

Summary
Inflammation during prenatal development can be detrimental to neurodevelopmental processes, increasing the risk of neuropsychiatric disorders. Prenatal exposure to maternal viral infection during pregnancy is a leading environmental risk factor for manifestation of these disorders. Preclinical animal models of maternal immune activation (MIA), established to investigate this link, have revealed common immune and microbial signaling pathways that link mother and fetus and set the tone for prenatal neurodevelopment. In particular, maternal intestinal T helper 17 cells, educated by endogenous microbes, appear to be key drivers of effector IL‐17A signals capable of reaching the fetal brain and causing neuropathologies. Fetal microglial cells are particularly sensitive to maternally derived inflammatory and microbial signals, and they shift their functional phenotype in response to MIA. Resulting cortical malformations and miswired interneuron circuits cause aberrant offspring behaviors that recapitulate core symptoms of human neurodevelopmental disorders. Still, the popular use of “sterile” immunostimulants to initiate MIA has limited translation to the clinic, as these stimulants fail to capture biologically relevant innate and adaptive inflammatory sequelae induced by live pathogen infection. Thus, there is a need for more translatable MIA models, with a focus on relevant pathogens like seasonal influenza viruses.
Keywords: influenza virus, maternal immune activation, microbial signaling, microglia, neurodevelopment, TH17 cells
1. MATERNAL IMMUNE ACTIVATION DURING PREGNANCY
In 1988, S. Mednick's group published a seminal paper connecting influenza virus infection during gestation with later development of schizophrenia. 1 In the decades since, numerous epidemiological studies have solidified the link between maternal viral infections and increased risk of offspring neurodevelopmental disorders (NDDs), 2 particularly schizophrenia (SZ) 3 , 4 , 5 , 6 , 7 , 8 and autism spectrum disorders (ASD). 9 , 10 , 11 , 12 , 13 , 14 To investigate the mechanisms underlying this link, preclinical animal models of maternal immune activation (MIA) have been established, wherein various immune stimulants are used to initiate an inflammatory response in the pregnant female. 15 , 16 These models produce offspring that recapitulate many of the behavioral and pathophysiological hallmarks of human NDDs, 17 , 18 indicating that maternal inflammation is disrupting fetal neurodevelopmental processes, ultimately leading to the manifestation of neuropsychiatric disorders postnatally.
The immature fetal brain is especially vulnerable to MIA insult in part because homeostatic neurodevelopment relies on innate immune signaling molecules, including inflammatory cytokines. 19 Thus, a disrupted balance in the prenatal cytokine milieu can negatively impact brain development trajectories. 20 While tissues of the maternal‐fetal interface serve as both physical and immunological barriers against vertical transmission of pathogens, 21 , 22 maternal inflammation can still lead to adverse or stunted offspring development. 22 , 23 , 24 Thus, it is not the pathogen that serves as a teratogen, but the activation of the maternal immune system. 25 Viral pathogens—including non‐vertically transmitted influenza viruses—are still the leading environmental cause of MIA. 26
Historical outbreaks of influenza infection, dating back almost a century, provide population‐based epidemiologic data that support the connection between maternal infection and offspring NDDs. 4 , 8 To this day, influenza viruses are considered prototypical re‐emerging global pathogens, as they are stably adapted to and transmitted within multiple host species. 27 Domestic livestock hosts (birds and swine) and human hosts serve as natural reservoirs for circulating influenza viruses, which continuously mutate and form new antigenic variants. 28 Across the globe, outbreaks of new variants consistently cause epidemics of respiratory disease in 5%‐15% of the human population every year. While there are four distinct influenza viruses (A, B, C, and D), influenza A viruses (IAV) infect the widest range of species and are the only type known to cause pandemics. 29 The prevalence and severity of seasonal IAV epidemics 28 and continuous danger of spillover events from livestock 30 make this virus a primary public health concern. Notably, pregnant women infected with IAV experience increased hospitalization and fatality rates, and their infants are at a greater risk of adverse outcomes—low birth weights, premature delivery, and stillbirth—even in the absence of transplacental viral transmission. 31 , 32 , 33 , 34 Instead, the inflammatory mediators produced by the maternal immune system to protect the mother from respiratory disease appear to be the very same mediators that increase the risk of offspring NDDs.
Through decades of MIA research, inflammatory cytokines IL‐6 and IL‐17A have been singled out as primary actors evoking neurodevelopmental abnormalities during MIA. Most of this evidence comes from studies that utilize synthetic viral mimetic polyinosinic:polycytidylic acid (poly I:C) to induce MIA. Increases in circulating IL‐6 during mimetic‐induced MIA 35 have been shown to initiate an inflammatory cascade within the fetal compartment due to binding of IL‐6 to receptors on the placenta, ultimately leading to neuropathologies and abnormal behaviors. 36 More recent evidence has also documented increased production of IL‐17A by maternal T helper lymphocytes (TH17 cells), leading to a measurable increase in this effector cytokine in maternal circulation during MIA, which results in fetal cortical malformations and aberrant behaviors. 37 Administration of IL‐6 to unchallenged pregnant dams, or of IL‐17A into the developing fetal brain, recapitulates the adverse offspring outcomes observed during poly I:C‐induced MIA. 35 , 37 Conversely, administration of antibodies that block either cytokine during MIA is sufficient to rescue offspring deficits. 35 , 37 , 38 As IL‐6 has a known role in promoting differentiation of TH17 cells, 39 the possibility that IL‐6 may be acting upstream of IL‐17A during MIA has been raised. 17 , 37 Thus, increasing attention has fallen on maternal TH17 cells as major players mediating offspring psychiatric risk during MIA (as reviewed elsewhere 40 ). Decades of work in this area, however, has also confirmed that not all immune stimulants are created equal, 41 , 42 , 43 especially when comparing the highly‐popular synthetic poly I:C to a clinically relevant live viral infection. 44
While mounting evidence supports the involvement of maternally derived IL‐6 and IL‐17A in offspring NDDs, exactly which fetal brain cells are responding to these cytokines, resulting in the manifestation of neuropathologies, is still up for debate. Some studies suggest that neurons or neural progenitor cells respond directly to maternal inflammation, 45 , 46 , 47 while others indicate that immune cells—microglia 48 , 49 , 50 or macrophages 51 —are orchestrating these changes. The wide range of different MIA modeling techniques and assessment end points has led to contradictory conclusions about the role microglia play. 52 Yet, as appreciation for the integral involvement of microglial cells during healthy neurodevelopment continues to mount, 53 , 54 the possibility that prenatal perturbation of these cells could have widespread consequences for neuronal development and circuitry has gained more traction. There is also evidence to suggest that microglia are involved in pathologies relevant to both ASD 55 , 56 , 57 , 58 , 59 and SZ. 60 , 61 , 62 , 63 , 64 Overall, the extent to which dysregulated microglia may contribute to neuronal disruption during MIA is incompletely understood.
In this review, we will address how major progress in the field of immunostimulant‐induced MIA can (or cannot) be translated to influenza virus‐induced MIA. In particular, we focus on mechanistic pathways underlying MIA‐induced pathologies: pathogen recognition pathways, downstream innate and adaptive immune responses, and altered phenotypes and functions of offspring microglial cells. Overall, we aim to highlight the major differences between poly I:C and live IAV infection and dissect the evidence placing prenatal microglial cells at the center of altered neurodevelopmental trajectories during MIA.
2. MATERNAL IMMUNE RESPONSES TO LIVE VIRUS VERSUS IMMUNOSTIMULANTS
Before poly I:C‐induced MIA modeling took over in popularity, one group in particular should be credited with performing the most extensive investigations using mouse‐adapted IAV. In the 1990s, Fatemi et al 65 developed a mouse model of prenatal exposure to a neurotropic strain of influenza A/NWS/33 (H1N1). In a series of experiments, they revealed dysregulated corticogenesis, excitatory‐inhibitory imbalances, and neuropathology consistent with that found in ASD and SZ patients, as well as behavioral deficits in exposed offspring that persisted into adulthood. 65 , 66 , 67 , 68 , 69 , 70 The cause of these abnormalities was determined to be from the virus‐induced maternal inflammatory response rather than direct transmission of the virus from mother to fetus. 71 In fact, many behavioral abnormalities found in adult offspring from influenza‐infected mice are recapitulated by “sterile” immunostimulants alone 72 (as reviewed in Ref. 73). Today, the current understanding of MIA‐driven neurodevelopmental abnormalities can largely be credited to studies conducted with pathogen mimetics, such as bacterial endotoxin lipopolysaccharide (LPS) or synthetic viral double‐stranded RNA (dsRNA) poly I:C. These toll‐like receptor agonists induce a controlled 24‐48 hours innate immune response that allows researchers to target specific fetal developmental periods. While these models are fundamental in elucidating potential disease etiologies, mimetics fail to accurately recreate pathological conditions. For example, live viruses like IAV actively replicate within infected tissue, eliciting a complex cascade of inflammatory responses involving innate and adaptive immune signaling pathways over a one‐to‐two‐week duration, a significantly longer and more complex process compared with poly I:C. If mimetic‐induced MIA models fail to replicate these clinically relevant immune phenotypes, they may also fail to capture critical downstream neurodevelopmental disease etiologies.
2.1. Toll‐like receptor recognition of poly I:C and IAV
The innate immune system recognizes a broad range of pathogen‐associated molecular patterns (PAMPs) through various pattern recognition receptors (PRRs). Amongst the most well‐characterized PRRs are toll‐like receptors (TLRs; as reviewed in Ref. 74, 75). For the purpose of this review, we focus primarily on endosomal TLRs that recognize viral PAMPs. Poly I:C, a dsRNA synthetic viral analog, is a potent inducer of endosomal TLR3. Ligand binding to TLR3 activates the TIR‐domain‐containing adaptor‐inducing interferon‐β (TRIF) pathway, which further activates transcriptional regulators interferon regulatory factor 3 (IRF3) and nuclear factor kappa B (NFκB) to mediate production of type I antiviral interferons (IFNs) and pro‐inflammatory cytokines, respectively 74 (Figure 1A). This results in acute inflammation lasting 24‐48 hours on average. IAV, which is a single‐stranded RNA (ssRNA) virus, activates TLR3 through unidentified dsRNA structures present in dying influenza‐infected cells. 76 Viral ssRNA is directly detected by endosomal TLR7 (mice and humans) and TLR8 (humans). Ligand binding of TLR7/8 activates the myeloid differentiation primary response 88 (MyD88) pathway that further activates transcriptional regulators IRF7 and NFκB to regulate production of type I interferons and pro‐inflammatory cytokines 77 (Figure 1B). While endosomal TLR activation ultimately produces comparable end products, the variation in upstream pathways could be sufficient to result in significant differences between MIA models that involve activation of distinct TLRs.
FIGURE 1.
