

Abstract
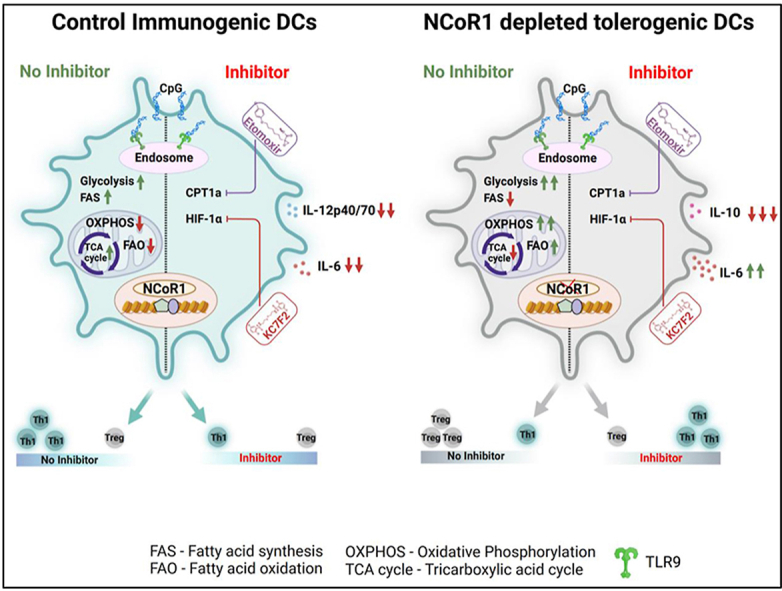
Dendritic cells (DCs) undergo rapid metabolic reprogramming to generate signal-specific immune responses. The fine control of cellular metabolism underlying DC immune tolerance remains elusive. We have recently reported that NCoR1 ablation generates immune-tolerant DCs through enhanced IL-10, IL-27 and SOCS3 expression. In this study, we did comprehensive metabolic profiling of these tolerogenic DCs and identified that they meet their energy requirements through enhanced glycolysis and oxidative phosphorylation (OXPHOS), supported by fatty acid oxidation-driven oxygen consumption. In addition, the reduced pyruvate and glutamine oxidation with a broken TCA cycle maintains the tolerogenic state of the cells. Mechanistically, the AKT-mTOR–HIF–1α-axis mediated glycolysis and CPT1a-driven β-oxidation were enhanced in these tolerogenic DCs. To confirm these observations, we used synthetic metabolic inhibitors and found that the combined inhibition of HIF-1α and CPT1a using KC7F2 and etomoxir, respectively, compromised the overall transcriptional signature of immunological tolerance including the regulatory cytokines IL-10 and IL-27. Functionally, treatment of tolerogenic DCs with dual KC7F2 and etomoxir treatment perturbed the polarization of co-cultured naïve CD4+ T helper (Th) cells towards Th1 than Tregs, ex vivo and in vivo. Physiologically, the Mycobacterium tuberculosis (Mtb) infection model depicted significantly reduced bacterial burden in BMcDC1 ex vivo and in CD103+ lung DCs in Mtb infected NCoR1DC−/−mice. The spleen of these infected animals also showed increased Th1-mediated responses in the inhibitor-treated group. These findings suggested strong involvement of NCoR1 in immune tolerance. Our validation in primary human monocyte-derived DCs (moDCs) showed diminished NCOR1 expression in dexamethasone-derived tolerogenic moDCs along with suppression of CD4+T cell proliferation and Th1 polarization. Furthermore, the combined KC7F2 and etomoxir treatment rescued the decreased T cell proliferative capacity and the Th1 phenotype. Overall, for the first time, we demonstrated here that NCoR1 mediated control of glycolysis and fatty acid oxidation fine-tunes immune tolerance versus inflammation balance in murine and human DCs.
Keywords: NCoR1, Glycolysis, OXPHOS, HIF-1α, FAO, Th1, Tregs
Graphical abstract
Highlights
-
•
AKT-mTOR–HIF–1α mediated glycolysis and CPT1a driven β-oxidation drives tolerance in NCoR1 depleted tolerogenic DCs.
-
•
Truncated TCA cycle with reduced pyruvate and glutamine oxidation facilitates tolerance.
-
•
Combined perturbation of HIF-1α and CPT1a, shifts the balance of tolerogenic DCs towards inflammatory.
-
•
Treatment of Mtb infected animals with inhibitors resulted in decreased Mtb load in CD103+ lung DCs.
-
•
Dexamethasone-induced moDCs depicted reduced NCOR1 expression, increased proliferation and Th1 responses with inhibitors.
Abbreviations
- BMDCs
Bone marrow derived dendritic cells
- DCs
Dendritic cells
- Dex
Dexamethasone
- DEGs
Differentially expressed genes
- ECAR
Extracellular acidification rate
- ETC
Electron transport chain
- FAO
Fatty acid oxidation
- mtDNA
Mitochondrial DNA
- NCoR1
Nuclear receptor co-repressor 1
- moDCs
Monocyte derived dendritic cells
- Mtb
Mycobacterium tuberculosis
- OXPHOS
Oxidative phosphorylation
- OCR
Oxygen consumption rate
- ROS
Reactive oxygen species
- TLR9
Toll-like receptor 9
- TMRM
Tetramethylrhodamine, methyl ester
1. Introduction
The process of DC activation and cytokine production is highly coordinated. Activation also necessitates significant energy needs by undergoing rapid metabolic reprogramming events. Metabolic reprogramming processes in addition to signalling cascades, transcription factors, and co-regulators, play pivotal roles in fine-tuning the balance between inflammation and tolerance [1]. DCs differentiated with either GM-CSF or FLT3 show differential metabolic adaptations upon TLR specific activation [2]. Among which, early and late activation profiles are also distinct. Under steady-state conditions, OXPHOS dominates in DCs, reducing power being generated primarily by fatty acid oxidation (FAO) and the glutaminolysis-driven TCA cycle [1,[3], [4], [5], [6]]. These steady state adaptations are regulated by the AMP/ATP sensor AMPK and maintain the metabolic homeostasis of the cell. Early activation of immunogenic DCs upon TLR engagement alters the metabolic profile to glycolytic with stable OXPHOS. Such metabolic energy needs in inflammatory DCs are primarily controlled via the kinases TBK1, IKKε, and HK2 [7] whereas, sustained activation leads to decreased OXPHOS and a dominant glycolytic profile mediated by the mTOR/HIF-1α axis [8,9]. These events are crucial regarding DC migration, up-regulation of co-stimulatory or inhibitory molecules and secretion of cytokines. Such evidence is also being drawn from the 2-DG inhibitor studies [[10], [11], [12]]. Another important aspect of DC biology is to prevent hyperactivation and auto-immune diseases by inducing immune-tolerance. Tolerogenic DCs assist in overcoming such conditions by secreting increased levels of IL-10, IL-27, and TGF-β cytokines resulting in increased frequency of Treg induction than Th1 cells [13,14]. These DCs are conventionally attained by Vitamin D3 (VD3), and Dexamethasone (Dex), either individually or in combination. Whereas antigen presenting cell (APC) mediated Treg induction is obtained by IL-10, retinaldehyde dehydrogenase (RALDH), and indoleamine 2,3-dioxygenase (IDO-1) [15,16].
Metabolism in tolerized DCs is governed by increased OXPHOS, FAO and glycolytic capacity [[17], [18], [19], [20], [21], [22], [23]]. However, the transcriptional and molecular cues underpinning such metabolic activities that generates tolerance in DCs is still lacking. Also, metabolic events impacted by such a transcriptional network are unexplored. Global co-regulators such as nuclear receptor co-repressor 1 (NCoR1) and silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) are well characterized for their metabolic functions. They're recognized to be important in maintaining homeostasis and regulating systemic metabolic processes [24,25]. Among these, NCoR1 has diverse metabolic roles in muscle cells and adipocytes [26,27]. The preferential implication of NCoR1 is also being established in energy metabolism with respect to autophagy and atherosclerosis [[28], [29], [30]]. In the context of regulation of inflammatory versus tolerogenic immune response, our earlier functional and epigenomics studies on type I conventional DCs have demonstrated that NCoR1 depletion polarizes DCs towards tolerance that in turn shifts the balance of naive CD4+ co-cultured T cells towards Tregs [31,32]. However, the metabolic adjustments of these tolerogenic DCs is largely unknown, hence, in this study we focused to understand the control of various metabolic pathways required to maintain a fine balance of inflammation and tolerance in pursuit of energy metabolism in both murine and human DCs.
In this study, we performed comprehensive molecular and metabolic characterization of NCoR1 depleted immune-tolerant DCs in-vitro (CD8α+ DC line) and ex vivo (bone marrow-derived dendritic cells from NCoR1DC−/− mice). DCs were activated by ligating TLR9 using unmethylated CpG (CPG-B) as an immunological stimulant. Characteristic pathways specific for energy metabolism were profiled by employing extracellular flux assays, transcriptome analysis and T cell polarization. In addition, primary human DCs and Mtb infection model substantiated the key metabolic adaptations required to fine-tune immunogenic versus tolerogenic balance.
2. Results
2.1. NCoR1 depleted tolerogenic DCs depicted enhanced glycolysis and OXPHOS
To decipher the metabolic adaptations controlled by NCoR1 leading to strong immune-tolerant state in conventional Type-I DCs (cDC1), we have used a stable Ncor1 gene knock-down (KD) cDC1 mutu-DC line where the mean Ncor1 gene KD efficiency of ∼80% at transcript level was confirmed by RT-qPCR (Fig. S1A). We have already confirmed in details the tolerogenic phenotype of NCoR1 depleted cDC1 in our earlier studies [31]. Now to investigate the real-time metabolic changes of these NCoR1 depleted and control cDC1 in unstimulated (Uns) and CpG challenged conditions, extracellular flux assays were performed using Seahorse flux analyzer. The CpG challenged NCoR1 deficient tolerogenic DCs demonstrated significantly enhanced glycolysis and glycolytic capacity (Fig. 1A and Fig. S1B). Along with it, increased extracellular lactate build-up and decreased pH complemented the same (Fig. 1B and Fig. S1C). Furthermore, glycolytic ATP determined with and without 2-DG (2-Deoxy-D-glucose) also showed a similar pattern (Fig. S1D). To further confirm this phenotype, we looked into the transcript expression of the key characteristic glycolytic genes in the RNAseq data and found that genes like Pkm, Pfkp, Pfkfb3, Hk2, and Ldhal6b were upregulated in the NCoR1 KD CpG condition compared to control (Fig. 1C). In addition, metabolic pathways such as “Respiratory electron transport” and “Pyruvate metabolism” were top enriched pathways in the differentially expressed genes (DEGs) of 6h CpG activated NCoR1 depleted DCs (Fig. 1D). Following which we also performed RT-qPCR and specifically examined the relative mRNA expression of glucose transporter Slc2a1 and rate-limiting enzymes like Hk2 and Pkm2, along with Ldha. The mRNA levels were found to be significantly higher in stimulated NCoR1 KD DCs compared to controls (Fig. 1E). Increased expression of the glut transporter 1 and rate-limiting enzymes confirmed enhanced glucose uptake and its metabolization through the glycolytic pathway. At the metabolite level, quantitation by Gas chromatography-mass spectrometry (GC-MS) depicted significantly increased levels of pyruvic acid, lactic acid, and alanine reassuring similar bioenergetic profiles (Fig. 1F). Increased lactic acid and alanine hint towards a decreased TCA cycle activity and acetyl Co-A conversion [33]. Furthermore, NCoR1 ChIPseq data depicted that the rate limiting glycolytic genes such as Pfkb and Pfkfb3 are directly bound and regulated by NCoR1 in control Uns and 6h CpG condition (Fig. 1G). This implies that when the co-repressor is removed, Pfk's gene expression increases and thus glycolysis. Next, we focussed on a major parallel pathway of glycolysis called pentose phosphate pathway (PPP) since it is important for regulating redox homeostasis and biosynthesis. We first checked the transcript level expression (log2 FC) of the oxidative and non-oxidative PPP genes in our RNAseq data and found them to be downregulated except Glucose 6 phosphate dehydrogenase 2 (G6pd2), an important rate-limiting enzyme of PPP, although not significant (Fig. S1E). Further to confirm, we analysed the protein level expression of G6PD, and found it to be profoundly reduced (Fig. S1F) supporting that PPP might not be playing a pivotal role in metabolizing Glucose 6-phosphate. Therefore, the above results confirmed enhanced glycolysis to be a distinctive feature of NCoR1 depleted immune-tolerant DCs upon CpG stimulation.
Fig. 1.

NCoR1 depleted tolerogenic cDC1 DCs depicted enhanced glycolysis and OXPHOS as compared to control cells
A. Representative line graph and scatter plots showing the glyco-stress parameters (glycolysis and glycolytic capacity) in unstimulated and 6h CpG activated control and NCoR1 depleted tolerogenic DCs, as measured using Seahorse extracellular flux analyzer. (n = 6)
B. Scatter plot depicting the extracellular lactate accumulation in the culture supernatants of unstimulated and 6h CpG activated control and NCoR1 depleted tolerogenic DCs at a dilution of 1:10. (n = 5)
C. Heatmap showing the differentially expressed genes (DEGs) enriched for the glycolytic pathway. RNAseq of unstimulated and 6h CpG activated NCoR1 KD as compared to control DCs were used to determine the DEGs. (n = 2)
D. Bar graphs depicting the top enriched metabolic pathways from the DEGs in NCoR1 KD CpG activated tolerogenic DCs as compared to controls, using Reactome Pathway Database. (n = 2)
E. RT-qPCR analysis showing the relative transcript expression of glut transporter Slc2a1, and glycolytic genes Hk2, Pkm2 and Ldha in unstimulated and 6h CpG activated NCoR1 KD tolerogenic DCs. (n = 5–7)
F. Bar graphs demostrating the relative intensities of intracellular metabolites of NCoR1 KD CpG activated DCs as compared to controls as quantified using GC-MS. (n = 3)
G. IGV snapshots showing the ChIPseq binding of NCoR1 at the Pfkp and Pfkfb3 gene loci in unstimulated and 6h CpG stimulated control DCs.
H. Representative line graph and scatter plots showing the mito-stress parameters (basal respiration, maximal respiration, coupled ATP and spare respiratory capacity) in unstimulated and 6h CpG activated control and NCoR1 depleted tolerogenic DCs as measured using an extracellular flux analyzer. (n = 8)
I. Box and whisker plots showing the min-max values for extracellular nitrite in the culture supernatant collected from unstimulated and 6h CpG activated control and NCoR1 depleted tolerogenic DCs. (n = 8)
J. Box and whisker plots depicting the min-max values for mitochondrial DNA copies (Nd-1/Hk-II) quantified by RT-qPCR in unstimulated and 6h CpG activated control and NCoR1 KD DCs. (n = 5)
K. Bar graphs with dots depicting the MFI levels of mitochondrial SOX, (n = 6) and cellular ROS, (n = 5) in unstimulated and 6h CpG stimulated control and NCoR1 KD DCs as quantified from flow cytometry.
L. Bar graphs with dots showing the MFI levels of TMRM measured for mitochondrial potential estimation in unstimulated and 6h CpG activated control and NCoR1 KD DCs. (n = 6)
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed paired student’s t-test. Data shown in figure is combined from 3 independent experiments [A] and from 4 independent experiments [H]. Error bars represent SEM, except I and J, horizontal lines depicting mean.
We then investigated the OXPHOS levels, since a fine-balance of glycolysis and OXPHOS in response to activation are reported to be important for inflammation and immune-tolerance. Tolerogenic DCs are known to exhibit higher levels of OXPHOS [18,34]. To decipher the OXPHOS levels in control and NCoR1 KD DCs, first we checked the expression of genes belonging to complex I–V of the electron transport chain (ETC) using RNAseq data. The ETC genes were enriched for the NCoR1 ablated DCs both in Uns and CpG condition (Fig. S1G). Further, transcript expressions of the nuclear respiratory factor-1 (Nrf-1) and Complex-IV gene Cox5a were found to behave in a similar manner (Fig. S1H). Moreover, oxygen consumption rate (OCR) with regard to basal respiration, maximal respiration, coupled ATP, and spare respiratory capacity were found to be significantly higher in NCoR1 KD cells both in Uns and CpG conditions with respect to controls. The activated states depicted significantly lowered OCR levels in control and a decreasing trend in NCoR1 KD DCs (Fig. 1H and Fig. S1I). This suggested an inclination of stimulated NCoR1 KD cells towards glycolysis for energy requirement. Also, the coupled ATP from OXPHOS differed from the total intracellular ATP levels, which points towards the direct generation of ATP from glycolysis (Fig. S1J). To substantiate the preceding assertion regarding OXPHOS, we measured the extracellular nitrite levels. Nitrite and nitrate, stable end products of Nitric Oxide (NO), impact mitochondrial respiration inversely [35,36]. Since NCoR1 KD cells showed higher mitochondrial respiration, their extracellular nitrite levels were found to be significantly lower (Fig. 1I). Other important parameters like mitochondrial DNA (mtDNA) copy number can also facilitate OXPHOS proportionately [37]. Therefore, mtDNA copy numbers were determined taking the ratio of Nd-1/Hk-II (mitochondrial/nuclear) [38]. NCoR1 KD cells had significantly more mtDNA copies and gave a plausible explanation for the enhanced mitochondrial respiratory parameters (Fig. 1J). Besides nitrite and mtDNA copy numbers, pivotal aspects impacting respiratory levels are mitochondrial potential and reactive oxygen species. Mitochondrial SOX and cellular ROS levels were estimated and found to be significantly reduced in CpG-stimulated NCoR1 KD tolerant DCs (Fig. 1K and Fig. S1K), distinctive features of inflammatory DCs. On the other side, increased TMRM (Tetramethylrhodamine, methyl ester) levels in NCoR1 KD DCs showed enhanced mitochondrial potential, supporting elevated OXPHOS (Fig. 1L and Fig. S1L). The median levels for Mitochondrial SOX, cellular ROS and TMRM were determined by flow cytometry and uniform gating strategies were used (Fig. S1M). Overall, the tolerogenic NCoR1 KD DCs exhibited, although not significantly, higher OXPHOS levels in Uns condition than in CpG. In comparison to control cells, the levels were elevated and significant. The relative increase in OXPHOS in NCoR1 depleted DCs hinted towards increased bioenergetic pathways fuelling the electron transport chain (ETC). This also suggested that regulation of stimulation-dependent tolerogenic cytokine secretion is an integral part of metabolic pathways.
