

Abstract
Inflammation and coagulation are the critical responses to infection that include leukocytes, platelets, and vascular endothelial cells responding in concert to eradicate the invading pathogen. In sepsis, a variety of cell surface receptors, including toll‐like receptors, Fcγ‐receptors, G‐protein‐coupled receptors, and adhesion receptors, detect the pathogens and elicit thromboinflammatory responses. Concurrently, the molecular patterns released from host damaged cells accelerate the immune responses through binding to the same pattern recognition receptors. Cytokines, chemokines, and extracellular vesicles are important mediators for amplifying the responses to distant cells as part of the systemic response to infections. At the same time, cells communicate with each other via direct contact, adhesion molecules, paracrine mediators, and tunneling nanotubes, which are important for regulating inflammation and thrombus formation. Despite increasing attention to immunothrombosis in sepsis, these close communication systems are less understood but play a critical role in host defense mechanisms. In this review, cellular activation and direct intercellular communication systems in sepsis with a focus on the coagulation response will be considered.
Keywords: adhesion molecule, endothelial cell, immunothrombosis, platelet, sepsis, thromboinflammation
1. INTRODUCTION
Inflammation and coagulation are the first‐line defense systems against infection. 1 Leukocytes, platelets, erythrocytes, and vascular endothelial cells are at the frontline in the acute response to invading pathogens. 2 Hyperinflammation upregulates coagulation, and as a result, disseminated intravascular coagulation (DIC)/sepsis‐induced coagulopathy (SIC) frequently complicates severe infection. 3 , 4 Immunothrombosis occurs to immobilize and prevent the systemic spread of pathogens, a response that sacrifices tissue circulation with microthrombosis and localized injury. 5 Therefore, precise and integrated regulation to maintain the blood flow and performance of innate immunity is important. The monitoring of thromboinflammation continues to be determined. Although the monitoring of platelet count and some coagulation markers are not direct indicators of thromboinflammation, they are routine for clinical management of patients with sepsis. The Japanese guidelines for sepsis management recommend the frequent assessment of coagulation abnormalities and the treatment with anticoagulants for septic patients with DIC. 6
Cells signal distant cells as part of a systemic response by secreting cytokines, alarmins, and extracellular vesicles, and are important in the pathophysiologic mechanisms of host defense as well as sepsis‐induced organ dysfunction. 7 , 8 , 9 In addition to this distant and systemic signaling, cells communicate and interact with neighboring cells by direct contact and short‐range cell‐to‐cell signaling by mechanisms often not considered. As a result, in this commentary, we will review how inflammation and coagulation are regulated using elaborate direct and short‐range communication systems in sepsis.
2. CELLULAR ACTIVATION IN SEPSIS
2.1. Leukocyte activation
Leukocytes are the most important cellular components against infection, and express various surface receptors such as toll‐like receptors (TLRs), Fcγ‐receptors, G‐protein‐coupled receptors, adhesion receptors, and cytokine receptors to sense pathogen invasion. 10 TLRs play key roles in mediating systemic host responses to pathogens by binding to pathogen‐associated molecular patterns (PAMPs). Since TLRs are a class of pattern recognition molecules, they also sense host‐derived stress molecules called damage‐associated molecular patterns (DAMPs). 11 For example, other than the well known activation mechanism by lipopolysaccharide, high mobility group box 1 (HMGB1), a nuclear protein released from host necrotic cells, activates monocytes by binding to the same receptor, that is, toll‐like receptor 4. After the subsequent complex multi‐step signal transduction, monocytes express proinflammatory cytokines, chemokines, and adhesion molecules. In addition, the ‘danger signals’ transduced by TLRs stimulate the expression of tissue factor and phosphatidylserine on the cell membrane, thereby upregulating the coagulation systems. 12 HMGB1 is also known to bind to the receptor for advanced glycation end products (RAGE) and induce similar monocyte reactions. 13
The Fcγ receptor is a receptor for immune complexes that facilitates microbe opsonization by immunoglobulin G (IgG). The expression of Fcγ receptors on monocytes is upregulated in sepsis, described as ‘angry macrophages’, and this triggers the increased production of proinflammatory cytokines. 14 The inflammatory signals transduced through Fcγ receptor binding can upregulate the procoagulant activity of monocytes by increased tissue factor expression. 15
Protease activated receptors (PARs), G‐protein‐coupled transmembrane receptors, are also important receptors that mediate inflammation and coagulation in sepsis. Monocytes mainly express PAR‐1 and PAR‐3 that are activated through proteolytic cleavage by their tethered ligands following binding to thrombin. 16 Thrombin is an important proinflammatory stimulus that also activates platelets, leukocytes, and endothelial cells by binding to PAR‐1, 17 and contributes to facilitating the vicious cycle of inflammation and coagulation in sepsis.
The adhesion molecules represented by integrins and immunoglobulin superfamily adhesion molecules are also known to regulate innate immunity, that is, complement‐driven phagocytosis, and cellular interactions in sepsis. Integrin αMβ2 (macrophage‐1 antigen, Mac‐1, CD11b/CD18) on monocyte and other phagocytes acts as the receptor for complement component C3b and namely complement receptor (CR) 3, which synergistically functions with Fcγ receptors. 18 (Figure 1). Zhong et al. 19 demonstrated that the mechanism of intercellular cell adhesion molecule‐1 (ICAM‐1)‐mediated macrophage phagocytosis depends on TLR4‐mediated reactive oxygen species production.
FIGURE 1.