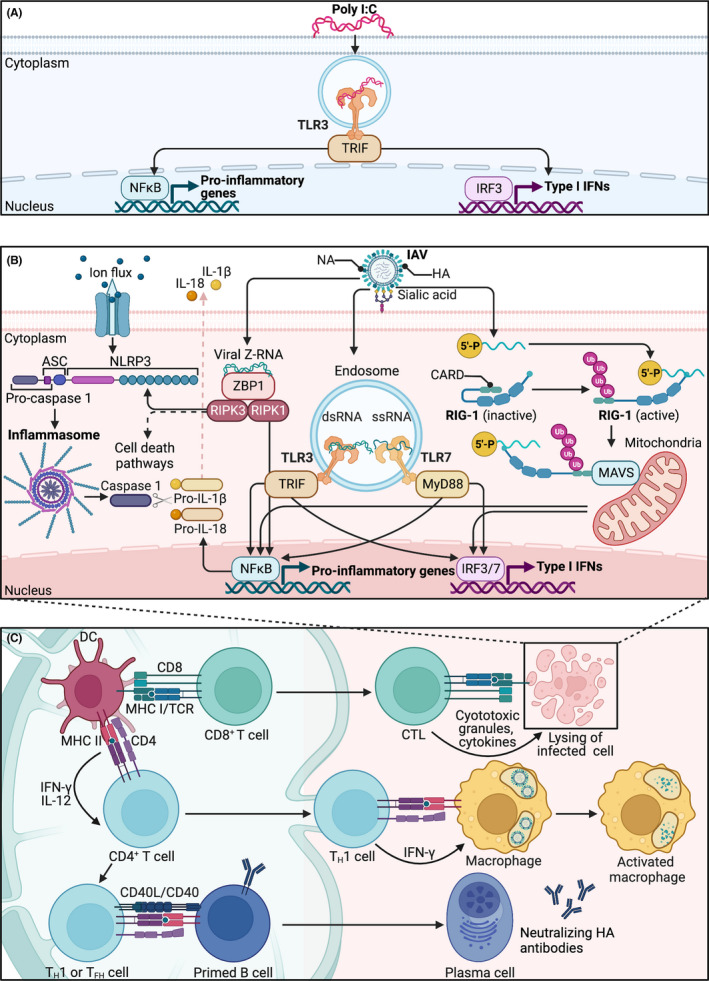
Influenza A virus initiates a more complex immune cascade than poly I:C. (A) Poly I:C activates the innate immune system by binding to cells that express endosomal toll‐like receptor 3 (TLR3). Recognition of poly I:C activates TIR‐domain‐containing adaptor‐inducing interferon‐β (TRIF), resulting in activation of interferon regulatory factor 3 (IRF3) and nuclear factor kappa B (NFκB). These transcriptional regulators mediate production of type I antiviral interferons (IFNs) and pro‐inflammatory cytokines. (B) Influenza A virus (IAV) initiates a more complex innate response in infected respiratory epithelial cells. IAV enters cells by binding of hemagglutinin (HA) glycoprotein to sialic acid residues. ssRNA from the virus itself, along with unidentified dsRNA from dying cells, activate TLR7 and TLR3, respectively. TLR7 signals through myeloid differentiation primary response 88 (Myd88), where it further activates IRF7 and NFκB. Unmodified 5′ triphosphate ends of ssRNA are recognized by retinoic acid‐inducible gene 1 (RIG‐I). Binding of ssRNA to RIG‐1 exposes the caspase activating and recruitment domain (CARD) to recruit polyubiquitinated (Ub) chains. Polyubiquitinated CARDs bind mitochondrial antiviral‐signaling proteins (MAVS), which ultimately activate IRF3 and NFκB pathways. NFκB produces pro‐IL‐18 and pro‐IL‐1β, which prime the nod‐like receptor family pyrin domain containing 3 (NLRP3) inflammasome. A second signal, such as ion flux, allows NLRP3 to form oligomers with adaptor protein ASC. ASC interacts with pro‐caspase 1 through CARD for cleavage of pro‐caspase 1 into active caspase 1. Cleaved caspase 1 then cleaves pro‐IL‐18 and pro‐IL‐1β into active IL‐18 and IL‐1β. Binding of viral Z‐RNA to Z‐DNA binding protein 1 (ZPB1) activates programmed cell death pathways and NFκB via receptor‐interacting protein kinases (RIPK) 1 and 3. The infected cell alerts surrounding immune cells to prime the (C) adaptive immune response. Respiratory dendritic cells (DCs) migrate to draining lymph nodes to present viral peptides via major histocompatibility complex (MHC) class I to naive CD8+ T cells, which differentiate into effector cytotoxic T cells (CTLs) that kill IAV‐infected cells. DCs also present viral peptides via MHC class II to naive CD4+ T cells. IFN‐γ and IL‐12 promote differentiation of T helper (TH) 1 cells. These effector TH1 cells produce IFN‐γ, which activates alveolar macrophages for direct lyses of virus. CD4+ T follicular helper (TFH) cells or TH1 cells activate primed B cells through antigen recognition and binding of CD40L/CD40. B cells differentiate into plasma cells that produce antibodies against HA glycoprotein. dsRNA, double‐stranded RNA; NA, neuraminidase; ssRNA, single‐stranded RNA; TCR, T‐cell receptor
A recent study demonstrated that prenatal immune activation via TLR7 generated contrasting behavioral abnormalities in offspring compared with poly I:C‐induced MIA. 43 Notably, poly I:C‐induced MIA is known to cause deficits in social behavior and increased anxiety‐like symptoms in exposed offspring. 78 Missig et al, 43 however, revealed that inoculation with a TLR7 agonist reduced anxiety‐like behavior and caused fragmentation of social behavior, leading to general hyperresponsivity of stimuli in exposed offspring. Additional work using TLR7 and TLR3 agonists showed differences in cytokine and chemokine expression in the placenta and fetal brain that was dependent on the specific TLR activated. 42 If maternal immune activation by TLR3 compared with TLR7 is sufficient to induce contrasting behavioral changes in offspring, it is safe to postulate that IAV could also produce differing MIA phenotypes due to its wide range of inflammatory pathways. While early studies have evaluated behavioral outcomes of IAV exposed offspring, 72 direct comparisons between IAV and poly I:C have not been extensively performed. One study reported distinct embryonic brain transcriptome clustering between poly I:C, IL‐6, and influenza‐induced MIA, with notable overlap in certain gene subsets. 44 While Garbett et al 44 believe that their findings support a theory wherein similar canonical pathways are altered among the three MIA groups, they also concede that differences in timing and molecular action of the stimulants play a major role. It is difficulties like these, highlighted in the sections below, that make direct comparisons between IAV and poly I:C quite complex in the context of MIA modeling.
2.2. Innate and adaptive immune responses of influenza A virus
Immunostimulants are largely restricted to TLR activation, whereas live viruses activate a plethora of immune‐related pathways. The following sections describe how influenza viruses, which infect host respiratory cells by binding hemagglutinin (HA) to sialic acid residues, 79 activate specific innate and adaptive immune responses.
2.2.1. Antiviral pathways of the infected cell
The respiratory epithelium is highly susceptible to inhaled antigens like IAV. Upon infection, these respiratory epithelial cells initiate intracellular innate defense mechanisms, which includes activation of TLRs (Figure 1B). It is important to note that TLR3 is constitutively expressed in alveolar and bronchial epithelial cells 80 whereas TLR7 is only expressed in bronchial epithelial cells. 81 In addition to TLRs, another PRR in the influenza cascade is retinoic acid‐inducible gene 1 (RIG‐I), which is a member of the RIG‐I‐like receptors (RLRs). RIG‐I resides intracellularly and binds to viral ssRNA by sensing unmodified 5′‐triphosphate ends. 82 Once ssRNA binds to RIG‐I, a conformational change exposes the caspase activating and recruitment domain (CARD) to recruit E3 ligases for binding of K63 polyubiquitinated chains. Polyubiquitinated CARDs then bind to mitochondrial antiviral‐signaling proteins (MAVS), which act through a signaling cascade to activate IRF3 and NFκB pathways. 83 Pro‐IL‐1β and pro‐IL‐18 from RLR and TLR activation prime the nod‐like receptor family pyrin domain containing 3 (NLRP3) inflammasome. 84 A second signal, such as ion flux through ion channels, allows NLRP3 to form oligomers with adaptor protein ASC, which then interacts with pro‐caspase I through CARD for cleavage of pro‐caspase I into active caspase 1. 77 Cleaved caspase I can then cleave pro‐IL‐1β and pro‐IL‐18 into inflammatory cytokines to induce pyroptosis of the infected cell. 85
Actively replicating IAV can also be sensed by Z‐DNA‐Binding Protein 1 (ZBP1), which acts as a central regulator of programmed cell death pathways and lung inflammation. 86 Upon recognition of IAV ligands—viral Z‐RNA (left‐handed Z‐form RNA) and ribonucleoprotein complex subunits—ZBP1 interacts with receptor‐interacting protein kinase (RIPK) 3 to induce programmed cell death pathways (apoptosis and necroptosis) and NLRP3 inflammasome‐dependent production of IL‐1β and IL‐18 and pyroptosis. 87 , 88 , 89 , 90 Active ZBP1 also interacts with RIPK1 to regulate cytokine production through NFκB and RIPK3‐independent apoptosis 87 , 91 (Figure 1B). As ZBP1 gene expression is induced by type I IFNs, 87 ZBP1 signaling is dependent upon both IAV replication and TLR and RIG‐I activation, 89 and therefore, occurs further along the infection timeline. Thus, influenza‐mediated activation of endosomal TLRs and intracellularly located PRRs (RIG‐I, ZBP1) in the infected cell contribute to a robust innate immune response. It is important to note that intracellularly complexed poly I:C, but not pure poly I:C, can stimulate both RLRs—such as RIG‐I and melanoma differentiation associated gene 5 (MDA‐5)—and NLRP3. 92 , 93 , 94 , 95 Since the majority of MIA models inoculate with pure poly I:C, it is safe to postulate that RLRs and NLRP3 play a negligible role in poly I:C‐initiated MIA. Furthermore, it has been demonstrated that poly I:C is not recognized by ZBP1, 87 further highlighting the unique ability of IAV to induce robust intracellular antiviral pathways that are not observed during poly I:C stimulation.
2.2.2. Adaptive immunity at the site of infection
Respiratory epithelial cells infected with influenza virus alert surrounding innate immune cells—such as alveolar macrophages, circulating monocytes, respiratory dendritic cells (DCs), natural killer cells (NKs), and innate lymphoid cells (ILCs)—via a complex cascade of inflammatory pathways (as reviewed in Ref. 77). Antiviral and pro‐inflammatory signals from infected and innate immune cells prime the adaptive immune response (Figure 1C). During IAV infection, respiratory DCs located at the mucosal surface migrate to draining lymph nodes where they present endogenous viral peptides via major histocompatibility complex (MHC) class I to naive CD8+ T cells. 96 Antigen presentation and co‐stimulatory signals from activated DCs promote differentiation and proliferation of CD8+ T cells into cytotoxic T lymphocytes (CTLs). These CTLs are further shaped by the inflammatory milieu of the lung where they kill IAV‐infected respiratory cells by releasing cytotoxic granules and inflammatory cytokines. 97 , 98 , 99 Migratory respiratory DCs also present phagocytosed viral peptides in draining lymph nodes via MHC class II to naive CD4+ T cells. 100 Antigen recognition, along with IFN‐γ production from innate cells and IL‐12 production from dendritic cells, promotes differentiation of CD4+ T cells into TH1 cells. 101 , 102 Effector TH1 cells produce copious amounts of IFN‐γ, which activates alveolar macrophages for direct lysis of the virus. 103 Another subset of CD4+ T cells in the lymph nodes are T follicular helper (TFH) cells, which activate B cells already primed with antigen for antibody production against the HA glycoprotein. 104 Recent studies indicate that TH1 cells also drive production of IAV antibodies, even in the absence of germinal center TFH cells. 105 While TH1 cells of type I immunity are thought to be the primary immune response in eliminating viral infections, TH17 cells of type III immunity also play an important role in influenza infections due to their residence in mucosal tissue. 106
2.3. Mucosal adaptive immunity: the common mucosal immune system theory
Interestingly, IL‐17‐producing TH17 cells have also been implicated in MIA‐mediated fetal brain abnormalities. 37 , 47 , 107 This work has shown that intraperitoneal injection of poly I:C in gestating dams leads to activation of pre‐existing intestinal TH17 cells, resulting in increased maternal production of IL‐17A over the course of 48 hours, subsequently resulting in fetal cortical malformations. 37 , 107 This group revealed that IL‐6, which was first implicated in abnormal development of MIA offspring in 2007, 35 is required for and precedes production of IL‐17A in poly I:C‐challenged dams. 37 They later demonstrated that segmented filamentous bacteria (SFB), which are known to promote TH17 cell development, 108 are required for MIA production of IL‐17A and subsequent offspring phenotypes, 107 which has since been replicated by others. 38 Work done by this group and others has shifted attention in the field towards IL‐17A‐mediated developmental abnormalities in offspring of poly I:C‐challenged dams.
Notably, IL‐17‐producing intestinal TH17 cells have also been identified as potential drivers of influenza‐mediated intestinal injury. 109 During poly I:C‐induced MIA, intestinal DCs detect poly I:C directly through TLR3 and induce IL‐17A production from pre‐existing intestinal TH17 cells 107 (ie, innate immune mechanisms; Figure 2A). Conversely, models of IAV infection suggest that adaptive mechanisms are initiated, involving misguided homing of activated TH1 cells to the intestine instead of the lungs, potentially through common mucosal pathways; this is known as the Common Mucosal Immune System Theory. IFN‐γ‐producing CCR9+CD4+ T cells disrupt intestinal microbial communities, which leads to antimicrobial signaling by intestinal epithelial cells (involving IL‐15), ultimately promoting TH17 cell polarization from resident naive CD4+ T cells and prompting production of IL‐17A 109 (Figure 2B). Notably, this occurs much later (6‐7 days post‐IAV inoculation) compared with 12‐48 hours post‐poly I:C injection. 37
FIGURE 2.
