

Abstract
Objective
Dravet syndrome (DS) and Lennox–Gastaut syndrome (LGS) are rare treatment‐resistant childhood epilepsies classed as developmental and epileptic encephalopathies. ELEKTRA investigated the efficacy and safety of soticlestat (TAK‐935) as adjunctive therapy in children with DS or LGS (NCT03650452).
Methods
ELEKTRA was a phase 2, randomized, double‐blind, placebo‐controlled study of soticlestat (≤300 mg twice daily, weight‐adjusted) in children (aged 2–17 years) with DS, demonstrating three or more convulsive seizures/month, or with LGS, demonstrating four or more drop seizures/month at baseline. The 20‐week treatment period comprised an 8‐week dose‐optimization period and a 12‐week maintenance period. Efficacy endpoints included change from baseline in seizure frequency versus placebo. Safety assessments included incidence of treatment‐emergent adverse events (TEAEs).
Results
ELEKTRA enrolled 141 participants; 126 (89%) completed the study. The modified intent‐to‐treat population included 139 participants who received one or more doses of study drug and had one or more efficacy assessments (DS, n = 51; LGS, n = 88). ELEKTRA achieved its primary endpoint: the combined soticlestat‐treated population demonstrated a placebo‐adjusted median reduction in seizure frequency of 30.21% during the maintenance period (p = .0008, n = 139). During this period, placebo‐adjusted median reductions in convulsive and drop seizure frequencies of 50.00% (p = .0002; patients with DS) and 17.08% (p = .1160; patients with LGS), respectively, were observed. TEAE incidences were similar between the soticlestat (80.3%) and placebo (74.3%) groups and were mostly mild or moderate in severity. Serious TEAEs were reported by 15.5% and 18.6% of participants receiving soticlestat and placebo, respectively. TEAEs reported in soticlestat‐treated patients with ≥5% difference from placebo were lethargy and constipation. No deaths were reported.
Significance
Soticlestat treatment resulted in statistically significant, clinically meaningful reductions from baseline in median seizure frequency (combined patient population) and in convulsive seizure frequency (DS cohort). Drop seizure frequency showed a nonstatistically significant numerical reduction in children with LGS. Soticlestat had a safety profile consistent with previous studies.
Keywords: cholesterol 24‐hydroxylase, developmental and epileptic encephalopathies, Dravet syndrome, Lennox–Gastaut syndrome, soticlestat, TAK‐935
Key Points.
DS and LGS are rare childhood epilepsies that are often resistant to current treatment
Soticlestat, a first‐in‐class inhibitor of cholesterol 24‐hydroxylase, is being developed as a potential treatment for DS and LGS
Soticlestat treatment in ELEKTRA significantly reduced the placebo‐adjusted median seizure frequency in the combined patient population
Soticlestat treatment reduced plasma 24HC levels compared with placebo, and no new safety signals were identified
Results from ELEKTRA support investigation of soticlestat in phase 3 studies and of plasma 24HC as a biomarker for soticlestat activity
1. INTRODUCTION
Dravet syndrome (DS) and Lennox–Gastaut syndrome (LGS) are developmental and epileptic encephalopathies (DEEs), a group of conditions characterized by developmental impairment and frequent epileptic activity that is associated with further slowing of development. 1 Both the early onset treatment‐resistant seizures and the nonseizure symptoms associated with DS and LGS are not fully controlled by the current standard of care and contribute significantly to poor quality of life for patients and carers. 2 , 3 , 4 , 5 , 6 , 7 , 8
DS accounts for approximately 3%–8% of all children who experience seizures in the first year of life 9 and involves multiple seizure types, including tonic–clonic seizures, bilateral clonic seizures, focal clonic and other focal seizures, myoclonic seizures, and atypical absence seizures. 3 Children usually experience a convulsive seizure first, which can be associated with fever or vaccination. 3 The stabilization stage of DS typically occurs from 5 years of age, with convulsive seizures becoming less frequent and occurring mainly during sleep, and other seizure types persisting, becoming less frequent, or disappearing. 3 Adults experience less frequent but persistent seizures that usually include focal and/or convulsive seizures. 3 , 10 DS in adulthood is also associated with persistent motor and cognitive dysfunction. 2
LGS accounts for approximately 4% of all childhood epilepsies 11 and includes multiple seizure types, of which tonic seizures are the most characteristic 4 , 12 , 13 ; however, typically, drop seizures are the first sign in young children. 14 , 15 The onset of LGS is most commonly between 3 and 5 years of age and usually occurs before 8 years of age, although it may occur later in some cases. 14 , 15 The number and variety of seizures usually decrease over time, but tonic seizures tend to persist, particularly during sleep. 16 LGS does persist into adolescence and adulthood, with 80%–90% of individuals continuing to have seizures after childhood. 14 , 15 , 17
As a result of the treatment resistance associated with DS and LGS, polytherapy and multiple treatment changes over time are common in both conditions. 6 , 15 , 18 However, polytherapy can be associated with tolerability issues caused by additive adverse effects, such as worsening behavioral problems and sedation. 15 , 19 Together with the poor control of symptoms, these drawbacks highlight an unmet need that exists for effective treatments that have a novel mechanism of action, compared with existing antiseizure therapies, and a favorable side‐effect profile. 6 , 20
Soticlestat (TAK‐935) is a first‐in‐class, selective inhibitor of cholesterol 24‐hydroxylase (CH24H; also known as CYP46A1) 21 that is currently under development as a potential treatment for DS and LGS. CH24H plays a role in cholesterol homeostasis in the brain by catabolizing it to 24S‐hydroxycholesterol (24HC). 21 Preclinical findings showed that soticlestat treatment lowered brain 24HC levels and also suppressed elevation of extracellular glutamate levels, resulting in reduced hyperexcitability. 21 24HC is also known for various neuromodulatory activities, including positive allosteric modulation of N‐methyl‐D‐aspartate receptors and inflammatory signaling. As such, inhibition of CH24H could potentially decrease glutamatergic signaling via multimodal mechanisms. 21 In phase 1 clinical studies, soticlestat dose‐dependently decreased plasma 24HC levels in healthy volunteers. 22 , 23 Additionally, in a phase 1b/2a clinical trial in adults with DEEs, soticlestat as adjunctive therapy resulted in a safety profile that was consistent with that observed in studies of healthy volunteers; it also decreased plasma 24HC levels, compared with placebo, and was associated with a reduction in median seizure frequency over the study duration. 24
Here, we report results from ELEKTRA, a phase 2, multicenter clinical study investigating the efficacy, safety, and tolerability of soticlestat as adjunctive therapy in children with DS or LGS (ClinicalTrials.gov identifier: NCT03650452).