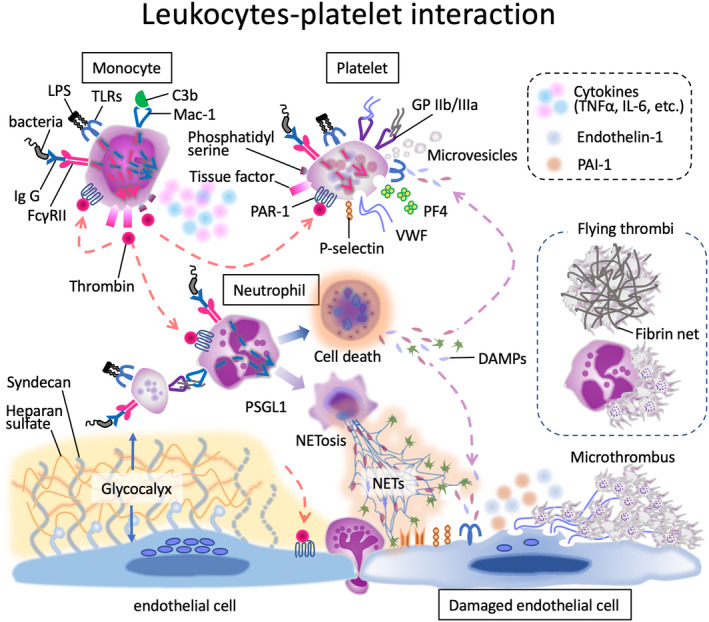
Microthrombus formation in sepsis. In sepsis, signals transduced through various receptors on the surface of monocytes, neutrophils, and platelets induce the cellular responses of these cells. The representative receptors are toll‐like receptors (TLRs), Fcγ receptor (FcγR), adhesion receptor macrophage‐1 antigen (Mac‐1, complement receptor 3), and protease activated receptor (PAR)‐1. Activated monocytes express tissue factor and phosphatidylserine on the surface, which initiate coagulation cascades. The responses also include the release proinflammatory cytokines such as tumor necrosis factor α (TNFα) and interleukin (IL)‐6. Activated neutrophils eject neutrophil extracellular traps (NETs) and release various damage‐associated molecular patterns (DAMPs) along with cell death. Platelets release procoagulant microvesicles and prothrombotic substances such as von Willebrand factor (VWF) and platelet factor 4 (PF4). Blue dashed arrow: proinflammatory reaction; red dashed arrow: prothrombotic reaction; LPS: lipopolysaccharide; Ig: immunoglobulin; GP: glycoprotein; C3b: complement fragment 3b; PAI‐1: plasminogen activator inhibitor 1.
Similar to monocytes, neutrophils express a large number of common receptors, but their expression and cellular responses can be different as monocytes act as conductors of inflammation while neutrophils attack and immobilize pathogens directly. Although the receptors are the same, complex and delicate regulation systems should be present. 20 A novel approach of adsorptive carrier‐based activated granulocyte and monocyte apheresis for the treatment of inflammatory diseases such as ulcerative colitis is used clinically where Fcγ receptors and CR 3 ligands of the activated leukocytes are targeted for adsorption. 21
2.2. Platelet activation
Thrombocytopenia is frequently observed in patients with sepsis and is reported to be a strong and independent predictor of poor outcomes in sepsis. 22 , 23 Thrombocytopenia results from massive platelet activation and consumption. Platelets release a variety of inflammatory and hemostatic mediators such as platelet factor 4, von Willebrand factor (VWF), factor VIII, and P‐selectin. In addition to platelet aggregation, these mediators also activate leukocytes, and endothelial cells, form conjugates with leukocytes, and bridges between leukocytes and endothelium. These important cellular interactions also lead to functional changes in the cells, as shown in Figure 2. When patients with sepsis develop severe thrombocytopenia, thrombotic microangiopathy (TMA) should be considered. 24 TMA includes various diseases such as thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, and other TMAs. For some of these diseases, specific treatments are available.
FIGURE 2.
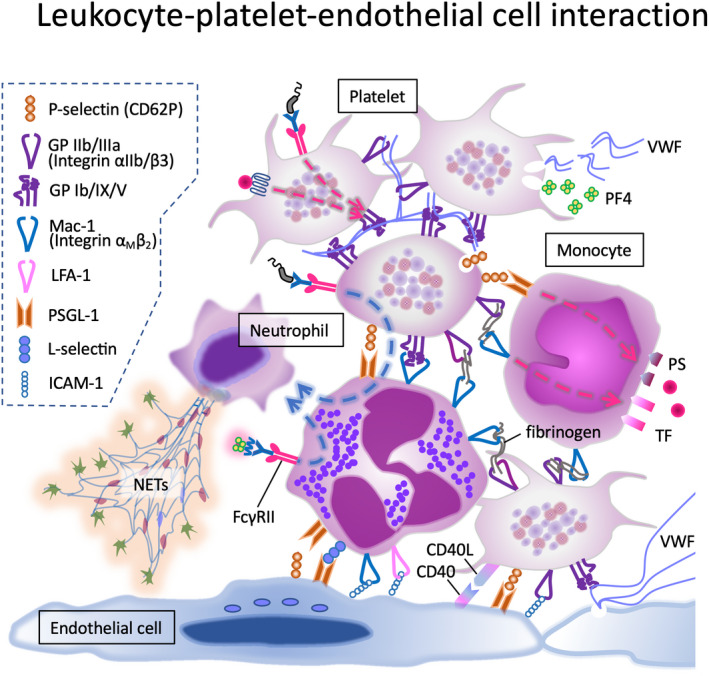
Cooperative interplay between cellular adhesion molecules. Interactions between the platelets, platelets and leukocytes, platelets and endothelial cells, and leukocytes and endothelial cells are shown. Interactions occur via direct contact between cell surface adhesion molecules, or via binding to the intermediate substances such as von Willebrand factor and fibrinogen. These interplays are either the result or cause of activation of related cells.
Numerous adhesion molecules also contribute to the interactions with monocytes, neutrophils, and endothelial cells. As with other hemostatic components, platelets are important participants in inflammation and thrombosis. 25 Platelet factor 4, a member of the CXC chemokine family, is stored in α‐granules, and released during platelet activation. 26 The biological role of platelet factor 4 remains to be elucidated, but it has an anti‐pathogenic effect, and the level increases significantly in sepsis. 27 Positively charged platelet factor 4 further facilitates bacteria‐killing by binding to the bacteria charge dependently (opsonization) as part of host defense. 28 Platelet factor 4 is important in the pathogenesis of heparin‐induced thrombocytopenia (HIT) and vaccine‐induced immune thrombotic thrombocytopenia (VITT), also known as thrombosis with thrombocytopenia syndrome (TTS). 29 In these cases, immunospecific IgG for anti‐platelet factor 4/polyanion antibody induces platelet activation and aggregation by linking to Fcγ receptor IIA on platelets and leads to thrombosis. 30 Moreover, this immune complex can induce neutrophil extracellular trap (NET) ejection via binding to Fcγ receptor IIA on neutrophils. 28 Since NETs are a strong initiator of thrombosis, neutrophil activation via the Fcγ receptor should be carefully assessed.