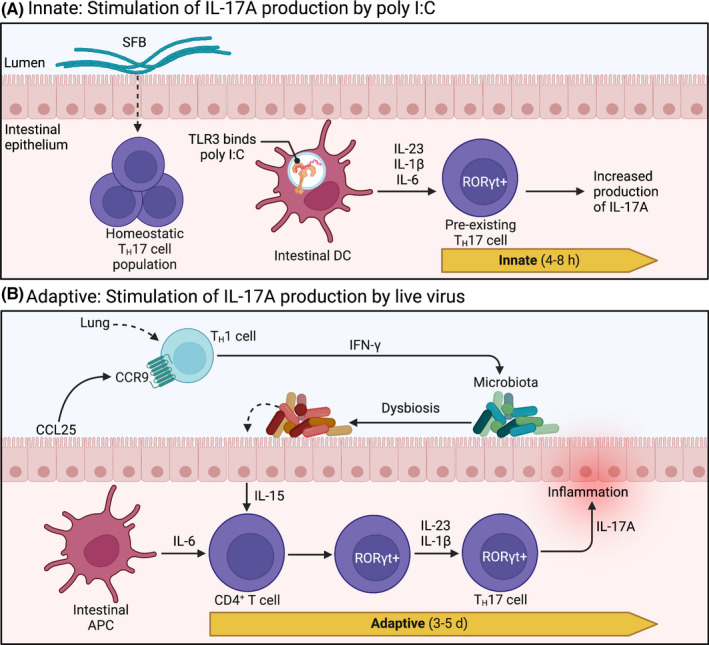
Intestinal TH17 cells in poly I:C and influenza models. (A) RORγt+ TH17 cells are constitutively expressed in the intestine and rely on commensal segmented filamentous bacteria (SFB) for homeostatic regulation. Upon intraperitoneal administration of poly I:C, intestinal dendritic cells (DC) detect poly I:C directly through TLR3, initiating production of IL‐1β, IL‐6, and IL‐23. These secreted cytokines stimulate pre‐existing resident TH17 cells to produce IL‐17A. This innate immune signaling pattern is initiated quickly, within 4‐8 h, resulting in a measurable accumulation of IL‐17A in maternal circulation at 48 h. (B) During respiratory influenza A virus (IAV) infection, CCR9+CD4+ TH1 cells—stimulated in the lungs—are recruited to the uninfected intestine by chemokine CCL25, where they produce copious amounts of IFN‐γ. Subsequent disruption of endogenous gut microbes (ie, dysbiosis) stimulates intestinal epithelial cells to produce IL‐15. In conjunction with stimulation by intestinal antigen presenting cells (APC), cytokines IL‐15, IL‐6, IL‐23, and IL‐1β prompt naive CD4+ T cells to express transcription factor RORγt, promoting polarization towards TH17 lineage. Polarized effector TH17 cells then produce IL‐17A, which has been linked to intestinal injury during IAV infection. This adaptive immune response takes up to 5 d, resulting in measurable increases in intestinal TH17 cells within 6‐7 d after IAV infection. d, days; h, hours; RORγt, retinoic acid receptor‐related orphan receptor γt; TLR, toll‐like receptor. Images were adapted from Cua and Tato (2010), 110 and the description of the common mucosal immune response to IAV was informed by Wang et al (2014). 109
We have established a clinically relevant model of maternal IAV infection to determine whether a similar adaptive response drives IL‐17A production from TH17 cells during gestation. 111 IAV‐infected gestating dams upregulated intestinal RORγt (retinoic acid receptor‐related orphan nuclear receptor gamma t)—the master regulator of IL‐17 producing cells 112 —seven days post‐inoculation, indicating a potential increase in numbers of TH17 cells. However, this was not accompanied by enhanced IL‐17A production. 111 This finding does not exclude the possibility that maternal IL‐17A production might be enhanced at earlier time points following infection. Furthermore, it is possible that a more severe IAV infection would further enhance propagation and/or activation of intestinal RORγt+ cells beyond what we observed with a moderately pathogenic IAV strain. It is also important to note that cells other than TH17 cells express RORγt and can produce IL‐17A. Other RORγt+ IL‐17‐producing cells include innate γδ T cells, ILC3s, and invariant natural killer T (iNKT) cells; non‐lymphoid Paneth cells and neutrophils can also produce IL‐17, but do not express RORγt. 110 While the IL‐17 family of cytokines includes IL‐17A through F, IL‐17A is the predominant effector molecule released by all IL‐17‐producing cells and primarily functions to recruit neutrophils and enhance mucosal barrier function. 110
Altogether, sufficient evidence indicates that gestational IAV infection, like poly I:C‐induced MIA, can lead to heightened levels of maternal IL‐17A, though further studies are needed. Moreover, the signaling mechanisms preceding an augmentation of IL‐17A production between the two models involve disparate pathways: while poly I:C initiates an immediate innate immune signaling cascade, IAV infection induces adaptive immune mechanisms that involve a disruption in endogenous microbes (ie, dysbiosis; Figure 2B). This gut dysbiosis appears to be at the crux of IAV‐induced intestinal inflammation (as reviewed in Ref. 113). Although the virus does not infect intestinal tissue, gastroenteritis‐like symptoms and disrupted intestinal microbial communities are often evident during respiratory IAV infection. 114 , 115 These shifts in the gut microbiota, 116 accompanied by altered production of antimicrobial peptides, 115 , 117 promote intestinal inflammation. Importantly, depleting gut microbes through antibiotic treatment prior to IAV infection protects against intestinal injury and inhibits IL‐17A production. 109
Recent studies implicate the gut microbiota, and specifically, the distinction between commensal and pathogenic microbes, in shaping homeostatic versus inflammatory TH17 subtypes. 118 Whether intestinal microbial shifts caused by respiratory IAV infection induce a more inflammatory TH17 cell phenotype needs to be tested. It is also possible that inflammatory conditions unique to IAV infection drive specific TH17 cell subtypes. It was previously thought that TGF‐β and IL‐6 were both necessary to promote differentiation of naive T cells into TH17 cells 112 ; however, other studies have shown that IL‐6 (in conjunction with IL‐23 and IL‐1β) in the absence of TGF‐β leads to a more pathogenic TH17 phenotype. 119 , 120 In our model of IAV‐induced MIA, we observe an upregulation of IL‐6 and RORγt mRNA transcripts in the ileum and colon of IAV‐infected dams at 2 days post‐infection with no changes in TGF‐β transcripts (unpublished data). This could indicate a pathogenic TH17 subtype not seen in poly I:C models due to IAV‐specific gut dysbiosis. Overall, the evidence suggests that microbial disruption precedes intestinal inflammation during respiratory IAV infection. More importantly, complex infection‐induced dysbiosis is a crucial and clinically relevant phenotype that is not properly recapitulated in poly I:C MIA models.
While increasing evidence links maternal inflammation—and inflammation‐mediated dysbiosis—with fetal brain abnormalities, the mechanisms behind these neurodevelopmental perturbations have yet to be fully elucidated. In the following section, we emphasize the role of microglia, the innate immune cells of the brain, in homeostatic fetal brain development and describe how disrupting these cells results in aberrant neurological and behavioral phenotypes that resemble NDD pathologies. We also highlight the potential crosstalk between embryonic microglia and the maternal microbiome during pregnancy.
3. EMBRYONIC MICROGLIA AS CENTRAL PLAYERS IN MIA PHENOTYPES
As the primary resident central nervous system (CNS) macrophages, microglial cells are uniquely positioned to bind and respond to an extensive repertoire of signaling molecules, 121 , 122 , 123 resulting in a keen sensitivity to dynamic changes in their environment. Thus, they are often identified as the most likely cell type within the fetal brain to respond to MIA‐induced signaling. We hypothesize that immune perturbations originating within maternal tissues during gestation can disrupt the physiological functioning of fetal microglial cells, with lasting consequences for neural circuitry formation and function. Exactly which cellular functions are shifted during MIA, and how, has yet to be clarified.
Beginning in early embryonic development, yolk sac‐derived microglial precursors invade the brain parenchyma, differentiate into immature microglia, and migrate throughout the brain in synchronized time‐ and location‐dependent patterns. 124 , 125 During this colonization period, microglia sequentially populate the deeper cortical layers while also congregating at specific brain areas (eg, axonal tracts, neurogenic niches) where they support and regulate neurogenesis. 50 , 126 , 127 , 128 , 129 As microglial colonization occurs concurrently with neurogenesis and prior to the generation of other glial cells (astrocytes and oligodendrocytes), microglia are unique in their ability to aid and direct prenatal neurodevelopmental processes. 130 Providing support through production and release of neurotrophic and immune regulatory factors, 131 microglia interact closely with neural precursors and radial glia across multiple embryonic brain regions. 49 , 129 , 132 In this early period, they promote neural precursor cell proliferation, axonal outgrowth, and interneuron wiring, 127 , 133 , 134 followed by selective phagocytosis of excess precursors. 50 Notably, MIA has been shown to perturb each of these microglial‐mediated neurodevelopmental processes 50 , 127 , 133 and to interrupt microglial proliferation, maturation, and motility 122 , 135 , 136 , 137 (Figure 3).
FIGURE 3.
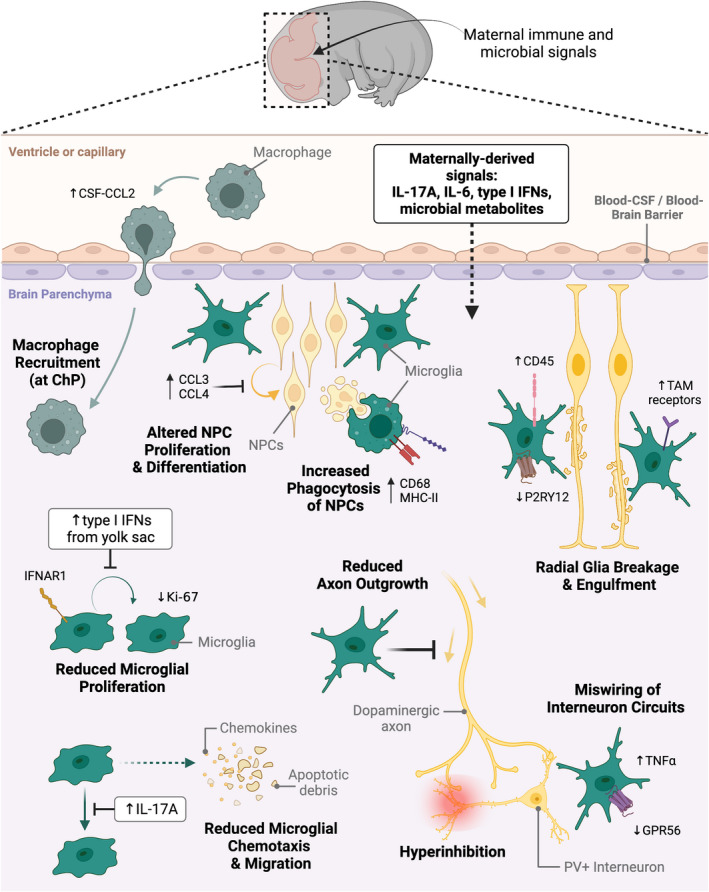
Embryonic microglia as central players in maternal immune activation neuropathologies. Maternally derived immune and microbial signals direct neurodevelopmental trajectories during embryonic development. These signals are believed to cross the immature blood‐brain barrier and enter the brain parenchyma. Once there, they are thought to bind receptors on various CNS cell types, including microglia. A multitude of studies have revealed microglial‐directed alterations to various neurodevelopmental processes—including neural progenitor cell (NPC) proliferation and differentiation, radial glia scaffold extension, dopaminergic axonal outgrowth, and wiring of inhibitory interneuron circuits—resulting from abnormal microglial behaviors. Gestational insult leads to increased production of chemokines CCL3 and CCL4 by a subpopulation of microglia adjacent to NPCs, altering the proliferative capacity and maturation fate of these cells. Additionally, microglial upregulation of phagocytic markers CD68 and MHCII in response to maternally derived factors, including IL‐17A, may result in exaggerated engulfment of NPCs. Gestational insult may also lead to a more activated microglial phenotype through upregulation of CD45 and downregulation of purinergic receptor P2RY12. Neuroprotective TAM receptor signaling on microglia allows them to sense disruptions in projecting radial glia, resulting in increased degeneration and phagocytosis of radial glia cells following insult. MIA has also been shown to reduce microglial proliferation, measured in part by reduced Ki‐67 expression. This is driven by increased type I interferon (IFN) production originating in the yolk sac, which binds interferon alpha receptor 1 (IFNAR1) on microglia. Impaired chemotactic ability of microglia following MIA may also be related to IL‐17A‐specific inhibition of microglial migratory capacity, which alters their localization in the developing brain. IL‐17A has also been shown to direct microglial expression of G Protein‐Coupled Receptor 56 (GPR56), which, in conjunction with increased TNFα, appears to impair parvalbumin‐positive (PV+) interneuron generation. This impairment ultimately leads to miswiring of inhibitory interneuron circuits, triggering hyperinhibition in the neocortex. MIA‐induced inflammation in the embryonic brain is also propagated by increased production of CCL2 in cerebral spinal fluid (CSF), prompting macrophage recruitment and entry into the choroid plexus (ChP). Figure and legend creation was informed by data cited in the main text. TAM, family of receptor tyrosine kinases TYRO, AXL, and MERTK
Microglial elimination studies indicate that these cells are essential for establishing brain circuitry, such that excessive growth and distribution of neurons occurs in their absence. 127 Other evidence supports the idea that microglia communicate bi‐directionally with neurons and neural precursor cells, allowing for microglia‐regulated neuronal elimination and survival. 49 , 132 , 138 Several studies demonstrate that prenatal inflammatory insults impact how embryonic microglia regulate neural progenitor cell populations 49 , 50 and monitor radial glia projections. 132 In some cases, eliminating embryonic microglia during prenatal insult is sufficient to rescue neuronal or behavioral phenotypes, indicating that microglia are the primary mediators of these altered phenotypes during development. 49 Intriguingly, some groups have described overlap between the dysfunctional neuronal circuit phenotypes that arise during microglial elimination and those that are produced by MIA, 127 , 133 suggesting that MIA may in some way prevent fetal microglia from fulfilling their usual neurotrophic roles during development. Indeed, transient depletion of microglia in CX3CR1 knock‐out mice results in disrupted synaptic pruning, decreased functional brain connectivity, social deficits, and increased restricted repetitive behaviors each indicative of NDDs. 139 Disruption of microglial autophagy also leads to excess synapse accumulation, causing decreased sociability in mice. 140 , 141 The integrity of synapses and connectivity networks is also compromised in offspring exposed to MIA. 127 , 142 , 143 , 144 , 145 , 146
Disrupted neurodevelopmental circuitries and synapse integrity can manifest in various ways in the context of neuropathology. In particular, ASD brains are often characterized by altered cortical thickness/layering 147 and cell patterning, 148 altered neurogenesis, 147 , 149 and both over‐ and under‐connectivity. 150 , 151 , 152 Evidence of similar alterations in schizophrenic patients also exists. 61 , 153 Specifically, it appears that insufficiencies in interneuron development, which lead to an altered balance of excitation and inhibition, are hallmark to both MIA animal models 45 , 48 , 133 , 154 , 155 and human neurodevelopmental disorders. 156 , 157 One group has consistently demonstrated microglial involvement in interneuron positioning and density in the developing brain, 127 which results in altered temporal regulation of inhibitory circuit wiring. 133 Another recent study demonstrates that microglia might regulate MIA‐induced deficits in interneuron development through G protein‐coupled receptor 56 (GPR56). 48 Whether MIA induces a predictable dysregulated microglial phenotype, however, is still up for debate.
3.1. Microglia priming theory in MIA
One popular theory describing the involvement of microglia in psychiatric illness surrounds the concept of “microglia priming” or “psychological immune memory”, wherein a psychological or immune stressor early in life results in an over‐activation of microglia. This leads to prolonged hyper‐activation in adolescent and adult brains, even after the stressor is removed. 158 , 159 However, conclusions drawn from MIA models examining persistent hyper‐activation of microglial cells in juvenile and adult animals are broadly inconclusive, primarily because most studies use different rodent species or strains and/or different MIA induction time points. 62 , 64 , 136 , 160 , 161 , 162 , 163 , 164 Transcriptional analysis of microglia from MIA‐challenged mice revealed that differential expression in developmental genes was much more evident at the early microglia stage compared with the adult stage, suggesting that microglia phenotype might realign to normal by adulthood. 122 Several studies indicate that cellular and inflammatory CNS markers, 160 and microglia density and amoeboid morphology, 165 are transiently elevated due to MIA but resolve shortly after birth. This is in agreement with clinical data on patients with neuropsychiatric disorders who do not display heightened inflammatory markers at birth or in early life. 166
We have previously shown, using a swine model of viral‐induced MIA, that prenatal microglia display altered transcripts that are temporally regulated and sexually dimorphic. These temporal changes in microglial transcription coincided with alterations in fetal microglial phenotype (expression of MHCII and CD68), function (phagocytosis and chemotaxis), and cell density, but not cell morphology. 167 Notably, the stunted phagocytic and chemotactic activity, as well as increased amygdalar microglial density observed 7 days after maternal viral infection was almost completely resolved 2 weeks later (21 days post‐infection), supporting the idea that fetal microglia may be transiently perturbed by viral‐induced MIA. Furthermore, we demonstrated that postnatal microglia, isolated from weanling MIA piglets, produced normal levels of inflammatory signaling molecules when stimulated in vitro, despite measurable changes in social behavior at this time point. 168 Collectively, our data indicate that viral‐induced MIA acutely perturbs fetal microglial transcripts and functional profiles, and that the resulting phenotypes are brain region‐ and sex‐dependent. This transient perturbation may be sufficient to disrupt neuronal circuit formation and/or function—at least as measured by the manifestation of aberrant social behaviors—despite a lack of microglial “priming”.