2.2. FLT3L differentiated primary BMDCs from NCoR1DC−/− mice depicted similar bioenergetics as mutu-cDC1 line
We further confirmed the glycolytic and mitochondrial bioenergetic parameters observed in the mutu-cDC1 line in primary BMDCs generated from NCoR1DC−/− mice. First genotyping was performed to confirm homozygous NCoR1DC−/− progenies (Fig. S2A). In addition, the NCoR1 ablation in BMDCs was confirmed by RT-qPCR with respect to control CD11c-Cre BMDCs (Fig. S2B). Furthermore, cellular homeostasis of FLT3L differentiated DCs at day 9 (CD11c+MHCII+) in NCoR1DC−/− and CD11c-Cre control mice were confirmed by flow cytometry (Fig. S2C). No disparity in percentage of DC numbers were observed between the NCoR1DC−/− and CD11c-Cre control mice. Unlike the DC line, extracellular bioenergetic parameters in BMDCs were determined at Uns and CpG 18h, since a prolonged and sustained activation was required for primary DCs to generate effective immune responses [39,40]. Real-time metabolic changes pertaining to glycolysis and glycolytic capacity were significantly increased in NCoR1DC−/− BMDCs upon stimulation (Fig. 2A). Extracellular lactate levels also complemented the enhanced glycolytic levels (Fig. 2B). The relative mRNA expression of the glucose transporter Slc2a1 and rate-limiting enzymes like Hk2 and Pkm2, along with Ldha were elevated as in the DC line and confirmed the enhanced glycolytic rate in NCoR1DC−/− BMDCs at CpG 18h (Fig. 2C). Further, mitochondrial energy parameters like basal respiration, maximal respiration, and spare respiratory capacity also showed a similar trend in NCoR1DC−/− and control CD11c-Cre BMDCs as observed in the DC line (Fig. 2D). Parameters regulating mitochondrial respiration and inflammation like cellular ROS were found to be significantly decreased in the NCoR1DC−/− BMcDC1s (Fig. 2E and Fig. S2D). Whereas TMRM mediated mitochondrial potential was high in both Uns and 18h CpG stimulation (Fig. 2F and Fig. S2E). Uniform gating strategies were used for depicting the median levels of cellular ROS and TMRM in BMcDC1s (Fig. S2F). Overall, the tolerogenic NCoR1DC−/− BMDCs displayed similar bioenergetic profiles as mutu-cDC1 line both in Uns and CpG activated conditions.
Fig. 2.

FLT3L differentiated primary BMDCs from NCoR1DC−/− mice showed enhanced glycolysis and OXPHOS as observed in mutu-cDC1 line
A. Representative line graph and scatter plots showing the glyco-stress parameters (glycolysis and glycolytic capacity) in unstimulated and 18h CpG activated control CD11c-Cre and NCoR1 ablated FLT3 differentiated BMDCs on day 9, measured using Seahorse extracellular flux analyzer. (n = 11)
B. Scatter plot depicting the levels of extracellular lactate accumulated in the culture supernatants of control CD11c-Cre and NCoR1DC−/− BMDCs before and after 18h CpG activation, at a dilution of 1:5. (n = 6)
C. Bar graphs showing the relative transcript expression of glucose transporter Slc2a1, and glycolytic genes, Hk2, Pkm2 and Ldha in unstimulated and 18h CpG activated CD11c-Cre and NCoR1DC−/− BMDCs. (n = 4–5)
D. Representative line graph and scatter plots showing the mito-stress parameters (basal respiration, maximal respiration, coupled ATP and spare respiratory capacity) in unstimulated and 18h CpG activated control CD11c-Cre and NCoR1DC−/− BMDCs on day 9 as measured using Seahorse extracellular flux analyzer. (n = 6)
E. Bar graphs with dots depicting the MFI levels of cellular ROS in unstimulated and 18h CpG activated CD11c-Cre control and NCoR1DC−/− BMcDC1s on day 9 as measured in F4/80−CD11c+CD24+ gated cells using flow cytometry. (n = 12)
F. Bar graphs with dots showing the MFI levels of mitochondrial potential upon TMRM staining in CD11c-Cre control and NCoR1DC−/− BMcDC1s on day 9 before and after 18h CpG stimulation as measured in F4/80−CD11c+CD24+ gated cells using flow cytometry. (n = 7)
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed unpaired student’s t-test. Data shown in figure is combined from 4 independent experiments [A] and from 3 independent experiments [D]. Error bars represent SEM.
2.3. AKT/mTOR induced HIF-1α expression regulates glycolysis and secretion of tolerogenic cytokines
To gain insight into how NCoR1 depletion in cDC1 impacted glycolytic metabolic reprogramming, we assessed the signalling events crucial for DC metabolic responses. The TBK1/IKK/AKT/HK-II axis and the AMPK versus mTOR/HIF-1α pathway is known to regulate early and late glycolytic induction in DCs, respectively [7,41,42]. We focussed on 6h CpG activated control and NCoR1 KD cDC1 and thus considered the late glycolytic induction signalling cascade. NCoR1 depleted 6h CpG challenged cDC1 depicted enhanced phosphorylated levels of AKT and mTORC1 in western blotting (Fig. 3A). Increased levels of AKT and mTORC1 in concert essentially stabilised the HIF-1α expression (Fig. 3A), responsible for the upregulation of glucose transporters like Slc2a1 and glycolytic enzymes like PFK, PKM2, LDHa, etc. [[43], [44], [45]]. The relative mRNA expression of the Hif-1α gene was also significantly upregulated (Fig. 3B). The above findings imply that in CpG challenged NCoR1-deficient DCs, a signal transduction circuit including AKT-mTORC1-HIF-1α axis coordinated increased glycolysis. From RNAseq data, we found that DEGs of the HIF-1α pathway genes were significantly upregulated upon CpG stimulation in NCoR1 KD cells (Fig. 3C). Mechanistically, ChIPseq data revealed direct NCoR1 binding peaks at the distal regions of the Hif-1α gene body, exhibiting direct regulation of Hif-1α expression by NCoR1 (Fig. 3D). Further, to confirm how mTORC1 and HIF-1α modulates glycolytic function, extracellular acidification rate (ECAR) was measured after treatments with mTOR inhibitor rapamycin and the HIF-1α inhibitor KC7F2. We performed cell viability assay at the concentrations of rapamycin and KC7F2 used for experiments and more than 90% viability was observed (Fig. S3A). We observed that control and NCoR1 KD DCs responded differentially upon inhibitor treatments. For confirmation of the potent dysregulation of the HIF-1α molecule upon KC7F2 treatment, we also performed RT-qPCR and looked into the transcript level expression of Hif-1α in both control and NCoR1 KD DCs upon stimulation with CpG for 6 h (Fig. S3B). Glycolysis and glycolytic capacity levels were significantly impaired in CpG stimulated NCoR1 depleted cells with respect to controls. Moreover, KC7F2 inhibition depicted robust perturbation of ECAR levels in comparison to rapamycin, which suggested a strong dependence of NCoR1 KD cells on HIF-1α signalling axis for enhanced glycolytic function (Fig. 3E). Further, to know how rapamycin and KC7F2 treatments impacted the balance between tolerance and inflammation, we determined the levels of tolerogenic cytokines like IL-10 and IL-27 along with inflammatory ones like IL-6 and IL-12p40/70 by flow cytometry. We found a significant reduction in the percent positive cells as well as in MFI shifts for tolerogenic cytokines upon KC7F2 treatment in NCoR1 KD condition (Fig. 3F and Fig. S3C). No noticeable changes were observed in the expression of inflammatory cytokines, whereas control cells depicted a significant reduction in MFI levels of IL-12p40/70 upon KC7F2 treatment (Fig. S3D). Overall, the results confirmed an essential role of HIF-1α in regulating glycolysis and its importance in inducing immune-tolerance in NCoR1 KD DCs.
Fig. 3.

AKT-mTOR–HIF–1α axis in regulating glycolysis and tolerogenic cytokine production in NCoR1 depleted DCs
A. Representative western blots showing the phosphorylated and total protein levels of AKT, mTOR and HIF-1α in control and NCoR1 depleted DCs after 6h CpG stimulation. Ratios of phosphorylated to total protein levels were further normalized to the housekeeping control β-actin. Corresponding bar plots depicted the densitometry analysis of the western blots. (n = 3–4)
B. Bar graph with dots showing the relative mRNA expression of Hif-1α in 6h CpG activated control and NCoR1 depleted tolerogenic DCs. (n = 7)
C. Heatmap showing the differentially expressed genes of HIF-1α pathway observed in the RNAseq data of control and NCoR1 KD 6h CpG stimulated DCs. (n = 3)
D. IGV snapshot depicting the NCoR1 binding at the distal genomic regions of Hif-1α gene loci in unstimulated and 6h CpG activated control DCs.
E. Line graph and box and whisker plots with dots depicting the levels of ECAR along with glycolysis and glycolytic capacity respectively upon pre-injection with either rapamycin (20nM) or KC7F2 (10μM) in control and NCoR1 KD DCs after 6h CpG activation, as measured using Seahorse extracellular flux analyzer. (n = 6)
F. Scatter plots (percent positive cells) and bar graphs (MFI) showing the cytokine levels of IL-10 and IL-27 upon treatment with rapamycin (20nM) and KC7F2 (10μM) in CpG activated control and NCoR1 depleted tolerogenic DCs. (n = 7–8)
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed paired student’s t-test. Error bars represent SEM.
2.4. Reduced pyruvate and glutamine oxidation facilitates immune tolerance
Pyruvate oxidation, glutaminolysis and FAO replenish the TCA cycle and generate reducing powers to be used in the ETC for OXPHOS [46]. To decipher how active OXPHOS levels were maintained in NCoR1 depleted cells under CpG activation, we did a comprehensive characterization of the aforementioned pathways and its impact on tolerance. As noted earlier, the enhanced ECAR facilitated lactic acid and alanine production and hinted towards the non-incorporation of pyruvate into the mitochondrial tricarboxylic acid (TCA) cycle. Reduced pyruvate dehydrogenase (PDH) expression at the protein level in NCoR1 KD cDC1 confirmed the same (Fig. 4A). Furthermore, pyruvate and glutamine oxidation measured after using mitochondrial pyruvate carrier (MPC-1) inhibitor UK-5099 along with BPTES, the glutaminase1 (GLS1) inhibitor and etomoxir that inhibits carnitine palmitoyltransferase 1 (CPT1a), we found a significant reduction in percent dependencies of NCoR1 depleted cells on these pathways (Fig. 4B and Fig. S3E). Further, the DEGs of the TCA cycle pertaining to pyruvate oxidation like Pdhb showed reduced expression (Fig. 4C). In addition, transcript levels of the sequential enzymes of TCA cycle from citrate to isocitrate (Aco2), isocitrate to α-ketoglutarate (Idh2/3a/3b/3g), α-ketoglutarate to succinate (Ogdh/Ogdhl) and succinate to fumarate (Sdha/b) were also found to be decreased (Fig. 4C). The above data gave us an insight into the broken TCA cycle being operated in the NCoR1 KD cells. Functionally, to know the balance between tolerance and inflammation, flow cytometry was used to determine the secretory levels of tolerogenic and inflammatory cytokines upon treatment with UK-5099, BPTES and glutamine analogue DON. No significant changes in the cell viability were observed upon treatment with these inhibitors (Fig. S3F). In addition, further reduction in pyruvate and glutamine oxidation significantly increased IL-10 levels both in control and NCoR1 depleted DCs (Fig. 4D and Fig. S3G). On the other hand, decreased IL-12p40/70 percent positive cells were obtained for both control and NCoR1 KD cells upon UK-5099 treatment (Fig. S3H). BPTES and DON impacted the MFI shifts of IL-6 and IL-12p40/70 insignificantly (Fig. S3H). Taken together, the aforesaid findings suggested that lowered levels of pyruvate and glutamine oxidation play crucial roles in maintaining immune tolerance in NCoR1 depleted DCs. Interestingly, potent disruption of glutaminolysis and pyruvate oxidation render control DCs towards tolerance.
Fig. 4.

Mitochondrial fuel/flux (Pyruvate oxidation, Glutamine oxidation and FAO) characteristics regulates tolerogenicity in NCoR1 depleted DCs
A. Representative Western blot showing the protein level expression of pyruvate dehydrogenase (PDH) in control and NCoR1 depleted tolerogenic DCs after 6h CpG activation. The protein level was normalized to the housekeeping control β-tubulin. Corresponding bar graph depicted the densitometric analysis of the Western blot bands. (n = 4)
B. Plots demonstrating the percent dependencies of 6h CpG activated control and NCoR1 depleted DCs on pyruvate [(first injection- UK-5099-2μM and second a combination of etomoxir (4μM)+BPTES (3μM) together] and glutamine oxidation [first injection - BPTES (3μM) and second a combination of UK-5099 (2μM) + etomoxir (4μM) together], measured from an extracellular flux analyzer. (n = 6)
C. Heatmap showing the differentially expressed genes of TCA cycle observed in the RNAseq of unstimulated and 6h CpG activated control and NCoR1 depleted tolerogenic DCs. (n = 2)
D. Scatter plots (percent positive cells) and bar graphs (MFI) demonstrating the cytokine levels of IL-10 and IL-27 upon treatment with UK-5099 (5μM), BPTES (5μM) and DON (5μM) in control and NCoR1 depleted tolerogenic DCs after 6h CpG stimulation. (n = 6)
E. Line graph and scatter plot showing the FAO index in control and NCoR1 KD DCs after 6h CPG activation upon treatment with etomoxir (50μM) and Oligomycin (2μM), as measured using Seahorse Extracellular flux analyzer. (n = 6)
F. Bar graph depicting the relative mRNA expression of Cpt1a in 6h CpG activated control and NCoR1 depleted tolerogenic DCs quantified using RT-qPCR. (n = 3)
G. Heatmap showing the differentially expressed genes of FAO pathway in control and NCoR1 depleted DCs before and after 6h CpG activation using RNAseq data. (n = 2)
H. Scatter plots (percent positive cells) and bar graphs (MFI) showing the cytokine levels of IL-10 and IL-27 upon treatment with etomoxir (50μM) in 6h CpG activated control and NCoR1 depleted DCs. (n = 6–7)
I. Bar plots showing the normalized counts for Acly from RNAseq data and relative mRNA expression of Acaca and Fasn using RT-qPCR in 6h CpG stimulated control and NCoR1 KD DCs. (n = 3 for Acly and n = 5 for Acaca and Fasn)
J. Representative microscopic images showing the Oil red-O stained neutral lipids in the control and NCoR1 KD DCs after 6h CpG activation. Corresponding dot plot depicted the quantification of the oil-red-O stain using absorbance at 500nm. (n = 6)
K. Bar plot showing the normalized counts for Ppar-γ in the RNAseq data of 6h CpG activated control and NCoR1 depleted tolerogenic DCs. (n = 3)
L. IGV snapshot showing the direct binding of NCoR1 at Ppar-γ gene loci in cDC1 before and after 6h CpG activation.
M. Schematic illustration showing the non-incorporation of pyruvate into the TCA cycle, accumulation of citrate, reduced expression of TCA cycle genes and decreased glutamine oxidation in 6h CpG activated NCoR1 KD DCs.
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed paired student’s t-test. Data shown in figure is combined from 3 independent experiments [B]. Error bars represent SEM. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
2.5. Active OXPHOS driven by FAO regulates immune tolerance in NCoR1 KD DCs
The above results indicate that the active state of OXPHOS in NCoR1 depleted DCs is driven by pathways other than pyruvate and glutamine oxidation. We speculated β-oxidation of fatty acids might facilitate the same. We found that at 6h CpG activation, oxygen consumption mediated FAO using etomoxir resulted in a significantly higher FAO index in NCoR1 KD DCs (Fig. 4E and Fig. S3I). The relative mRNA expression of the rate-limiting FAO gene Cpt1a was also found to be significantly elevated (Fig. 4F). In accordance with the FAO index, DEGs pertaining to β-oxidation pathways showed enhanced expression of genes like Pex7, Acoxl, Acadl, Phyh, Acsl1, Adipor1, Por, and Acaa1a in RNAseq data of CpG stimulated NCoR1 KD DCs (Fig. 4G). Since unstimulated NCoR1 depleted DCs showed higher OXPHOS compared to stimulated DCs, we could speculate that the pattern of gene expression with regard to β-oxidation behaved similarly (Fig. 4G). Functionally, modulation of immune response cytokines upon inhibition of β-oxidation pathway using etomoxir were also analyzed. Etomoxir did not hamper the cell viability levels (Fig. S3J). Etomoxir treatment showed significant reduction in the secretory levels of tolerogenic cytokines IL-10 and IL-27 (Fig. 4H and Fig. S3K), while inflammatory cytokines IL-12p40/70 and IL-6 remain unchanged in NCoR1 depleted condition (Fig. S3L). Next, we asked how fatty acid oxidation is being facilitated in NCoR1 KD DCs? It is known that β-oxidation in cells is aided by de novo fatty acid synthesis (FAS) via citrate, intracellular fatty acid reserves and exogenous fatty acid imports via CD36 [[47], [48], [49]]. Moreover, de novo FAS via citrate is characteristic of immunogenic DCs [7]. Although, NCoR1 depleted DCs produced more of citric acid (Oxaloacetic acid generated from anaplerotic reaction of aspartic acid and acetyl Co-A from β-oxidation), but its sequential conversion to produce free FAs and its re-entry into mitochondria through CPT1a to undergo oxidation was hampered due to decreased expression of Acly, Acaca and Fasn (Fig. 4I and Fig. S3M). Moreover, reduced Cd36 normalized expression further opposed the notion of extracellular FA import (Fig. S3N). However, oil-red-O staining exhibited more neutral lipid deposits in NCoR1 depleted cells, suggesting the role of intracellular fatty acid reserves in mediating β-oxidation [28] (Fig. 4J). At the same time, lipid storage and fatty acid biosynthesis require NADPH to supply reducing equivalents. As we showed above that the PPP is downregulated in NCoR1 depleted DCs, we also looked into the ME1 and IDH1 related pathways. Since, G6PD decrease results in NADP production, which induces compensatory increases in IDH1 and ME1 [50,51]. Pyruvate shuttle into malate for converting into oxaloacetate might be one of the crucial routes for TCA cycle replenishment and NADP+ production or decarboxylation of malate into pyruvate for effective NADPH production for fatty acid synthesis. For this, we looked into the metabolite levels of Malic acid in our cellular metabolite analysis data of Mass Spec and found its levels to be profoundly higher in the NCoR1 depleted DC line (Fig. S3O), suggesting that active enzyme activity of Me1 appears to be involved in fatty acid biosynthesis or lipid storage in NCoR1 depleted DCs. Further, Idh1 transcript levels didn’t show any significant changes upon stimulation (Fig. S3P).
Now, the obvious question was how FAO is being regulated? Several reports have suggested that there is a direct role of PPARs in regulating mitochondrial β-oxidation [52,53]. From the RNAseq data we found an enhanced Ppar-γ expression in NCoR1 DCs (Fig. 4K). At the same time ChIPseq depicted direct binding of NCoR1 at Ppar-γ in cis-regulatory regions suggesting its direct control by NCoR1 (Fig. 4L). Possibly a feed-forward loop is generated through mTORC1 and PPAR-γ to facilitate FAO in CpG activated NCoR1 KD DCs [54]. Though this requires further work for its confirmation. Overall, the metabolic state of NCoR1 KD DCs demonstrated accumulation of citrate, lowered expression of TCA cycle genes and enhanced FAO index along with reduced pyruvate and glutamine oxidation (Fig. 4M). Thus, tolerogenic immune responses generated in NCoR1 ablated DCs are majorly contributed by CPT1a mediated β-oxidation along with HIF-1α driven glycolysis.