Other than platelet factor 4–anti‐platelet factor 4 IgG antibody complexes, IgG‐coated antigens and microbes also bind to this receptor, and this observation further supports platelet involvement in innate immunity. Immunothrombosis induced through this mechanism is usually beneficial for the host defense; however, if the platelet/leukocyte responses are disproportionately strong, unfavorable circulatory disturbance and subsequent organ damage ensue. 31
Similar to the Fcγ receptor, platelets express common receptors as leukocytes, such as TLRs and PARs. This is likely due to the role of platelets in host defense, inflammatory, and immune reactions with leukocytes. For example, platelets recognize lipopolysaccharide via TLR4 and upregulate cytokine synthesis, initiate aggregation, and release granule contents in response to the gram‐negative bacteria infection. 32 PARs are the key factors in orchestrating the interactions between coagulation and inflammation. Prothrombotic and proinflammatory reactions of platelets are mediated by the signals from PARs. Nevertheless, whether the expression of PAR‐1 on platelets is upregulated in sepsis is unclear. Raque et al. 33 reported increased PAR‐1‐positive platelets in patients with sepsis. By contrast, Reiter et al. 34 showed a decrease in PAR‐1 on platelets. Since the key mediator thrombin crosslinks to both thrombotic and inflammatory pathways, understanding the regulation of its receptor PAR‐1 is important.
CD40 ligand, a transmembrane protein of platelets that structurally mimics TNF‐α, is a thromboinflammatory molecule. 35 CD40 ligand’s receptor CD40 is present on monocytes/macrophages and endothelial cells, suggesting that these cells collaborate with activated platelets to build thrombi. Other than CD40, CD40 ligand also binds to integrin αIIbβ3 and integrin αMβ2 and upregulates cellular adhesion. 36
An additional anti‐inflammatory receptor, the C‐type lectin‐like receptor 2 (CLEC‐2), is a podoplanin receptor on platelets that is thought to attenuate inflammation in sepsis. Although its physiological role continues to be defined, the mouse model of lipopolysaccharide‐induced peritonitis reported macrophage podoplanin with platelet CLEC‐2 limits macrophage recruitment to the infection site, whereas reduction of CLEC‐2 increased macrophage proinflammatory cytokine release. 37 In addition, CLEC‐2 is also known to regulate endothelial permeability and prevent microcirculatory bleeding during inflammation. 38
As previously mentioned, platelet count correlates with the severity of infection; however, it is noteworthy that the increased functional activity and the decrease in platelet count are not always parallel. Even if the platelet count is maintained, platelet aggregation and thrombogenicity can be increased, and COVID‐19 is an important example. 39 Insoluble fibrin is efficiently formed by the conversion of soluble fibrinogen on the surface of the activated platelets, suggesting that platelet aggregation and coagulation can be regulated separately. Therefore, not only the platelet count but other indicators of platelet activity should be monitored.
Although cohort studies report the potential survival merit of antiplatelet therapy for patients with sepsis, the effect has not been confirmed in randomized controlled studies, although future studies are needed. 40 , 41
2.3. Endothelial cell activation
Vascular endothelial cells interact with leukocytes and platelets and actively participate in immunothrombosis, 42 proving a systemic response to infections to limit pathogen spreading. These complex immune and thrombotic responses are complex, providing procoagulant, proadhesive, and proinflammatory conditions producing glycocalyx damage, upregulating adhesion molecules, releasing VWF, and vascular tone impairment, all pathophysiologic responses that facilitate immunothrombus formation. 43
Vascular endothelial cells also express innate immune receptors, such as TLRs and protease‐activated receptors. TLRs activate intracellular inflammatory pathways mediated through myeloid differentiation factor (MyD88) that leads to the early activation of nuclear factor‐κB (NF‐κB) and mitogen‐activated protein kinase (MAP kinase). 44 TLR agonists, including lipopolysaccharide, lipoteichoic acid, and peptidoglycans, upregulate endothelial cell expression of inflammatory and procoagulant mediators. 45 Furthermore, activation of TLRs modulates microvascular permeability and adhesion molecule expression. In this scenario, microthrombi trap microorganisms, thereby limiting the dissemination of pathogens. However, disorganized activation of endothelial cells leads to coagulopathy and tissue edema, which promote organ malcirculation. 46
Thrombin activation of PAR‐1 induces disruption of endothelial barrier function and dysregulated vascular permeability through multiple pathways. The mechanistic details of AR signaling are still the subject of current investigation. 47 PAR‐1 activation promotes the functional changes of endothelial cells toward a proinflammatory and procoagulant direction, results in increased permeability, induces the expression of prothrombotic proteins, and stimulates the secretion of inflammatory cytokines, which mediate the local accumulation of leukocytes and platelets. 48 Lastly, fibrinolytic suppression also affects thrombus formation. Disorganized endothelial production of plasminogen activator inhibitor 1 (PAI‐1) and thrombin‐mediated activation of thrombin‐activatable fibrinolysis inhibitor suppress the fibrinolytic function and facilitate thromboinflammation and potential procoagulant effects in sepsis. 49 These changes consequently lead to microcirculatory thrombosis, cellular injury, and tissue hypoxia. Taken together, all the functional changes of endothelial cells are viewed as an integral component of host defense systems.
2.4. Erythrocyte activation
Unlike other blood cells, active receptors for inflammation are not recognized on the erythrocyte membrane. Nevertheless, erythrocytes also participate in the thrombus formation during sepsis. Eryptosis/erythroptosis, an apoptotic process of erythrocytes, is characterized by phosphatidylserine expression on the cellular membrane. 50 Since phosphatidylserine acts as an ‘eat‐me signal’, eryptosis may be the mechanism of removal of defective erythrocytes by the phagocytes to prevent unfavorable hemolysis. 51 However, phosphatidylserine‐positive erythrocytes are increased in sepsis, and these erythrocytes may have an active role in facilitating coagulation and thrombus formation. 52 Lang et al. 50 reported phosphatidylserine‐positive erythrocytes are capable of adhering to the endothelial cells by binding to CXC‐Motiv‐chemokine‐16/Scavenger‐receptor for phosphatidylserine and oxidized low‐density lipoprotein (CXCL16). Oxidative stress under septic conditions accelerates the above process and leads to the further interference of microcirculation. 53 Although the significance of erythrocyte activation is not well characterized, since erythrocytes are components of thrombus, they appear to have an active role in host defense.