Here, it is important to note that the phenomenon of microglia priming also relies on the concept of microglial cells being long‐lived. Early reports suggested that population turnover was slow, though even at the time it was acknowledged that methodological limitations likely resulted in an underestimation of microglia proliferation. 169 Askew et al 170 revisited this concept, discovering that microglial turnover rates in the adult brain are actually much higher than expected, and result in a complete renewal and comprehensive restructuring of the microglial landscape approximately every 96 days. Furthermore, microglial renewal appears to vary by location, 171 suggesting that there is functional significance in the disparate turnover rates between specific brain regions. Therefore, reevaluation of the theory of microglial‐specific “memory” in the context of this new backdrop is required.
A more updated perspective points towards the possibility of differing levels of resilience and susceptibility among MIA offspring. 172 , 173 Indeed, evidence from both humans and rodents supports the concept that only a subgroup of individuals, those classified as having a “high immune profile”, present with microglial anomalies. 174 In this backdrop, it is possible that a primed microglial phenotype exists in only a subset of MIA offspring, those classified as susceptible, with high baseline inflammation. Future studies that are properly powered to detect these subgroup differences will be required to further investigate this possibility.
3.2. Microbes, IL‐17 signaling, and microglia
Several studies have shown an alleviation of offspring behavioral and neuropathological phenotypes after postnatal administration of minocycline, 163 , 175 , 176 , 177 a potent inhibitor of microglial activation. While these studies suggest that MIA offspring display heightened baseline microglial immunoreactivity, it is unclear whether the therapeutic effect of minocycline in this context is mostly due to its direct anti‐inflammatory properties versus indirect reduction in low‐grade stimulation from exogenous factors—namely, microbes or microbial products. As minocycline is a tetracycline antibiotic, intraperitoneal administration of this drug over the course of several weeks may also be altering the endogenous intestinal microbial population. The ability of endogenous microbes to regulate peripheral and central immune responses in MIA offspring has recently been described, 178 , 179 , 180 and a thorough coverage of these data is beyond the scope of this review. However, a critical component of microbial effector potential is the bi‐directional communication with microglia, likely through metabolite and immune signaling pathways.
Expression of microglia “sensome” genes, a collection of transcripts encoding receptors and proteins that sense endogenous and exogenous ligands, 121 begins prenatally. 123 Early expression of these genes likely explains why microglia are sensitive to shifts in microbiome diversity, 123 , 181 , 182 even during the prenatal period (as reviewed in Ref. 183). While much is known about the impact of endogenous microbes on early postnatal brain development, recent evidence has shed light on how maternal microbes, mostly through metabolite production, shape development in utero. Critical neurodevelopmental processes, such as axonal outgrowth, are intrinsically tied to microbiota‐dependent metabolites produced by the maternal gut microbiome during gestation. 184 Altered composition of the maternal intestinal and vaginal microbiome during pregnancy has also been directly implicated in offspring brain and behavioral deficits. 38 , 107 , 178 , 185 , 186 , 187 Furthermore, germ‐free mice display disrupted embryonic blood‐brain barrier (BBB) formation and increased vascular permeability, 188 as well as region‐specific alterations in perinatal microglial colonization and activation patterns. 189 Thus, the interplay between inflammatory signaling and microbial dysbiosis during maternal infection represents a novel pathway by which MIA may be altering fetal microglial function.
As discussed above, a common thread in recent MIA studies surrounds the ability of maternal microbes to induce production of effector cytokine IL‐17A, which disrupts offspring brain and immune development. 37 , 38 , 47 , 107 , 180 In a series of elegant experiments, G. Choi and J. Huh's group demonstrated that MIA induces neuronal abnormalities in specific cortical regions—the primary somatosensory cortex (S1) and the temporal association cortex (TeA)—through induction of IL‐17 receptor subunit A (IL‐17RA) on postmitotic neurons. 37 , 47 These data indicate that an imbalance in excitation and inhibition in this discrete neural circuit is involved in the disruption of social behaviors, though how IL‐17RA activation leads to cortical malformations, and the specific cell types involved in this pathology, is still not entirely clear. 47 Microglia, which express IL‐17RA 190 and are responsive to IL‐17 in vitro, 191 have been shown to increase their proliferation, trafficking, and activation in response to sustained production of IL‐17A during pathological states. 192 Thus, it follows that fetal microglia, in addition to other IL‐17RA+ cells, would be sensitive to increases in IL‐17A. Indeed, not only does intraventricular administration of recombinant IL‐17A (rIL‐17A) into fetal brains recapitulate cortical malformations induced by poly I:C‐MIA, 37 but recent studies confirm that microglia are also affected. When Sasaki et al 193 injected E14.5 embryos with rIL‐17A, they observed distinct clustering of microglial cells in the subventricular medial cortex, and increased numbers of microglia expressing CD68. These findings, while mostly observational, could indicate that microglia are not only more pro‐phagocytic in response to IL‐17A, but also that their migratory patterns are disrupted, which is likely to alter the cells' ability to support healthy corticogenesis. Yu et al 48 recently demonstrated that the ability of microglia to provide neurotrophic support to interneuron progenitors is in fact IL‐17A dependent. Their data bolster the hypothesis that specific receptors on microglia are indeed molecular targets of MIA, and that regulation of these receptors is downstream of IL‐17A signaling.
Critically, the potential hematogenous transfer of maternally derived IL‐17A into fetal circulation, versus MIA‐induced amplification of placental‐ or fetal‐derived IL‐17A production, has yet to be clarified. A specific facet of our future work is focused on tracing maternally derived molecules across fetal tissue barriers in order to answer this question. Altogether, we believe that there is sufficient evidence to suggest that maternally derived microbial metabolites and immune signaling molecules are collectively binding receptors on fetal microglia during MIA, ultimately redirecting these cells from their neurotrophic roles. We believe that this shift in microglial function, while not solely responsible for MIA‐induced neuropathologies, is nonetheless a significant contributor.
Finally, the overwhelming lack of studies investigating microglial responses to live virus‐induced MIA makes it extremely challenging to determine whether these cells are in fact consistently disrupted. Our investigations using maternal viral infection in swine indicate that fetal microglia are indeed sensitive to inflammatory conditions resulting from respiratory disease in pregnant sows, and that their phenotypic changes are fluid. Only by expanding the use of live viruses in MIA modeling can more concrete conclusions be drawn regarding microglial involvement in MIA‐induced neuropathologies.
4. FUTURE PERSPECTIVES AND CONSIDERATIONS FOR MIA MODELING
The various complexities which arise when using live viruses to model MIA can introduce considerable variability in offspring outcomes. This inherent variability, however, is necessary for capturing the full spectrum of viral‐induced NDD pathologies and is a critical step toward developing clinically translatable therapies. Thus, it is important to consider a multitude of parameters when designing preclinical MIA experiments. Here, we briefly review the impacts of gestational timing in MIA modeling and emphasize the need for a reliable and replicable IAV‐induced MIA phenotype.
4.1. Timing of prenatal immune activation
The timing of prenatal exposure to maternal inflammation plays an important role in the characteristics of neuroanatomical and the behavioral abnormalities that manifest. 45 , 69 , 194 , 195 , 196 , 197 One of the first epidemiological studies linking gestational influenza infection with increased risk of schizophrenia in offspring found that infection during the first trimester was associated with a higher risk of schizophrenia development in offspring than infection during the second and third trimester. 3 , 198 These findings are supported by a series of experiments conducted by U. Meyer and colleagues demonstrating that gestational timing of poly I:C exposure leads to distinct behavioral and neuropathological phenotypes in offspring. Namely, gestational day (GD)9‐ but not GD17‐exposed offspring showed increased instances of anxiety‐like behavior, impaired sensorimotor gating, and reduction of dopamine D1 receptors in the medial prefrontal cortex. 194 , 195 Contrastingly, GD17‐ but not GD9‐exposed offspring showed increased instances of preservative behavior, impaired spatial working memory, an increase in apoptotic cells in the hippocampus, and a reduction in NMDA receptor subunit NR1 in the dorsal hippocampus. 194 , 195 In the 2006 study, neuropathology of GD9‐exposed offspring showed reduction of reelin, which is crucial for proper neuronal migration and positioning during corticogenesis, 65 in the hippocampus. However, the 2008 study failed to recapitulate this result and rather showed reduction of reelin‐positive cells in the medial prefrontal cortex, not the hippocampus, at both time points. The difference in results indicates postnatal timing could also play a factor in neuropathology.
More recent studies continue to highlight the importance of prenatal timing in poly I:C‐induced MIA models. 45 , 196 , 197 A central theme in these studies is the temporally regulated impairment of GABAergic interneuron subtypes. Origination and migration of cortical interneurons takes place over the course of approximately 9 days in the healthy embryonic mouse brain. Maternal poly I:C injections given at three different gestational time points along this trajectory produces distinct proliferative outcomes among interneuron subtypes, which align with earlier or later time points during which each interneuron subtype originates. 45 While it is crucial to continue to identify and characterize MIA‐induced abnormalities in interneuron development, the effects of IAV infection are likely to have drastically different consequences. In this case, the maternal inflammatory response to IAV would extend across the 9‐day interneuron development period, in contrast to the acute inflammatory response to poly I:C, which allowed certain interneuron subtypes to be spared. This example is one of many that epitomizes the need for live virus‐induced MIA modeling.
While limited, some evidence does indicate that gestational timing of influenza exposure during pregnancy results in disparate fetal outcomes. Maternal influenza infection on GD9 versus GD18 results in neuropathological phenotypes unique to each time point. 69 However, behavioral outcomes were not assessed in this study, demonstrating the need to reinstate the use of virus‐induced MIA models to compare them more accurately to poly I:C‐induced MIA models. In our IAV‐initiated MIA model, we inoculate dams with influenza on GD9.5 based on epidemiological data 3 and because it is around the time microglia migrate from the yolk sac to the fetal brain to aid in a multitude of neurogenic processes. 199 A caveat of IAV‐initiated MIA rodent models is that inoculation at later time points (as done in poly I:C models) might not be sufficient to capture the complete innate and adaptive immune landscape in utero, as the murine gestational period is so brief.
Notably, the time points discussed here refer to murine gestation, where GD9‐10 represents approximately the end of the first trimester in humans and GD16‐18 represents the end of the second trimester in humans. 200 Other studies have evaluated timing of MIA induction in rats 201 , 202 , 203 ; however, comparatively little is known beyond rodent models. Utilizing swine and non‐human primate models more accurately captures the human pregnancy timeline and will be influential in understanding just how gestational timing affects offspring development. Critically, the timing of prenatal insult could indicate why certain neurodevelopmental disorders (eg, schizophrenia, autism, obsessive compulsive disorder) develop over others.
4.2. Reproducibility and variability of MIA modeling
The reproducibility of MIA phenotypes is not only contingent upon timing of infection but also the dosage given, routes of administration, animal vendor, and in cases of viral infection, type and strain used. For thorough review on the reproducibility of immunostimulant‐induced MIA see Kentner et al 15
Since the advent of the poly I:C MIA model, the potency of the maternal immune response has significantly decreased due to variance in molecular weights between and within manufacturers. 204 , 205 Estes et al 205 showed that the standard 20 mg/kg intraperitoneal (i.p.) injection of poly I:C failed to produce the high levels (>10 000 pg/mL) of IL‐6 in maternal circulation previously seen at the field's inception. 35 Route of poly I:C administration is also subject to variability and is largely underappreciated in the MIA space. For instance, intravenous (i.v.) injection of poly I:C is much more lethal than i.p. injection 206 because it immediately enters systemic circulation whereas i.p. inoculants first travel through the peritoneal cavity and peritoneal lymphatic system before entering circulation. 207 While this is accounted for by administering poly I:C intravenously at roughly a fivefold lower dose than when administered peritoneally, 208 it does not take into account the pharmacokinetics of the stimulus and could produce differences in maternal inflammation. It is also important to note that live respiratory viruses like IAV enter intranasally, further adding to potential differences in systemic responses. Furthermore, recent work has shown that even the same strain of mice from different animal vendors influences MIA phenotypes. Work by G. Choi and J. Huh's group demonstrated that C57BL/6 mice from Taconic Biosciences, which naturally harbor intestinal SFB, produce robust levels of IL‐17A after poly I:C administration, and those from Jackson Laboratory, which do not have SFB, do not. 37 , 107 Subsequent studies have verified poly I:C‐induced upregulation of IL‐17A production is unique to SFB+ dams and that MIA in these mice preferentially leads to autism‐like phenotypes not seen in SFB− mice. 38 Thus, mouse vendor or SFB status should also be taken into account when designing MIA experiments.