2. MATERIALS AND METHODS
2.1. Ethics
The study was conducted in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice (E6), as well as with current ethics guidelines, including the Declaration of Helsinki and the International Ethical Guidelines of the Council for International Organizations of Medical Sciences. All other applicable laws and local regulations were followed. Written informed consent was obtained from each patient, or his/her legally authorized representative, prior to the patient entering the study. In addition, documented assent was obtained from the patient if applicable. This study was conducted in compliance with the institutional review boards or independent ethics committees of each study location.
2.2. Patients
Patients aged 2–17 years with a clinical diagnosis of DS or LGS and weighing ≥10 kg at the screening visit were eligible for the study. Patients were also accepted into the trial if they had a history of inadequate response to at least two antiseizure medications (ASMs) and if, at baseline, they were receiving one to four ASMs at a stable dose and were experiencing at least three convulsive seizures (DS) or four drop seizures (LGS) per month. Concomitant use of perampanel throughout the study (from screening through follow‐up) was prohibited because of a potential pharmacodynamic drug–drug interaction with soticlestat. Patients who had been admitted to a medical facility and intubated for treatment of status epilepticus two or more times in the 3 months preceding the screening visit were not eligible for the study.
2.3. Study design
ELEKTRA was a phase 2, multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study conducted across 35 sites in Australia, Canada, China, Israel, Poland, Portugal, Spain, and the USA. Following a screening and baseline period (4–6 weeks), patients were randomized 1:1 to soticlestat or placebo by a computer‐generated random sequence, using an interactive web response system. The full 20‐week treatment period comprised an 8‐week dose‐optimization period followed by a 12‐week maintenance period (Figure 1A). Soticlestat or placebo treatments were administered orally or via gastrostomy tube/percutaneous endoscopic gastrostomy tube twice daily (BID) with or without food during the treatment period. For patients weighing ≥60 kg, soticlestat was titrated up to 300 mg BID, with weight‐based dosing used for those weighing <60 kg. During the dose‐optimization period, patients received gradually increasing doses of soticlestat up to the appropriate maximum dose, while being monitored for drug safety and tolerability. The dose could be changed within the dose‐optimization period, following which the dose was kept constant throughout the maintenance period (unless a dose decrease was required for reasons of safety or tolerability). Following study completion, patients were given the option to enroll in an open‐label extension study (ENDYMION; ClinicalTrials.gov identifier: NCT03635073) or to enter a double‐blind taper period (maximum 14 days).
FIGURE 1.
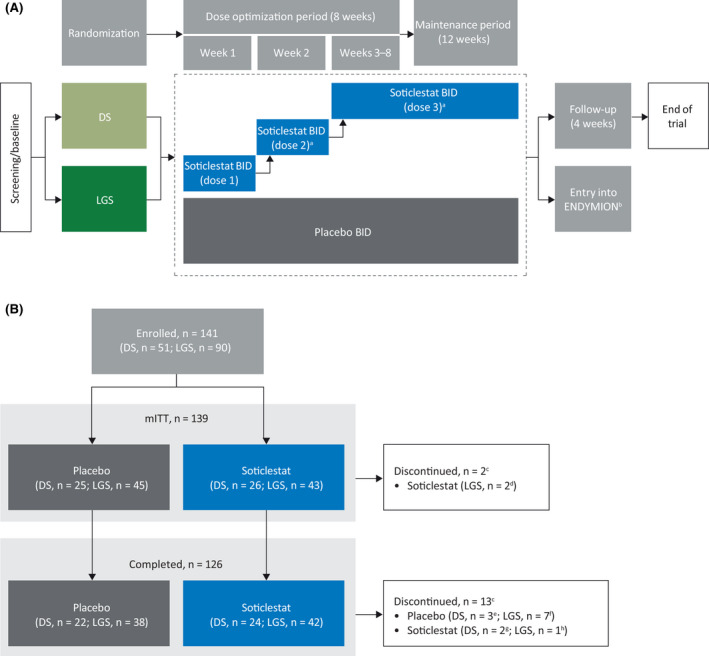
Study flow charts showing the study design (A) and patient disposition (B). aDose changes were permitted in the first 6 weeks of dose optimization according to the judgment of the investigator and with approval of the medical monitor, after which the final dose was maintained throughout the maintenance period, unless it needed to be lowered for safety or tolerability reasons. bOpen‐label extension study. cReceived at least one dose of study drug and had at least one efficacy assessment. dNo seizure diary records during treatment period. eAdverse event (AE; n = 1), withdrawal by parent or guardian (n = 1), or other reason (n = 1; completed minimum requirement and entered ENDYMION open‐label extension study [ClinicalTrials.gov identifier: NCT03635073]). fAE (n = 2), withdrawal by parent or guardian (n = 2), or other reason (n = 3; completed minimum requirement and entered ENDYMION open‐label extension study). gAE (n = 1) or withdrawal by parent or guardian (n = 1). hAE. BID, twice daily; DS, Dravet syndrome; LGS, Lennox–Gastaut syndrome; mITT, modified intent‐to‐treat.
2.4. Quantification of plasma 24HC levels
Plasma 24HC levels were determined by high‐performance liquid chromatography with tandem mass spectrometry, using samples acquired at baseline and at specified time points throughout the study.
2.5. Efficacy endpoints
The primary endpoint was the change from baseline in frequency of convulsive seizures (DS) or drop seizures (LGS) in patients receiving soticlestat in the combined patient population during the maintenance period, compared with those receiving placebo. Secondary efficacy endpoints included the change from baseline in seizure frequency in the combined patient population during the full 20‐week treatment period, and the change from baseline in convulsive and drop seizure frequencies in the DS and LGS patient populations, respectively, during both the maintenance and the full treatment periods.