2.6. Dual inhibition of HIF-1α and FAO tweaked immune-tolerance to inflammation in NCoR1 depleted tolerogenic DCs
Individual inhibitions of HIF-1α-induced glycolysis and CPT1a-mediated FAO inhibited the levels of tolerogenic cytokines. It showed the importance of HIF-1α and CPT1a mediated metabolic changes in the modulation of tolerogenic response genes in NCoR1 KD DCs. Furthermore, to acquire potent inhibition of tolerance, we performed dual inhibition studies. Combined KC7F2 and etomoxir usage had no major effects on cell viability levels (Fig. S4A). We profiled the major tolerogenic cytokines IL-10 and IL-27 by flow cytometry and found their levels to be significantly impaired in NCoR1 depleted conditions when treated with the combined inhibitors (Fig. 5A and Fig. S4B). On the other hand, the levels of inflammatory cytokines were impaired significantly in control cells, suggesting that optimal glycolysis levels are important for induction of immunogenic response (Fig. S4C). At the same time, an increase in glycolysis above a threshold perturbs the immunogenic balance towards tolerance. NCoR1 depleted cells showed more dependency in terms of IL-10 and IL-27 secretion upon combined inhibition than individual perturbation with inhibitors (Fig. S4D). Furthermore, to get a global insight into the tolerogenic versus inflammatory responses, we performed RNAseq to investigate the DEGs, segregating with and without combined inhibition of HIF-1α and CPT1a. The principal component analysis of the 4 samples (1; Control cells activated with CpG, 2; Control cells activated with CpG and treated with KC7F2 + etomoxir, 3; NCoR1 KD cells activated with CpG, and 4; NCoR1 KD cells activated with CpG and treated with KC7F2 + etomoxir) depicted separate clusters, which indicated distinct transcriptome profiles (Fig. S4E). Then, we looked into the genes and their regulators pertaining to glycolysis and FAO. NCoR1 KD DCs at CpG 6h depicted enhanced expression of glycolytic rate-limiting genes like Hk2, Pfkfb3, Ldha and Hk3 (Fig. 5B), and important regulators like Stat3, which is known for transcriptional up-regulation of HIF-1α and underlying glycolytic genes [55,56]. Stat3 expression is significantly upregulated in NCoR1 perturbed cells than control [57]. Furthermore, FAO genes like Acsl5, Acox3 and Hpgds also showed significant enrichment (Fig. 5B). Upon combined inhibition, the expression levels of glycolytic and FAO genes were drastically reduced depicting the impact of combined effects of KC7F2 and etomoxir (Fig. 5B). To check the impact of inhibitor treatment on inflammatory and tolerogenic genes, we first identified unique or common genes differentially expressed upon inhibitor treatment at 6h CpG activated control and NCoR1 KD DCs. We found 381 genes to be significantly upregulated upon NCoR1 depletion in DCs compared to control, that in turn were found to be downregulated after combined inhibitor treatment (Fig. S4F). On the other hand, 1142 genes significantly upregulated in CpG activated NCoR1 KD DCs compared to control cells did not show any change upon treatment with inhibitors (Fig. S4F). Next, we performed gene ontology for biological process enrichment analysis of the above two gene sets using cluster Profiler. We plotted significantly enriched GO terms associated with inflammatory and tolerogenic response and the log2 fold change of genes in NCoR1 KD + inhibitor treated condition. Characteristic pathways such as tolerance induction, IL-27 mediated signalling pathway, Indole containing compound catabolic process, negative regulation of inflammatory response, and their associated genes like Cd274, Ido1, Il10, Socs1, Tnfaip3, Socs3, Ido2, Kmo, Ebi3, Il27, Stat3 involved in tolerogenic function were significantly downregulated upon combined inhibitor treatment (Fig. 5C). On the other hand, inflammatory response genes (Il6 and Il12b) or pathways such as dendritic cell cytokine production, TLR signalling pathway, positive regulation of inflammatory response, etc. did not show any significant change upon inhibitor treatment (Fig. 5C). Following this, we also plotted the distribution of z-scores of all the tolerogenic and inflammatory genes across different conditions. The median expression of the genes enriched for the tolerogenic pathway showed higher levels in NCoR1 KD, which significantly reduced after inhibitor treatment (Fig. 5D). On the other hand, the genes enriched for inflammatory pathways showed no significant difference after inhibitor treatment (Fig. 5D). We also investigated the normalized expression of a few candidate genes such as Il10, Il27, Il12, Il6, Ido1, Ctla4, Cd274, and Socs3. Upon inhibitor treatment, the tolerogenic genes that showed enhanced expression in CpG activated NCoR1 KD DCs depicted substantial impairment in expression, however, no significant changes were observed in the inflammatory genes (Fig. 5E and Fig. S4G). Furthermore, specific target genes of HIF-1α were also analyzed from the Harmonizome database that has a curated list from chromatin enrichment analysis (CHEA). We found that out of a total of 341 target genes, 40 genes were differentially expressed after inhibitor treatment in control DCs and 17 genes were differentially expressed after inhibitor treatment in NCoR1 KD DCs. Three different clusters were obtained for these differentially expressed target genes (Fig. S4H). Cluster 1 showed upregulation of DEGs upon inhibitor treatment, possibly the negative regulators whereas cluster 2 mainly depicted Hif-1α targeted metabolic genes. These were significantly upregulated in the NCoR1 KD condition and showed diminished or reduced expression upon inhibitor treatment. Cluster 3 didn’t show any major change. Overall, combined inhibition of HIF-1α and CPT1a displayed potent dysregulation of glycolytic and FAO pathways that significantly impacted the levels of tolerogenic genes with either little or no significant changes in inflammatory ones. Functionally, to delve into the T cell response induced by these DCs upon combined inhibitor treatment, we performed DC-T cell co-culture experiments using CD4+ T helper cells from OT-II transgenic mice. Control and NCoR1 KD cDC1 were pulsed with OT-II peptide (ISQAVHAAHAEINEAGR) overnight followed by 1 h of pre-treatment with inhibitor (combined KC7F2 + etomoxir) before 6h of CpG activation and incubation with FACS sorted CD3+CD4+ OT-II T cells (Fig. 5F). The sorted CD4+ T cells showed more than 90% purity (Fig. S4I). The proliferation of CD4+ T cells was determined using the eF670 dye after an incubation of 72 h. Combined inhibitor treatment suppressed the CD4+ T cell proliferation in control cells whereas no change was observed in the proliferative capacity in the NCoR1 KD conditions (Fig. 5G). Following proliferation, the profiling of these co-cultured CD4+ T cells into Th1 and Treg subtypes was performed. Control and NCoR1 KD cells behaved conversely with combined inhibition. In the control DC with inhibitor treatment conditions, we observed a significantly decreased percentages of IFN-γ+ and T-bet+ populations (Th1 phenotype) as compared to untreated cells (Fig. 5H). On the contrary, in NCoR1 depleted and combined inhibitor treated conditions the frequency of Th1 populations were significantly upregulated, when compared to untreated NCoR1 KD DCs (Fig. 5H). These results complemented the cytokine and transcriptomic data obtained with the combined inhibition. Further, to affirm the observed inflammatory phenotype in NCoR1 KD DCs, CD25+FOXP3+ (Tregs) populations were also identified. Control cells displayed non-significant changes in this population whereas, NCoR1 KD condition showed significant lower percentages of Tregs (Fig. 5I), which provided functional evidence of depleted tolerogenic cytokines with an intact inflammatory response upon dual inhibitor treatment. A similar experiment was performed using FLT3L differentiated primary NCoR1DC−/− BMDCs. On day 9 of differentiation, cells were treated with OT-II peptide overnight, followed by 1h of combined KC7F2+etomoxir treatment and CpG stimulation for 18h. The profiling data showed similar results as the cDC1 line (Fig. S4J). To further negate experimental artefacts obtained during ex vivo experiments, we performed an in vivo study, where NCoR1 perturbed DCs (5 × 106) with and without dual perturbation of HIF-1α and CPT-1a were injected intraperitoneally into OT-II transgenic mice. Ova incubation, inhibitor treatment, and CpG stimulation were done in vitro for these cells as explained before (Fig. 5J). On day 5, draining lymph nodes from animals were harvested and Th1 and Treg populations were determined by flow cytometry. The in vivo results were complementary to the ex vivo ones, which further substantiated the observed phenotype (Fig. 5K-L and Fig. S4K-M). Overall, T cell polarization studies confirmed the functional aspect of the impaired inflammatory and tolerogenic responses obtained in control and NCoR1 depleted DCs respectively.
Fig. 5.

Global transcriptomic changes and T cell polarization upon combined inhibition of HIF-1α and CPT1a
A. Scatter plots (percent positive cells) and bar graphs (MFI) showing the cytokine levels of IL-10 and IL-27 upon combined treatment with etomoxir (50μM) + KC7F2 (10μM) in 6h CpG activated control and NCoR1 depleted tolerogenic DCs. (n = 5)
B. Heatmap showing the differentially expressed genes of glycolytic and FAO pathway and their regulators in the RNAseq data before and after treatment with combined inhibitors KC7F2 and etomoxir in 6h CpG stimulated control and NCoR1 KD DCs. (n = 3)
C. Heatmap with bar plots showing the enriched GO terms associated with genes differentially regulated in 6h CpG activated NCoR1 KD before and after combined inhibition with HIF-1α and CPT1a. The asterisk in the heatmap denotes the significantly up or down regulated genes (p-adjusted <0.05) from DESeq2 results in CpG activated NCoR1 KD + Inhibitor treated as compared to only NCoR1 KD conditions. (n = 3)
D. Box plots depicting the distribution of gene expression (z-score) belonging to tolerogenic and inflammatory pathways with and without combined inhibition of HIF-1α and CPT1a in 6 h CpG activated control and NCoR1 KD DCs. (n = 3)
E. Bar graphs showing the normalized counts (DESeq2) of Il10, Il27, Ido1, Ctla4, Cd274 and Socs3 in 6h CpG stimulated control and NCoR1 KD DCs before and after combined inhibition of HIF-1α and CPT1a. (n = 3)
F. Schematic outline showing the control and NCoR1 KD DCsT cell co-culture experiment with the naive CD4+T cells isolated from OT-II transgenic mice ex vivo in the presence and absence of combined inhibitors KC7F2 and etomoxir.
G. Bar graph and histogram plot showing MFI levels and shifts respectively of eF670 labelled CD4+ T cells upon co-culturing with 6h CpG activated control and NCoR1 KD DCs before and after treatment with combined inhibitors of HIF-1α and CPT1a. Changes in the cell proliferation levels were measured for CD3+CD4+CD44+ gated effector T cells by flow cytometry after 72 h of co-culture. (n = 6)
H. Scatter plots (percent positive cells) and bar graphs (MFI) showing the IFN-γ+ and T-bet+ cells gated on CD3+CD4+CD44+ effector T cells upon co-culturing with control and NCoR1 KD DCs, with and without combined inhibition of HIF-1α and CPT1a. Percent positive cells and MFI levels were analyzed by flow cytometry after 96 h of co-culture. (n = 3)
I. Scatter plot showing the percent CD25+ and FOXP3+ double positive cells (Tregs) gated on CD3+CD4+CD44+ effector T cells upon co-culturing with control and NCoR1 KD DCs, with and without combined inhibition of HIF-1α and CPT1a. (n = 3)
J. Schematic showing the outline of an in vivo experiment for polarization of naive T cells upon adoptive transfer of NCoR1 KD DCs intraperitoneally in OT-II transgenic mice with and without combined inhibition of HIF-1α and CPT1a. Nearest draining lymph nodes (DLNs) were harvested after 96 h to profile the Th1 and Tregs.
K. Scatter plots (percent positive cells) and bar graphs (MFI) showing the IFN-γ+ and T-bet+ cells, gated on CD3+CD4+CD44+ effector T cells in DLNs after 96 h, representing the Th1 subtype in OT-II mice upon adoptive transfer of NCoR1 KD DCs with and without combined inhibition of HIF-1α and CPT1a. (n = 7)
L. Scatter plot showing the percent CD25+ and FOXP3+ double positive cells (Tregs), gated on CD3+CD4+CD44+ effector T cells in DLNs after 96 h, representing the Treg subtype in the OT-II mice upon adoptive transfer of NCoR1 KD DCs, with and without combined inhibition of HIF-1α and CPT1a. (n = 7).
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed paired [A, G, H and I] and unpaired [K and L] student’s t-test. Error bars represent SEM.
2.7. Combined KC7F2+etomoxir treatment facilitates Mtb (H37Rv) clearance in NCoR1 depleted DCs ex vivo and in vivo
Studies have reported on the complex immune regulatory networks perturbed by Mtb infection to increase its survival. Mtb infection up-regulates mTOR activity and upon treatment with rapamycin, IL-12 production and Th1 response goes high to clear the pathogen [58]. The former results exhibited active perturbation of HIF-1α and CPT1a in NCoR1 depleted DCs promoted a Th1 response via reduction of IL-10 to IL-12 cytokine balance. To further elucidate the physiological relevance of the immunostimulatory capacity of NCoR1 KD DCs with combined inhibition, we infected NCoR1DC−/− BMDCs with mCherry tagged Mtb (H37Rv strain) and harvested the cells at 2 h and 18 h post infection (Fig. 6A). Combined inhibition significantly reduced the bacterial load both at percent positive and MFI levels from the F4/80−CD11c+CD24+ NCoR1DC−/− BMcDC1s at 2 h and 18 h post-infection (Fig. 6B). Further, to validate our results in vivo, we infected NCoR1DC−/− KO mice with 1 × 106 CFUs of GFP expressing Mtb (H37Rv strain) intranasally. Mice in the control group were infected with only Mtb whereas, the drug treated group received treatment of KC7F2+etomoxir at day 3, day 8, day 12, and day 16 intraperitoneally after Mtb infection. Animals were sacrificed on day 21 to determine the bacterial burden in lung DCs and Th1 response in the Spleen (Fig. 6C). The lung CD11c+CD103+ DCs (cDC1 counterpart in lungs) showed significantly reduced Mtb burden upon combined inhibition (Fig. 6D and Figs. S5A–B) however, the overall bacterial burden in the lungs was found to be increased (Fig. S5C). A plausible explanation for the same can be drawn from hypoxia mediated studies for Mtb survival in lungs [59]. On the other hand, increased Th1 response (IFN-γ+ and T-bet+ effector T cells) were observed in splenic cells with no significant changes in the FOXP3+ cells (Fig. 6E and Figs. SD–E). Since, Mtb infection modulates the host immune response towards tolerance for survival, our results on the combined KC7F2+etomoxir treated NCoR1DC−/− BMcDC1s and lung CD103+ DCs demonstrated increased Th1 responses and pathogen clearance.
Fig. 6.

Mtb disease burden upon dual inhibition of HIF-1α and CPT1a in NCoR1DC−/− mice ex vivo and in vivo
A. Schematic showing the mCherry-Mtb (H37Rv) infection in primary NCoR1DC−/− BMcDC1s gated on F4/80−CD11c+CD24+ cells with and without combined treatment with KC7F2 and etomoxir. The cells were harvested after 2h and 18h post infection (hpi) and analyzed by flow cytometry.
B. Representative FACS contour plot depicting gating of Mtb infected cells, along with scatter plot and bar graph showing the cumulative percent infection and change in MFI levels with and without combined treatment with KC7F2 and etomoxir at 2h and 18h post infection in NCoR1DC−/− BMcDC1s. (n = 6)
C. Schematic outline showing the GFP-Mtb (H37Rv) infection in vivo in NCoR1DC−/− mice with (inhibitors were injected intraperitoneally at day 3, day 8, day 12 and day 16 post infection) and without combined inhibition with KC7F2 and etomoxir. Mice were sacrificed on day 21 to check bacterial burden in CD103+ lung DCs (cDC1 counterpart in lungs) and Th cell phenotype in spleen.
D. Scatter plot and bar graph showing the cumulative percent infection and change in Mtb burden (MFI) in lung DCs (gated on CD11c+CD103+ cells) in NCoR1 DC−/− mice at day 21 with and without combined treatment with KC7F2 and etomoxir. (n = 8)
E. Representative FACS dot plots depicting the gating of IFN-γ+ and T-bet+ cells gated on CD3+CD4+CD44+ effector T cells in the Spleen of treated and untreated mice. Scatter plots and bar graphs showed the cumulative percent infection and change in MFI levels at day 21 with and without combined treatment with KC7F2 and etomoxir in NCoR1DC−/− mice. (n = 5)
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed unpaired student’s t-test. Error bars represent SEM.
2.8. Human tolerogenic moDCs showed reduced NCOR1 expression and increased Th1 phenotype upon combined KC7F2+etomoxir treatment
To establish a relationship between NCOR1 and tolerance, we sought to investigate the correlation of NCOR1 expression with tolerogenic genes in primary human moDCs differentiated with GM-CSF and IL-4. From a publicly available dataset [60], we observed a negative correlation of NCOR1 with IL10 (p = 0.079) and SOCS3 (p = 0.038) in mature moDCs (Fig. 7A). This suggested an inverse relationship between NCOR1 and tolerance. Human conventional DCs treated individually or in combination with Dexamethasone (Dex), VD3, and rapamycin show characteristic tolerogenic features [61]. Therefore to further confirm this assertation, we also checked the normalized expression of NCOR1 (n = 4) between mature and other tolerogenic groups [60] and observed a significantly decreased expression in the Dex treated group compared to untreated mature moDCs (Fig. 7B). Next, to validate this observation, we derived CD14+ monocytes from PBMCs of healthy individuals (n = 5) followed by its differentiation into moDCs using GM-CSF and IL-4. The differentiated moDCs were divided into two groups, 1) Dex induced tolerance, and 2) mature control moDC group. To induce tolerance and activation in these moDCs, mature moDCs were treated with Dex for 5 days followed by 24 h of PolyIC (pIC) stimulation (Fig. 7C). Since human moDCs lack TLR9 expression, potent activation was attained using pIC [62]. The relative mRNA expression of NCOR1 was found to be significantly downregulated with an upregulation of tolerogenic gene IL-10 and downregulation of inflammatory gene IL-12 in the Dex-treated tolerogenic group (Fig. 7D). Further, glycolytic genes like HK2 and LDHA also showed enhanced expression (Fig. 7D). Next to confirm the NCOR1 expression at protein level, we performed confocal microscopy. The Dex-treated tolerogenic group showed significantly reduced NCOR1 protein levels as compared to the untreated mature moDCs (Fig. 7E). The above results suggested a strong inverse relationship of NCOR1 with tolerance in human moDCs. A previous study on human moDCs has shown that tolerogenic cells are dependent on glycolysis, glycolytic capacity, and FAO [17,18]. Therefore, we hypothesised that combined treatment of KC7F2+etomoxir could restore the inflammatory phenotype of the tolerogenic moDCs. To validate the effect of combined inhibition, we co-cultured the Dex-treated tolerogenic moDCs with primary CD4+ T cells. We checked the proliferation and polarization of these T cells co-cultured with and without inhibitor treated moDCs (Fig. 7F). The CD4+ T cells conjugated with the eF670 proliferation dye showed reduced proliferation as evident from decreased P2 population and increased MFI shifts for the Dex-treated tolerogenic group. However, with the combined inhibition of KC7F2+etomoxir, the proliferative capacity of the CD4+ T cells were rescued (Fig. 7G). Furthermore, Th1 response cytokine IFN-γ and transcription factor T-bet gated on CD3+CD4+ T cells showed reduced expression in the tolerogenic group that was found to be restored upon combined treatment (Fig. 7H). Overall, our findings suggested a direct role of NCoR1 in regulation of immune tolerance in human moDCs which could be reversed in terms of proliferation and Th1 response upon combined inhibition of glycolysis and FAO using KC7F2 and etomoxir, respectively.