Similar discrepancies exist in influenza‐mediated MIA models, albeit to an even greater extent due to the variation of virus subtype and strain used. It is important to note that the majority of murine influenza models come from human influenza viruses that have been mouse‐adapted through serial passage in the mouse lung (as reviewed in Ref. 209). Some of the earliest gestational influenza models determined sublethal dosage of influenza type A subtype H1N1 (A/H1N1) strain NWS/33 as 105.25 median tissue culture infectious dose (TCID50). 69 Another study surveyed the effects of the 2009 influenza A/H1N1 pandemic during gestation using 2x106 plaque‐forming units (PFU) of wild‐type virus and 150 PFU of mutant‐type virus. 210 To understand the effects of gestational influenza infection with low pathogenicity, one group used endemic influenza A/H1N1 strain Brisbane/59/07 at 155 PFU. 24 Until recently, little was known about gestational influenza A subtype H3N2 despite it being just as prevalent as H1N1. 211 We recently demonstrated that a moderately pathogenic strain of influenza A/H3N2, X31 (IAV‐X31), at 103 TCID50 failed to induce fetal neuroinflammatory transcripts commonly seen in other MIA models. 111 Notably, this dose is roughly 10× lower than another study that used IAV‐X31 at 104 PFUs, 23 which could indicate that a threshold of infection exists in MIA models.
Using a wide variety of influenza subtypes and strains more accurately reflects the virus's seasonality; however, this same variability makes it difficult to establish a robust IAV‐induced MIA phenotype. Furthermore, it is difficult to determine how comparable murine infectious doses are to human infections. The complexity of variation in live viruses has made the use of immunostimulants, such as poly I:C, an attractive option. However, recreating the clinical condition with live viruses is crucial in understanding disease etiologies of neurodevelopmental disorders in humans.
5. CONCLUSIONS
An immense amount of progress has been made in the MIA field in the past several decades. Yet, the predominant theories in the field still come from animal models that rely on non‐pathogenic viral or bacterial mimics to induce acute and predictable immune responses. Instead, we aim to emphasize the need for more clinically relevant live pathogen MIA models to confirm and expand upon the current findings. In particular, the potential involvement of maternal TH17 cells and IL‐17A signaling needs to be examined and defined in models of live viral infection. Furthermore, fetal microglial functions and phenotypes in response to viral‐induced MIA need to be flushed out, along with the up‐ and down‐stream signaling pathways leading to and resulting from possible dysregulated microglial activity. Overall, dysfunction in microglia during this critical developmental window could be the missing link between inflammatory signaling and disrupted brain development.
Exactly how maternal intestinal immune cells ultimately trigger disrupted brain development is still uncertain, especially because maternally derived effector molecules must trespass the tissues of the maternal‐fetal interface before they can access the fetal brain. Therefore, it is also important to understand the role of the placenta, an organ unique to pregnancy that acts as a physical and immune barrier between dam and fetus, in the context of MIA. 212 , 213 We and others have observed changes in placental weights and transcriptional profiles (including altered inflammatory and antimicrobial responses) during maternal IAV infection, suggesting a partial breakdown of placental barrier integrity and perfusion. 23 , 44 , 111 Remarkably, the fetal neuroimmune landscape appeared to be protected during moderate IAV infection. 111 This work raised the question of whether there may be an infection severity threshold beyond which placental integrity is critically compromised, resulting in fetal brain abnormalities. Although controversial, the placenta may also mediate the vertical transmission of microbes between mother and fetus. 214 , 215 Overall, our understanding of how maternal endogenous microbes contribute to the fetal microenvironment, especially in the context of MIA, is at a relatively nascent stage.
The emergence of a global coronavirus pandemic has highlighted the urgency to dissect the pathways driving MIA‐mediated developmental abnormalities. 216 Unfortunately, newly emerging preliminary evidence links maternal SARS‐CoV‐2 infection with increased instances of NDDs in the first year of life. 217 The continuous threat of re‐merging global pathogens like IAV and coronaviruses epitomizes the necessity of using these live pathogens in preclinical MIA modeling to better evaluate complete inflammatory cascades and improve translation to the clinic.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
We thank the members of the Antonson Developmental Neuroimmunology Laboratory for valuable discussion throughout the process of writing this manuscript, from conception to finalization. A multitude of past and current laboratory members, mentors, and collaborators contributed to our ideas and knowledge, and we are grateful to all. Images created with BioRender.com.
Otero AM, Antonson AM. At the crux of maternal immune activation: Viruses, microglia, microbes, and IL‐17A . Immunol Rev. 2022;311:205‐223. doi: 10.1111/imr.13125
This article is part of a series of reviews covering Neuroimmunology appearing in Volume 311 of Immunological Reviews.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Mednick SA. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry. 1988;45(2):189. doi: 10.1001/archpsyc.1988.01800260109013 [DOI] [PubMed] [Google Scholar]
- 2. Al‐Haddad BJS, Jacobsson B, Chabra S, et al. Long‐term risk of neuropsychiatric disease after exposure to infection in utero. JAMA Psychiatry. 2019;76(6):594. doi: 10.1001/jamapsychiatry.2019.0029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brown AS, Begg MD, Gravenstein S, et al. Serologic evidence of prenatal influenza in the Etiology of schizophrenia. Arch Gen Psychiatry. 2004;61(8):774. doi: 10.1001/archpsyc.61.8.774 [DOI] [PubMed] [Google Scholar]
- 4. Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167(3):261‐280. doi: 10.1176/appi.ajp.2009.09030361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miller BJ, Culpepper N, Rapaport MH, Buckley P. Prenatal inflammation and neurodevelopment in schizophrenia: a review of human studies. Prog Neuro‐Psychopharmacol Biol Psychiatry. 2013;42:92‐100. doi: 10.1016/j.pnpbp.2012.03.010 [DOI] [PubMed] [Google Scholar]
- 6. Khandaker GM, Zimbron J, Lewis G, Jones PB. Prenatal maternal infection, neurodevelopment and adult schizophrenia: a systematic review of population‐based studies. Psychol Med. 2013;43(2):239‐257. doi: 10.1017/S0033291712000736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheslack‐Postava K, Brown AS. Prenatal infection and schizophrenia: a decade of further progress. Schizophr Res. 2021;44(2):245‐258. doi: 10.1016/j.schres.2021.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kępińska AP, Iyegbe CO, Vernon AC, Yolken R, Murray RM, Pollak TA. Schizophrenia and influenza at the centenary of the 1918‐1919 Spanish influenza pandemic: mechanisms of psychosis risk. Front Psych. 2020;11:72. doi: 10.3389/fpsyt.2020.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brown AS, Sourander A, Hinkka‐Yli‐Salomäki S, McKeague IW, Sundvall J, Surcel HM. Elevated maternal C‐reactive protein and autism in a national birth cohort. Mol Psychiatry. 2014;19(2):259‐264. doi: 10.1038/mp.2012.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang HY, Xu LL, Shao L, et al. Maternal infection during pregnancy and risk of autism spectrum disorders: a systematic review and meta‐analysis. Brain Behav Immun. 2016;58:165‐172. doi: 10.1016/j.bbi.2016.06.005 [DOI] [PubMed] [Google Scholar]
- 11. Hornig M, Bresnahan MA, Che X, et al. Prenatal fever and autism risk. Mol Psychiatry. 2018;23(3):759‐766. doi: 10.1038/mp.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patel S, Masi A, Dale RC, et al. Social impairments in autism spectrum disorder are related to maternal immune history profile. Mol Psychiatry. 2018;23(8):1794‐1797. doi: 10.1038/mp.2017.201 [DOI] [PubMed] [Google Scholar]
- 13. Atladóttir HÓ, Thorsen P, Østergaard L, et al. Maternal infection requiring hospitalization during pregnancy and autism Spectrum disorders. J Autism Dev Disord. 2010;40(12):1423‐1430. doi: 10.1007/s10803-010-1006-y [DOI] [PubMed] [Google Scholar]
- 14. Patel S, Dale RC, Rose D, et al. Maternal immune conditions are increased in males with autism spectrum disorders and are associated with behavioural and emotional but not cognitive co‐morbidity. Transl Psychiatry. 2020;10(1):286. doi: 10.1038/s41398-020-00976-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kentner AC, Bilbo SD, Brown AS, et al. Maternal immune activation: reporting guidelines to improve the rigor, reproducibility, and transparency of the model. Neuropsychopharmacology. 2019;44(2):245‐258. doi: 10.1038/s41386-018-0185-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meyer U, Feldon J, Fatemi SH. In‐vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009;33(7):1061‐1079. doi: 10.1016/j.neubiorev.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 17. Estes ML, McAllister AK. Maternal immune activation: implications for neuropsychiatric disorders. Science. 2016;353(6301):772‐777. doi: 10.1126/science.aag3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. 2012;26(4):607‐616. doi: 10.1016/j.bbi.2012.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zengeler KE, Lukens JR. Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat Rev Immunol. 2021;21(7):454‐468. doi: 10.1038/s41577-020-00487-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meyer U, Feldon J, Yee BK. A review of the Fetal brain cytokine imbalance hypothesis of schizophrenia. Schizophr Bull. 2009;35(5):959‐972. doi: 10.1093/schbul/sbn022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ander SE, Diamond MS, Coyne CB. Immune responses at the maternal‐fetal interface. Sci Immunol. 2019;4(31):eaat6114. doi: 10.1126/sciimmunol.aat6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harding AT, Goff MA, Froggatt HM, Lim JK, Heaton NS. GPER1 is required to protect fetal health from maternal inflammation. Science. 2021;371(6526):271‐276. doi: 10.1126/science.aba9001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liong S, Oseghale O, To EE, et al. Influenza A virus causes maternal and fetal pathology via innate and adaptive vascular inflammation in mice. Proc Natl Acad Sci USA. 2020;117(40):24964‐24973. doi: 10.1073/pnas.2006905117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Littauer EQ, Esser ES, Antao OQ, Vassilieva EV, Compans RW, Skountzou I. H1N1 influenza virus infection results in adverse pregnancy outcomes by disrupting tissue‐specific hormonal regulation. PLoS Pathog. 2017;13(11):e1006757. doi: 10.1371/journal.ppat.1006757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yockey LJ, Iwasaki A. Interferons and proinflammatory cytokines in pregnancy and fetal development. Immunity. 2018;49(3):397‐412. doi: 10.1016/j.immuni.2018.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Massrali A, Adhya D, Srivastava DP, Baron‐Cohen S, Kotter MR. Virus‐induced maternal immune activation as an environmental factor in the etiology of autism and schizophrenia. Front Neurosci. 2022;16. doi: 10.3389/fnins.2022.834058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taubenberger JK, Kash JC. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe. 2010;7(6):440‐451. doi: 10.1016/j.chom.2010.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petrova VN, Russell CA. The evolution of seasonal influenza viruses. Nat Rev Microbiol. 2018;16(1):47‐60. doi: 10.1038/nrmicro.2017.118 [DOI] [PubMed] [Google Scholar]
- 29. Long JS, Mistry B, Haslam SM, Barclay WS. Host and viral determinants of influenza A virus species specificity. Nat Rev Microbiol. 2019;17(2):67‐81. doi: 10.1038/s41579-018-0115-z [DOI] [PubMed] [Google Scholar]
- 30. Moore TC, Fong J, Rosa Hernández AM, Pogreba‐Brown K. CAFOs, novel influenza, and the need for one health approaches. One Health. 2021;13:100246. doi: 10.1016/j.onehlt.2021.100246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rasmussen SA, Jamieson DJ, Uyeki TM. Effects of influenza on pregnant women and infants. Am J Obstet Gynecol. 2012;207(3 Suppl):S3‐S8. doi: 10.1016/j.ajog.2012.06.068 [DOI] [PubMed] [Google Scholar]
- 32. Jamieson DJ, Honein MA, Rasmussen SA, et al. H1N1 2009 influenza virus infection during pregnancy in the USA. Lancet. 2009;374(9688):451‐458. doi: 10.1016/S0140-6736(09)61304-0 [DOI] [PubMed] [Google Scholar]
- 33. Matsuda S. Pregnancy and infection. Asian Med J. 1986;29(5):291‐298. doi: 10.5005/jp/books/12390_4 [DOI] [Google Scholar]
- 34. Holstein R, Dawood FS, O'Halloran A, et al. Characteristics and outcomes of hospitalized pregnant women with influenza, 2010 to 2019. Ann Intern Med. 2021;175(2):149‐158. doi: 10.7326/M21-3668 [DOI] [PubMed] [Google Scholar]
- 35. Smith SEP, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters Fetal brain development through interleukin‐6. J Neurosci. 2007;27(40):10695‐10702. doi: 10.1523/JNEUROSCI.2178-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu WL, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH. The placental interleukin‐6 signaling controls fetal brain development and behavior. Brain Behav Immun. 2017;62:11‐23. doi: 10.1016/j.bbi.2016.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Choi GB, Yim YS, Wong H, et al. The maternal interleukin‐17a pathway in mice promotes autism‐like phenotypes in offspring. Science. 2016;351(6276):933‐939. doi: 10.1126/science.aad0314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lammert CR, Frost EL, Bolte AC, et al. Cutting edge: critical roles for microbiota‐mediated regulation of the immune system in a prenatal immune activation model of autism. J Immunol. 2018;201(3):845‐850. doi: 10.4049/jimmunol.1701755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou L, Ivanov II, Spolski R, et al. IL‐6 programs TH‐17 cell differentiation by promoting sequential engagement of the IL‐21 and IL‐23 pathways. Nat Immunol. 2007;8(9):967‐974. doi: 10.1038/ni1488 [DOI] [PubMed] [Google Scholar]