Patients and/or their caregivers were given a paper seizure diary to record the type and number of all seizures experienced by the patient every day throughout the study, starting at the screening visit, in accordance with the updated seizure classification system of the International League Against Epilepsy. 25 The convulsive seizures recorded for DS included generalized tonic–clonic, focal to bilateral tonic–clonic, hemiclonic, bilateral clonic (generalized clonic), and convulsive status epilepticus seizures. Drop seizures were defined as seizures that involved the entire body, trunk, or head, and that either: led to the patient falling, becoming injured, slumping in a chair, or hitting their head on a surface; or could have led to the patient falling or becoming injured, depending on their position at the time of the attack. Seizure classification and primary seizure types at screening, as well as new seizures occurring after randomization, were confirmed by the Epilepsy Study Consortium.
Treatment response, defined as a reduction in seizure frequency of ≥50%, ≥75%, or 100% from baseline during the treatment period, was also characterized.
Global functioning was evaluated using the investigator‐ and caregiver‐reported Clinical Global Impression of Change (CGI‐C and Care GI‐C, respectively), assessed on Study Day 1, at Visits 4 and 5, and at the end of the study.
2.6. Safety endpoints
Safety assessments included the incidence of treatment‐emergent adverse events (TEAEs) and evaluation of the percentage of myoclonic and atypical absence seizure‐free days during the maintenance period. TEAEs were defined as any adverse event that started or increased in severity during or after the first dose of study treatment and within 15 days of the last dose of study treatment. The percentage of days free from myoclonic and atypical absence seizures was assessed to determine whether these seizure types worsened during treatment with soticlestat.
2.7. Data analysis
Sample size calculations were based on unadjusted Mann–Whitney (Wilcoxon rank‐sum) tests, with randomization stratified by DS and LGS diagnosis. The two patient populations were combined for the primary endpoint analysis to increase the power of the study. Assuming a mean difference in percentage change from baseline between the soticlestat and placebo treatment arms of 29% (standard deviation [SD] = 42.3%) in the combined patient population, with 56 patients per treatment group, the power to test treatment difference would be 92% at a two‐sided .05 level of significance. Assuming a 10% dropout rate, 63 patients would be randomized per arm. Approximately 23 patients with DS and 40 patients with LGS were planned to be randomized to each treatment group. Descriptive statistics were used for summaries of continuous variables, and frequency and percentage were used for categorical and ordinal variables. For all analyses, “baseline” refers to the prospective 4‐week baseline period. To assess changes in seizure frequency, the frequencies of convulsive seizures (DS) or drop seizures (LGS) per 28 days were determined for the baseline, maintenance, and full treatment periods, with the outputs used to calculate percentage change in seizure frequency from baseline. Rank‐transformed analysis of covariance was used to compare percentage changes in seizure frequency from baseline in the soticlestat and placebo treatment groups, with adjustments for baseline seizure frequency and indication. Additionally, the median treatment difference and confidence interval (CI) between soticlestat and placebo were estimated by the Hodges–Lehmann method based on unadjusted rank statistics. Changes in the CGI‐C and Care GI‐C scores on the impression of the efficacy and tolerability of treatment were summarized descriptively, and the soticlestat and placebo treatment groups were compared using a generalized linear mixed model.
The analysis sets reported here include the modified intent‐to‐treat (mITT) and safety analysis sets. The mITT analysis set included all randomized patients who had received at least one dose of study medication and had been assessed for ≥1 day in the treatment period. The safety analysis set included all randomized patients who had received at least one dose of study medication.
An efficacy analysis set, comprising a subset of the mITT analysis set, was specified in the study statistical analysis plan for the primary endpoint analysis. Given that it is a more inclusive population, we report findings from the mITT analysis set for all analyses, including the primary endpoint.
Treatment‐emergent adverse events were divided by treatment group and summarized using descriptive statistics. Days free from myoclonic and atypical absence seizures were summarized using descriptive statistics.
All statistical analyses were performed using SAS version 9.4.
3. RESULTS
3.1. Patient disposition
In total, 141 patients were randomized, of whom 139 were included in the mITT population; 126 patients completed the study (Figure 1B). Two patients did not have any seizure diary records during the treatment period and were not included in the mITT population. The mITT population included 51 patients with DS and 88 patients with LGS.
3.2. Baseline demographics and clinical characteristics
Patient demographics were generally similar between the soticlestat and placebo groups (Table 1). Patients had a mean age of 9.5 years, and 64% were male. The baseline convulsive seizure frequency in patients with DS was similar between the soticlestat and placebo groups, with mean frequencies (per 28 days) of 13.77 (SD = 11.017) and 13.19 (SD = 23.914), respectively (Table 1). However, the baseline frequency for drop seizures in patients with LGS was more varied, with mean frequencies in the soticlestat and placebo groups of 440.98 (SD = 1133.542) and 150.02 (SD = 203.822), respectively (Table 1). Both groups of patients reported a wide range of seizure frequencies at baseline, although this was especially pronounced in patients with LGS for both the total seizure frequency (6.5–8042.3) and the drop seizure frequency (4.0–5187.7; Table 1).
TABLE 1.
Baseline demographics and clinical characteristics in the modified intent‐to‐treat analysis set (n = 139)
| Characteristic | DS | LGS | ||
|---|---|---|---|---|
| Soticlestat, n = 26 | Placebo, n = 25 | Soticlestat, n = 43 | Placebo, n = 45 | |
| Age, years | ||||
| Mean (SD) | 8.7 (3.92) | 8.8 (4.50) | 10.0 (4.19) | 9.8 (3.58) |
| Minimum, maximum | 4, 17 | 2, 16 | 2, 17 | 3, 17 |
| Sex, n (%) | ||||
| Male | 17 (65.4) | 14 (56.0) | 30 (69.8) | 28 (62.2) |
| Female | 9 (34.6) | 11 (44.0) | 13 (30.2) | 17 (37.8) |
| Race, n (%) | ||||
| Asian | 11 (42.3) | 6 (24.0) | 11 (25.6) | 16 (35.6) |
| Black or African American | 0 | 0 | 0 | 1 (2.2) |
| White | 15 (57.7) | 19 (76.0) | 32 (74.4) | 28 (62.2) |
| Ethnicity, n (%) | ||||
| Hispanic or Latino | 2 (7.7) | 6 (24.0) | 8 (18.6) | 4 (8.9) |
| Not Hispanic or Latino | 24 (92.3) | 19 (76.0) | 35 (81.4) | 41 (91.1) |
| Weight, kg, mean (SD) | 29.1 (10.74) | 30.1 (15.10) | 36.0 (17.04) | 34.2 (16.42) |
| Number of ASMs, n (%) a | ||||
| 1 | 0 | 1 (4.0) | 2 (4.7) | 1 (2.2) |
| 2 | 11 (42.3) | 6 (24.0) | 12 (27.9) | 10 (22.2) |
| ≥3 | 15 (57.7) | 18 (72.0) | 29 (67.4) | 34 (75.6) |
| Seizures of interest frequency b , c | ||||
| Mean (SD) | 13.77 (11.017) | 13.19 (23.914) | 440.98 (1133.542) | 150.02 (203.822) |
| Median | 9.05 | 6.00 | 67.30 | 89.80 |
| Minimum, maximum | 2.6, 40.3 | 2.5, 125.0 | 8.1, 5187.7 | 4.0, 1040.1 |
| Total seizures frequency b , d | ||||
| Mean (SD) | 210.70 (546.987) | 55.10 (145.301) | 662.75 (1513.043) | 400.91 (667.045) |
| Median | 22.98 | 18.67 | 159.68 | 153.52 |
| Minimum, maximum | 4.1, 2073.0 | 2.5, 737.1 | 8.1, 8042.3 | 6.5, 3629.3 |
Abbreviations: ASM, antiseizure medication; DS, Dravet syndrome; LGS, Lennox–Gastaut syndrome.