Fig. 7.

NCOR1 expression and T cell polarization in human PBMCs derived moDCs
A. Scatter plot showing the correlation between NCOR1 and tolerogenic genes IL-10 and SOCS3 from a publicly available RNAseq dataset. (n = 5)
B. Violin plot showing the normalized counts of NCOR1 in human conventional DCs in immature, mature, and in different tolerogenic setup (Dex, VD3 and rapamycin treated groups), from publicly available RNAseq datasets. (n = 4)
C. Schematic showing the experimental outline used for differentiation of mature and tolerogenic (Dex-treated) moDCs to check the mRNA (RT-qPCR) and protein level expression of NCOR1.
D. Bar plots depicting the relative mRNA expression of NCOR1, IL-10, IL-12, HK-2 and LDHA in mature and tolerogenic moDCs after 24h pIC stimulation. (n = 4–5)
E. Representative microscopic images showing the NCOR1 protein levels (red) in mature and tolerogenic moDCs after 24h pIC stimulation. Corresponding bar graph depicted the quantification of the NCOR1 intensities in the microscopic images. (n = 5)
F. Schematic showing the CD4 T cell proliferation and T cell subtype profiling after 24h pIC stimulation in the co-culture of CD4+ T cells with mature and tolerogenic moDCs with and without KC7F2 and etomoxir treatment.
G. Representative FACS contour plots showing the gating of P1 and P2 populations of eF670 stained CD3+CD4+ T cells upon co-culture with mature and tolerogenic moDCs with and without combined treatment with KC7F2 and etomoxir after 72h after 24h pIC stimulation. Corresponding bar graph depicted the cumulative MFI shifts calculated from independent biological replicates and histogram showed the representative MFI shifts observed. (n = 5)
H. Representative FACS contour plot showing the gating of IFN-γ+ and T-bet+ populations gated on CD3+CD4+T cells upon co-culturing with moDCs for 96 h. Scatter plots and bar graphs depicted the cumulative percent positive and MFI shifts of IFN-γ+ and T-bet+ cells in mature and tolerogenic moDCs conditions with and without combined treatment with KC7F2 and etomoxir at 24h pIC stimulation.(n = 5)
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed unpaired student’s t-test. Error bars represent SEM. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3. Discussion
DC activation profiles converge into an abundance of descending signals that strategize various molecular cues at the transcriptional and translational level to elicit an antigen-specific immunological responses [63]. Alteration in cellular metabolism forms an important aspect of immune response in DCs. DCs when activated, they orchestrate metabolic reprogramming processes and shift towards a glycolytic profile with altered TCA cycle activity, and lower OXPHOS [4]. Several types of research have focused on the pathways that initiate DC metabolism, however limited studies have shown how it is being negatively regulated. The interplay between anabolic and catabolic processes is pivotal for a fine-balance of immunogenicity and tolerogenicity in DCs. It has been observed that anabolic metabolism in general is associated with the induction of immunogenic responses in DCs, whereas the catabolic pathways and active mitochondrial respiration are important for regulating tolerogenic function [11,13]. Hence, our study primarily focused on the catabolic routes of metabolism to understand the molecular mechanisms underlying tolerance in NCoR1-depleted DCs. We have recently identified that NCoR1 depletion induces strong immune tolerance in DCs [31]. Since the NCoR1 co-repressor complex regulates the functionality of mitochondrial metabolism in fat and muscle cells, we hypothesised a potential link of NCoR1 in regulating a fine-balance of immunogenic and tolerogenic responses in DCs through energy metabolism [24,26]. Murine and human DCs have been demonstrated to drive T cell polarization through distinct metabolic changes. Metabolic adaptations to OXPHOS and FAO help DCs meet their metabolic needs in the steady-state and TLR ligation with specific cytokine milieu maintains the cells in an immature and tolerogenic condition [2]. NCoR1 depleted tolerogenic DCs depicted enhanced glycolysis, glycolytic capacity, extracellular lactate and direct binding at the genomic regions of pfkp and pfkfb3. The role of glycolysis in regulating DC inflammation is well acknowledged. It does, however, play a crucial role in immunological tolerance also. Tolerogenic DCs have been known to show increased glycolysis [64]. Increased glycolysis results in greater lactate buildup and extracellular media acidification. In co-culture experiments with naive CD4+ T cells, DCs with substantial lactate accumulation limit proliferation and polarize towards Tregs. This occurs as a result of T cells intaking lactate and thereby reducing their glycolysis and proliferation. VD3, a synthetic anti-inflammatory inducer, also depicts similar profiles [18,65]. Sustained activation in murine and human tolerogenic DCs regulate glucose metabolism and increased glycolysis by PI3K/AKT/mTOR axis by stabilising HIF-1α expression [66,67]. This signalling axis similarly favoured the enhanced glycolytic rate in our NCoR1 ablated tolerogenic DC model. Several other groups have also reported the functioning of the former signalling axis greatly in DCs [17,[68], [69], [70]]. HIF-1α perturbation led to decreased glycolysis and reduced secretion of tolerogenic cytokines in NCoR1 KD cells remarkably. Dependency on HIF-1α proved to be vital and more dominant than mTOR. Moreover, NCoR1 binding peaks at the Hif-1α genomic region are reasoned enough for direct regulation of glycolytic genes through NCoR1 [71].
On the other hand, mitochondrial respiration in control and NCoR1 depleted tolerogenic DCs depicted a similar pattern as reported earlier [2]. We also showed that mitochondrial SOX and cellular ROS levels were significantly downregulated in NCoR1 depleted DCs both in the DC line and in cDC1 BMDCs upon CpG stimulation (Fig. 1, Fig. 2E), despite the fact that NCoR1 depleted cells depicted enhanced glycolysis and OXPHOS. This can be because the immunogenic DCs are remarkable in producing ROS to elicit inflammatory responses, whereas, tolerogenic ones show subdued ROS levels, which may be the case for NCoR1-depleted ones [5,6,72]. The next important question was how NCoR1 depleted tolerogenic DCs had higher levels of OXPHOS even after CpG activation than control cells. We focussed on the three important routes generating reducing powers to facilitate TCA cycle and thereby OXPHOS [73,74]. In NCoR1 KD cells, DEGs of the TCA cycle for pyruvate and glutamine oxidation were downregulated, which was substantiated by the observed decreased dependence. Further suppression of pyruvate oxidation decreased IL-12p40/70 secretion while increasing IL-10. This metabolic node turned out to be a crucial feature for inflammation. Whereas inhibition of glutaminolysis with BPTES and DON, considerably enhanced IL-10 secretion in both control and NCoR1 KD DCs while having no effect on IL-12p40/70 levels, implying a crucial relationship between tolerance and glutaminolysis. Such intricate balance is also depicted in human pDCs under TLR stimulation and glutaminolysis inhibition [75]. The third important node which modulates inflammation versus tolerance is fatty acid oxidation. Increased expression of the rate-limiting β-oxidation gene Cpt1a along with DEGs in NCoR1 KD cells resulted in a higher OCR dependent FAO index. This provided evidence for active OXPHOS being operated through β-oxidation under TLR9 specific CpG stimulation. PPAR-γ mediated intracellular fatty acid reserves fuelled the FAO index and NCoR1 binding at the genomic regions of PPAR-γ further validated the whole cyclical aspect being operated to drive active OXPHOS in these cells. Deposition of neutral lipids in NCoR1 KD cells suggests the presence of increased lipid synthesis which requires NADPH. Because NADPH can be generated via the PPP, IDH1, and ME1 related pathways. Increased malic acid with reduced PPP and non-significant Idh1 probably explains the accumulation of lipids and decreased ROS levels within the NCoR1 depleted DCs [76,77]. Maintenance of tolerance primarily through glycolysis and FAO is well established. But until now no study has depicted combinatorial effects of dual inhibition of HIF-1α and CPT1a on immune-tolerance and T cell responses. Combined inhibition changed the global transcriptomic landscape of tolerogenic response genes for NCoR1 KD cells. While inflammatory response genes were largely unchanged. Co-culture studies further affirmed the intact inflammation by showing increased Th1 response ex vivo and in vivo. On the contrary, control DCs showed reduced secretion of IL-6 and IL-12p40/70 levels and subdued Th1 response. This depicts an intricate fine balance of metabolic pathways for inflammation and tolerance in control and NCoR1 KD DCs. Physiologically, NCoR1 depleted DCs effectively cleared Mtb burden upon treatment with combined inhibitors. Although bacterial burden was decreased in lung DCs, there was an overall increase in Mtb survival in lungs, suggesting decreased HIF-1α expression as a prominent feature of Mtb survival [59]. Modulation of inflammatory versus tolerogenic behaviour in Mtb infected host cells is a pivotal immune response mechanism regulated both by Mtb and host factors. Rewiring of host metabolic machinery by Mtb for increased survival is also conjugated with the defence mechanism [78]. Further, to develop a correlation between NCoR1 with tolerance in human moDCs, a Dex-treated tolerogenic state was attained. Significant reduction in the expression levels of NCoR1 in mRNA and protein established a strong relationship between them. The differential regulation of energy metabolism of tolerogenic human moDCs upon dual impairment appeared to demonstrate a similar bioenergetic pattern to the murine ones. Increased proliferative capacity and Th1 response corroborated the same.
In conclusion, correlation of NCoR1 with tolerance in human moDCs and can act as a potential biomarker for diseases encompassing inflammatory versus tolerogenic states like in IBDs (inflammatory bowel diseases). Subsequently, HIF-1α and FAO inhibitors at optimal doses can mitigate inflammatory responses in murine DCs, its validation in human setup needs further investigation.
4. Limitations of the study
We have used mutu-cDC1 lines to perform initial experiments, since homogenous primary CD8α+ cells are difficult to isolate, and they undergo activation during the purification process. It is also reported that metabolic experiments performed on sorted and magnetic selected cells alter redox state and cellular metabolome [79]. Hence, we considered total FLT3 differentiated BMDCs for extracellular flux assays. Since, both cDC1 and cDC2 cell types depict upregulation of tolerogenic genes in NCoR1 depleted conditions [31].
Contributions
Conceptualization, K.S., R.P., G.P.M., and S.K.R.; Methodology, K.S., R.P., A.J., G.P.M., S.P1., S.C., S.S., S.P2., A.S., M.S., R.K.N. and S.K.R.; Investigation, K.S., R.P., A.J., G.P.M., S.C., R.K.N and S.K.R.; Writing – Original Draft, K.S., and S.K.R.; Data Curation, G.P.M and S.K.R.; Formal Analysis, G.P.M.; Supervision, S.K.R., Visualization, K.S., R.P., A.J., G.P.M., and S.K.R.; Funding Acquisition, S.K.R.
Declaration of completing interest
The authors declare no competing interests.
Acknowledgements
We would like to thank Professor Johan Auwerx, EPFL, Lausanne for providing us with the NCoR1fl/fl mice. Core BSL-3 and ABSL-3 facilities at ILS for Mtb related work. ILS Animal facility for providing us with breeding and maintenance of control CD11c Cre, NCoR1DC−/− KO and OT-II transgenic mice. We would also like to thank ILS sequencing and High-performance computing facility for NGS and data analysis respectively.
K.S. is supported by UGC-SRF, R.P is supported by DBT-RA, A.J is supported by ILS fellowship, G.P.M is supported by DBT-BINC, S.P1 is supported by UGC-SRF, S.C is supported by CSIR-SPM, S.P2 is supported by ICMR-SRF and A.S is supported by CSIR-SRF. Grants - SERB-India (EMR/2016/000717), DBT-India (BT/PR15908/MED/12/725/2016), supported this study; ILS provided intramural grants and infrastructure. R.K.N. acknowledges core support from ICGEB, New Delhi.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102575.
Materials and Methods
Mice
CD11c specific homozygous NCoR1 KO (Knockout) mice were used for performing the ex vivo and in vivo experiments. Mice generation and selection of homozygous progenies were carried out as reported elsewhere [31]. For KO confirmation, genotyping was done from DNA extracted from ear tissue. Briefly, tissue samples were treated with 50mM NaOH and heated at 95°C for 35 min followed by 1 M Tris-Cl (pH-8.0) treatment and centrifuged at 12,000 rpm for 5 min at RT. DNA was processed for PCR using NCoR1 loxP and CD11c Trans gene specific forward and reverse primers. The primer sequences are listed in the key resource table. PCR products were loaded onto a 0.8% agarose gel for confirming the presence or absence of NCoR1 loxP and CD11c Trans gene bands.
OT-II (gifted by Prof. Hans Acha-Orbea) and FLT3 (gifted by Ton Rolink) transgenic mice were transported from Switzerland. CD11c specific NCoR1 KO, CD11c Cre, OT-II, FLT3, and C57BL/6 mice were bred and maintained at the ILS animal facility.
Dendritic Cell (DC) Culture
The CD8ɑ+ murine tumour cDC1 DC line was gifted by Prof. Hans Acha-Orbea. The DC line showed characteristic phenotypic and functional resemblance to type-I splenic CD8α conventional DCs [80]. The cell line was cultured in IMDM-glutamax media supplemented with 10% FBS, PenStrep, Sodium bicarbonate, HEPES buffer, and β-mercaptoethanol, and further maintained in a humidified incubator at 37 °C with 5% CO2. For invitro experiments, potent activation was attained when challenged with TLR9 specific CpG-B at a concentration of 1 μg/ml for 6 h.
Generation of Stable NCoR1 KD CD8α+ DC line
Stable control and NCoR1 KD DC lines were generated using pre-cloned shRNA constructs under the pLKO.1 lentiviral vector backbone (Sigma). Three different shRNAs targeting complementary determining sequence (CDS) and 3′UTR regions were selected for transduction. The mean KD efficiency with shRNA 2 was highest (∼80%) and hence chosen for experiments. Transfection of HEK (293T), generation of viral particles, and transduction in the DC line was followed as reported earlier [31]. Stable cells were selected using Puromycin. The KD efficiency was quantified using a RT-qPCR primer specific to the mouse Ncor1 gene. The primer sequences are listed in the key resource table.
RNA isolation and RT-qPCR
The extraction of RNA was done using Trizol reagent (RNA isoplus). Briefly, cells at specific densities (DC line - 1 × 106 and BMDCs - 3 × 106 cells/well) in a 6 well plate were treated with 500μl of Trizol reagent and kept at −80 °C until processing. During processing 100μl of chloroform was added and vortexed vigorously for 30 s. The Trizol-chloroform mixture was centrifuged at 11,000 g for 10 min at 4°C. After centrifugation, the aqueous phase of the mixture was taken, and an equivalent amount of isopropanol was mixed and incubated for 5 min. After incubation, the mixture was again centrifuged at 11,000 g for 10 min at 4 °C. Following this 300μl of ethanol was added and centrifuged at a maximum speed in a table top centrifuge at 4°C. The supernatant thus obtained was discarded and the pellet was kept for air drying. Finally, resuspension was done in nuclease-free water (NFW) and the amount was quantified using nanodrop (Thermo). Then 1μg of total RNA was used to prepare cDNA using high-capacity cDNA Reverse Transcriptase kit (Applied Biosystems). Quantitative PCR was performed using Power SYBR Green (Applied Biosystems) and PCR amplification was monitored in real-time using Quant studio 6 Instrument (Applied biosystems). Primer pairs used are listed in the key resource table.
Generation of Bone Marrow-Derived DCs (BMDCs) for ex vivo Studies
Male or female mice of age 6–8 weeks were sacrificed by CO2 inhalation, and tibias and femurs were surgically removed under aseptic and sterile conditions. The bones were cut at an angle of 45° at the ends and marrow was flushed out in RPMI-1640 media with the help of a 5 ml syringe. Single-cell suspension was prepared, and clumps were removed with the help of a 70μm cell strainer. The cells were centrifuged at 400g for 5 min at 4°C. The pellet was suspended with a 1x RBC lysis buffer (Tonbo) for 5 min on ice. The bone marrow cells were washed as previously and counted for seeding. The cells were plated in a 6 well having 6 × 106 cells/well with FLT3 sera (from FLT3 transgenic mice). The cells were cultured at 37°C in an incubator containing 5% CO2 and left untouched for 9 days. Loosely adherent differentiated cells were collected, counted, and re-plated for experiments.
Inhibitor treatment and Flow Cytometry (FACS)
DC line and BMDCs were processed with 1 h pre-incubation with inhibitor treatments with rapamycin (20nM), KC7F2 (10μM), UK-5099 (5μM), BPTES (5μM), DON (5μM), and etomoxir (50μM) followed by CpG stimulation (6 h in DC line and 18 h in BMDCs) and BFA treatment for 4 h. Combined inhibitor studies with KC7F2 and etomoxir were performed in a similar manner. Thereafter cells were harvested and processed with flow cytometry. Surface and intracellular (IC) staining were done sequentially for experiments. In the DC line and BMDCs, control and NCoR1 perturbed cells were processed for surface staining for 30 min at 4°C in 1x FACS buffer (3% FCS in 1x PBS, 5 mM EDTA) followed by intracellular staining upon fixation and permeabilization with 2% PFA and 1x Perm buffer respectively. The IC staining cocktail was prepared in a Perm buffer and incubated with cells for 45 min at room temperature in the dark. During the estimation of Mitochondrial SOX (5μM), cellular ROS (according to manufacturer’s recommendations) and mitochondrial potential with TMRM (20nM), cells were incubated with corresponding dyes in the culture medium for 20 min at 37°C in a 5% CO2 incubator and proceeded for flow cytometry after washing with 1x FACS buffer. The acquisition was done using an LSRII fortessa flow cytometer (BD Biosciences). The acquired data were analysed using FlowJo-X software (Treestar). Antibodies and dyes used for flow cytometry experiments are listed in the key resource table.
DC - T cell co-culture experiments
For the ex vivo DC-T cell co-culture experiment, mutuDC line and FLT3 differentiated BMDCs (10,000 cells/condition) were pulsed with OVA peptide (323–339)/OT-II at 200nM concentration overnight. Thereafter cells were washed and combined inhibitor treatment with KC7F2 (10μM) and etomoxir (50μM) for 1 h and CpG stimulation for 6h in the DC line and 18h for BMDCs were done. Sorted CD3+CD4+ T cells were added to the DCs at a ratio of 1:10. Following this, T cell proliferation was determined using amine-based dye (eFlour670-APC-1μM) after 72h. Whereas, T cell differentiation into Th1 (T-bet+ and IFN-γ+) and Tregs (CD25+ and FoxP3+) were determined by flow cytometry upon restimulation with PMA (10 ng/ml), Ionomycin (500 ng/ml) and Brefeldin A (2.5 μg/ml) together for 5h after 96h. Effector T cells were identified using CD44 as a marker. For the in vivo experiment, NCoR1 KD DCs were OVA pulsed, treated with dual inhibitors and CpG stimulated invitro. These cells (5 × 106 cells/mice) were injected intraperitoneally (I.P) into OT-II transgenic mice and nearest draining lymph nodes were collected after 4 days for determining the Th1 vs Treg subtype. Cells thus obtained from draining lymph nodes were stimulated for 5h with PMA/Iono/BFA before proceeding for flow cytometry. Antibodies used for flow cytometry experiments are listed in the key resource table.