3. CELLULAR INTERACTION
3.1. Cellular adhesion
Besides the primary role in hemostasis, platelets are important components in the process of restoration of endothelium and regulation of inflammation in sepsis. 25 The term thromboinflammation was first used in the early 2000s to describe platelet–leukocyte interaction via sequential reactions initiated from P‐selectin (CD62P) and its ligand P‐selectin glycoprotein ligand‐1 (PSGL‐1). 54 Currently, it is accepted that cell adhesion contributes to cell binding but also transfers signals from one cell to another cell. Endotoxin cannot stimulate NETs release by itself, but the release is induced upon interaction with lipopolysaccharide‐activated P‐selectin expressing platelets. 55 Platelet–leukocyte interaction is known to be stimulated by the signals from the aforementioned multiple receptors, including TLRs and PARs. 56 The memorial work in this field was done by Engelmann and Massberg 57 in 2013, and they describe an innate immune response induced by intravascular thrombus formation as immunothrombosis (Figure 3). Besides, recent studies demonstrated the acceleration of platelet aggregation by the complement anaphylatoxins C3a and C5a contributed to immunothrombosis in sepsis. 58 In sepsis, the transmigration and increased cell death in apoptosis of the neutrophils are reported. In this scenario, PAR‐1 agonists such as thrombin upregulate cell adhesion molecule expression, apoptosis, and cytokine production. However, the regulation is complex, and low‐dose thrombin or PAR‐2 agonists such as serine proteases are reported to reduce transendothelial migration and suppress apoptosis. 59 , 60 Platelets interact with monocytes/neutrophils via the activation of various adhesion molecules. P‐selectin is expressed on the platelet surface upon activation. Platelets stick to monocytes by P‐selectin‐PSGL‐1 linkage. 61 Other than this mechanism, platelet’s integrin αIIb/β3 (GP IIb/IIIa) binds Mac‐1 (GP αM/β2, CD11b/CD18) on monocytes via fibrinogen. 62 The latter, in turn, is bound to activated integrin αIIb/β3 on the platelet surface. 63 Furthermore, Mac‐1 on leukocytes interacts with GP Ib on the platelets because Mac‐1 domain is homologous to the A1 domain of VWF. 64 Various adhesion molecules are also utilized for platelet aggregation, and platelet−VWF interactions are involved in aggregate formation. Activation of endothelial cells induces the release of VWF from the Weibel‐Palade bodies as well as the release of VWF and platelet factor 4 from α‐granule of the platelets. P‐selectin, GP Ib/IX/V, and integrin αIIb/β3 are utilized for platelet–platelet interaction. 5
FIGURE 3.
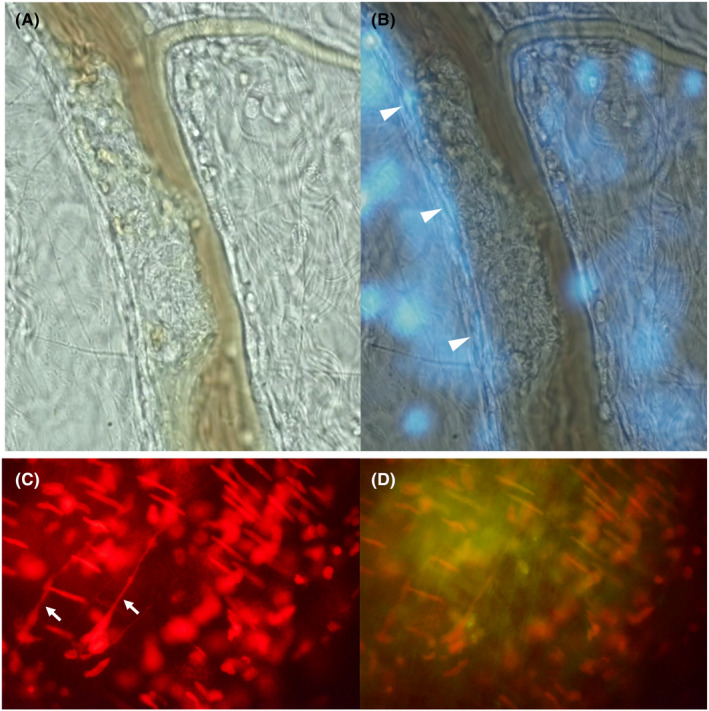
Immunothrombus formation in sepsis model. Sepsis model of rat was made by intravenous injection of lipopolysaccharide. Six hours later, micro thrombus formed of leukocyte–platelet aggregates was observed in the mesenteric venule under the intravital microscope (A). The nuclei of the severely damaged endothelial cells where the thrombus was formed were stained by DAPI (B). DNA component in neutrophil extracellular traps (NETs) (white arrows) were visualized by the immunofluorescence (Nuclear‐ID Red stain) (C). Platelets staining by anti‐CD 41 antibody (yellow) was overlayed (D).
Vascular endothelial cells regulate leukocyte trafficking through the expression of adhesion molecules, predominantly in the postcapillary venules. First, P‐ and E‐selectin, selectin family adhesion molecules, on endothelial cells mediates tethering and rolling. Second, immunoglobulin superfamily adhesion molecules such as intercellular cell adhesion molecule‐1 (ICAM‐1) and vascular cell adhesion molecule‐1 (VCAM‐1) orchestrate the adhesion of the leukocytes. 65 As for the platelet adhesion to endothelium, P‐selectin‐PSGL‐1 linking is the first step for adhesion, followed by the integrin‐platelet endothelial cell adhesion molecule (PECAM‐1) link that contributes to scaffolding and induces various signal transduction and cellular responses. 66 Consequently, these multistep cell adhesion pathways may be responsible for the biological and pathophysiological functions such as thrombosis and inflammation in sepsis.
As for the treatment targeting cell adhesion, since endothelial glycocalyx degradation increased the availability of adhesion molecules on the endothelium, glycocalyx preservation by heparanase or glucuronidase inhibition is expected. 67
3.2. Paracrine communication
Other than the hemostasis, platelets are now recognized as important components in the process of endothelium restoration, inflammation, and thrombus formation during sepsis. 25 The lipid mediators, including prostaglandins, leukotrienes, platelet‐activating factors, and lysophospholipid mediators are released from activated platelets and act as autocrine and/or paracrine mediators which affect neighboring cells such as leukocytes, endothelial cells, and platelets. In sepsis, bioactive lipids have been shown to play important roles in hemostasis/thrombosis, maintenance of vascular kinetics, and regulation of inflammation. 68 Other than lipid mediators, serotonin, adenosine diphosphate (ADP), and growth factors from platelets stimulate the release of vasoactive substances such as nitric oxide, prostaglandins, and endothelin, and these substances exert the effects in a paracrine and autocrine fashion. 69
Endothelial cells also produce paracrine mediators such as lipid mediators, nitric oxide, hydrogen peroxide, hydrogen sulfide, and growth factors. 70 Paracrine signaling from endothelial cells can stimulate surrounding cells and monocytes to react to inflammatory situations with an expression of tissue factor.