Ongoing ASMs were included if the start date was before the first dose date or was missing.
Seizure frequency per 28 days.
Convulsive seizures for DS and drop seizures for LGS.
All seizure types. For uncountable cluster seizures, a seizure count of 1 was assigned.
Most patients were using more than one ASM at baseline, with 96 (69%) taking at least three ASMs. The top three ASMs used by patients with DS were stiripentol, clobazam, and valproic acid, and the top three used by patients with LGS were valproic acid, clobazam, and lamotrigine (Table S1).
3.3. Treatment exposure
Overall, 80.3% of patients receiving soticlestat were included in Dose Level 3 (weight‐adjusted equivalent of adult 300 mg BID) at the end of the maintenance period, 9.9% were included in Dose Level 2 (weight‐adjusted equivalent of adult 200 mg BID), and 7.0% were included in Dose Level 1 (weight‐adjusted equivalent of adult 100 mg BID; Table S2). A high adherence rate was observed in both treatment groups (Table S2).
3.4. Plasma 24HC levels
In the combined patient population receiving soticlestat, plasma 24HC levels decreased over the first 2 weeks of treatment and then remained relatively stable at the reduced levels throughout the full treatment period (Figure 2A), with similar results observed between the DS and LGS patient groups (Figure 2B). Conversely, plasma 24HC levels in patients receiving placebo remained similar to those recorded at baseline.
FIGURE 2.
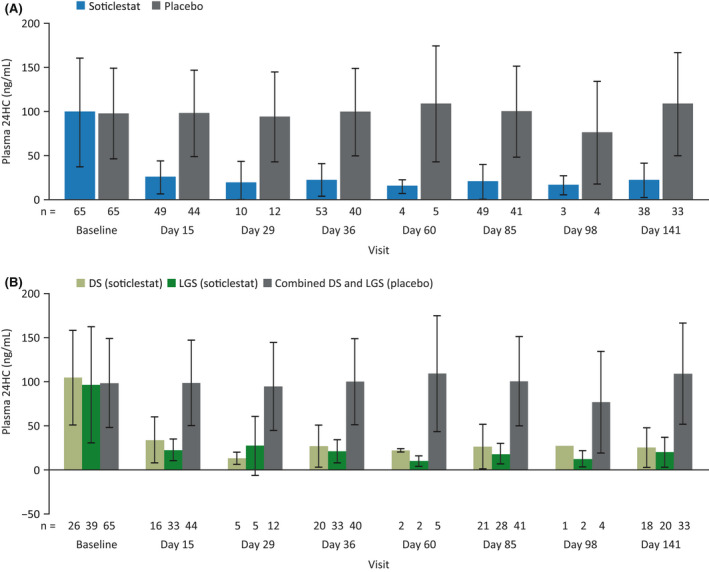
Mean plasma 24S‐hydroxycholesterol (24HC) levels (±SD) at baseline and over the full treatment period in the combined patient population (A) and in the Dravet syndrome (DS) and Lennox–Gastaut syndrome (LGS) populations (B) in the modified intent‐to‐treat analysis set. Placebo values are from the combined patient population in the safety analysis set.
3.5. Efficacy
3.5.1. Changes in seizure frequency
For the primary endpoint, patients in the combined patient population who received soticlestat demonstrated a placebo‐adjusted median reduction in seizure frequency of 30.21% (95% CI = −45.97% to −13.88%) during the maintenance period (p = .0008; Figure 3A). Similar reductions were observed over the full 20‐week treatment period, with soticlestat‐treated patients demonstrating a placebo‐adjusted median reduction in seizure frequency of 25.06% (95% CI = −42.48% to −10.69%, p = .0024; Figure 3A).
FIGURE 3.
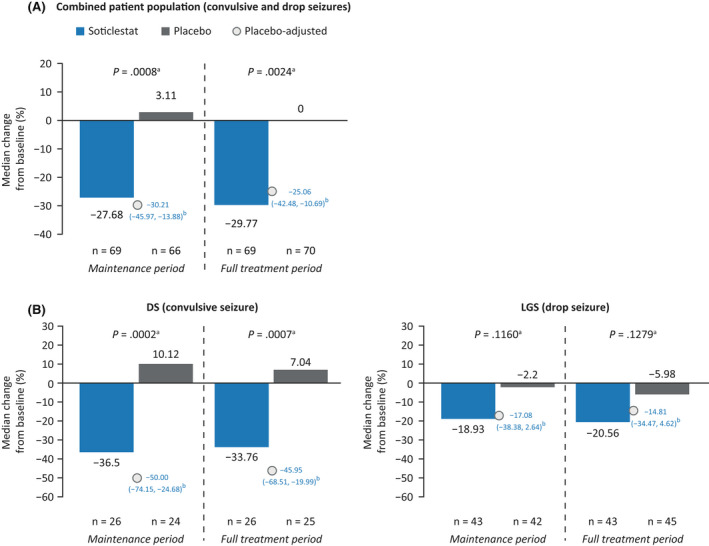
Changes in seizure frequency from baseline in the combined patient population (A) and in the Dravet syndrome (DS) and Lennox–Gastaut syndrome (LGS) populations (B) in the modified intent‐to‐treat analysis set. Patients who withdrew from the study before the maintenance period were excluded from the maintenance period analyses: four patients receiving placebo in A, one patient with DS receiving placebo in B, and three patients with LGS receiving placebo in B. aRank‐transformed analysis of covariance, adjusting for baseline seizure frequency and protocol amendment cohort. bHodges–Lehmann estimate (95% confidence interval) of the median treatment difference (percentage change from baseline with soticlestat vs. percentage change from baseline with placebo).