- 40. Kwon HK, Choi GB, Huh JR. Maternal inflammation and its ramifications on fetal neurodevelopment. Trends Immunol. 2022;43(3):230‐244. doi: 10.1016/j.it.2022.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smolders S, Smolders SMT, Swinnen N, et al. Maternal immune activation evoked by polyinosinic: Polycytidylic acid does not evoke microglial cell activation in the embryo. Front Cell Neurosci. 2015;9:301. doi: 10.3389/fncel.2015.00301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kwon J, Suessmilch M, McColl A, Cavanagh J, Morris BJ. Distinct trans‐placental effects of maternal immune activation by TLR3 and TLR7 agonists: implications for schizophrenia risk. Sci Rep. 2021;11(1):23841. doi: 10.1038/s41598-021-03216-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Missig G, Robbins JO, Mokler EL, et al. Sex‐dependent neurobiological features of prenatal immune activation via TLR7. Mol Psychiatry. 2020;25(10):2330‐2341. doi: 10.1038/s41380-018-0346-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garbett KA, Hsiao EY, Kálmán S, Patterson PH, Mirnics K. Effects of maternal immune activation on gene expression patterns in the fetal brain. Transl Psychiatry. 2012;2(4):e98. doi: 10.1038/tp.2012.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vasistha NA, Pardo‐Navarro M, Gasthaus J, et al. Maternal inflammation has a profound effect on cortical interneuron development in a stage and subtype‐specific manner. Mol Psychiatry. 2020;25(10):2313‐2329. doi: 10.1038/s41380-019-0539-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reed MD, Yim YS, Wimmer RD, et al. IL‐17a promotes sociability in mouse models of neurodevelopmental disorders. Nature. 2020;577(7789):249‐253. doi: 10.1038/s41586-019-1843-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shin Yim Y, Park A, Berrios J, et al. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature. 2017;549(7673):482‐487. doi: 10.1038/nature23909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu D, Li T, Delpech JC, et al. Microglial GPR56 is the molecular target of maternal immune activation‐induced parvalbumin‐positive interneuron deficits. Sci Adv. 2022;8(18):eabm2545. doi: 10.1126/sciadv.abm2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rosin JM, Sinha S, Biernaskie J, Kurrasch DM. A subpopulation of embryonic microglia respond to maternal stress and influence nearby neural progenitors. Dev Cell. 2021;56(9):1326‐1345.e6. doi: 10.1016/j.devcel.2021.03.018 [DOI] [PubMed] [Google Scholar]
- 50. Cunningham CL, Martinez‐Cerdeno V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013;33(10):4216‐4233. doi: 10.1523/JNEUROSCI.3441-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cui J, Shipley FB, Shannon ML, et al. Inflammation of the embryonic choroid plexus barrier following maternal immune activation. Dev Cell. 2020;55(5):617‐628.e6. doi: 10.1016/j.devcel.2020.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smolders S, Notter T, Smolders SMT, Rigo JM, Brône B. Controversies and prospects about microglia in maternal immune activation models for neurodevelopmental disorders. Brain Behav Immun. 2018;73:51‐65. doi: 10.1016/j.bbi.2018.06.001 [DOI] [PubMed] [Google Scholar]
- 53. Low D, Ginhoux F. Recent advances in the understanding of microglial development and homeostasis. Cell Immunol. 2018;330:68‐78. doi: 10.1016/j.cellimm.2018.01.004 [DOI] [PubMed] [Google Scholar]
- 54. Eyo UB, Dailey ME. Microglia: key elements in neural development, plasticity, and pathology. J Neuroimmune Pharmacol. 2013;8(3):494‐509. doi: 10.1007/s11481-013-9434-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Depino AM. Peripheral and central inflammation in autism spectrum disorders. Mol Cell Neurosci. 2013;53:69‐76. doi: 10.1016/j.mcn.2012.10.003 [DOI] [PubMed] [Google Scholar]
- 56. Goines P, Van De Water J. The immune system's role in the biology of autism. Curr Opin Neurol. 2010;23(2):111‐117. doi: 10.1097/WCO.0b013e3283373514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Morgan JT, Chana G, Pardo CA, et al. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry. 2010;68(4):368‐376. doi: 10.1016/j.biopsych.2010.05.024 [DOI] [PubMed] [Google Scholar]
- 58. Morgan JT, Chana G, Abramson I, Semendeferi K, Courchesne E, Everall IP. Abnormal microglial‐neuronal spatial organization in the dorsolateral prefrontal cortex in autism. Brain Res. 2012;1456:72‐81. doi: 10.1016/j.brainres.2012.03.036 [DOI] [PubMed] [Google Scholar]
- 59. Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57(1):67‐81. doi: 10.1002/ana.20315 [DOI] [PubMed] [Google Scholar]
- 60. Monji A, Kato TA, Mizoguchi Y, et al. Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog Neuro‐Psychopharmacol Biol Psychiatry. 2013;42:115‐121. doi: 10.1016/j.pnpbp.2011.12.002 [DOI] [PubMed] [Google Scholar]
- 61. Na KS, Jung HY, Kim YK. The role of pro‐inflammatory cytokines in the neuroinflammation and neurogenesis of schizophrenia. Prog Neuro‐Psychopharmacol Biol Psychiatry. 2014;48:277‐286. doi: 10.1016/j.pnpbp.2012.10.022 [DOI] [PubMed] [Google Scholar]
- 62. Van Den Eynde K, Missault S, Fransen E, et al. Hypolocomotive behaviour associated with increased microglia in a prenatal immune activation model with relevance to schizophrenia. Behav Brain Res. 2014;258:179‐186. doi: 10.1016/j.bbr.2013.10.005 [DOI] [PubMed] [Google Scholar]
- 63. Roumier A, Pascual O, Béchade C, et al. Prenatal activation of microglia induces delayed impairment of glutamatergic synaptic function. PLoS ONE. 2008;3(7):e2595. doi: 10.1371/journal.pone.0002595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hui CW, St‐Pierre A, El Hajj H, et al. Prenatal immune challenge in mice leads to partly sex‐dependent behavioral, microglial, and molecular abnormalities associated with schizophrenia. Front Mol Neurosci. 2018;11:13. doi: 10.3389/fnmol.2018.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Fatemi SH, Emamian ES, Kist D, et al. Defective corticogenesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol Psychiatry. 1999;4(2):145‐154. doi: 10.1038/sj.mp.4000520 [DOI] [PubMed] [Google Scholar]
- 66. Fatemi SH, Earle J, Kanodia R, et al. Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Cell Mol Neurobiol. 2002;22(1):25‐33. doi: 10.1023/A:1015337611258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Fatemi SH, Emamian ES, Sidwell RW, et al. Human influenza viral infection in utero alters glial fibrillary acidic protein immunoreactivity in the developing brains of neonatal mice. Mol Psychiatry. 2002;7(6):633‐640. doi: 10.1038/sj.mp.4001046 [DOI] [PubMed] [Google Scholar]
- 68. Fatemi SH, Pearce DA, Brooks AI, Sidwell RW. Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: a potential animal model for schizophrenia and autism. Synapse. 2005;57(2):91‐99. doi: 10.1002/syn.20162 [DOI] [PubMed] [Google Scholar]
- 69. Fatemi SH, Reutiman TJ, Folsom TD, et al. Maternal infection leads to abnormal gene regulation and brain atrophy in mouse offspring: implications for genesis of neurodevelopmental disorders. Schizophr Res. 2008;99(1–3):56‐70. doi: 10.1016/j.schres.2007.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shi L, Smith SEP, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun. 2009;23(1):116‐123. doi: 10.1016/j.bbi.2008.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shi L, Tu N, Patterson PH. Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. Int J Dev Neurosci. 2005;23(2–3):299‐305. doi: 10.1016/j.ijdevneu.2004.05.005 [DOI] [PubMed] [Google Scholar]
- 72. Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23(1):297‐302. doi: 10.1523/JNEUROSCI.23-01-00297.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Meyer U. Prenatal poly(I:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry. 2014;75(4):307‐315. doi: 10.1016/j.biopsych.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 74. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol. 2010;11(5):373‐384. doi: 10.1038/ni.1863 [DOI] [PubMed] [Google Scholar]
- 75. Moresco EMY, LaVine D, Beutler B. Toll‐like receptors. Curr Biol. 2011;21(13):R488‐R493. doi: 10.1016/j.cub.2011.05.039 [DOI] [PubMed] [Google Scholar]
- 76. Schulz O, Diebold SS, Chen M, et al. Toll‐like receptor 3 promotes cross‐priming to virus‐infected cells. Nature. 2005;433(7028):887‐892. doi: 10.1038/nature03326 [DOI] [PubMed] [Google Scholar]
- 77. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14(5):315‐328. doi: 10.1038/nri3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Meyer U, Feldon J, Schedlowski M, Yee BK. Towards an immuno‐precipitated neurodevelopmental animal model of schizophrenia. Neurosci Biobehav Rev. 2005;29(6):913‐947. doi: 10.1016/j.neubiorev.2004.10.012 [DOI] [PubMed] [Google Scholar]
- 79. Ibricevic A, Pekosz A, Walter MJ, et al. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J Virol. 2006;80(15):7469‐7480. doi: 10.1128/JVI.02677-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Le Goffic R, Pothlichet J, Vitour D, et al. Cutting edge: influenza A virus activates TLR3‐dependent inflammatory and RIG‐I‐dependent antiviral responses in human lung epithelial cells. J Immunol. 2007;178(6):3368‐3372. doi: 10.4049/jimmunol.178.6.3368 [DOI] [PubMed] [Google Scholar]
- 81. Cherfils‐Vicini J, Platonova S, Gillard M, et al. Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J Clin Invest. 2010;120(4):1285‐1297. doi: 10.1172/JCI36551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pichlmair A, Schulz O, Tan CP, et al. RIG‐I‐mediated antiviral responses to single‐stranded RNA bearing 5′‐phosphates. Science. 2006;314(5801):997‐1001. doi: 10.1126/science.1132998 [DOI] [PubMed] [Google Scholar]
- 83. Kowalinski E, Lunardi T, McCarthy AA, et al. Structural basis for the activation of innate immune pattern‐recognition receptor RIG‐I by viral RNA. Cell. 2011;147(2):423‐435. doi: 10.1016/j.cell.2011.09.039 [DOI] [PubMed] [Google Scholar]
- 84. Zheng D, Liwinski T, Elinav E. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov. 2020;6(1):36. doi: 10.1038/s41421-020-0167-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206(1):79‐87. doi: 10.1084/jem.20081667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Basavaraju S, Mishra S, Jindal R, Kesavardhana S. Emerging role of ZBP1 in Z‐RNA sensing, influenza virus‐induced cell death, and pulmonary inflammation. mBio. 2022;13(3):e00401‐22. doi: 10.1128/mbio.00401-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kuriakose T, Man SM, Subbarao Malireddi RK, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1(2):aag2045. doi: 10.1126/sciimmunol.aag2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Thapa RJ, Ingram JP, Ragan KB, et al. DAI senses influenza a virus genomic RNA and activates RIPK3‐dependent cell death. Cell Host Microbe. 2016;20(5):674‐681. doi: 10.1016/j.chom.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kesavardhana S, Kuriakose T, Guy CS, et al. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J Exp Med. 2017;214(8):2217‐2229. doi: 10.1084/jem.20170550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhang T, Yin C, Boyd DF, et al. Influenza virus Z‐RNAs induce ZBP1‐mediated necroptosis. Cell. 2020;180(6):1115‐1129.e13. doi: 10.1016/j.cell.2020.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Rebsamen M, Heinz LX, Meylan E, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF‐κB. EMBO Rep. 2009;10(8):916‐922. doi: 10.1038/embor.2009.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Miranda J, Yaddanapudi K, Hornig M, Lipkin WI. Astrocytes recognize intracellular polyinosinic‐polycytidylic acid via MDA‐5. FASEB J. 2009;23(4):1064‐1071. doi: 10.1096/fj.08-121434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Rajan JV, Warren SE, Miao EA, Aderem A. Activation of the NLRP3 inflammasome by intracellular poly I:C. FEBS Lett. 2010;584(22):4627‐4632. doi: 10.1016/j.febslet.2010.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sha W, Mitoma H, Hanabuchi S, et al. Human NLRP3 inflammasome senses multiple types of bacterial RNAs. Proc Natl Acad Sci USA. 2014;111(45):16059‐16064. doi: 10.1073/pnas.1412487111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Dauletbaev N, Cammisano M, Herscovitch K, Lands LC. Stimulation of the RIG‐I/MAVS pathway by Polyinosinic:Polycytidylic acid upregulates IFN‐β in airway epithelial cells with minimal Costimulation of IL‐8. J Immunol. 2015;195(6):2829‐2841. doi: 10.4049/jimmunol.1400840 [DOI] [PubMed] [Google Scholar]