Neutrophils respond by expelling NETs and inducing coagulatory responses. This may cause the formation of microthrombi designated as immunothrombosis. 71 During sepsis, activated leukocytes also release CXC chemokines and DAMPs to the neighboring cells and adherent endothelium, which amplifies the inflammatory signals and increases oxidative stress in a paracrine fashion. 72 The approach to managing the paracrine signal is likely to develop new pathways for treating the clinical problem of sepsis.
3.3. Gap junctions
The smooth and antithrombotic lining of the endothelium is required to avoid unexpected thrombus formation. The integrity of inter‐endothelial junctions is essential for the maintenance of endothelial barrier function, vascular permeability, and cell adhesion and extravasation. Endothelial cells are connected by three junction complexes, that is, adherens junction, tight junction, and gap junction. 73 Adherens junction serves mechanical anchorage and mechanotransduction, and tight junction seals the intercellular space to limit paracellular permeability. 74 Gap junction is a channel‐like structure that connects the cytoplasm of adjacent cells and allows the movement of small molecules between the cells. Under steady conditions, endothelial cells can regulate the fluid exchange between intra‐ and extra‐vascular lumen on purpose, and the well‐known regulators of vascular permeability are prostaglandins, nitric oxide, prostacyclin, vascular growth factor, and cytokines. In sepsis, the contact systems can easily be damaged and result in gap formation, which increases permeability and finally leads to vascular collapse and tissue edema. 75
Endothelial‐cadherin is the transmembrane component of the endothelial adherens junction. Cytokine‐induced phosphorylation of the cytoplasmic domain was reported to trigger cleavage of its extracellular domain, which affects the junctional strength and leads to increased vascular permeability in sepsis. 76
The endothelial gap junction is formed by transmembrane adhesive molecules linked to the networks of cytoplasmic and cytoskeletal proteins. Connexin is a constituent structural unit of gap junction channels and is required for the coupling of endothelial cells. Bolon et al. 77 reported lipopolysaccharide, hypoxia, and reoxygenation decrease the electrical coupling between microvascular endothelial cells by damaging connexin in sepsis. In addition, another study revealed that thrombin decreased transendothelial resistance in a similar manner. 78 Meanwhile, non‐junctional connexin hemichannels are reported to be opened by PAMPs and facilitate the release of DAMPs in sepsis. 79 , 80 The components of adherens and gap junctions are critical for maintaining the endothelial barrier and permeability, however there have still been a limited number of studies addressing the role of contact communication, and such research is warranted.
3.4. Tunneling nanotubes
In contrast to contact cell communication, various cells release extracellular vesicles by shedding or exocytosis to deliver cytosolic components such as proteins, micro‐, messenger‐RNAs, and cellular debris to the distant cells. These ‘shipping containers’ can also carry receptors, ligands, procoagulant membrane surface phospholipids, and proteins such as tissue factor. 81 Thus, the role of extracellular vesicles in the propagation of inflammation and coagulation is quite important. 9 Besides extracellular vesicles, mesenchymal cells are also known to use unique systems for cell‐to‐cell communication. Cytoplasmic extensions to the other cells comprising open‐ended channels connected solidly by actin filament termed tunneling nanotubes potentiate the exchange of cytoplasmic materials. Unlike other communication systems, large materials, including organelles, cytosolic vesicles, and even pathogens, can be transported through this string‐like canal (Figure 4). This functional connectivity between immune cells may help the cells to resist infections. Endothelial cells also adopt this system for connecting to the circulating blood cells and transfer various biomolecules such as signaling mediators, proteins, lipids, and RNAs. 82 The role of the tunneling nanotube is not fully unveiled, however Jackson et al. 83 reported improved macrophage phagocytic activity by mitochondrial transfer from mesenchymal stromal cells by this system in the sepsis model. This trafficking system may be used to damage cell recovery and to promote tissue regeneration. 84 Further, dendritic cells serve nanotube‐like structures as a normal component of their function and apply it for adaptive immunity. Zaccard et al. 85 reported dendritic cells acquired a unique modality by rapidly forming intercellular communications using this structure upon antigen‐driven interaction with helper T cells. After all, host cells utilize diverse modalities for maintaining cellular homeostasis. However, ironically, this system is also available for pathogen transmission in diseases like COVID‐19. 86 The export of toxic substances and the recharge of energy by importing mitochondria from neighboring cells with this system will rescue the cells from cell death.
FIGURE 4.
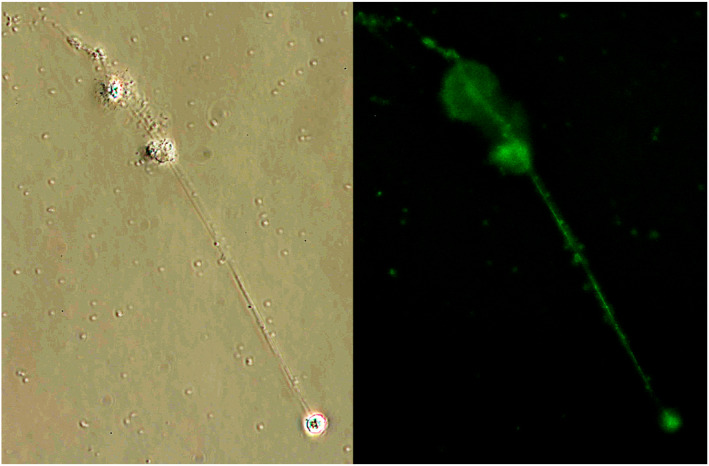
Tunneling nanotubes. Phase‐contrast view (left) and anti‐CD11b immunofluorescent stain (right) of the tunneling nanotubes. Leukocytes were co‐cultured with Escherichia coli for 4 h. Leukocytes are connected by string‐like tunneling nanotubes. Since the tube is formed of the cytoplasmic membrane, it is stained by anti‐CD 11b antibody.
4. CONCLUSIONS
Inflammation and coagulation are the essential responses to infection, and inflammatory thrombosis is the current topic of research. The inflammatory signals are transduced through the various receptors such as TLR, Fcγ‐receptors, G‐protein‐coupled receptors, and adhesion receptors on the immune cells. Tight communication between leukocytes and platelets in consort with vascular endothelial cells is essential for the immunothrombosis that leads to the deterioration of organ function.