Patients with DS who received soticlestat demonstrated a statistically significant placebo‐adjusted median reduction in convulsive seizure frequency of 50.00% (95% CI = −74.15% to −24.68%, p = .0002) over the maintenance period and of 45.95% (95% CI = −68.51% to −19.99%, p = .0007) over the full treatment period (Figure 3B). Patients with LGS who received soticlestat demonstrated a nonstatistically significant placebo‐adjusted median reduction in seizure frequency of 17.08% (95% CI = −38.38% to 2.64%, p = .1160) over the maintenance period and of 14.81% (95% CI: −34.47% to 4.62%, p = .1279) over the full treatment period.
3.5.2. Treatment response
Over the full treatment period, a greater proportion of patients receiving soticlestat than of those receiving placebo experienced at least a 50% reduction from baseline in seizure frequency (Figure 4A). This reduction was observed in 30.8% of patients with DS who received soticlestat versus 0% of those who received placebo, and in 16.3% of patients with LGS who received soticlestat versus 13.3% of those who received placebo. Results from the maintenance period were similar to those reported during the full treatment period (Figure S1).
FIGURE 4.
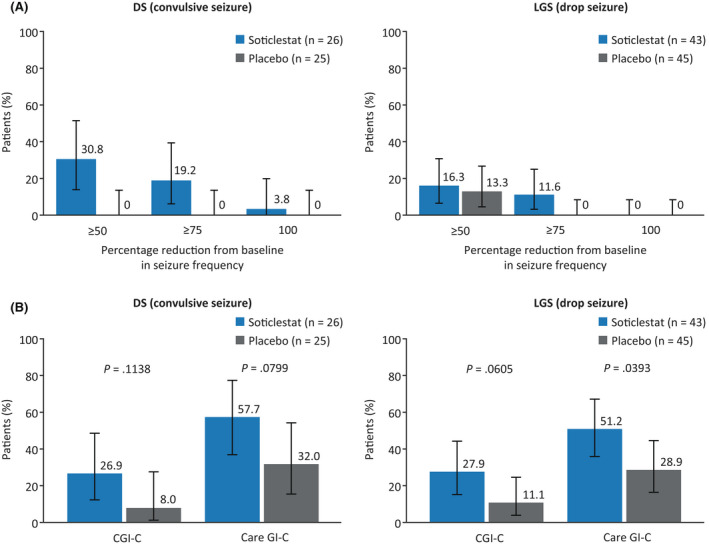
Proportions of treatment responders over the full treatment period (A) and proportions of patients with any improvement in investigator‐ and caregiver‐reported Clinical Global Impression of Change (CGI‐C and Care GI‐C, respectively) scores at the last visit (B) in the modified intent‐to‐treat analysis set. (A) Error bars represent the 95% confidence interval. (B) Investigator‐reported improvement included “marked improvement and no side effects” and “marked improvement and minimal side effects”; caregiver‐reported improvement included “slightly improved,” “much improved,” and “very much improved.” The p‐value of the difference between soticlestat and placebo (B) was computed using a generalized linear mixed model; the CIs plotted are the Wilson score intervals with continuity correction. DS, Dravet syndrome; LGS, Lennox–Gastaut syndrome.
3.5.3. Global functioning
At the last study visit, CGI‐C responses that indicated marked improvement were reported for greater proportions of patients with DS and patients with LGS who received soticlestat than of those who received placebo (26.9% vs. 8.0% and 27.9% vs. 11.1%, respectively; Figure 4B). These results were reflected by caregivers, with Care GI‐C responses that indicated improvement being reported for greater proportions of patients with DS and patients with LGS who received soticlestat than of those who received placebo (57.7% vs. 32.0% and 51.2% vs. 28.9%, respectively).
3.6. Safety and tolerability
In total, 109 patients (77.3%) experienced a TEAE, with most being mild (n = 94, 66.7%) or moderate (n = 44, 31.2%; Table 2). Lethargy (n = 5) and constipation (n = 4) both occurred with a difference of ≥5% compared with placebo.
TABLE 2.
Summary of TEAEs (safety analysis set, n = 141)
| Soticlestat, n = 71 | Placebo, n = 70 | |
|---|---|---|
| Patients with any TEAEs a | 57 (80.3) | 52 (74.3) |
| Mild | 47 (66.2) | 47 (67.1) |
| Moderate | 25 (35.2) | 19 (27.1) |
| Severe | 10 (14.1) | 11 (15.7) |
| Treatment‐related TEAEs | 38 (53.5) | 26 (37.1) |
| Withdrawal due to TEAEs | 4 (5.6) | 3 (4.3) |
| DS | 1 | 1 |
| LGS | 3 | 2 |
| Serious TEAEs | 11 (15.5) | 13 (18.6) |
| Related | 2 (2.8) | 1 (1.4) |
| Deaths | 0 | 0 |
| Common TEAEs b | ||
| Upper respiratory tract infection | 13 (18.3) | 12 (17.1) |
| Pyrexia | 11 (15.5) | 8 (11.4) |
| Seizure (worsening or new) | 6 (8.5) | 9 (12.9) |
| Nasopharyngitis | 6 (8.5) | 6 (8.6) |
| Decreased appetite | 6 (8.5) | 5 (7.1) |
| Vomiting | 6 (8.5) | 4 (5.7) |
| Somnolence | 6 (8.5) | 3 (4.3) |
| Diarrhea | 5 (7.0) | 4 (5.7) |
| Lethargy c | 5 (7.0) | 0 |
| Fatigue | 4 (5.6) | 3 (4.3) |
| Pneumonia | 4 (5.6) | 2 (2.9) |
| Irritability | 4 (5.6) | 2 (2.9) |
| Constipation c | 4 (5.6) | 0 |
Note: Values are n (%).