Mitochondrial DNA copy number estimation
In 12 well plates, control and NCoR1 KD DCs were grown, and roughly 0.5 × 106 cells were collected per condition. Cells were centrifuged and resuspended in 300μl of Tissue and cell lysis solution (Lucigen) containing proteinase K and mixed thoroughly. Next, samples were incubated at 65°C for 15 min and vortexed every 5 min. After which samples were cooled to 37°C and RNase A was added. Next, they were placed on ice for 3–5 min and proceeded with DNA precipitation. During this step 150μl of Protein Precipitation Reagent (PPR-Lucigen) was added to the 300μl of lysed samples and vortexed vigorously for 10 s. Thereafter, samples were centrifuged at 4°C for 10mins at 10,000g, supernatant was collected and pellets were discarded. 500μl of isopropanol was added, mixed and centrifuged to collect the DNA pellet. Final resuspension was done in 30μl of TE buffer and quantified. The DNA thus obtained, were processed for RT-qPCR with Nd-1 and Hk-II primer pairs along with Power SYBR green. Ct of Hk-II - Ct of Nd-1 gave the dCT value. Mitochondrial DNA copies were calculated as 2*2^ (dCT).
Extracellular Nitrite assay
Control and NCoR1 KD DCs were cultured in 6 well plates and culture supernatants of 2.5 × 106 cells were harvested per condition. In a 5% phosphoric acid solution, 100μl of culture supernatant was combined with an equivalent volume of 0.1% naphthylethylenediamine (NEDA) and 1% sulphanalic acid. Absorbance was measured at 550 nm. Known concentrations of sodium nitrite were used for preparation of standard.
Intracellular ATP extraction and determination
Intracellular ATP was extracted using boiling distilled water. Control and NCoR1 KD DCs (0.5 × 106 cells) were seeded in 12 well plates and harvested with and without CpG stimulation. Samples were centrifuged and boiling water was put for 15min and vortexed every 5min. Following which they were incubated in 4°C for 15min and centrifuged again to collect the supernatant for ATP determination. ATP determination was carried out using a specific solution containing recommended proportions of dH2O, reaction buffer, dTT, d-luciferin and luciferase enzyme using an ATP measurement kit (Invitrogen, A22066). The standard curve of ATP was obtained by serial dilutions of 10μM ATP solution. For determining glycolytic ATP, cells were treated with 2-DG (10mM) pre-treatment for 1h. Total intracellular ATP - ATP of 2-DG treated cells gave an approximation of the glycolytic ATP. Luminescence was captured in a multimode reader (PerkinElmer).
Metabolite sample preparation for GC-MS analysis
Control and NCoR1 KD DCs were cultured in 10 cm plates and approximately 10 × 106 cells were harvested per condition. For metabolite extraction, cells were washed twice with 0.9% sodium chloride (NaCl) solution in minimum possible time and chilled methanol (800μl, stored at −20 °C) was added for quenching the metabolic reactions followed by Milli-Q water (800μl) in each well. 10μl of internal standard (2mg/ml Ribitol) was further supplemented in each sample. Following which cells were scraped and collected in 15 ml centrifuge tubes. To each tube, chloroform was added (1.6ml) and vortexed at 4°C for 30min. Cells were centrifuged at 10,000g for 10min at room temperature and the aqueous phase (methanolic) samples were collected and stored at −20 °C until further processing. For GC-MS, 300μl of aqueous samples were taken and dried using speed vac. MS data acquisition of these extracted molecules were carried out using LC-MS as it is or in derivatized form using Methoxamine hydrochloride (MeOX-HCl) (2%) followed by N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) before running in GC-MS.
Extracellular lactate assay and pH measurement
Extracellular lactate was determined from 30,000 cells/condition from culture supernatants (1:10 dilution) in DC line and 0.1 × 106 (1:5 dilution) cells in BMDCs both for control and NCoR1 depleted DCs. According to manufacturer’s recommended protocol (J5021-Promega), supernatant was diluted in 1x PBS before mixing to an equivalent volume of reaction solution (reductase, reductase substrate, luciferin detection solution, NAD, and lactate dehydrogenase). The standard curve was obtained by serial dilutions of 400μM of lactate solution. Luminescence was captured in a multimode reader (PerkinElmer). Change in pH was also determined from culture media to validate the observed lactate measurements.
Extracellular flux (Seahorse) assay
Control and NCoR1 KD DC line (30,000 cells/well) and BMDCs (0.1 × 106 cells/well) were seeded in culture miniplates for the assay. Mito-stress test was performed using serial injections of Oligomycin (2μM), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (CCCP-2μM) and Rotenone+Antimycin A (0.5μM each). The WAVE software was used to calculate respiratory characteristics such as baseline oxygen consumption rate (OCR), Coupled ATP, maximal respiratory capacity, and spare respiratory capacity. Serial doses of Glucose (10mM), Oligomycin (2μM), and 2-DG (50mM) were used in the Glyco-stress test. A pre-injection ECAR experiment was performed to determine the dependency of stimulated control and NCoR1 KD cells on mTOR and HIF-1α using rapamycin (20nM) and KC7F2 (10μM) respectively. ECAR values were determined similar to a mito-stress test using the WAVE software report generator. To establish pyruvate and glutamine dependency, the Mito fuel flex test was used. During pyruvate oxidation measurement, the first injection consisted of UK-5099 (2μM) and the second a combination of etomoxir (4μM) + BPTES (3μM). In a similar way, glutamine oxidation was measured with sequential injections of BPTES (3μM) followed by UK-5099 (2μM) + etomoxir (4μM) together. All the chemicals were prepared according to the manufacturer’s protocol (Seahorse bioscience, Agilent technologies). Fatty acid oxidation index was calculated from control and NCoR1 KD CpG samples by sequential injections of etomoxir (50μM) and Oligomycin (2μM).
Oil red - O staining and quantification
Neutral lipid accumulation in control and NCoR1 depleted DCs were visualized by Oil Red O staining. Cells were cultivated on microscope coverslips, placed in 12 well cell culture plates, followed by stimulation with CpG for 6h. After the incubation period, cells were washed with 1x PBS followed by treatment with 1ml of formalin (10%) in 1x PBS for 5min at RT. After that, formalin was discarded, fresh formalin was added and incubated for at least 1h. Before staining, cells were washed thrice with Milli-Q water for the complete removal of formalin and washed once with 60% isopropanol. Then coverslips were dried completely. Staining was performed with 1ml of oil red O staining solution for 10 min followed by intense washing with Milli-Q water. Following which, cells were visualized using ZEISS Apotome microscope. Further the dye was eluted with isopropanol (100%) for 10min and quantified absorbance was noted at 500 nm.
Western blotting
Control and NCoR1 KD whole cell lysates were prepared using RIPA buffer (0.5 M EDTA, 1 M Tris-Cl pH7.5, 1 M NaCl, 200 mM, Roche protease inhibitor) at 6h CpG stimulation. HIF-1α specific cell lysates were treated with 100μM cobalt chloride for 24h before harvesting. After harvesting, samples were processed for sonication in a bioruptor (Diagenode) and protein estimation was done using BCA reagent (Pierce BCA Protein Assay Kit) and absorbance was measured at 562nM. SDS–PAGE was used to separate the cell lysates, which were then transferred to nitrocellulose membranes. Thereafter, membranes were probed with primary antibodies (p-AKT, T-AKT, p-mTOR, T-mTOR, HIF-1α, PDH and G6PD). Next, HRP conjugated secondary antibodies were added to obtain signals by adding ECL solution onto an x-ray film. We probed the membrane with phospho-antibodies first, stripped and re-probed the same membrane with corresponding total antibodies for western blotting of phospho and its respective total protein. Then we normalized the phosphorylated form with their respective loading controls before performing densitometric analysis. For its comparable total protein, a similar procedure was used. Finally the ratio of normalized values were plotted as relative intensity. Antibodies used for western blotting are listed in the key resource table.
Mtb culture
Middlebrook 7H9 broth (BD Difco, Becton Dickinson) supplemented with 10% ADC (Becton Dickinson), 0.4% Glycerol, and 0.05% Tween-80 was used to grow Mycobacterium sp. GFP and mCherry tagged M. tuberculosis were used for flow cytometry.
Mtb infection model ex vivo and in vivo
For ex vivo experiments, FLT3 differentiated CD11c specific NCoR1 KO BMDCs were either processed for combined inhibition with KC7F2 (10μM) and etomoxir (50μM) for 1h or left untreated. Infection was carried out using mCherry tagged Mtb (H37Rv) at a MOI 10 for 2h and 18h. Following which cells were harvested and flow cytometry was performed to check bacterial burden in F4/80−CD11c+CD24+ cells.
For the in vivo experiment, 1 × 106 Mtb-GFP (H37Rv) in logarithmic phase (OD600 = 0.5–0.6) in 20μl 1x PBS were given intranasally into mice anaesthetized with Ketamine-Xylazine. On day 3, 8, 12, and 16 one set of NCoR1 KO mice were injected with KC7F2 (5mg/kg/BW) and etomoxir (10mg/kg/BW) intraperitoneally. After 21 days, Mtb burden was determined in lungs by using flow cytometry in the CD11c+CD103+ DCs. Splenic cells were processed for RBC lysis and stained with surface and IC antibodies to determine the CD4+ effectors, Th1 and Treg population (gated on effector cells) upon PMA (10ng/ml), Ionomycin (500ng/ml) and Brefeldin A (2.5μg/ml) treatment for 5h. Antibodies used for flow cytometry are listed in the key resource table.
RNAseq library preparation
DC line samples (Control CpG 6h, Control CpG 6h+KC7F2+etomoxir, NCoR1 KD CpG 6h and NCoR1 KD CpG 6h+KC7F2+etomoxir) were processed for RNA extraction and library preparation. RNA extraction was performed using a kit from Macherey-Nagel. Briefly, samples were lysed with 350 μl of LBP buffer, following which genomic DNA was removed using a nucleospin gDNA removal column. The flow-through was mixed with 100μl of the binding solution, centrifuged at 11,000×g for 15s, and washed using wash buffer 1 and 2 before eluting RNA in 20μl of RNAse-free water. For library preparation, magnetic beads and an mRNA isolation kit were used to isolate mRNA from 1μg of total RNA (Poly-A mRNA isolation module, NEB). NEB's mRNA library preparation kit Ultra II was later used to prepare RNAseq libraries according to the manufacturer's instructions. Qubit 2.0 (Invitrogen) was used to determine library concentrations, and Bio-analyzer was used to examine fragment sizes (Agilent).
Nucleic acid sequencing protocol
cDNA samples were denatured using 0.2N NaOH at RT for 5min. Further, 10mM Tris-Cl pH-8.5 was used to neutralize the effect of NaOH. 4nM cDNA libraries were diluted with HT1 buffer for a final loading concentration of 1.6 pM. Following that, the libraries were sequenced on the Illumina NextSeq 550 platform at in-house ILS Sequencing facility.
RNAseq analysis
Paired end raw reads were first checked for quality control using FASTQC and then aligned to mouse reference genome (mm10) using hisat2 [81]. Gene count was then extracted using feature Counts function from RSubRead R package [82]. Principal component analysis and differential gene expression analysis were performed using the DESeq2 package [83].GO term enrichment analysis were performed using cluster Profiler, GO (biological processes) terms downloaded from MSigDb [84].
ChIPseq for NCoR1-NGS library preparation and analysis
NCoR1 ChIPseq data in control Uns and at 6h CpG stimulation has been utilized from our previous study [31] to denote NCoR1 binding peaks at the genomic regions of Pfkfb3, Pfkp, Hif-1α and Ppar-γ.
Immunofluorescence
NCOR1 expression in mature and tolerogenic human moDCs was determined by immunofluorescence staining. Briefly, human moDCs with and without Dex treatment were cultured on glass coverslips in a 12-well cell culture plate followed by stimulation with pIC for 24h. After the incubation period, cells were fixed in 4% paraformaldehyde for 30min at RT, permeabilized, and blocked for 20min in 0.1% Triton X-100 and 2% goat serum. Following this, fixed cells were washed and incubated overnight with an anti-NCOR1 primary antibody (1:100; CST). After the primary antibody incubation, cells were incubated with a fluorescence-conjugated secondary antibody F(ab') 2-Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594) for 120min and mounted with ProLong™ Gold antifade mountant with DAPI (Invitrogen). Finally the cells were observed under a confocal microscope (Leica). Antibodies used are listed in the resource table.
PBMCs isolation, moDC differentiation and primary CD4 T-Co-culture
Blood samples were drawn from 5 healthy individuals for PBMCs isolation. PBMCs were isolated using density gradient centrifugation (2000 rpm, brakes-0 for 20 min) utilizing lymphoprep. Cells were washed with 1x PBS and magnetic based negative selection of CD14+ monocytes (Stem cell technologies) were done. To differentiate them into moDCs, cells were treated with hu-GMCSF (50ng/ml) and hu-IL-4 (20ng/ml) for a period of 6 days. On the other hand, for acquiring tolerogenic moDCs, they were additionally supplemented with Dex at 1μM concentration. Additional supplementation was done on day 3. On day 6, the inhibitor group received 1h pre-treatment of KC7F2 (10μM) and etomoxir (50μM). Following which, all sample groups were stimulated with pIC at 10μg/ml concentration for 24h. After activation, cells were washed thoroughly and incubated with sorted CD4+T cells from 5 other healthy individuals (mixed) at a ratio of 1:10. Proliferation group received eF670 dye stained CD4+T cells whereas, profiling group received cells as it is. After 72h and 96h post incubation, T cells were analyzed for proliferation and Th1 profiling respectively. Reagents and antibodies used are listed in the key resource table.
Ethics statement
In accordance with the necessary ethical approval, human primary PBMCs were extracted from 5ml of venous blood from 10 adult healthy control donors at the Institute of Life Sciences in Bhubaneswar, Odisha, India, for the ex vivo experiment. Table S1 contains the details of the donors in an anonymous form. The entire process complied fully with the Declaration of Helsinki 2013, which sets forth the moral standards for medical research involving human subjects. The participants provided written informed consent prior to the collection of PBMCs for ex vivo studies, and all subject data was anonymized.
Statistical analysis
Data are presented as mean values ± SEM, except where stated categorically. Statistical comparisons are drawn using a two-tailed Student's t-test. Differences were considered significant if P ≤ 0·05.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
NCoR1 depletion in cDC1 and bioenergetics of cDC1 DCs (related to Fig. 1)
A. Bar graph showing the relative mRNA expression of Ncor1 in control shRNA versus NCoR1 shRNA transduced mutu-cDC1 line. (n = 3)
B. An idealistic glyco-stress line graph showing glycolysis, glycolytic capacity and glycolytic reserve upon sequential injections with glucose, oligomycin and 2-DG.
C. Scatter plot representing the change in pH in extracellular media in the control and NCoR1 KD tolerogenic DCs before and after 6h CpG stimulation. (n = 6)
D. Scatter plot depicting the total intracellular ATP levels in unstimulated and 6h CpG activated control and NCoR1 KD tolerogenic DCs with and without treatment with 2-DG (10mM). Corresponding bar graph depicted the differences in the ATP levels. (n = 4)
E. Heat map depicting the log2 fold change expression of PPP genes in NCoR1 KD versus control DCs, * depicts significance and NS denotes non-significant change. (n = 3)
F. Western blot and its densitometric analysis showing the decreased protein expression of PPP rate-limiting enzyme G6PD in NCoR1 KD as compared to control DCs. (n = 4)
G. Heatmap showing the differentially expressed genes of complex-I to complex-V for the electron transport chain (ETC) from RNAseq data of unstimulated and 6h CpG activated control and NCoR1 KD tolerogenic DCs. (n = 2)
H. Bar graph demonstrating the relative mRNA expression of Nrf-1 and Cox5a in unstimulated and 6h CpG activated control and NCoR1 KD tolerogenic DCs. (n = 6)
I. An idealistic mito-stress line graph showing basal respiration, maximal respiration, coupled ATP and spare respiratory capacity upon sequential injections of oligomycin, CCCP and Rot/AA in Seahorse extracellular flux analyzer.
J. Scatter plot depicting the total intracellular ATP levels in unstimulated and 6h CpG activated control and NCoR1 KD tolerogenic DCs. (n = 3)
K-L Histogram plots depicting the MFI shifts for mitochondrial SOX, cellular ROS and TMRM (mitochondrial potential) for control and NCoR1 KD tolerogenic DCs before and after 6h CpG activation.
M. Dot plots showing the back gating strategies to identify viable cells in cDC1 mutu-DC line by flow cytometry.
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed paired student’s t-test. Error bars represent SEM.
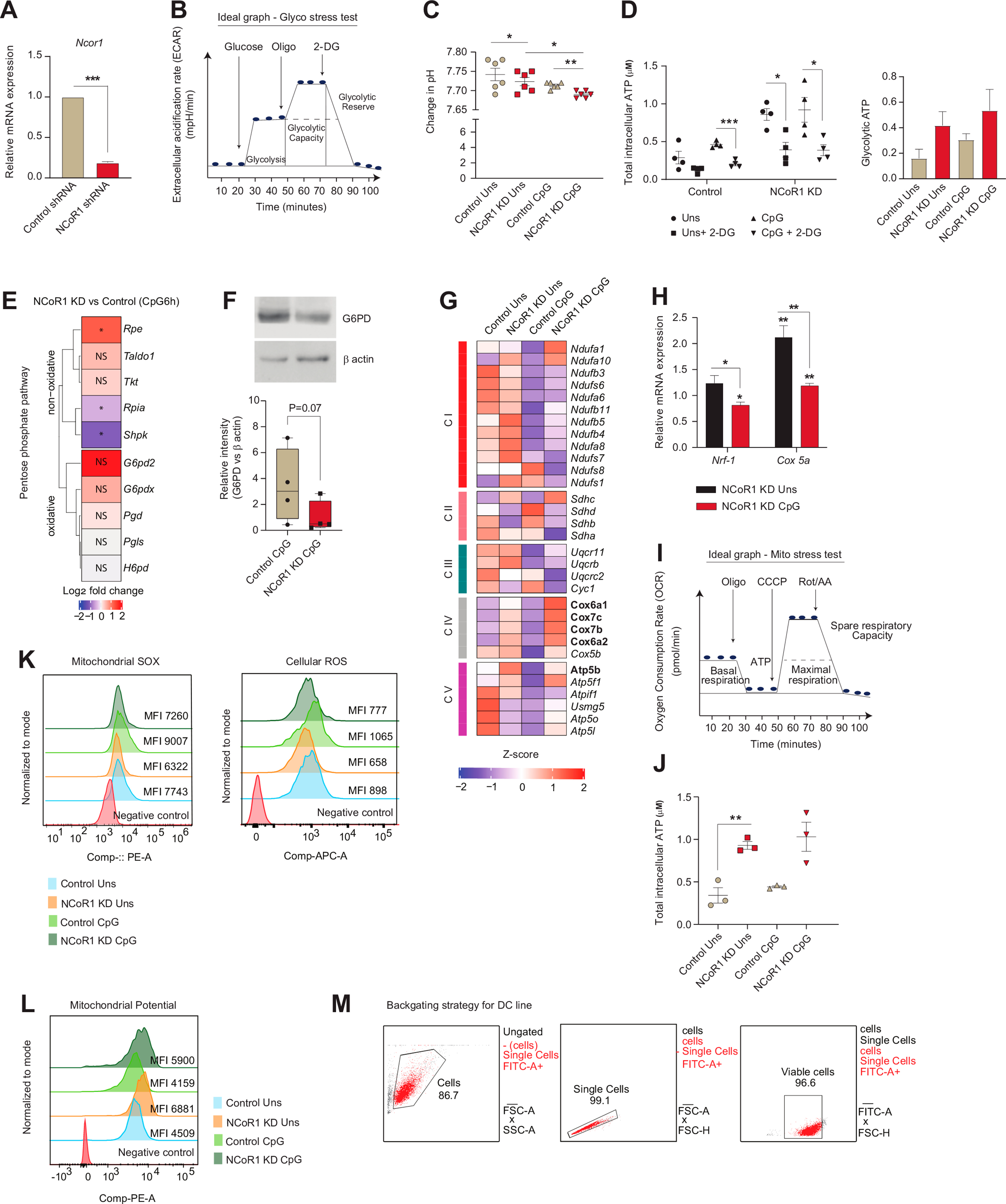{kind=link}
Mouse genotyping, cellular homeostasis and gating strategies used in BMDCs ex vivo flow cytometry experiments (related to Fig. 2)
A. Mouse genotyping - representative agarose gel image showing the NCoR1 Lox-P bands in Wild Type (WT) and NCoR1DC−/− mice, whereas the CD11c trans gene band is specific to NCoR1DC−/− mice only.