Complex and highly integrated regulation of inflammation and coagulation serves as efficient protection against infections. However, dysregulated, excessive, or inadequate immune responses harm hosts and lead to organ dysfunction and/or DIC/SIC. An extensive number of clinical trials have been performed to examine the effects of anti‐inflammation or anticoagulation therapies, however none has shown success in terms of improving survival. It is regrettable to say that except for source management and antimicrobial treatment, there is still no established therapy for reducing the immunothrombotic complications of sepsis. Receptors for PAMPs and DAMPs, short‐range intercellular communication systems, and transportation systems such as the gap junction and tunneling nanotube are especially important for collaborative work. We hope that a better understanding of the mechanisms for cellular activation and intercellular communication will provide the insight to develop new therapeutic approaches and mitigate the deadly insult of sepsis.
AUTHOR CONTRIBUTIONS
TI and JHL wrote the draft. ML reviewed and revised the manuscript.
FUNDING INFORMATION
This work was supported in part by a Grant‐in‐Aid for Scientific Research C Grant Number JP19K09424.
CONFLICT OF INTEREST
TI has received a research grant from Japan Blood Products Organization and Asahi Kasei Pharmaceuticals. ML has received grants and has participated in advisory boards of NovoNordisk, Eli Lilly, Asahi Kasei Pharmaceuticals America, and Johnson & Johnson. JHL serves on the Steering Committees for Instrumentation Laboratories, Merck, and Octapharma.
Iba T, Levi M, Levy JH. Intracellular communication and immunothrombosis in sepsis. J Thromb Haemost. 2022;20:2475‐2484. doi: 10.1111/jth.15852
REFERENCES
- 1. van der Poll T, Levi M. Crosstalk between inflammation and coagulation: the lessons of sepsis. Curr Vasc Pharmacol. 2012;10(5):632‐638. [DOI] [PubMed] [Google Scholar]
- 2. Iba T, Levy JH. Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J Thromb Haemost. 2018;16(2):231‐241. [DOI] [PubMed] [Google Scholar]
- 3. Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers. 2016;2:16037. [DOI] [PubMed] [Google Scholar]
- 4. Iba T, Levy JH. Sepsis‐induced coagulopathy and disseminated intravascular coagulation. Anesthesiology. 2020;132(5):1238‐1245. [DOI] [PubMed] [Google Scholar]
- 5. Martinod K, Deppermann C. Immunothrombosis and thromboinflammation in host defense and disease. Platelets. 2021;32(3):314‐324. [DOI] [PubMed] [Google Scholar]
- 6. Egi M, Ogura H, Yatabe T, et al. The Japanese clinical practice guidelines for Management of Sepsis and Septic Shock 2020 (J‐SSCG 2020). J Intensive Care. 2021;9(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent JL. Sepsis and septic shock. Nat Rev Dis Primers. 2016;2:16045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis. 2017;8(5):e2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Iba T, Ogura H. Role of extracellular vesicles in the development of sepsis‐induced coagulopathy. J Intensive Care. 2018;6:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Futosi K, Fodor S, Mócsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. 2013;17(3):638‐650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salomão R, Martins PS, Brunialti MK, et al. TLR signaling pathway in patients with sepsis. Shock. 2008;30(Suppl 1):73‐77. [DOI] [PubMed] [Google Scholar]
- 12. Gould TJ, Lysov Z, Swystun LL, et al. Canadian critical care translational biology group. Extracellular histones increase tissue factor activity and enhance thrombin generation by human blood monocytes. Shock. 2016;46(6):655‐662. [DOI] [PubMed] [Google Scholar]
- 13. Li W, Sama AE, Wang H. Role of HMGB1 in cardiovascular diseases. Curr Opin Pharmacol. 2006;6(2):130‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schinkel C, Sendtner R, Zimmer S, Faist E. Functional analysis of monocyte subsets in surgical sepsis. J Trauma. 1998;44(5):743‐748. [DOI] [PubMed] [Google Scholar]
- 15. Kasthuri RS, Glover SL, Jonas W, et al. PF4/heparin‐antibody complex induces monocyte tissue factor expression and release of tissue factor positive microparticles by activation of FcγRI. Blood. 2012;119(22):5285‐5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thaler B, Hohensinner PJ, Baumgartner J, et al. Protease‐activated receptors 1 and 3 are differentially expressed on human monocyte subsets and are upregulated by lipopolysaccharide ex vivo and in vivo. Thromb Haemost. 2019;119(9):1394‐1402. [DOI] [PubMed] [Google Scholar]
- 17. Gudmundsdóttir IJ, Megson IL, Kell JS, et al. Direct vascular effects of protease‐activated receptor type 1 agonism in vivo in humans. Circulation. 2006;114(15):1625‐1632. [DOI] [PubMed] [Google Scholar]
- 18. Lukácsi S, Nagy‐Baló Z, Erdei A, Sándor N, Bajtay Z. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement‐mediated phagocytosis and podosome formation by human phagocytes. Immunol Lett. 2017;189:64‐72. [DOI] [PubMed] [Google Scholar]
- 19. Zhong H, Lin H, Pang Q, et al. Macrophage ICAM‐1 functions as a regulator of phagocytosis in LPS induced endotoxemia. Inflamm Res. 2021;70(2):193‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hanna MOF, Abdelhameed AM, Abou‐Elalla AA, Hassan RM, Kostandi I. Neutrophil and monocyte receptor expression in patients with sepsis: implications for diagnosis and prognosis of sepsis. Pathog Dis. 2019;77(6):ftz055. [DOI] [PubMed] [Google Scholar]
- 21. Saniabadi AR, Hanai H, Takeuchi K, et al. Adacolumn, an adsorptive carrier based granulocyte and monocyte apheresis device for the treatment of inflammatory and refractory diseases associated with leukocytes. Ther Apher Dial. 2003;7(1):48‐59. [DOI] [PubMed] [Google Scholar]
- 22. Venkata C, Kashyap R, Farmer JC, Afessa B. Thrombocytopenia in adult patients with sepsis: incidence, risk factors, and its association with clinical outcome. J Intensive Care. 2013;1(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Levi M. Platelets in critical illness. Semin Thromb Hemost. 2016;42(3):252‐257. [DOI] [PubMed] [Google Scholar]