Abbreviations: DS, Dravet syndrome; LGS, Lennox–Gastaut syndrome; PT, preferred term; TEAE, treatment‐emergent adverse event.
Patients with TEAEs of the same PTs are counted once at the highest severity. Patients with TEAEs of different PTs of the same highest severity level are counted once. Patients with TEAEs of different PTs and different highest severity are counted separately for each of the highest severity levels.
Occurring in ≥5% of patients in either treatment group.
TEAEs occurring with a difference of ≥5% over placebo.
Serious TEAEs were experienced by similar proportions of patients in the soticlestat and placebo arms (15.5% and 18.6%, respectively) and were reported by 24 patients in total (17.0%).
Sixty‐four patients (45.4%) reported TEAEs that were considered by the investigator to be study‐drug related, three of whom experienced serious adverse events (soticlestat, n = 2; placebo, n = 1). Of the two patients receiving soticlestat, one experienced two serious TEAEs of speech disorder and seizure, and one experienced a serious TEAE of septic shock; the patient receiving placebo experienced two serious TEAEs of seizure cluster and seizure. No deaths were reported in this study.
TEAEs led to dose modification in 18 patients, interrupted dosing in two patients, and discontinuation in seven patients (soticlestat, n = 4; placebo, n = 3). Of the four patients receiving soticlestat, three discontinued treatment owing to TEAEs considered to be possibly related to treatment: the patient who reported two serious TEAEs of speech disorder and increased seizure frequency; one patient who reported a moderate TEAE of somnolence; and one patient who reported a severe TEAE of apathy. In addition, one patient reported a serious TEAE of feeding difficulties considered to be unrelated to treatment. Three patients receiving placebo discontinued treatment because of TEAEs considered to be possibly related to treatment: the patient who reported two serious TEAEs of seizure cluster and seizure; one patient who reported a moderate TEAE of decreased appetite; and one patient who reported a moderate TEAE of seizure.
Similar mean proportions of days free from myoclonic and atypical absence seizures were reported by the soticlestat and placebo groups (73.26% and 72.07%, respectively; Table S3).
4. DISCUSSION
ELEKTRA achieved its primary endpoint, with adjunctive soticlestat treatment resulting in a statistically significant median reduction from baseline in seizure frequency compared with placebo in the combined DS and LGS patient population during the 12‐week maintenance period. Furthermore, in patients with DS, soticlestat treatment was associated with a statistically significant and clinically meaningful median reduction in convulsive seizure frequency. In patients with LGS, a nonstatistically significant numerical reduction in drop seizure frequency compared with placebo was observed. Global functioning was shown to improve with soticlestat treatment, with investigators and caregivers reporting a greater improvement with soticlestat than with placebo. These observed improvements in seizures in patients with DS and in functioning in both patient groups were accompanied by a reduction in plasma 24HC levels in patients receiving soticlestat. Soticlestat had a safety profile that was consistent with previous studies, and similar rates of TEAEs were reported in the soticlestat and placebo treatment arms. Furthermore, no new safety signals were identified. Taken together, these findings support the further evaluation of soticlestat as a potential therapy for DS and LGS in phase 3 clinical studies.
Statistically significant reductions in seizure frequency were observed in patients with DS, but not in those with LGS. LGS is known to be a complex condition with a heterogenous etiology and clinical features that may change with time. 14 , 15 The resistance of LGS‐associated seizures to pharmacological treatment is also well known, with one study reporting that, among patients with DEEs, the largest proportion of treatment‐resistant individuals have a diagnosis of LGS (90%). 15 , 18 Furthermore, drop seizures can be difficult to define because their presentation and clinical importance vary in terms of characteristics (e.g., the patient may not fall if seated in a wheelchair or lying down) and body involvement (whole body, head only, or other body areas). 26 , 27 ELEKTRA used the definition of drop seizure that was used in previous studies with clobazam 28 and cannabidiol. 29 However, the observed differences in baseline seizure frequency, in otherwise similar populations as defined by inclusion/exclusion criteria, suggest a difference in how the definition of drop seizure was implemented. Clarifying which seizure types can result in drop seizures may have been a more appropriate approach. These complexities are important to consider when assessing treatments in clinical trials or real‐world settings, and they highlight that a personalized approach, tailored to the individual symptoms and responses of the patient, is particularly important. 30
Although seizure frequency in patients with LGS was not significantly reduced, similar proportions of patients with DS or LGS were classified as having improved global functioning, according to CGI‐C and Care GI‐C responses. For both groups of patients, clinicians and caregivers reported a greater improvement in global functioning following treatment with soticlestat than with placebo. The CGI‐C and Care GI‐C assessments are designed to assess global functioning, and they take into consideration knowledge of the patient's history, psychosocial circumstances, symptoms, and behavior, and the impact of the symptoms on the patient's ability to function. As such, changes in factors beyond seizures can be recorded to provide a broader picture of potential treatment effects, as illustrated by these findings.
In line with previous results, treatment with soticlestat resulted in a reduction of plasma 24HC levels, which was maintained throughout the full treatment period. These findings indicate engagement of soticlestat with CH24H, which is predominantly expressed in the brain, resulting in the observed systemic reductions of 24HC. 22 , 23 , 24 Although soticlestat treatment reduced plasma 24HC levels in both the DS and the LGS patient groups, a significant reduction in seizure frequency was only observed in patients with DS. Further studies are planned to assess the efficacy of soticlestat in patients with LGS, which may help to explain these differences in seizure reduction despite similar reductions in 24HC levels in both ELEKTRA patient groups. The results reported here build on those reported from clinical studies in healthy volunteers and adults with DEEs to support the use of 24HC as a peripheral biomarker for soticlestat activity in the brain. 22 , 23 , 24
Safety findings from ELEKTRA were in line with those reported in previous studies, with no new safety signals being identified. 22 , 23 , 24 TEAEs were generally mild to moderate in severity and occurred at similar frequencies with soticlestat and placebo treatment, except for lethargy and constipation, which occurred more often with soticlestat than with placebo. Patients in the soticlestat and placebo groups experienced similar proportions of days free from myoclonic and atypical absence seizures, suggesting that soticlestat treatment did not exacerbate these seizure types.