B. Bar graph depicting the relative mRNA expression of NCoR1 in control CD11c-Cre and NCoR1DC−/− BMDCs. (n = 3)
C. FACS plot showing the gating of percent double positive CD11c+MHCII+ cells in control CD11c-Cre and NCoR1DC−/− BMDCs at day 9 of differentiation. Dot plots showing the back gating strategy and scatter plot depicting the cumulative data. (n = 4)
D-E. Histogram plots depicting the MFI shifts of cellular ROS and TMRM (mitochondrial potential) in control CD11c-Cre and NCoR1DC−/− BMcDC1s before and after 18h CpG activation.
F. Dot plots showing the back gating strategy to identify F4/80−CD11c+CD24+ BMcDC1s at day 9 of FLT3L differentiated mouse bone marrow cells.
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed unpaired student’s t-test. Error bars represent SEM.
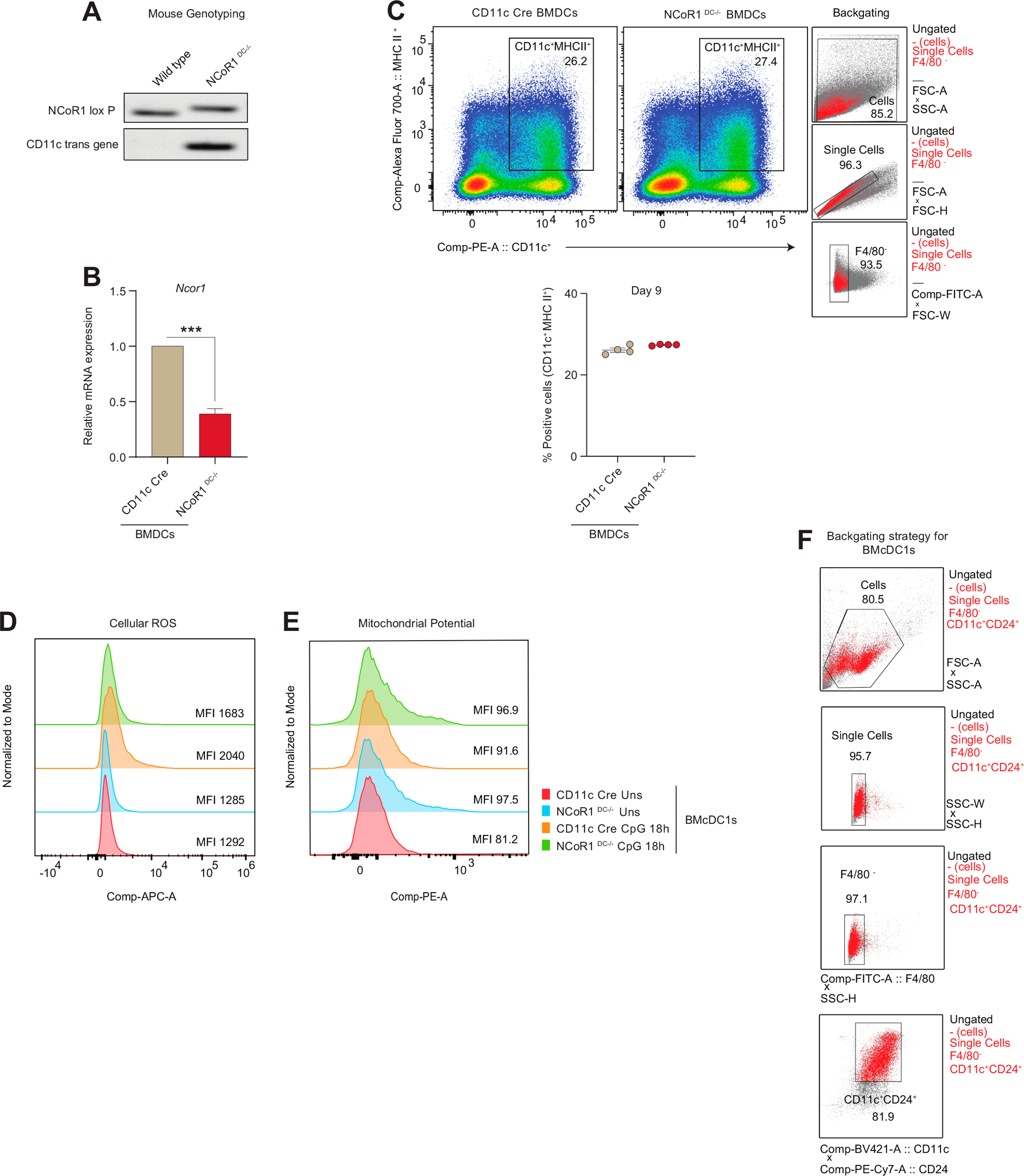{kind=link}
Impact of mTOR, HIF-1α, pyruvate, glutamine and fatty acid oxidation perturbation on inflammatory cytokine secretion (related to Fig. 3, Fig. 4)
A. Bar graphs showing the normalized cell viability levels (>90%) upon rapamycin (20nM) and KC7F2 (10μM) treatment in Control and NCoR1 KD DCs. (n = 10)
B. RT-qPCR depicting the transcript expression of Hif-1α gene before and after treatment with KC7F2 in control and NCoR1 KD cDC1 DCs. (n = 4)
C. Histogram plots showing the MFI shifts of IL-10 and IL-27 upon rapamycin and KC7F2 treatment.
D. Scatter plot and bar graphs showing the cytokine levels of IL-6 and IL-12p40/70 in 6h CpG activated cells, after treatment with rapamycin and KC7F2. (n = 7–8)
E. An idealistic pyruvate and glutamine oxidation dependency graph from Seahorse extracellular flux analyzer. Experimental line graphs showing the pyruvate and glutamine dependency in terms of OCR when specific inhibitors were used either singly or in combinations. Dependency percentage is calculated as baseline OCR - target inhibitor OCR / baseline OCR - all inhibitors OCR * 100.
F. Bar graphs depicting the normalized cell viability levels (>95%) upon UK-5099 (5μM), BPTES (5μM) and DON (5μM) treatment in control and NCoR1 KD DCs. (n = 4)
G. Histogram plots showing the MFI shifts of IL-10 and IL-27 upon UK-5099, BPTES and DON treatment in control and NCoR1 KD DCs.
H. Scatter plot and bar graphs displaying the cytokine levels of IL-6 and IL-12p40/70 in 6h CpG activated Control and NCoR1 KD DCs treated with UK-5099, BPTES and DON. (n = 6)
I. A representative seahorse line graph for determining FAO index in control and NCoR1 KD DCs. FAO index for control is shown as C4 – C3 whereas, for NCoR1 KD as N4 – N3.
J. Bar graphs showing the normalized cell viability levels (>90%) upon etomoxir (50μM) treatment in control and NCoR1 KD DCs. (n = 5)
K. Histogram plots showing the MFI shifts of IL-10 and IL-27 upon etomoxir treatment in control and NCoR1 KD DCs.
L. Scatter plot and bar graphs displaying the cytokine levels of IL-6 and IL-12p40/70 in 6h CpG activated control and NCoR1 KD DCs treated with etomoxir. (n = 4)
M. Bar graphs showing the relative intensities of metabolites (aspartic acid and citric acid) in 6h CpG activated control and NCoR1 KD DCs as quantified by GC-MS. (n = 3)
N. Bar graph displaying the normalized counts of Cd36 in 6h CpG stimulated control and NCoR1 KD DCs from RNAseq data. (n = 3)
O. Bar graphs showing the relative intensity of metabolite (malic acid) in 6h CpG activated control and NCoR1 KD DCs as quantified by GC-MS. (n = 3)
P. Normalized expression (DESeq2) of Idh1 in Unstimulated and CpG activated control and NCoR1 KD DCs. (n = 2)
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed paired student’s t-test. Error bars represent SEM.
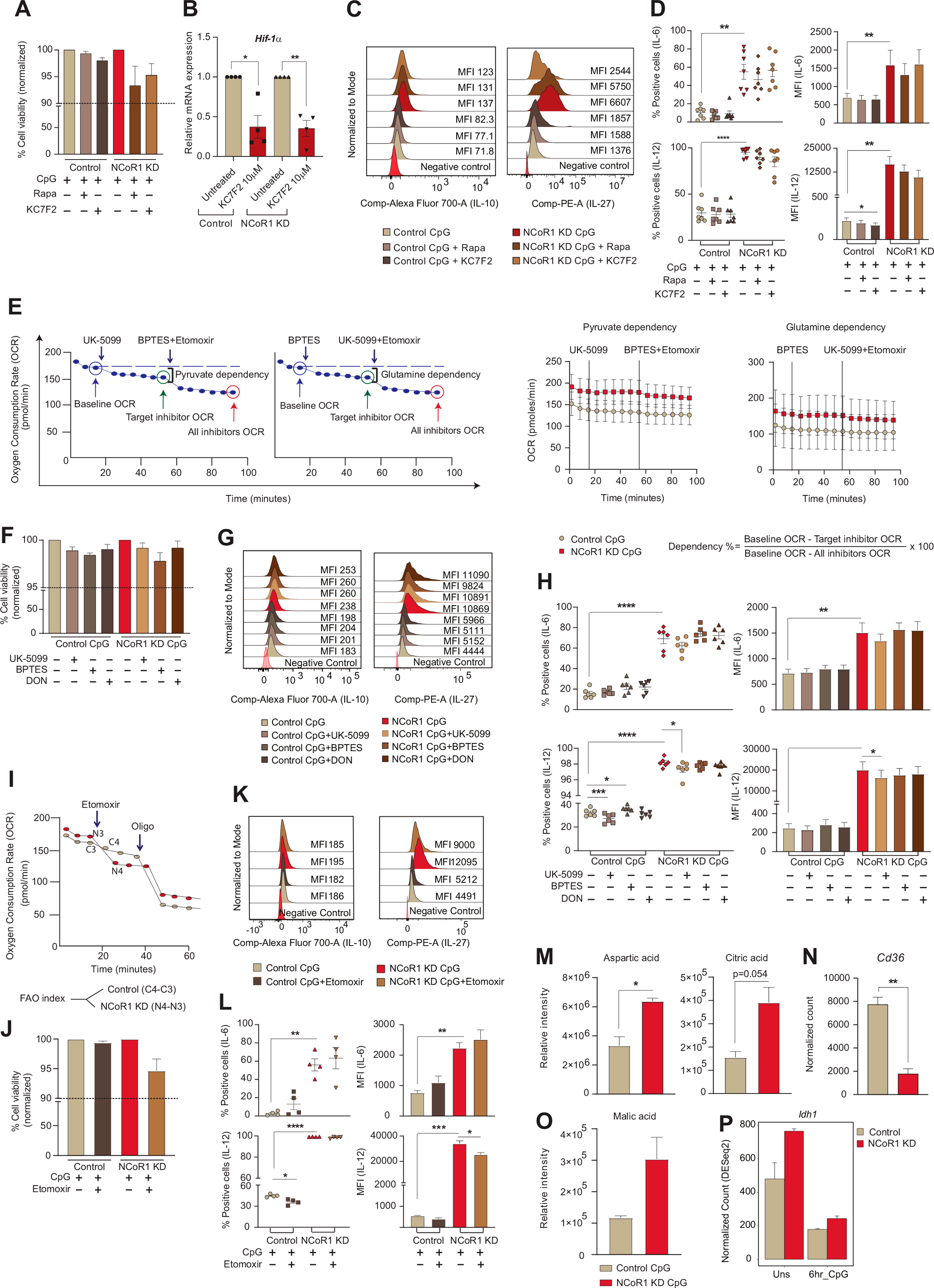{kind=link}
Impact of dual inhibition of HIF-1α and FAO on inflammatory cytokine secretion and gating strategies to identify Th1 and Treg population in DC-T cell co-culture experiments (related to Fig. 5)
A. Bar graph showing the normalized cell viability levels (>95%) upon combined treatment with KC7F2 (10μM) and etomoxir (50μM) in control and NCoR1 KD DCs. (n = 5)
B. Histogram plots showing the MFI shifts of IL-10 and IL-27 upon combined KC7F2 + etomoxir treatment in control and NCoR1 KD DCs.
C. Scatter plot and bar graphs displaying the cytokine levels of IL-6 and IL-12p40/70 in 6h CpG activated control and NCoR1 KD tolerogenic DCs treated with combined KC7F2 + etomoxir. (n = 3–4)
D. Line graph showing the percent dependency of NCoR1 KD DCs for IL-10 and IL-27 secretion upon individual and combined metabolic perturbations with KC7F2 and etomoxir. Differences in MFI levels with and without inhibitors are plotted as percentages.
E. PCA plot showing specific cluster of samples (Control CpG 6h, Control CpG 6h treated with KC7F2 + etomoxir, NCoR1 KD CpG 6h and NCoR1 KD CpG 6h treated with KC7F2 + etomoxir) from RNAseq data. (n = 3)
F. Upset plot depicting the unique or common genes differentially expressed (Up or downregulated) in NCoR1 KD, inhibitor treatment and NCoR1 KD + inhibitor treatment. (n = 3)
G. Bar graphs showing the relative mRNA expression of NCoR1 upon combined inhibition and normalized counts of Il12b and Il6 in different conditions (Control CpG 6h, Control CpG 6h treated with KC7F2 + etomoxir, NCoR1 KD CpG 6h and NCoR1 KD CpG 6h treated with KC7F2 + etomoxir) from RNAseq data.
H. Impact of KC7F2 on specific Hif-1α target genes in control and NCoR1 KD cDC1 DCs before and after treatment. (n = 3)
I. FACS dot plot and bar graph showing the purity of CD3+CD4+ T cells from OT-II mice before and after sorting for DC-T cell co-culture experiments. (n = 3)
J. Schematic outline depicting the co-culture of FLT3L differentiated BMDCs from NCoR1DC−/− mice with CD4+ T cells sorted from OT-II mice upon combined inhibitor treatment with KC7F2 and etomoxir. Scatter plots displaying the cumulative percent of IFN-γ+, T-bet+ and CD25+ & FOXP3+ double positive cells together gated on CD3+CD4+CD44+ effector T cells. (n = 3)
K. Dot plots showing the back gating strategy to denote effector T cells in draining lymph nodes (DLNs) in OT-II mice.
L. Contour plots depicting the gating of IFN-γ+ and T-bet+ populations from respective FMO controls in NCoR1 KD and NCoR1 KD DCs treated with KC7F2 + etomoxir treatment.
M. Contour plots depicting the gating of double positive CD25+ and FOXP3+ populations from respective FMO controls in NCoR1 KD and NCoR1 KD DCs treated with KC7F2 + etomoxir treatment.
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed paired [C and G] and unpaired [I & J] student’s t-test. Error bars represent SEM.
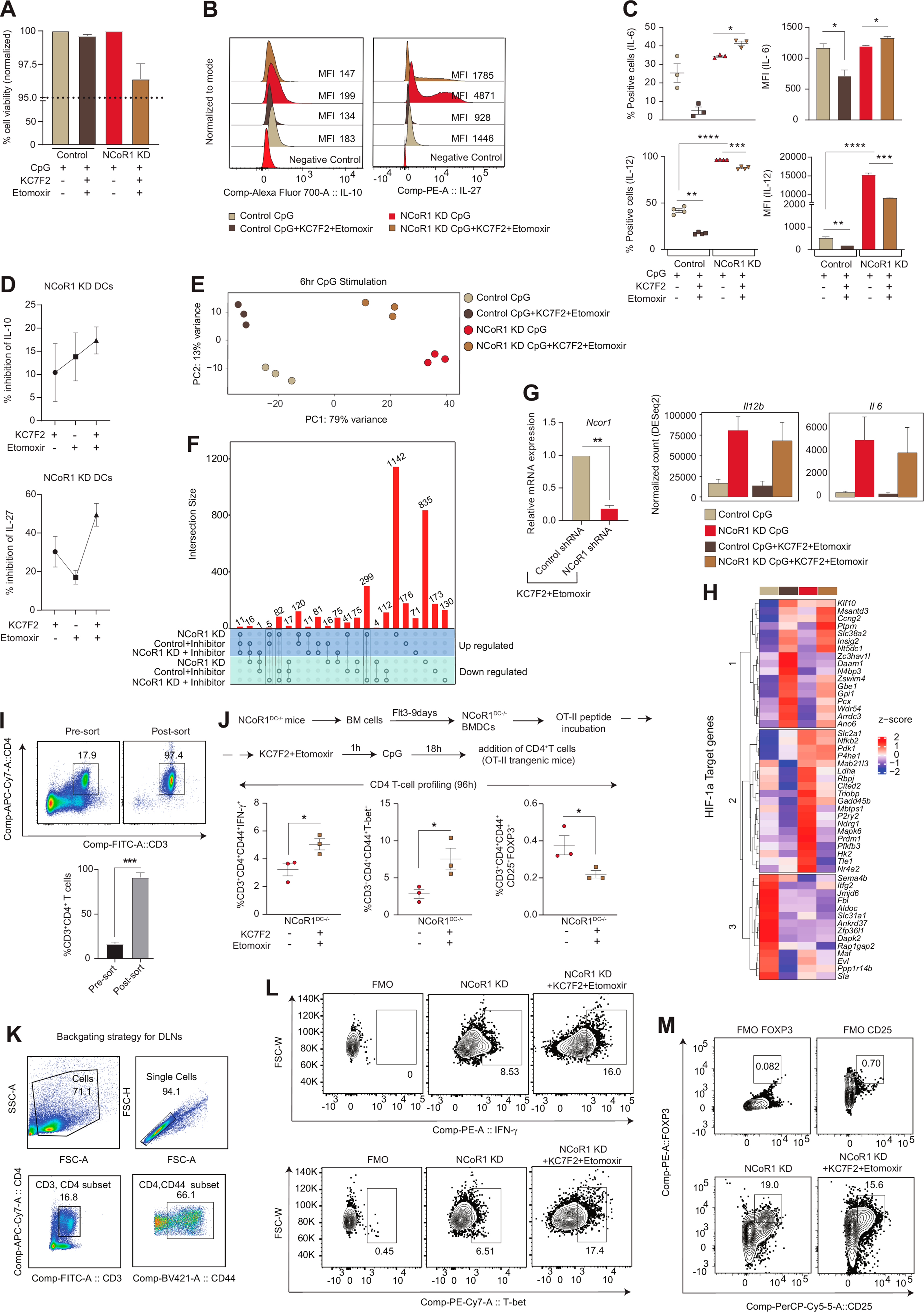{kind=link}
Gating strategies to demarcate DCs and effector T cells in the lungs and spleen of Mtb infected mice, respectively (related to Fig. 6)
A. Contour plots depicting the gating of live cells and back gating strategy to identify CD11c+ and CD103+ cells using respective FMO controls, in Mtb infected lungs in NCoR1DC−/− mice.