- 24. Iba T, Levy JH, Wada H, Thachil J, Warkentin TE, Levi M. Differential diagnoses for sepsis‐induced disseminated intravascular coagulation: communication from the SSC of the ISTH. J Thromb Haemost. 2019;17(2):415‐419. [DOI] [PubMed] [Google Scholar]
- 25. Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost. 2015;114(3):449‐458. doi: 10.1160/TH14-12-1067 [DOI] [PubMed] [Google Scholar]
- 26. Kowalska MA, Rauova L, Poncz M. Role of the platelet chemokine platelet factor 4 (PF4) in hemostasis and thrombosis. Thromb Res. 2010;125(4):292‐296. [DOI] [PubMed] [Google Scholar]
- 27. Maharaj S, Chang S. Anti‐PF4/heparin antibodies are increased in hospitalized patients with bacterial sepsis. Thromb Res. 2018;171:111‐113. [DOI] [PubMed] [Google Scholar]
- 28. Palankar R, Kohler TP, Krauel K, Wesche J, Hammerschmidt S, Greinacher A. Platelets kill bacteria by bridging innate and adaptive immunity via platelet factor 4 and FcγRIIA. J Thromb Haemost. 2018;16(6):1187‐1197. [DOI] [PubMed] [Google Scholar]
- 29. Iba T, Levy JH. Thrombosis and thrombocytopenia in COVID‐19 and after COVID‐19 vaccination. Trends Cardiovasc Med. 2022;32(5):249‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Althaus K, Möller P, Uzun G, et al. Antibody‐mediated procoagulant platelets in SARS‐CoV‐2‐vaccination associated immune thrombotic thrombocytopenia. Haematologica. 2021;106(8):2170‐2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Patel P, Michael JV, Naik UP, McKenzie SE. Platelet FcγRIIA in immunity and thrombosis: adaptive immunothrombosis. J Thromb Haemost. 2021;19(5):1149‐1160. [DOI] [PubMed] [Google Scholar]
- 32. Panzer S. Differential response to LPS isotypes induced platelet activation mediated by toll‐like receptor (TLR)‐4. Clin Immunol. 2013;146(1):13‐14. [DOI] [PubMed] [Google Scholar]
- 33. Raque VX, Carlos SJ, Eduardo RR, et al. Modification of immunological features in human platelets during sepsis. Immunol Invest. 2018;47(2):196‐211. [DOI] [PubMed] [Google Scholar]
- 34. Reiter R, Derhaschnig U, Spiel A, et al. Regulation of protease‐activated receptor 1 (PAR1) on platelets and responsiveness to thrombin receptor activating peptide (TRAP) during systemic inflammation in humans. Thromb Haemost. 2003;90(5):898‐903. [DOI] [PubMed] [Google Scholar]
- 35. Henn V, Slupsky JR, Gräfe M, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391(6667):591‐594. [DOI] [PubMed] [Google Scholar]
- 36. Kojok K, Akoum SE, Mohsen M, Mourad W, Merhi Y. CD40L priming of platelets via NF‐κB activation is CD40‐ and TAK1‐dependent. J Am Heart Assoc. 2018;7(23):e03677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bourne JH, Beristain‐Covarrubias N, Zuidscherwoude M, et al. CLEC‐2 prevents accumulation and retention of inflammatory macrophages during murine peritonitis. Front Immunol. 2021;12:693974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rayes J, Lax S, Wichaiyo S, et al. The podoplanin‐CLEC‐2 axis inhibits inflammation in sepsis. Nat Commun. 2017;8(1):2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Iba T, Helms J, Levi M, Levy JH. The role of platelets in heat‐related illness and heat‐induced coagulopathy. Thromb Res. 2022;S0049‐3848(22)00342‐5. Epub ahead of print. doi: 10.1016/j.thromres.2022.08.009 [DOI] [PubMed] [Google Scholar]
- 40. Tsai MJ, Ou SM, Shih CJ, et al. Association of prior antiplatelet agents with mortality in sepsis patients: a nationwide population‐based cohort study. Intensive Care Med. 2015;41(5):806‐813. [DOI] [PubMed] [Google Scholar]
- 41. Kiers D, van der Heijden WA, van Ede L, et al. A randomised trial on the effect of anti‐platelet therapy on the systemic inflammatory response in human endotoxaemia. Thromb Haemost. 2017;117(9):1798‐1807. [DOI] [PubMed] [Google Scholar]
- 42. Joffre J, Hellman J, Ince C, Ait‐Oufella H. Endothelial responses in sepsis. Am J Respir Crit Care Med. 2020;202(3):361‐370. [DOI] [PubMed] [Google Scholar]
- 43. Iba T, Levy JH. Derangement of the endothelial glycocalyx in sepsis. J Thromb Haemost. 2019;17(2):283‐294. [DOI] [PubMed] [Google Scholar]
- 44. Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;86(1):9‐22. [DOI] [PubMed] [Google Scholar]
- 45. Kirschning CJ, Bauer S. Toll‐like receptors: cellular signal transducers for exogenous molecular patterns causing immune responses. Int J Med Microbiol. 2001;291(4):251‐260. [DOI] [PubMed] [Google Scholar]
- 46. Travassos LH, Girardin SE, Philpott DJ, et al. Toll‐like receptor 2‐dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 2004;5(10):1000‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rezaie AR. Protease‐activated receptor signalling by coagulation proteases in endothelial cells. Thromb Haemost. 2014;112(5):876‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alberelli MA, De Candia E. Functional role of protease activated receptors in vascular biology. Vascul Pharmacol. 2014;62(2):72‐81. [DOI] [PubMed] [Google Scholar]
- 49. Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis, thrombosis and organ dysfunction. Thromb Res. 2012;129(3):290‐295. [DOI] [PubMed] [Google Scholar]
- 50. Lang KS, Lang PA, Bauer C, et al. Mechanisms of suicidal erythrocyte death. Cell Physiol Biochem. 2005;15(5):195‐202. [DOI] [PubMed] [Google Scholar]
- 51. Briglia M, Rossi MA, Faggio C. Eryptosis: ally or enemy. Curr Med Chem. 2017;24(9):937‐942. [DOI] [PubMed] [Google Scholar]
- 52. Kempe DS, Akel A, Lang PA, et al. Suicidal erythrocyte death in sepsis. J Mol Med (Berl). 2007;85(3):273‐281. [DOI] [PubMed] [Google Scholar]
- 53. Lang F, Abed M, Lang E, Föller M. Oxidative stress and suicidal erythrocyte death. Antioxid Redox Signal. 2014;21(1):138‐153. [DOI] [PubMed] [Google Scholar]
- 54. Tanguay JF, Geoffroy P, Sirois MG, et al. Prevention of in‐stent restenosis via reduction of thrombo‐inflammatory reactions with recombinant P‐selectin glycoprotein ligand‐1. Thromb Haemost. 2004;91(6):1186‐1193. [DOI] [PubMed] [Google Scholar]