Both the nonseizure symptoms and the seizures that characterize DS and LGS have a profound effect on the affected individuals. Although early and effective intervention may potentially improve long‐term developmental outcomes by reducing epileptic activity, treatment goals should also go beyond seizure control and aim to address the nonseizure symptoms as well, especially given the low health‐related quality of life reported by affected individuals and their caregivers. 5 , 31 However, even seizure control can be a difficult target to reach. Despite often using polytherapy, including both pharmacological and nonpharmacological treatments, patients rarely achieve control of seizures. 15 , 32 At least two ASMs had failed for all the patients enrolled in the current study, and most patients were using more than one ASM at baseline, supporting the ongoing need for more effective treatment options for individuals with DS or LGS. 6 , 20 , 30 , 33 , 34
Despite patients in this study having a history of inadequate response to multiple ASMs, efficacy signals were seen with soticlestat for DS, and potentially for LGS, along with the sustained reductions in plasma 24HC levels. The reduction of 24HC via inhibition of CH24H has been shown to reduce glutamatergic signaling in mice and potentially to reduce inflammation, which may have downstream effects on seizure susceptibility. 21 Furthermore, maximal CH24H inhibition in wild‐type mice did not lead to any notable effects on motor coordination or spontaneous locomotor activity, suggesting that CH24H inhibition can tip the neural excitatory/inhibitory balance without indiscriminately dampening neural excitation. 21 As a first‐in‐class selective inhibitor of CH24H, soticlestat has the potential to provide therapy via a different mechanism of action from existing available therapies. 21
Strengths of the study include the multiple study sites across the eight countries involved, allowing for the enrollment of a relatively large number of participants. Additionally, this study provides evidence for the safety and tolerability of soticlestat in children, a population in which its use has not been investigated before. However, given the clinical trial setting, the results reported here may not be generalizable to a real‐world population.
5. CONCLUSIONS
In conclusion, soticlestat treatment resulted in a safety profile that was consistent with previous studies, was associated with a low rate of study discontinuation, and gave a signal of efficacy, with adjunctive soticlestat treatment resulting in a statistically significant median reduction in seizure frequency in the combined patient population. Additionally, further support was provided for the use of plasma 24HC levels as a biomarker for the activity of soticlestat. Taking these findings into consideration, individual phase 3 studies for DS and LGS are warranted to evaluate soticlestat treatment further in patients with these conditions.
AUTHOR CONTRIBUTIONS
All authors interpreted the results, contributed to the writing, and reviewed the manuscript. All authors approved the final version of the manuscript for submission.
CONFLICT OF INTEREST
D.A., S.H., and M.A. have been employees of Takeda Pharmaceutical Company Ltd and have owned stock or stock options. The following authors have received compensation for serving as consultants or speakers, or they or the institutions they work for have received research support or royalties from the companies or organizations indicated: C.D.H. (Greenwich Biosciences, Takeda, UCB), V.V. (Angelini Pharma, Bial, Eisai Inc., GW Pharmaceuticals, Novartis, Takeda, UCB, Zogenix), M.Z. (GW Pharmaceuticals, Marinus Pharmaceuticals, Takeda, Zogenix), D.D. (Aquestive Therapeutics, Biogen, BioPharm, Commonwealth of Pennsylvania Department of Health, Encoded Therapeutics, Greenwich Biosciences, Marinus Pharmaceuticals, Neurelis, Ovid Therapeutics, Pediatric Epilepsy Research Foundation, SK Life Science, Takeda, Epilepsy Study Consortium, UCB, Xenon Pharmaceuticals, Zogenix). Y.J. has no conflicts of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Figure S1
Tables S1–S3
ACKNOWLEDGMENTS
All studies were funded by the sponsor, Takeda Pharmaceutical Company Ltd. We thank the participants and investigators involved in the studies. We also thank Peter B. Forgacs and Yin Yang (Ovid Therapeutics Inc.) for their contribution to this study. Under the direction of the authors and funded by Takeda Pharmaceutical Company Ltd, Erin Aldera, PhD (https://orcid.org/0000‐0001‐7935‐1337) and Aimee Jones, DPhil (https://orcid.org/0000‐0003‐0279‐1814) of Oxford PharmaGenesis, Oxford, UK, provided writing assistance for this publication, in accordance with Good Publication Practice 3 guidelines (http://www.ismpp.org/gpp3). Editorial assistance in formatting, proofreading, copyediting, and fact‐checking was also provided by Oxford PharmaGenesis.
Hahn CD, Jiang Y, Villanueva V, Zolnowska M, Arkilo D & Hsiao S et al. A phase 2, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy and safety of soticlestat as adjunctive therapy in pediatric patients with Dravet syndrome or Lennox–Gastaut syndrome (ELEKTRA). Epilepsia. 2022;63:2671–2683. 10.1111/epi.17367
Dimitrios Arkilo was at Takeda Pharmaceutical Company at the time the study was conducted.