- 96. Kim TS, Braciale TJ. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus‐specific cytotoxic CD8+ T cell responses. PLoS ONE. 2009;4(1):e4204. doi: 10.1371/journal.pone.0004204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. McGill J, Van Rooijen N, Legge KL. Protective influenza‐specific CD8 T cell responses require interactions with dendritic cells in the lungs. J Exp Med. 2008;205(7):1635‐1646. doi: 10.1084/jem.20080314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Langlois RA, Legge KL. Plasmacytoid dendritic cells enhance mortality during lethal influenza infections by eliminating virus‐specific CD8 T cells. J Immunol. 2010;184(8):4440‐4446. doi: 10.4049/jimmunol.0902984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Duan S, Thomas PG. Balancing immune protection and immune pathology by CD8+ T‐cell responses to influenza infection. Front Immunol. 2016;7. doi: 10.3389/fimmu.2016.00025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Waithman J, Mintern JD. Dendritic cells and influenza A virus infection. Virulence. 2012;3(7):603‐608. doi: 10.4161/viru.21864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Komastu T, Ireland DDC, Reiss CS. IL‐12 and viral infections. Cytokine Growth Factor Rev. 1998;9(3–4):277‐285. doi: 10.1016/S1359-6101(98)00017-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445‐489. doi: 10.1146/annurev-immunol-030409-101212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Cline TD, Beck D, Bianchini E. Influenza virus replication in macrophages: balancing protection and pathogenesis. J Gen Virol. 2017;98(10):2401‐2412. doi: 10.1099/jgv.0.000922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yoo JK, Fish EN, Braciale TJ. LAPCs promote follicular helper T cell differentiation of Ag‐primed CD4+ T cells during respiratory virus infection. J Exp Med. 2012;209(10):1853‐1867. doi: 10.1084/jem.20112256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Miyauchi K, Sugimoto‐Ishige A, Harada Y, et al. Protective neutralizing influenza antibody response in the absence of T follicular helper cells. Nat Immunol. 2016;17(12):1447‐1458. doi: 10.1038/ni.3563 [DOI] [PubMed] [Google Scholar]
- 106. Guglani L, Khader SA. Th17 cytokines in mucosal immunity and inflammation. Curr Opin HIV AIDS. 2010;5(2):120‐127. doi: 10.1097/COH.0b013e328335c2f6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kim S, Kim H, Yim YS, et al. Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature. 2017;549(7673):528‐532. doi: 10.1038/nature23910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485‐498. doi: 10.1016/j.cell.2009.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wang J, Li F, Wei H, Lian ZX, Sun R, Tian Z. Respiratory influenza virus infection induces intestinal immune injury via microbiota mediated Th17 cell‐dependent inflammation. J Exp Med. 2014;211(12):2397‐2410. doi: 10.1084/jem.20140625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Cua DJ, Tato CM. Innate IL‐17‐producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10(7):479‐489. doi: 10.1038/nri2800 [DOI] [PubMed] [Google Scholar]
- 111. Antonson AM, Kenney AD, Chen HJ, Corps KN, Yount JS, Gur TL. Moderately pathogenic maternal influenza A virus infection disrupts placental integrity but spares the fetal brain. Brain Behav Immun. 2021;96:28‐39. doi: 10.1016/j.bbi.2021.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL‐17+ T helper cells. Cell. 2006;126(6):1121‐1133. doi: 10.1016/j.cell.2006.07.035 [DOI] [PubMed] [Google Scholar]
- 113. Chen CJ, Wu GH, Kuo RL, Shih SR. Role of the intestinal microbiota in the immunomodulation of influenza virus infection. Microbes Infect. 2017;19(12):570‐579. doi: 10.1016/j.micinf.2017.09.002 [DOI] [PubMed] [Google Scholar]
- 114. Hanada S, Pirzadeh M, Carver KY, Deng JC. Respiratory viral infection‐induced microbiome alterations and secondary bacterial pneumonia. Front Immunol. 2018;9:2640. doi: 10.3389/fimmu.2018.02640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Deriu E, Boxx GM, He X, et al. Influenza virus affects intestinal microbiota and secondary salmonella infection in the gut through type I interferons. PLoS Pathog. 2016;12(5):e1005572. doi: 10.1371/journal.ppat.1005572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Groves HT, Cuthbertson L, James P, Moffatt MF, Cox MJ, Tregoning JS. Respiratory disease following viral lung infection alters the murine gut microbiota. Front Immunol. 2018;9:182. doi: 10.3389/fimmu.2018.00182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Yildiz S, Mazel‐Sanchez B, Kandasamy M, Manicassamy B, Schmolke M. Influenza a virus infection impacts systemic microbiota dynamics and causes quantitative enteric dysbiosis. Microbiome. 2018;6(1):9. doi: 10.1186/s40168-017-0386-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Omenetti S, Bussi C, Metidji A, et al. The intestine harbors functionally distinct homeostatic tissue‐resident and inflammatory Th17 cells. Immunity. 2019;51(1):77‐89.e6. doi: 10.1016/j.immuni.2019.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ghoreschi K, Laurence A, Yang XP, et al. Generation of pathogenic TH17 cells in the absence of TGF‐β signalling. Nature. 2010;467(7318):967‐971. doi: 10.1038/nature09447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lee JY, Hall JA, Kroehling L, et al. Serum amyloid a proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell. 2020;180(1):79‐91.e16. doi: 10.1016/j.cell.2019.11.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hickman SE, Kingery ND, Ohsumi TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013;16(12):1896‐1905. doi: 10.1038/nn.3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Matcovitch‐Natan O, Winter DR, Giladi A, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353(6301):aad8670. doi: 10.1126/science.aad8670 [DOI] [PubMed] [Google Scholar]
- 123. Thion MS, Low D, Silvin A, et al. Microbiome influences prenatal and adult microglia in a sex‐specific manner. Cell. 2018;172(3):500‐516.e16. doi: 10.1016/j.cell.2017.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841‐845. doi: 10.1126/science.1194637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kierdorf K, Erny D, Goldmann T, et al. Microglia emerge from erythromyeloid precursors via Pu.1‐and Irf8‐dependent pathways. Nat Neurosci. 2013;16(3):273‐280. doi: 10.1038/nn.3318 [DOI] [PubMed] [Google Scholar]
- 126. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18(4):225‐242. doi: 10.1038/nri.2017.125 [DOI] [PubMed] [Google Scholar]
- 127. Squarzoni P, Oller G, Hoeffel G, et al. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014;8(5):1271‐1279. doi: 10.1016/j.celrep.2014.07.042 [DOI] [PubMed] [Google Scholar]
- 128. Swinnen N, Smolders S, Avila A, et al. Complex invasion pattern of the cerebral cortex bymicroglial cells during development of the mouse embryo. Glia. 2013;61(2):150‐163. doi: 10.1002/glia.22421 [DOI] [PubMed] [Google Scholar]
- 129. Barger N, Keiter J, Kreutz A, et al. Microglia: an intrinsic component of the proliferative zones in the fetal rhesus monkey (Macaca mulatta) cerebral cortex. Cereb Cortex. 2019;29(7):2782‐2796. doi: 10.1093/cercor/bhy145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Frost JL, Schafer DP. Microglia: architects of the developing nervous system. Trends Cell Biol. 2016;26(8):587‐597. doi: 10.1016/j.tcb.2016.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Thion MS, Ginhoux F, Garel S. Microglia and early brain development: an intimate journey. Science. 2018;362(6411):185‐189. doi: 10.1126/science.aat0474 [DOI] [PubMed] [Google Scholar]
- 132. Rosin JM, Marsters CM, Malik F, et al. Embryonic microglia interact with hypothalamic radial glia during development and upregulate the TAM receptors MERTK and AXL following an insult. Cell Rep. 2021;34(1):108587. doi: 10.1016/j.celrep.2020.108587 [DOI] [PubMed] [Google Scholar]
- 133. Thion MS, Mosser CA, Férézou I, et al. Biphasic impact of prenatal inflammation and macrophage depletion on the wiring of neocortical inhibitory circuits. Cell Rep. 2019;28(5):1119‐1126.e4. doi: 10.1016/j.celrep.2019.06.086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Antony JM, Paquin A, Nutt SL, Kaplan DR, Miller FD. Endogenous microglia regulate development of embryonic cortical precursor cells. J Neurosci Res. 2011;89(3):286‐298. doi: 10.1002/jnr.22533 [DOI] [PubMed] [Google Scholar]
- 135. Ben‐Yehuda H, Matcovitch‐Natan O, Kertser A, et al. Maternal type‐I interferon signaling adversely affects the microglia and the behavior of the offspring accompanied by increased sensitivity to stress. Mol Psychiatry. 2020;25(5):1050‐1067. doi: 10.1038/s41380-019-0604-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ozaki K, Kato D, Ikegami A, et al. Maternal immune activation induces sustained changes in fetal microglia motility. Sci Rep. 2020;10(1):21378. doi: 10.1038/s41598-020-78294-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhang J, Jing Y, Zhang H, Bilkey DK, Liu P. Maternal immune activation altered microglial immunoreactivity in the brain of postnatal day 2 rat offspring. Synapse. 2019;73(2):e22072. doi: 10.1002/syn.22072 [DOI] [PubMed] [Google Scholar]
- 138. Pierre WC, Smith PLP, Londono I, Chemtob S, Mallard C, Lodygensky GA. Neonatal microglia: the cornerstone of brain fate. Brain Behav Immun. 2017;59:333‐345. doi: 10.1016/j.bbi.2016.08.018 [DOI] [PubMed] [Google Scholar]
- 139. Zhan Y, Paolicelli RC, Sforazzini F, et al. Deficient neuron‐microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17(3):400‐406. doi: 10.1038/nn.3641 [DOI] [PubMed] [Google Scholar]
- 140. Kim HJ, Cho MH, Shim WH, et al. Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry. 2017;22(11):1576‐1584. doi: 10.1038/mp.2016.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lieberman OJ, McGuirt AF, Tang G, Sulzer D. Roles for neuronal and glial autophagy in synaptic pruning during development. Neurobiol Dis. 2019;122:49‐63. doi: 10.1016/j.nbd.2018.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Györffy BA, Gulyássy P, Gellén B, et al. Widespread alterations in the synaptic proteome of the adolescent cerebral cortex following prenatal immune activation in rats. Brain Behav Immun. 2016;56:289‐309. doi: 10.1016/j.bbi.2016.04.002 [DOI] [PubMed] [Google Scholar]
- 143. Pendyala G, Chou S, Jung Y, et al. Maternal immune activation causes behavioral impairments and altered cerebellar cytokine and synaptic protein expression. Neuropsychopharmacology. 2017;42(7):1435‐1446. doi: 10.1038/npp.2017.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Dickerson DD, Bilkey DK. Aberrant neural synchrony in the maternal immune activation model: using translatable measures to explore targeted interventions. Front Behav Neurosci. 2013;7:217. doi: 10.3389/fnbeh.2013.00217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Fernández de Cossío L, Guzmán A, van der Veldt S, Luheshi GN. Prenatal infection leads to ASD‐like behavior and altered synaptic pruning in the mouse offspring. Brain Behav Immun. 2017;63:88‐98. doi: 10.1016/j.bbi.2016.09.028 [DOI] [PubMed] [Google Scholar]
- 146. Cieślik M, Gassowska‐Dobrowolska M, Zawadzka A, et al. The synaptic dysregulation in adolescent rats exposed to maternal immune activation. Front Mol Neurosci. 2021;13. doi: 10.3389/fnmol.2020.555290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Amaral DG, Schumann CM, Nordahl CW. Neuroanatomy of autism. Trends Neurosci. 2008;31(3):137‐145. doi: 10.1016/j.tins.2007.12.005 [DOI] [PubMed] [Google Scholar]
- 148. Avino TA, Hutsler JJ. Abnormal cell patterning at the cortical gray‐white matter boundary in autism spectrum disorders. Brain Res. 2010;1360:138‐146. doi: 10.1016/j.brainres.2010.08.091 [DOI] [PubMed] [Google Scholar]
- 149. Mercadante MT, Cysneiros RM, Schwartzman JS, Arida RM, Cavalheiro EA, Scorza FA. Neurogenesis in the amygdala: a new etiologic hypothesis of autism? Med Hypotheses. 2008;70(2):352‐357. doi: 10.1016/j.mehy.2007.05.018 [DOI] [PubMed] [Google Scholar]
- 150. Belmonte MK, Allen G, Beckel‐Mitchener A, Boulanger LM, Carper RA, Webb SJ. Autism and abnormal development of brain connectivity. J Neurosci. 2004;24(42):9228‐9231. doi: 10.1523/JNEUROSCI.3340-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Just MA, Keller TA, Malave VL, Kana RK, Varma S. Autism as a neural systems disorder: a theory of frontal‐posterior underconnectivity. Neurosci Biobehav Rev. 2012;36(4):1292‐1313. doi: 10.1016/j.neubiorev.2012.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Holiga Š, Hipp JF, Chatham CH, et al. Patients with autism spectrum disorders display reproducible functional connectivity alterations. Sci Transl Med. 2019;11(481):eaat9223. doi: 10.1126/scitranslmed.aat9223 [DOI] [PubMed] [Google Scholar]
- 153. Ellman LM, Deicken RF, Vinogradov S, et al. Structural brain alterations in schizophrenia following fetal exposure to the inflammatory cytokine interleukin‐8. Schizophr Res. 2010;121(1–3):46‐54. doi: 10.1016/j.schres.2010.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Canetta S, Bolkan S, Padilla‐Coreano N, et al. Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol Psychiatry. 2016;21(7):956‐968. doi: 10.1038/mp.2015.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Wöhr M, Orduz D, Gregory P, et al. Lack of parvalbumin in mice leads to behavioral deficits relevant to all human autism core symptoms and related neural morphofunctional abnormalities. Transl Psychiatry. 2015;5(3):e525. doi: 10.1038/tp.2015.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Frankle WG, Cho RY, Prasad KM, et al. In vivo measurement of GABA transmission in healthy subjects and schizophrenia patients. Am J Psychiatry. 2015;172(11):1148‐1159. doi: 10.1176/appi.ajp.2015.14081031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron. 2015;87(4):684‐698. doi: 10.1016/j.neuron.2015.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Bilbo SD, Schwarz JM. Early‐life programming of later‐life brain and behavior: a critical role for the immune system. Front Behav Neurosci. 2009;3:14. doi: 10.3389/neuro.08.014.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Knuesel I, Chicha L, Britschgi M, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol. 2014;10(11):643‐660. doi: 10.1038/nrneurol.2014.187 [DOI] [PubMed] [Google Scholar]
- 160. Arsenault D, St‐Amour I, Cisbani G, Rousseau LS, Cicchetti F. The different effects of LPS and poly I: C prenatal immune challenges on the behavior, development and inflammatory responses in pregnant mice and their offspring. Brain Behav Immun. 2014;38:77‐90. doi: 10.1016/j.bbi.2013.12.016 [DOI] [PubMed] [Google Scholar]
