sci. USA. 2018;115:6572–6577. doi: 10.1073/pnas.1720770115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hong S.H., Hegde M., Kim J., Wang X., Jayaraman A., Wood T.K. Synthetic quorum-sensing circuit to control consortial biofilm formation and dispersal in a microfluidic device. Nat. Commun. 2012;3:613. doi: 10.1038/ncomms1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nguyen P.Q., Botyanszki Z., Tay P.K.R., Joshi N.S. Programmable biofilm-based materials from engineered curli nanofibres. Nat. Commun. 2014;5:4945. doi: 10.1038/ncomms5945. [DOI] [PubMed] [Google Scholar]
- 62.Gilbert C., Tang T.C., Ott W., Dorr B.A., Shaw W.M., Sun G.L., Lu T.K., Ellis T. Living materials with programmable functionalities grown from engineered microbial co-cultures. Nat. Mater. 2021;20:691–700. doi: 10.1038/s41563-020-00857-5. [DOI] [PubMed] [Google Scholar]
- 63.Bernstein H.C., Paulson S.D., Carlson R.P. Synthetic Escherichia coli consortia engineered for syntrophy demonstrate enhanced biomass productivity. J. Biotechnol. 2012;157:159–166. doi: 10.1016/j.jbiotec.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benedetti I., de Lorenzo V., Nikel P.I. Genetic programming of catalytic Pseudomonas putida biofilms for boosting biodegradation of haloalkanes. Metab. Eng. 2016;33:109–118. doi: 10.1016/j.ymben.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 65.Glass D.S., Riedel-Kruse I.H. A synthetic bacterial cell-cell adhesion toolbox for programming multicellular morphologies and patterns. Cell. 2018;174:649–658.e16. doi: 10.1016/j.cell.2018.06.041. [DOI] [PubMed] [Google Scholar]
- 66.Moore R.L., Marshall K.C. Attachment and rosette formation by hyphomicrobia. Appl. Environ. Microbiol. 1981;42:751–757. doi: 10.1128/aem.42.5.751-757.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Justice S.S., Hung C., Theriot J.A., Fletcher D.A., Anderson G.G., Footer M.J., Hultgren S.J. Differentiation and developmental pathways of uropathogenic Escherichia coli in urinary tract pathogenesis. Proc. Natl. Acad. Sci. USA. 2004;101:1333–1338. doi: 10.1073/pnas.0308125100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keim C.N., Martins J.L., Abreu F., Rosado A.S., de Barros H.L., Borojevic R., Lins U., Farina M. Multicellular life cycle of magnetotactic prokaryotes. FEMS Microbiol. Lett. 2004;240:203–208. doi: 10.1016/j.femsle.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 69.Anderson G.G., Palermo J.J., Schilling J.D., Roth R., Heuser J., Hultgren S.J. Intracellular bacterial biofilm-like pods in urinary tract infections. Science. 2003;301:105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 70.Schwartz D.J., Chen S.L., Hultgren S.J., Seed P.C. Population dynamics and niche distribution of uropathogenic Escherichia coli during acute and chronic urinary tract infection. Infect. Immun. 2011;79:4250–4259. doi: 10.1128/IAI.05339-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ulett G.C., Valle J., Beloin C., Sherlock O., Ghigo J.M., Schembri M.A. Functional analysis of antigen 43 in uropathogenic Escherichia coli reveals a role in long-term persistence in the urinary tract. Infect. Immun. 2007;75:3233–3244. doi: 10.1128/IAI.01952-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brunet T., King N. The origin of animal multicellularity and cell differentiation. Dev. Cell. 2017;43:124–140. doi: 10.1016/j.devcel.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tenaillon O., Skurnik D., Picard B., Denamur E. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 2010;8:207–217. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 74.Matz C., Kjelleberg S. Off the hook--how bacteria survive protozoan grazing. Trends Microbiol. 2005;13:302–307. doi: 10.1016/j.tim.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 75.Méric G., Kemsley E.K., Falush D., Saggers E.J., Lucchini S. Phylogenetic distribution of traits associated with plant colonization in Escherichia coli. Environ. Microbiol. 2013;15:487–501. doi: 10.1111/j.1462-2920.2012.02852.x. [DOI] [PubMed] [Google Scholar]
- 76.Ghigo J.M. Natural conjugative plasmids induce bacterial biofilm development. Nature. 2001;412:442–445. doi: 10.1038/35086581. [DOI] [PubMed] [Google Scholar]
- 77.Reisner A., Haagensen J.A.J., Schembri M.A., Zechner E.L., Molin S. Development and maturation of Escherichia coli K-12 biofilms. Mol. Microbiol. 2003;48:933–946. doi: 10.1046/j.1365-2958.2003.03490.x. [DOI] [PubMed] [Google Scholar]
- 78.Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zaslaver A., Bren A., Ronen M., Itzkovitz S., Kikoin I., Shavit S., Liebermeister W., Surette M.G., Alon U. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods. 2006;3:623–628. doi: 10.1038/nmeth895. [DOI] [PubMed] [Google Scholar]
- 80.Balleza E., Kim J.M., Cluzel P. Systematic characterization of maturation time of fluorescent proteins in living cells. Nat. Methods. 2018;15:47–51. doi: 10.1038/nmeth.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thomason L.C., Costantino N., Court D.L. E. coli genome manipulation by P1 transduction. Curr. Protoc. Mol. Biol. 2007:1.17.11–11.17.18. doi: 10.1002/0471142727.mb0117s79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All reported data in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.