REFERENCES
- 1. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anwar A, Saleem S, Patel UK, Arumaithurai K, Malik P. Dravet syndrome: an overview. Cureus. 2019;11(6):e5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011;52(Suppl 2):3–9. [DOI] [PubMed] [Google Scholar]
- 4. Asadi‐Pooya AA. Lennox‐Gastaut syndrome: a comprehensive review. Neurol Sci. 2018;39(3):403–14. [DOI] [PubMed] [Google Scholar]
- 5. Brunklaus A, Dorris L, Zuberi SM. Comorbidities and predictors of health‐related quality of life in Dravet syndrome. Epilepsia. 2011;52(8):1476–82. [DOI] [PubMed] [Google Scholar]
- 6. Lagae L, Brambilla I, Mingorance A, Gibson E, Battersby A. Quality of life and comorbidities associated with Dravet syndrome severity: a multinational cohort survey. Dev Med Child Neurol. 2018;60(1):63–72. [DOI] [PubMed] [Google Scholar]
- 7. Gallop K, Wild D, Verdian L, Kerr M, Jacoby A, Baker G, et al. Lennox‐Gastaut syndrome (LGS): development of conceptual models of health‐related quality of life (HRQL) for caregivers and children. Seizure. 2010;19(1):23–30. [DOI] [PubMed] [Google Scholar]
- 8. Gibson PA. Lennox‐Gastaut syndrome: impact on the caregivers and families of patients. J Multidiscip Healthc. 2014;7:441–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Millichap JJ, Koh S, Laux LC, Nordli DR Jr. Child neurology: Dravet syndrome: when to suspect the diagnosis. Neurology. 2009;73(13):e59–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and management of Dravet syndrome: recommendations from a North American consensus panel. Pediatr Neurol. 2017;68:18–34.e3. [DOI] [PubMed] [Google Scholar]
- 11. Trevathan E, Murphy CC, Yeargin‐Allsopp M. Prevalence and descriptive epidemiology of Lennox‐Gastaut syndrome among Atlanta children. Epilepsia. 1997;38(12):1283–8. [DOI] [PubMed] [Google Scholar]
- 12. He N, Li BM, Li ZX, Wang J, Liu XR, Meng H, et al. Few individuals with Lennox‐Gastaut syndrome have autism spectrum disorder: a comparison with Dravet syndrome. J Neurodev Disord. 2018;10(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim HJ, Kim HD, Lee JS, Heo K, Kim DS, Kang HC. Long‐term prognosis of patients with Lennox‐Gastaut syndrome in recent decades. Epilepsy Res. 2015;110:10–9. [DOI] [PubMed] [Google Scholar]
- 14. Cross JH, Auvin S, Falip M, Striano P, Arzimanoglou A. Expert opinion on the management of Lennox‐Gastaut syndrome: treatment algorithms and practical considerations. Front Neurol. 2017;8:505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Resnick T, Sheth RD. Early diagnosis and treatment of Lennox‐Gastaut syndrome. J Child Neurol. 2017;32(11):947–55. [DOI] [PubMed] [Google Scholar]
- 16. Kerr M, Kluger G, Philip S. Evolution and management of Lennox‐Gastaut syndrome through adolescence and into adulthood: are seizures always the primary issue? Epileptic Disord. 2011;13(Suppl 1):S15–26. [DOI] [PubMed] [Google Scholar]
- 17. Bourgeois BF, Douglass LM, Sankar R. Lennox‐Gastaut syndrome: a consensus approach to differential diagnosis. Epilepsia. 2014;55(Suppl 4):4–9. [DOI] [PubMed] [Google Scholar]
- 18. Chipaux M, Szurhaj W, Vercueil L, Milh M, Villeneuve N, Cances C, et al. Epilepsy diagnostic and treatment needs identified with a collaborative database involving tertiary centers in France. Epilepsia. 2016;57(5):757–69. [DOI] [PubMed] [Google Scholar]
- 19. Ostendorf AP, Ng YT. Treatment‐resistant Lennox‐Gastaut syndrome: therapeutic trends, challenges and future directions. Neuropsychiatr Dis Treat. 2017;13:1131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ben‐Menachem E. Medical management of refractory epilepsy—practical treatment with novel antiepileptic drugs. Epilepsia. 2014;55(Suppl 1):3–8. [DOI] [PubMed] [Google Scholar]
- 21. Nishi T, Kondo S, Miyamoto M, Watanabe S, Hasegawa S, Kondo S, et al. Soticlestat, a novel cholesterol 24‐hydroxylase inhibitor shows a therapeutic potential for neural hyperexcitation in mice. Sci Rep. 2020;10(1):17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang S, Chen G, Merlo Pich E, Affinito J, Cwik M, Faessel H. Safety, tolerability, pharmacokinetics, pharmacodynamics, bioavailability and food effect of single doses of soticlestat in healthy subjects. Br J Clin Pharmacol. 2021;87(11):4354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang S, Chen G, Pich EM, Affinito J, Cwik M, Faessel HM. Pharmacokinetics, pharmacodynamics and safety assessment of multiple doses of soticlestat in healthy volunteers. Br J Clin Pharmacol. 2022;88:2899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Halford JJ, Sperling MR, Arkilo D, Asgharnejad M, Zinger C, Xu R, et al. A phase 1b/2a study of soticlestat as adjunctive therapy in participants with developmental and/or epileptic encephalopathies. Epilepsy Res. 2021;174:106646. [DOI] [PubMed] [Google Scholar]
- 25. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):522–30. [DOI] [PubMed] [Google Scholar]
- 26. Conry JA, Ng YT, Paolicchi JM, Kernitsky L, Mitchell WG, Ritter FJ, et al. Clobazam in the treatment of Lennox‐Gastaut syndrome. Epilepsia. 2009;50(5):1158–66. [DOI] [PubMed] [Google Scholar]
- 27. Devinsky O, Patel AD, Cross JH, Villanueva V, Wirrell EC, Privitera M, et al. Effect of cannabidiol on drop seizures in the Lennox‐Gastaut syndrome. N Engl J Med. 2018;378(20):1888–97. [DOI] [PubMed] [Google Scholar]
- 28. Ng YT, Conry JA, Drummond R, Stolle J, Weinberg MA, OV‐1012 Study Investigators . Randomized, phase III study results of clobazam in Lennox‐Gastaut syndrome. Neurology. 2011;77(15):1473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thiele EA, Marsh ED, French JA, Mazurkiewicz‐Beldzinska M, Benbadis SR, Joshi C, et al. Cannabidiol in patients with seizures associated with Lennox‐Gastaut syndrome (GWPCARE4): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet. 2018;391(10125):1085–96. [DOI] [PubMed] [Google Scholar]
- 30. Strzelczyk A, Schubert‐Bast S. Expanding the treatment landscape for Lennox‐Gastaut syndrome: current and future strategies. CNS Drugs. 2021;35(1):61–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gallop K, Wild D, Nixon A, Verdian L, Cramer JA. Impact of Lennox‐Gastaut syndrome (LGS) on health‐related quality of life (HRQL) of patients and caregivers: literature review. Seizure. 2009;18(8):554–8. [DOI] [PubMed] [Google Scholar]
- 32. Wallace A, Wirrell E, Kenney‐Jung DL. Pharmacotherapy for Dravet syndrome. Paediatr Drugs. 2016;18(3):197–208. [DOI] [PubMed] [Google Scholar]
- 33. National Institute for Health and Care Excellence . Clinical Guideline 137. Epilepsies: diagnosis and management[cited 2021 Sep 29]. Available from: https://www.nice.org.uk/guidance/cg137/ [PubMed]
- 34. Knupp KG, Wirrell EC. Treatment strategies for Dravet syndrome. CNS Drugs. 2018;32(4):335–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Tables S1–S3