- 161. Giovanoli S, Notter T, Richetto J, et al. Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J Neuroinflammation. 2015;12(1):221. doi: 10.1186/s12974-015-0437-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Missault S, Van den Eynde K, Vanden Berghe W, et al. The risk for behavioural deficits is determined by the maternal immune response to prenatal immune challenge in a neurodevelopmental model. Brain Behav Immun. 2014;42:138‐146. doi: 10.1016/j.bbi.2014.06.013 [DOI] [PubMed] [Google Scholar]
- 163. Mattei D, Ivanov A, Ferrai C, et al. Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl Psychiatry. 2017;7(5):e1120. doi: 10.1038/tp.2017.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Giovanoli S, Weber‐Stadlbauer U, Schedlowski M, Meyer U, Engler H. Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain Behav Immun. 2016;55:25‐38. doi: 10.1016/j.bbi.2015.09.015 [DOI] [PubMed] [Google Scholar]
- 165. Ratnayake U, Quinn TA, Castillo‐Melendez M, Dickinson H, Walker DW. Behaviour and hippocampus‐specific changes in spiny mouse neonates after treatment of the mother with the viral‐mimetic Poly I:C at mid‐pregnancy. Brain Behav Immun. 2012;26(8):1288‐1299. doi: 10.1016/j.bbi.2012.08.011 [DOI] [PubMed] [Google Scholar]
- 166. Nielsen PR, Agerbo E, Skogstrand K, Hougaard DM, Meyer U, Mortensen PB. Neonatal levels of inflammatory markers and later risk of schizophrenia. Biol Psychiatry. 2015;77(6):548‐555. doi: 10.1016/j.biopsych.2014.07.013 [DOI] [PubMed] [Google Scholar]
- 167. Antonson AM, Lawson MA, Caputo MP, Matt SM, Leyshon BJ, Johnson RW. Maternal viral infection causes global alterations in porcine fetal microglia. Proc Natl Acad Sci USA. 2019;116(40):20190‐20200. doi: 10.1073/pnas.1817014116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Antonson AM, Radlowski EC, Lawson MA, Rytych JL, Johnson RW. Maternal viral infection during pregnancy elicits anti‐social behavior in neonatal piglet offspring independent of postnatal microglial cell activation. Brain Behav Immun. 2017;59:300‐312. doi: 10.1016/j.bbi.2016.09.019 [DOI] [PubMed] [Google Scholar]
- 169. Lawson LJ, Perry VH, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992;48(2):405‐415. doi: 10.1016/0306-4522(92)90500-2 [DOI] [PubMed] [Google Scholar]
- 170. Askew K, Li K, Olmos‐Alonso A, et al. Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep. 2017;18(2):391‐405. doi: 10.1016/j.celrep.2016.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Tay TL, Mai D, Dautzenberg J, et al. A new fate mapping system reveals context‐dependent random or clonal expansion of microglia. Nat Neurosci. 2017;20(6):793‐803. doi: 10.1038/nn.4547 [DOI] [PubMed] [Google Scholar]
- 172. Meyer U. Neurodevelopmental resilience and susceptibility to maternal immune activation. Trends Neurosci. 2019;42(11):793‐806. doi: 10.1016/j.tins.2019.08.001 [DOI] [PubMed] [Google Scholar]
- 173. Mueller FS, Scarborough J, Schalbetter SM, et al. Behavioral, neuroanatomical, and molecular correlates of resilience and susceptibility to maternal immune activation. Mol Psychiatry. 2021;26(2):396‐410. doi: 10.1038/s41380-020-00952-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Purves‐Tyson TD, Weber‐Stadlbauer U, Richetto J, et al. Increased levels of midbrain immune‐related transcripts in schizophrenia and in murine offspring after maternal immune activation. Mol Psychiatry. 2021;26(3):849‐863. doi: 10.1038/s41380-019-0434-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Xia Y, Zhang Z, Lin W, et al. Modulating microglia activation prevents maternal immune activation induced schizophrenia‐relevant behavior phenotypes via arginase 1 in the dentate gyrus. Neuropsychopharmacology. 2020;45(11):1896‐1908. doi: 10.1038/s41386-020-0743-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Zhu F, Zheng Y, Liu Y, Zhang X, Zhao J. Minocycline alleviates behavioral deficits and inhibits microglial activation in the offspring of pregnant mice after administration of polyriboinosinic–polyribocytidilic acid. Psychiatry Res. 2014;219(3):680‐686. doi: 10.1016/j.psychres.2014.06.046 [DOI] [PubMed] [Google Scholar]
- 177. Mattei D, Djodari‐Irani A, Hadar R, et al. Minocycline rescues decrease in neurogenesis, increase in microglia cytokines and deficits in sensorimotor gating in an animal model of schizophrenia. Brain Behav Immun. 2014;38:175‐184. doi: 10.1016/j.bbi.2014.01.019 [DOI] [PubMed] [Google Scholar]
- 178. Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, Costa‐Mattioli M. Microbial reconstitution reverses maternal diet‐induced social and synaptic deficits in offspring. Cell. 2016;165(7):1762‐1775. doi: 10.1016/j.cell.2016.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Hsiao EY, McBride SW, Hsien S, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155(7):1451‐1463. doi: 10.1016/j.cell.2013.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Kim E, Paik D, Ramirez RN, et al. Maternal gut bacteria drive intestinal inflammation in offspring with neurodevelopmental disorders by altering the chromatin landscape of CD4+ T cells. Immunity. 2022;55(1):145‐158.e7. doi: 10.1016/j.immuni.2021.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Lebovitz Y, Kowalski EA, Wang X, et al. Lactobacillus rescues postnatal neurobehavioral and microglial dysfunction in a model of maternal microbiome dysbiosis. Brain Behav Immun. 2019;81:617‐629. doi: 10.1016/j.bbi.2019.07.025 [DOI] [PubMed] [Google Scholar]
- 182. Erny D, De Angelis ALH, Jaitin D, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7):965‐977. doi: 10.1038/nn.4030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Abdel‐Haq R, Schlachetzki JCM, Glass CK, Mazmanian SK. Microbiome–microglia connections via the gut–brain axis. J Exp Med. 2019;216(1):41‐59. doi: 10.1084/jem.20180794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Vuong HE, Pronovost GN, Williams DW, et al. The maternal microbiome modulates fetal neurodevelopment in mice. Nature. 2020;586(7828):281‐286. doi: 10.1038/s41586-020-2745-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Jašarević E, Howerton CL, Howard CD, Bale TL. Alterations in the vaginal microbiome by maternal stress are associated with metabolic reprogramming of the offspring gut and brain. Endocrinology. 2015;156(9):3265‐3276. doi: 10.1210/en.2015-1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Jašarević E, Howard CD, Misic AM, Beiting DP, Bale TL. Stress during pregnancy alters temporal and spatial dynamics of the maternal and offspring microbiome in a sex‐specific manner. Sci Rep. 2017;7(1):44182. doi: 10.1038/srep44182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Jašarević E, Howard CD, Morrison K, et al. The maternal vaginal microbiome partially mediates the effects of prenatal stress on offspring gut and hypothalamus. Nat Neurosci. 2018;21(8):1061‐1071. doi: 10.1038/s41593-018-0182-5 [DOI] [PubMed] [Google Scholar]
- 188. Braniste V, Al‐Asmakh M, Kowal C, et al. The gut microbiota influences blood‐brain barrier permeability in mice. Sci Transl Med. 2014;6(263):263ra158. doi: 10.1126/scitranslmed.3009759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Castillo‐Ruiz A, Mosley M, George AJ, et al. The microbiota influences cell death and microglial colonization in the perinatal mouse brain. Brain Behav Immun. 2018;67:218‐229. doi: 10.1016/j.bbi.2017.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Das Sarma J, Ciric B, Marek R, et al. Functional interleukin‐17 receptor A is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis. J Neuroinflammation. 2009;6(1):14. doi: 10.1186/1742-2094-6-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Kawanokuchi J, Shimizu K, Nitta A, et al. Production and functions of IL‐17 in microglia. J Neuroimmunol. 2008;194(1–2):54‐61. doi: 10.1016/j.jneuroim.2007.11.006 [DOI] [PubMed] [Google Scholar]
- 192. Zimmermann J, Emrich M, Krauthausen M, et al. IL‐17A promotes granulocyte infiltration, myelin loss, microglia activation, and behavioral deficits during cuprizone‐induced demyelination. Mol Neurobiol. 2018;55(2):946‐957. doi: 10.1007/s12035-016-0368-3 [DOI] [PubMed] [Google Scholar]
- 193. Sasaki T, Tome S, Takei Y. Intraventricular IL‐17A administration activates microglia and alters their localization in the mouse embryo cerebral cortex. Mol Brain. 2020;13(1):93. doi: 10.1186/s13041-020-00635-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Meyer U. The time of prenatal immune challenge determines the specificity of inflammation‐mediated brain and Behavioral pathology. J Neurosci. 2006;26(18):4752‐4762. doi: 10.1523/JNEUROSCI.0099-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008;22(4):469‐486. doi: 10.1016/j.bbi.2007.09.012 [DOI] [PubMed] [Google Scholar]
- 196. Guma E, Bordeleau M, González Ibáñez F, et al. Differential effects of early or late exposure to prenatal maternal immune activation on mouse embryonic neurodevelopment. Proc Natl Acad Sci USA. 2022;119(12):e2114545119. doi: 10.1073/pnas.2114545119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Nakamura JP, Schroeder A, Gibbons A, Sundram S, Hill RA. Timing of maternal immune activation and sex influence schizophrenia‐relevant cognitive constructs and neuregulin and GABAergic pathways. Brain Behav Immun. 2022;100:70‐82. doi: 10.1016/j.bbi.2021.11.006 [DOI] [PubMed] [Google Scholar]
- 198. Brown AS, Patterson PH. Maternal infection and schizophrenia: implications for prevention. Schizophr Bull. 2011;37(2):284‐290. doi: 10.1093/schbul/sbq146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15(5):300‐312. doi: 10.1038/nrn3722 [DOI] [PubMed] [Google Scholar]
- 200. Kaufman MH, Baldock R, Bard JBL, Davidson D, Morriss‐Kay G, eds. Kaufma's Atlas of Mouse Development: With Coronal Sections. Supplement. Academic Press; 2016. [Google Scholar]
- 201. Duchatel RJ, Jobling P, Graham BA, et al. Increased white matter neuron density in a rat model of maternal immune activation—implications for schizophrenia. Prog Neuro‐Psychopharmacol Biol Psychiatry. 2016;65:118‐126. doi: 10.1016/j.pnpbp.2015.09.006 [DOI] [PubMed] [Google Scholar]
- 202. Duchatel RJ, Meehan CL, Harms LR, et al. Increased complement component 4 (C4) gene expression in the cingulate cortex of rats exposed to late gestation immune activation. Schizophr Res. 2018;199:442‐444. doi: 10.1016/j.schres.2018.03.035 [DOI] [PubMed] [Google Scholar]
- 203. Meehan C, Harms L, Frost JD, et al. Effects of immune activation during early or late gestation on schizophrenia‐related behaviour in adult rat offspring. Brain Behav Immun. 2017;63:8‐20. doi: 10.1016/j.bbi.2016.07.144 [DOI] [PubMed] [Google Scholar]
- 204. Kowash HM, Potter HG, Edye ME, et al. Poly(I:C) source, molecular weight and endotoxin contamination affect dam and prenatal outcomes, implications for models of maternal immune activation. Brain Behav Immun. 2019;82:160‐166. doi: 10.1016/j.bbi.2019.08.006 [DOI] [PubMed] [Google Scholar]
- 205. Estes ML, Prendergast K, MacMahon JA, et al. Baseline immunoreactivity before pregnancy and poly(I:C) dose combine to dictate susceptibility and resilience of offspring to maternal immune activation. Brain Behav Immun. 2020;88:619‐630. doi: 10.1016/j.bbi.2020.04.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Homan E. Studies on poly I:C toxicity in experimental animals. Toxicol Appl Pharmacol. 1972;23(4):579‐588. doi: 10.1016/0041-008X(72)90098-1 [DOI] [PubMed] [Google Scholar]
- 207. Al Shoyaib A, Archie SR, Karamyan VT. Intraperitoneal route of drug administration: should it be used in experimental animal studies? Pharm Res. 2020;37(1):12. doi: 10.1007/s11095-019-2745-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Haddad FL, Patel SV, Schmid S. Maternal immune activation by poly I:C as a preclinical model for neurodevelopmental disorders: a focus on autism and schizophrenia. Neurosci Biobehav Rev. 2020;113:546‐567. doi: 10.1016/j.neubiorev.2020.04.012 [DOI] [PubMed] [Google Scholar]
- 209. Bouvier NM, Lowen AC. Animal models for influenza virus pathogenesis and transmission. Viruses. 2010;2(8):1530‐1563. doi: 10.3390/v20801530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Chan KH, Zhang AJX, To KKW, et al. Wild type and mutant 2009 pandemic influenza A (H1N1) viruses cause more severe disease and higher mortality in pregnant BALB/c mice. PLoS ONE. 2010;5(10):e13757. doi: 10.1371/journal.pone.0013757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Jester BJ, Uyeki TM, Jernigan DB. Fifty years of influenza A(H3N2) following the pandemic of 1968. Am J Public Health. 2020;110(5):669‐676. doi: 10.2105/AJPH.2019.305557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Mor G, Aldo P, Alvero AB. The unique immunological and microbial aspects of pregnancy. Nat Rev Immunol. 2017;17(8):469‐482. doi: 10.1038/nri.2017.64 [DOI] [PubMed] [Google Scholar]
- 213. McCarthy MM. Placental signaling influences cerebellum development. Nat Neurosci. 2021;24(10):1343‐1345. doi: 10.1038/s41593-021-00918-1 [DOI] [PubMed] [Google Scholar]
- 214. Romano‐Keeler J, Weitkamp JH. Maternal influences on fetal microbial colonization and immune development. Pediatr Res. 2015;77(1–2):189‐195. doi: 10.1038/pr.2014.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Chen HJ, Gur TL. Intrauterine microbiota: missing, or the missing link? Trends Neurosci. 2019;42(6):402‐413. doi: 10.1016/j.tins.2019.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Reyes‐Lagos JJ, Abarca‐Castro EA, Echeverría JC, Mendieta‐Zerón H, Vargas‐Caraveo A, Pacheco‐López G. A translational perspective of maternal immune activation by SARS‐CoV‐2 on the potential prenatal origin of neurodevelopmental disorders: the role of the cholinergic anti‐inflammatory pathway. Front Psychol. 2021;12. doi: 10.3389/fpsyg.2021.614451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Edlow AG, Castro VM, Shook LL, Kaimal AJ, Perlis RH. Neurodevelopmental outcomes at 1 year in infants of mothers who tested positive for SARS‐CoV‐2 during pregnancy. JAMA Netw Open. 2022;5(6):e2215787. doi: 10.1001/jamanetworkopen.2022.15787 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.