B. Representative FACS dot plots showing the percent Mtb infection in NCoR1DC−/− mice gated on CD11c+CD103+ DCs in NCoR1DC−/− mice and NCoR1DC−/− treated with KC7F2 + etomoxir treated animals. (n = 8)
C. Bar graph with dots depicting overall Mtb burden (MFI levels) in lungs of NCoR1DC−/− mice with and without treatment with combined inhibitors KC7F2 and etomoxir. (n = 8)
D. FACS dot plots showing the back gating strategy to identify the live effector CD4+ T cells in Mtb infected Spleen of NCoR1DC−/− mice.
E. FACS dot plots displaying the gating of FOXP3 positive cells in the spleen of Mtb infected animals. Scatter plot and bar graph depicted the cumulative percent positive and MFI levels. (n = 5)
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed unpaired student’s t-test. Error bars represent SEM.
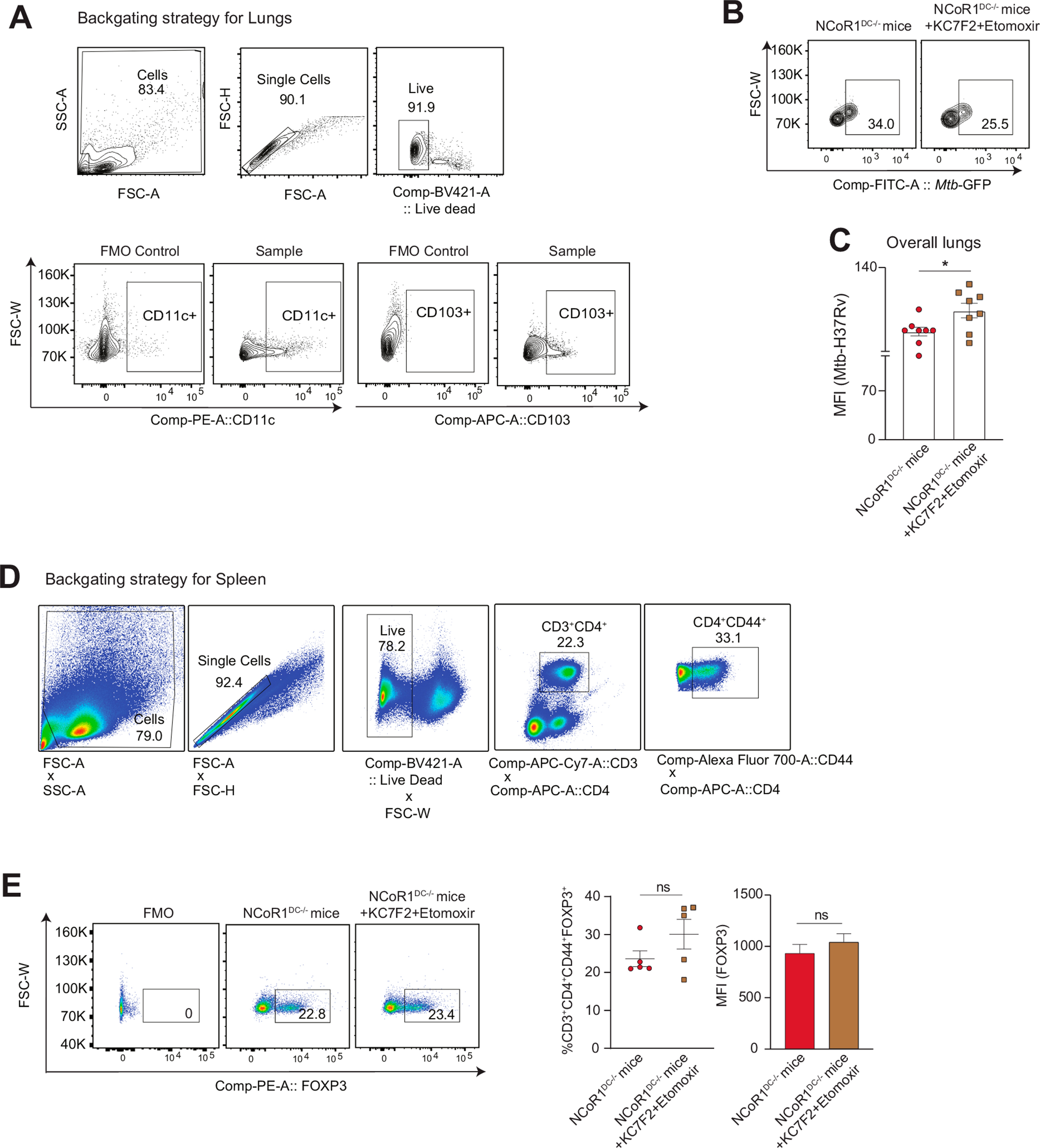{kind=link}
Data availability
Data will be made available on request.
References
- 1.O’Neill L.A.J., Pearce E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016;213:15–23. doi: 10.1084/jem.20151570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wculek S.K., Khouili S.C., Priego E., Heras-Murillo I., Sancho D. Metabolic control of dendritic cell functions: digesting information. Front. Immunol. 2019;10:775. doi: 10.3389/fimmu.2019.00775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pearce E.J., Everts B. Dendritic cell metabolism. Nat. Rev. Immunol. 2015;15:18–29. doi: 10.1038/nri3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly B., O’Neill L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–784. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Neill L.A.J., Kishton R.J., Rathmell J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016;16:553–565. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan D.G., O’Neill L.A.J. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 2017;591:2992–3006. doi: 10.1002/1873-3468.12744. [DOI] [PubMed] [Google Scholar]
- 7.Everts B., Amiel E., Huang S.C.-C., Smith A.M., Chang C.-H., Lam W.Y., Redmann V., Freitas T.C., Blagih J., van der Windt G.J.W., Artyomov M.N., Jones R.G., Pearce E.L., Pearce E.J. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKϵ supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014;15:323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jantsch J., Chakravortty D., Turza N., Prechtel A.T., Buchholz B., Gerlach R.G., Volke M., Gläsner J., Warnecke C., Wiesener M.S., Eckardt K.-U., Steinkasserer A., Hensel M., Willam C. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. Baltim. Md 1950. 2008;180:4697–4705. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- 9.Perrin-Cocon L., Aublin-Gex A., Diaz O., Ramière C., Peri F., André P., Lotteau V. Toll-like receptor 4-induced glycolytic burst in human monocyte-derived dendritic cells results from p38-dependent stabilization of HIF-1α and increased hexokinase II expression. J. Immunol. Baltim. Md 1950. 2018;201:1510–1521. doi: 10.4049/jimmunol.1701522. [DOI] [PubMed] [Google Scholar]
- 10.Guak H., Al Habyan S., Ma E.H., Aldossary H., Al-Masri M., Won S.Y., Ying T., Fixman E.D., Jones R.G., McCaffrey L.M., Krawczyk C.M. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat. Commun. 2018;9:2463. doi: 10.1038/s41467-018-04804-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Lima Thomaz L., Peron G., Oliveira J., da Rosa L.C., Thomé R., Verinaud L. The impact of metabolic reprogramming on dendritic cell function. Int. Immunopharm. 2018;63:84–93. doi: 10.1016/j.intimp.2018.07.031. [DOI] [PubMed] [Google Scholar]
- 12.Köhler T., Reizis B., Johnson R.S., Weighardt H., Förster I. Influence of hypoxia-inducible factor 1α on dendritic cell differentiation and migration. Eur. J. Immunol. 2012;42:1226–1236. doi: 10.1002/eji.201142053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sim W.J., Ahl P.J., Connolly J.E. Metabolism is central to tolerogenic dendritic cell function. Mediat. Inflamm. 2016:1–10. doi: 10.1155/2016/2636701. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takenaka M.C., Quintana F.J. Tolerogenic dendritic cells. Semin. Immunopathol. 2017;39:113–120. doi: 10.1007/s00281-016-0587-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Domogalla M.P., Rostan P.V., Raker V.K., Steinbrink K. Tolerance through education: how tolerogenic dendritic cells shape immunity. Front. Immunol. 2017;8:1764. doi: 10.3389/fimmu.2017.01764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodrigues C.P., Ferreira A.C.F., Pinho M.P., de Moraes C.J., Bergami-Santos P.C., Barbuto J.A.M. Tolerogenic Ido(+) dendritic cells are induced by PD-1-expressing mast cells. Front. Immunol. 2016;7:9. doi: 10.3389/fimmu.2016.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malinarich F., Duan K., Hamid R.A., Bijin A., Lin W.X., Poidinger M., Fairhurst A.-M., Connolly J.E. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J. Immunol. 2015;194:5174–5186. doi: 10.4049/jimmunol.1303316. [DOI] [PubMed] [Google Scholar]
- 18.Ferreira G.B., Vanherwegen A.-S., Eelen G., Gutiérrez A.C.F., Van Lommel L., Marchal K., Verlinden L., Verstuyf A., Nogueira T., Georgiadou M., Schuit F., Eizirik D.L., Gysemans C., Carmeliet P., Overbergh L., Mathieu C. Vitamin D3 induces tolerance in human dendritic cells by activation of intracellular metabolic pathways. Cell Rep. 2015;10:711–725. doi: 10.1016/j.celrep.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 19.Piemonti L., Monti P., Allavena P., Sironi M., Soldini L., Leone B.E., Socci C., Di Carlo V. Glucocorticoids affect human dendritic cell differentiation and maturation. J. Immunol. Baltim. Md 1950. 1999;162:6473–6481. [PubMed] [Google Scholar]
- 20.Xia C.-Q., Peng R., Beato F., Clare-Salzler M.J. Dexamethasone induces IL-10-producing monocyte-derived dendritic cells with durable immaturity. Scand. J. Immunol. 2005;62:45–54. doi: 10.1111/j.1365-3083.2005.01640.x. [DOI] [PubMed] [Google Scholar]
- 21.Bscheider M., Butcher E.C. Vitamin D immunoregulation through dendritic cells. Immunology. 2016;148:227–236. doi: 10.1111/imm.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adorini L., Penna G. Induction of tolerogenic dendritic cells by vitamin D receptor agonists. Handb. Exp. Pharmacol. 2009:251–273. doi: 10.1007/978-3-540-71029-5_12. [DOI] [PubMed] [Google Scholar]
- 23.Di Rosa M., Malaguarnera M., Nicoletti F., Malaguarnera L. Vitamin D3: a helpful immuno-modulator. Immunology. 2011;134:123–139. doi: 10.1111/j.1365-2567.2011.03482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mottis A., Mouchiroud L., Auwerx J. Emerging roles of the corepressors NCoR1 and SMRT in homeostasis. Genes Dev. 2013;27:819–835. doi: 10.1101/gad.214023.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ritter M.J., Amano I., Imai N., Soares De Oliveira L., Vella K.R., Hollenberg A.N. Nuclear Receptor CoRepressors, NCOR1 and SMRT, are required for maintaining systemic metabolic homeostasis. Mol. Metabol. 2021;53 doi: 10.1016/j.molmet.2021.101315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto H., Williams E.G., Mouchiroud L., Cantó C., Fan W., Downes M., Héligon C., Barish G.D., Desvergne B., Evans R.M., Schoonjans K., Auwerx J. NCoR1 is a conserved physiological modulator of muscle Mass and oxidative function. Cell. 2011;147:827–839. doi: 10.1016/j.cell.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeon Y.G., Lee J.H., Ji Y., Sohn J.H., Lee D., Kim D.W., Yoon S.G., Shin K.C., Park J., Seong J.K., Cho J.-Y., Choe S.S., Kim J.B. RNF20 functions as a transcriptional coactivator for PPARγ by promoting NCoR1 degradation in adipocytes. Diabetes. 2020;69:20–34. doi: 10.2337/db19-0508. [DOI] [PubMed] [Google Scholar]
- 28.Lima T.I., Valentim R.R., Araújo H.N., Oliveira A.G., Favero B.C., Menezes E.S., Araújo R., Silveira L.R. Role of NCoR1 in mitochondrial function and energy metabolism. Cell Biol. Int. 2018;42:734–741. doi: 10.1002/cbin.10973. [DOI] [PubMed] [Google Scholar]
- 29.Saito T., Kuma A., Sugiura Y., Ichimura Y., Obata M., Kitamura H., Okuda S., Lee H.-C., Ikeda K., Kanegae Y., Saito I., Auwerx J., Motohashi H., Suematsu M., Soga T., Yokomizo T., Waguri S., Mizushima N., Komatsu M. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 2019;10:1567. doi: 10.1038/s41467-019-08829-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geiger M.A., Guillaumon A.T., Paneni F., Matter C.M., Stein S. Role of the nuclear receptor corepressor 1 (NCOR1) in atherosclerosis and associated immunometabolic diseases. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.569358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahad A., Stevanin M., Smita S., Mishra G.P., Gupta D., Waszak S., Sarkar U.A., Basak S., Gupta B., Acha-Orbea H., Raghav S.K. NCoR1: putting the brakes on the dendritic cell immune tolerance. iScience. 2019;19:996–1011. doi: 10.1016/j.isci.2019.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mishra G.P., Jha A., Ahad A., Sen K., Sen A., Podder S., Prusty S., Biswas V.K., Gupta B., Raghav S.K. Epigenomics of conventional type-I dendritic cells depicted preferential control of TLR9 versus TLR3 response by NCoR1 through differential IRF3 activation. Cell. Mol. Life Sci. 2022;79:429. doi: 10.1007/s00018-022-04424-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gray L.R., Tompkins S.C., Taylor E.B. Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci. CMLS. 2014;71:2577–2604. doi: 10.1007/s00018-013-1539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhuang Q., Cai H., Cao Q., Li Z., Liu S., Ming Y. Tolerogenic dendritic cells: the pearl of immunotherapy in organ transplantation. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.552988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown G.C. Nitric oxide and mitochondrial respiration. Biochim. Biophys. Acta. 1999;1411:351–369. doi: 10.1016/s0005-2728(99)00025-0. [DOI] [PubMed] [Google Scholar]
- 36.Shiva S. Mitochondria as metabolizers and targets of nitrite, Nitric Oxide. Biol. Chem. 2010;22:64–74. doi: 10.1016/j.niox.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Filograna R., Mennuni M., Alsina D., Larsson N.-G. Mitochondrial DNA copy number in human disease: the more the better? FEBS Lett. 2021;595:976–1002. doi: 10.1002/1873-3468.14021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quiros P.M., Goyal A., Jha P., Auwerx J. Analysis of mtDNA/nDNA ratio in mice. Curr. Protoc. Mol. Biol. 2017;7:47–54. doi: 10.1002/cpmo.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim S.J., Diamond B. Generation and maturation of bone marrow-derived DCs under serum-free conditions. J. Immunol. Methods. 2007;323:101–108. doi: 10.1016/j.jim.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bode K.A., Schmitz F., Vargas L., Heeg K., Dalpke A.H. Kinetic of RelA activation controls magnitude of TLR-mediated IL-12p40 induction. J. Immunol. Baltim. Md 1950. 2009;182:2176–2184. doi: 10.4049/jimmunol.0802560. [DOI] [PubMed] [Google Scholar]
- 41.Møller S.H., Wang L., Ho P.-C. Metabolic programming in dendritic cells tailors immune responses and homeostasis, Cell. Mol. Immunol. 2022;19:370–383. doi: 10.1038/s41423-021-00753-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mishra A. Metabolic plasticity in dendritic cell responses: implications in allergic asthma. J. Immunol. Res. 2017;2017 doi: 10.1155/2017/5134760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kierans S.J., Taylor C.T. Regulation of glycolysis by the hypoxia‐inducible factor (HIF): implications for cellular physiology. J. Physiol. 2021;599:23–37. doi: 10.1113/JP280572. [DOI] [PubMed] [Google Scholar]
- 44.Porporato P.E., Dhup S., Dadhich R.K., Copetti T., Sonveaux P. Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front. Pharmacol. 2011;2 doi: 10.3389/fphar.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lum J.J., Bui T., Gruber M., Gordan J.D., DeBerardinis R.J., Covello K.L., Simon M.C., Thompson C.B. The transcription factor HIF-1α plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev. 2007;21:1037–1049. doi: 10.1101/gad.1529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goetzman E.S., Prochownik E.V. The role for myc in coordinating glycolysis, oxidative phosphorylation, glutaminolysis, and fatty acid metabolism in normal and neoplastic tissues. Front. Endocrinol. 2018;9:129. doi: 10.3389/fendo.2018.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wanders R.J.A., Visser G., Ferdinandusse S., Vaz F.M., Houtkooper R.H. Mitochondrial fatty acid oxidation disorders: laboratory diagnosis, pathogenesis, and the complicated route to treatment. J. Lipid Atheroscler. 2020;9:313–333. doi: 10.12997/jla.2020.9.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Souza-Mello V. Peroxisome proliferator-activated receptors as targets to treat non-alcoholic fatty liver disease. World J. Hepatol. 2015;7:1012–1019. doi: 10.4254/wjh.v7.i8.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corrales P., Vidal-Puig A., Medina-Gómez G. PPARs and metabolic disorders associated with challenged adipose tissue plasticity. Int. J. Mol. Sci. 2018;19:E2124. doi: 10.3390/ijms19072124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang H.-C., Yu H., Liu Y.-C., Chen T.-L., Stern A., Lo S.J., Chiu D.T.-Y. IDH-1 deficiency induces growth defects and metabolic alterations in GSPD-1-deficient Caenorhabditis elegans. J. Mol. Med. Berl. Ger. 2019;97:385–396. doi: 10.1007/s00109-018-01740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen L., Zhang Z., Hoshino A., Zheng H.D., Morley M., Arany Z., Rabinowitz J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 2019;1:404–415. [PMC free article] [PubMed] [Google Scholar]
- 52.Lamichane S., Dahal Lamichane B., Kwon S.-M. Pivotal roles of peroxisome proliferator-activated receptors (PPARs) and their signal cascade for cellular and whole-body energy homeostasis. Int. J. Mol. Sci. 2018;19:E949. doi: 10.3390/ijms19040949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao F., Xiao C., Evans K.S., Theivanthiran T., DeVito N., Holtzhausen A., Liu J., Liu X., Boczkowski D., Nair S., Locasale J.W., Hanks B.A. Paracrine wnt5a-β-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity. 2018;48:147–160. doi: 10.1016/j.immuni.2017.12.004. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blanchard P.-G., Festuccia W.T., Houde V.P., St-Pierre P., Brûlé S., Turcotte V., Côté M., Bellmann K., Marette A., Deshaies Y. Major involvement of mTOR in the PPARγ-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012;53:1117–1125. doi: 10.1194/jlr.M021485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jung J.E., Kim H.S., Lee C.S., Shin Y.J., Kim Y.N., Kang G.H., Kim T.Y., Juhnn Y.S., Kim S.J., Park J.W., Ye S.K., Chung M.H. STAT3 inhibits the degradation of HIF-1alpha by pVHL-mediated ubiquitination. Exp. Mol. Med. 2008;40:479–485. doi: 10.3858/emm.2008.40.5.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tošić I., Frank D.A. STAT3 as a mediator of oncogenic cellular metabolism: pathogenic and therapeutic implications. Neoplasia N. Y. N. 2021;23:1167–1178. doi: 10.1016/j.neo.2021.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jha A., Ahad A., Mishra G.P., Sen K., Smita S., Minz A.P., Biswas V.K., Tripathy A., Senapati S.B., Gupta B., Orbea H.A., Raghav S.K. NCoR1 and SMRT fine-tune inflammatory versus tolerogenic balance in dendritic cells by differentially regulating STAT3 signaling. Immunology. 2021 doi: 10.1101/2021.03.11.434976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Etna M.P., Severa M., Licursi V., Pardini M., Cruciani M., Rizzo F., Giacomini E., Macchia G., Palumbo O., Stallone R., Carella M., Livingstone M., Negri R., Pellegrini S., Coccia E.M. Genome-wide gene expression analysis of mtb-infected DC highlights the rapamycin-driven modulation of regulatory cytokines via the mTOR/GSK-3β Axis. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.649475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matta S.K., Kumar D. Hypoxia and classical activation limits Mycobacterium tuberculosis survival by Akt-dependent glycolytic shift in macrophages. Cell Death Dis. 2016;2 doi: 10.1038/cddiscovery.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Navarro-Barriuso J., Mansilla M.J., Naranjo-Gómez M., Sánchez-Pla A., Quirant-Sánchez B., Teniente-Serra A., Ramo-Tello C., Martínez-Cáceres E.M. Comparative transcriptomic profile of tolerogenic dendritic cells differentiated with vitamin D3, dexamethasone and rapamycin. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-33248-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim S.H., Jung H.H., Lee C.K. Generation, characteristics and clinical trials of ex vivo generated tolerogenic dendritic cells. Yonsei Med. J. 2018;59:807–815. doi: 10.3349/ymj.2018.59.7.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iwasaki A., Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 63.Cui J., Chen Y., Wang H.Y., Wang R.-F. Mechanisms and pathways of innate immune activation and regulation in health and cancer. Hum. Vaccines Immunother. 2014;10:3270–3285. doi: 10.4161/21645515.2014.979640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marin E., Bouchet-Delbos L., Renoult O., Louvet C., Nerriere-Daguin V., Managh A.J., Even A., Giraud M., Vu Manh T.P., Aguesse A., Bériou G., Chiffoleau E., Alliot-Licht B., Prieur X., Croyal M., Hutchinson J.A., Obermajer N., Geissler E.K., Vanhove B., Blancho G., Dalod M., Josien R., Pecqueur C., Cuturi M.-C., Moreau A. Human tolerogenic dendritic cells regulate immune responses through lactate synthesis. Cell Metabol. 2019;30:1075–1090. doi: 10.1016/j.cmet.2019.11.011. e8. [DOI] [PubMed] [Google Scholar]