- 55. Clark SR, Ma AC, Tavener SA, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463‐469. [DOI] [PubMed] [Google Scholar]
- 56. Blair P, Rex S, Vitseva O, et al. Stimulation of toll‐like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3‐kinase. Circ Res. 2009;104(3):346‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13(1):34‐45. [DOI] [PubMed] [Google Scholar]
- 58. Mollnes TE, Huber‐Lang M. Complement in sepsis‐when science meets clinics. FEBS Lett. 2020;594(16):2621‐2632. [DOI] [PubMed] [Google Scholar]
- 59. Bae JS, Kim YU, Park MK, Rezaie AR. Concentration dependent dual effect of thrombin in endothelial cells via par‐1 and Pi3 kinase. J Cell Physiol. 2009;219(3):744‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shpacovitch VM, Seeliger S, Huber‐Lang M, et al. Agonists of proteinase‐activated receptor‐2 affect transendothelial migration and apoptosis of human neutrophils. Exp Dermatol. 2007;16(10):799‐806. [DOI] [PubMed] [Google Scholar]
- 61. Gerrits AJ, Frelinger AL 3rd, Michelson AD. Whole blood analysis of leukocyte‐platelet aggregates. Curr Protoc Cytom. 2016;78:6.15.1‐6.15.10. [DOI] [PubMed] [Google Scholar]
- 62. Plescia J, Conte MS, VanMeter G, Ambrosini G, Altieri DC. Molecular identification of the cross‐reacting epitope on alphaM beta2 integrin I domain recognized by anti‐alphaIIb beta3 monoclonal antibody 7E3 and its involvement in leukocyte adherence. J Biol Chem. 1998;273(32):20372‐20377. [DOI] [PubMed] [Google Scholar]
- 63. Bennett JS. Structure and function of the platelet integrin alphaIIbbeta3. J Clin Invest. 2005;115(12):3363‐3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Simon DI, Chen Z, Xu H, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin mac‐1 (CD11b/CD18). J Exp Med. 2000;192(2):193‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118(26):6743‐6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gong N, Chatterjee S. Platelet endothelial cell adhesion molecule in cell signaling and thrombosis. Mol Cell Biochem. 2003;253(1–2):151‐158. [DOI] [PubMed] [Google Scholar]
- 67. Schmidt EP, Yang Y, Janssen WJ, et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat Med. 2012;18(8):1217‐1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vardon Bounes F, Mujalli A, Cenac C, et al. The importance of blood platelet lipid signaling in thrombosis and in sepsis. Adv Biol Regul. 2018;67:66‐73. [DOI] [PubMed] [Google Scholar]
- 69. Schiffrin EL. The endothelium and control of blood vessel function in health and disease. Clin Invest Med. 1994;17(6):602‐620. [PubMed] [Google Scholar]
- 70. Freed JK, Gutterman DD. Communication is Key: mechanisms of intercellular signaling in vasodilation. J Cardiovasc Pharmacol. 2017;69(5):264‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mussbacher M, Salzmann M, Brostjan C, et al. Cell type‐specific roles of NF‐κB linking inflammation and thrombosis. Front Immunol. 2019;10:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gomez H, Ince C, De Backer D, et al. A unified theory of sepsis‐induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock. 2014;41(1):3‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. 2008;1778(3):794‐809. [DOI] [PubMed] [Google Scholar]
- 74. Radeva MY, Waschke J. Mind the gap: mechanisms regulating the endothelial barrier. Acta Physiol. 2018;222(1):1‐20. [DOI] [PubMed] [Google Scholar]
- 75. Wautier JL, Wautier MP. Vascular permeability in diseases. Int J Mol Sci. 2022;23(7):3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Blaise S, Polena H, Vilgrain I. Soluble vascular endothelial‐cadherin and auto‐antibodies to human vascular endothelial‐cadherin in human diseases: two new biomarkers of endothelial dysfunction. Vasc Med. 2015;20(6):557‐565. [DOI] [PubMed] [Google Scholar]
- 77. Bolon ML, Peng T, Kidder GM, Tyml K. Lipopolysaccharide plus hypoxia and reoxygenation synergistically reduce electrical coupling between microvascular endothelial cells by dephosphorylating connexin40. J Cell Physiol. 2008;217(2):350‐359. [DOI] [PubMed] [Google Scholar]
- 78. O'Donnell JJ 3rd, Birukova AA, Beyer EC, Birukov KG. Gap junction protein connexin43 exacerbates lung vascular permeability. PLoS One. 2014;9(6):e100931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Perdomo J, Leung HHL, Ahmadi Z, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin‐induced thrombocytopenia. Nat Commun. 2019;10(1):1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Peng B, Xu C, Wang S, Zhang Y, Li W. The role of connexin hemichannels in inflammatory diseases. Biology. 2022;11(2):237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nawaz M, Fatima F. Extracellular vesicles, tunneling nanotubes, and cellular interplay: synergies and missing links. Front Mol Biosci. 2017;4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Charreau B. Secretome and tunneling nanotubes: a multilevel network for long range intercellular communication between endothelial cells and distant cells. Int J Mol Sci. 2021;22(15):7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jackson MV, Morrison TJ, Doherty DF, et al. Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ARDS. Stem Cells. 2016;34(8):2210‐2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Soundara Rajan T, Gugliandolo A, Bramanti P, Mazzon E. Tunneling nanotubes‐mediated protection of mesenchymal stem cells: an update from preclinical studies. Int J Mol Sci. 2020;21(10):3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zaccard CR, Watkins SC, Kalinski P, et al. CD40L induces functional tunneling nanotube networks exclusively in dendritic cells programmed by mediators of type 1 immunity. J Immunol. 2015;194(3):1047‐1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tiwari V, Koganti R, Russell G, Sharma A, Shukla D. Role of tunneling nanotubes in viral infection, neurodegenerative disease, and cancer. Front Immunol. 2021;14(12):680891. [DOI] [PMC free article] [PubMed] [Google Scholar]