- 65.Vanherwegen A.-S., Eelen G., Ferreira G.B., Ghesquière B., Cook D.P., Nikolic T., Roep B., Carmeliet P., Telang S., Mathieu C., Gysemans C. Vitamin D controls the capacity of human dendritic cells to induce functional regulatory T cells by regulation of glucose metabolism. J. Steroid Biochem. Mol. Biol. 2019;187:134–145. doi: 10.1016/j.jsbmb.2018.11.011. [DOI] [PubMed] [Google Scholar]
- 66.Everts B., Pearce E.J. Metabolic control of dendritic cell activation and function: recent advances and clinical implications. Front. Immunol. 2014;5:203. doi: 10.3389/fimmu.2014.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zeng Q., Mallilankaraman K., Schwarz H. Increased akt-driven glycolysis is the basis for the higher potency of cd137l-DCs. Front. Immunol. 2019;10:868. doi: 10.3389/fimmu.2019.00868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haidinger M., Poglitsch M., Geyeregger R., Kasturi S., Zeyda M., Zlabinger G.J., Pulendran B., Hörl W.H., Säemann M.D., Weichhart T. A versatile role of mammalian target of rapamycin in human dendritic cell function and differentiation. J. Immunol. Baltim. Md 1950. 2010;185:3919–3931. doi: 10.4049/jimmunol.1000296. [DOI] [PubMed] [Google Scholar]
- 69.Ohtani M., Hoshii T., Fujii H., Koyasu S., Hirao A., Matsuda S. Cutting edge: mTORC1 in intestinal CD11c+ CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL-10 production. J. Immunol. Baltim. Md 1950. 2012;188:4736–4740. doi: 10.4049/jimmunol.1200069. [DOI] [PubMed] [Google Scholar]
- 70.Tran C.W., Gold M.J., Garcia-Batres C., Tai K., Elford A.R., Himmel M.E., Elia A.J., Ohashi P.S. Hypoxia-inducible factor 1 alpha limits dendritic cell stimulation of CD8 T cell immunity. PLoS One. 2020;15 doi: 10.1371/journal.pone.0244366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McGettrick A.F., O’Neill L.A.J. The role of HIF in immunity and inflammation. Cell Metabol. 2020;32:524–536. doi: 10.1016/j.cmet.2020.08.002. [DOI] [PubMed] [Google Scholar]
- 72.Pantel A., Teixeira A., Haddad E., Wood E.G., Steinman R.M., Longhi M.P. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol. 2014;12 doi: 10.1371/journal.pbio.1001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martínez-Reyes I., Chandel N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020;11:102. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nolfi-Donegan D., Braganza A., Shiva S. Mitochondrial electron transport chain: oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020;37 doi: 10.1016/j.redox.2020.101674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Basit F., Mathan T., Sancho D., de Vries I.J.M. Human dendritic cell subsets undergo distinct metabolic reprogramming for immune response. Front. Immunol. 2018;9:2489. doi: 10.3389/fimmu.2018.02489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu Y.-X., Ju H.-Q., Liu Z.-X., Chen D.-L., Wang Y., Zhao Q., Wu Q.-N., Zeng Z.-L., Qiu H.-B., Hu P.-S., Wang Z.-Q., Zhang D.-S., Wang F., Xu R.-H. ME1 regulates NADPH homeostasis to promote gastric cancer growth and metastasis. Cancer Res. 2018;78:1972–1985. doi: 10.1158/0008-5472.CAN-17-3155. [DOI] [PubMed] [Google Scholar]
- 77.Liu C., Cao J., Lin S., Zhao Y., Zhu M., Tao Z., Hu X. Malic enzyme 1 indicates worse prognosis in breast cancer and promotes metastasis by manipulating reactive oxygen species. OncoTargets Ther. 2020;13:8735–8747. doi: 10.2147/OTT.S256970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu C.H., Liu H., Ge B. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell. Mol. Immunol. 2017;14:963–975. doi: 10.1038/cmi.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Llufrio E.M., Wang L., Naser F.J., Patti G.J. Sorting cells alters their redox state and cellular metabolome. Redox Biol. 2018;16:381–387. doi: 10.1016/j.redox.2018.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fuertes Marraco S.A., Grosjean F., Duval A., Rosa M., Lavanchy C., Ashok D., Haller S., Otten L.A., Steiner Q.-G., Descombes P., Luber C.A., Meissner F., Mann M., Szeles L., Reith W., Acha-Orbea H. Novel murine dendritic cell lines: a powerful auxiliary tool for dendritic cell research. Front. Immunol. 2012;3:331. doi: 10.3389/fimmu.2012.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NCoR1 depletion in cDC1 and bioenergetics of cDC1 DCs (related to Fig. 1)
A. Bar graph showing the relative mRNA expression of Ncor1 in control shRNA versus NCoR1 shRNA transduced mutu-cDC1 line. (n = 3)
B. An idealistic glyco-stress line graph showing glycolysis, glycolytic capacity and glycolytic reserve upon sequential injections with glucose, oligomycin and 2-DG.
C. Scatter plot representing the change in pH in extracellular media in the control and NCoR1 KD tolerogenic DCs before and after 6h CpG stimulation. (n = 6)
D. Scatter plot depicting the total intracellular ATP levels in unstimulated and 6h CpG activated control and NCoR1 KD tolerogenic DCs with and without treatment with 2-DG (10mM). Corresponding bar graph depicted the differences in the ATP levels. (n = 4)
E. Heat map depicting the log2 fold change expression of PPP genes in NCoR1 KD versus control DCs, * depicts significance and NS denotes non-significant change. (n = 3)
F. Western blot and its densitometric analysis showing the decreased protein expression of PPP rate-limiting enzyme G6PD in NCoR1 KD as compared to control DCs. (n = 4)
G. Heatmap showing the differentially expressed genes of complex-I to complex-V for the electron transport chain (ETC) from RNAseq data of unstimulated and 6h CpG activated control and NCoR1 KD tolerogenic DCs. (n = 2)
H. Bar graph demonstrating the relative mRNA expression of Nrf-1 and Cox5a in unstimulated and 6h CpG activated control and NCoR1 KD tolerogenic DCs. (n = 6)
I. An idealistic mito-stress line graph showing basal respiration, maximal respiration, coupled ATP and spare respiratory capacity upon sequential injections of oligomycin, CCCP and Rot/AA in Seahorse extracellular flux analyzer.
J. Scatter plot depicting the total intracellular ATP levels in unstimulated and 6h CpG activated control and NCoR1 KD tolerogenic DCs. (n = 3)
K-L Histogram plots depicting the MFI shifts for mitochondrial SOX, cellular ROS and TMRM (mitochondrial potential) for control and NCoR1 KD tolerogenic DCs before and after 6h CpG activation.
M. Dot plots showing the back gating strategies to identify viable cells in cDC1 mutu-DC line by flow cytometry.
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed paired student’s t-test. Error bars represent SEM.
Mouse genotyping, cellular homeostasis and gating strategies used in BMDCs ex vivo flow cytometry experiments (related to Fig. 2)
A. Mouse genotyping - representative agarose gel image showing the NCoR1 Lox-P bands in Wild Type (WT) and NCoR1DC−/− mice, whereas the CD11c trans gene band is specific to NCoR1DC−/− mice only.
B. Bar graph depicting the relative mRNA expression of NCoR1 in control CD11c-Cre and NCoR1DC−/− BMDCs. (n = 3)
C. FACS plot showing the gating of percent double positive CD11c+MHCII+ cells in control CD11c-Cre and NCoR1DC−/− BMDCs at day 9 of differentiation. Dot plots showing the back gating strategy and scatter plot depicting the cumulative data. (n = 4)
D-E. Histogram plots depicting the MFI shifts of cellular ROS and TMRM (mitochondrial potential) in control CD11c-Cre and NCoR1DC−/− BMcDC1s before and after 18h CpG activation.
F. Dot plots showing the back gating strategy to identify F4/80−CD11c+CD24+ BMcDC1s at day 9 of FLT3L differentiated mouse bone marrow cells.
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed unpaired student’s t-test. Error bars represent SEM.
Impact of mTOR, HIF-1α, pyruvate, glutamine and fatty acid oxidation perturbation on inflammatory cytokine secretion (related to Fig. 3, Fig. 4)
A. Bar graphs showing the normalized cell viability levels (>90%) upon rapamycin (20nM) and KC7F2 (10μM) treatment in Control and NCoR1 KD DCs. (n = 10)
B. RT-qPCR depicting the transcript expression of Hif-1α gene before and after treatment with KC7F2 in control and NCoR1 KD cDC1 DCs. (n = 4)
C. Histogram plots showing the MFI shifts of IL-10 and IL-27 upon rapamycin and KC7F2 treatment.
D. Scatter plot and bar graphs showing the cytokine levels of IL-6 and IL-12p40/70 in 6h CpG activated cells, after treatment with rapamycin and KC7F2. (n = 7–8)
E. An idealistic pyruvate and glutamine oxidation dependency graph from Seahorse extracellular flux analyzer. Experimental line graphs showing the pyruvate and glutamine dependency in terms of OCR when specific inhibitors were used either singly or in combinations. Dependency percentage is calculated as baseline OCR - target inhibitor OCR / baseline OCR - all inhibitors OCR * 100.
F. Bar graphs depicting the normalized cell viability levels (>95%) upon UK-5099 (5μM), BPTES (5μM) and DON (5μM) treatment in control and NCoR1 KD DCs. (n = 4)
G. Histogram plots showing the MFI shifts of IL-10 and IL-27 upon UK-5099, BPTES and DON treatment in control and NCoR1 KD DCs.
H. Scatter plot and bar graphs displaying the cytokine levels of IL-6 and IL-12p40/70 in 6h CpG activated Control and NCoR1 KD DCs treated with UK-5099, BPTES and DON. (n = 6)
I. A representative seahorse line graph for determining FAO index in control and NCoR1 KD DCs. FAO index for control is shown as C4 – C3 whereas, for NCoR1 KD as N4 – N3.
J. Bar graphs showing the normalized cell viability levels (>90%) upon etomoxir (50μM) treatment in control and NCoR1 KD DCs. (n = 5)
K. Histogram plots showing the MFI shifts of IL-10 and IL-27 upon etomoxir treatment in control and NCoR1 KD DCs.
L. Scatter plot and bar graphs displaying the cytokine levels of IL-6 and IL-12p40/70 in 6h CpG activated control and NCoR1 KD DCs treated with etomoxir. (n = 4)
M. Bar graphs showing the relative intensities of metabolites (aspartic acid and citric acid) in 6h CpG activated control and NCoR1 KD DCs as quantified by GC-MS. (n = 3)
N. Bar graph displaying the normalized counts of Cd36 in 6h CpG stimulated control and NCoR1 KD DCs from RNAseq data. (n = 3)
O. Bar graphs showing the relative intensity of metabolite (malic acid) in 6h CpG activated control and NCoR1 KD DCs as quantified by GC-MS. (n = 3)
P. Normalized expression (DESeq2) of Idh1 in Unstimulated and CpG activated control and NCoR1 KD DCs. (n = 2)
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed paired student’s t-test. Error bars represent SEM.
Impact of dual inhibition of HIF-1α and FAO on inflammatory cytokine secretion and gating strategies to identify Th1 and Treg population in DC-T cell co-culture experiments (related to Fig. 5)
A. Bar graph showing the normalized cell viability levels (>95%) upon combined treatment with KC7F2 (10μM) and etomoxir (50μM) in control and NCoR1 KD DCs. (n = 5)
B. Histogram plots showing the MFI shifts of IL-10 and IL-27 upon combined KC7F2 + etomoxir treatment in control and NCoR1 KD DCs.
C. Scatter plot and bar graphs displaying the cytokine levels of IL-6 and IL-12p40/70 in 6h CpG activated control and NCoR1 KD tolerogenic DCs treated with combined KC7F2 + etomoxir. (n = 3–4)
D. Line graph showing the percent dependency of NCoR1 KD DCs for IL-10 and IL-27 secretion upon individual and combined metabolic perturbations with KC7F2 and etomoxir. Differences in MFI levels with and without inhibitors are plotted as percentages.
E. PCA plot showing specific cluster of samples (Control CpG 6h, Control CpG 6h treated with KC7F2 + etomoxir, NCoR1 KD CpG 6h and NCoR1 KD CpG 6h treated with KC7F2 + etomoxir) from RNAseq data. (n = 3)
F. Upset plot depicting the unique or common genes differentially expressed (Up or downregulated) in NCoR1 KD, inhibitor treatment and NCoR1 KD + inhibitor treatment. (n = 3)
G. Bar graphs showing the relative mRNA expression of NCoR1 upon combined inhibition and normalized counts of Il12b and Il6 in different conditions (Control CpG 6h, Control CpG 6h treated with KC7F2 + etomoxir, NCoR1 KD CpG 6h and NCoR1 KD CpG 6h treated with KC7F2 + etomoxir) from RNAseq data.
H. Impact of KC7F2 on specific Hif-1α target genes in control and NCoR1 KD cDC1 DCs before and after treatment. (n = 3)
I. FACS dot plot and bar graph showing the purity of CD3+CD4+ T cells from OT-II mice before and after sorting for DC-T cell co-culture experiments. (n = 3)
J. Schematic outline depicting the co-culture of FLT3L differentiated BMDCs from NCoR1DC−/− mice with CD4+ T cells sorted from OT-II mice upon combined inhibitor treatment with KC7F2 and etomoxir. Scatter plots displaying the cumulative percent of IFN-γ+, T-bet+ and CD25+ & FOXP3+ double positive cells together gated on CD3+CD4+CD44+ effector T cells. (n = 3)
K. Dot plots showing the back gating strategy to denote effector T cells in draining lymph nodes (DLNs) in OT-II mice.
L. Contour plots depicting the gating of IFN-γ+ and T-bet+ populations from respective FMO controls in NCoR1 KD and NCoR1 KD DCs treated with KC7F2 + etomoxir treatment.
M. Contour plots depicting the gating of double positive CD25+ and FOXP3+ populations from respective FMO controls in NCoR1 KD and NCoR1 KD DCs treated with KC7F2 + etomoxir treatment.
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed paired [C and G] and unpaired [I & J] student’s t-test. Error bars represent SEM.
Gating strategies to demarcate DCs and effector T cells in the lungs and spleen of Mtb infected mice, respectively (related to Fig. 6)
A. Contour plots depicting the gating of live cells and back gating strategy to identify CD11c+ and CD103+ cells using respective FMO controls, in Mtb infected lungs in NCoR1DC−/− mice.
B. Representative FACS dot plots showing the percent Mtb infection in NCoR1DC−/− mice gated on CD11c+CD103+ DCs in NCoR1DC−/− mice and NCoR1DC−/− treated with KC7F2 + etomoxir treated animals. (n = 8)
C. Bar graph with dots depicting overall Mtb burden (MFI levels) in lungs of NCoR1DC−/− mice with and without treatment with combined inhibitors KC7F2 and etomoxir. (n = 8)
D. FACS dot plots showing the back gating strategy to identify the live effector CD4+ T cells in Mtb infected Spleen of NCoR1DC−/− mice.
E. FACS dot plots displaying the gating of FOXP3 positive cells in the spleen of Mtb infected animals. Scatter plot and bar graph depicted the cumulative percent positive and MFI levels. (n = 5)
*p ≤ 0.05, **p ≤ 0.01, ***p ≤0.001 and ****p ≤0.0001. p-value has been calculated using two tailed unpaired student’s t-test. Error bars represent SEM.
Data Availability Statement
Data will be made available on request.