

Abstract
Nitrogenase is the only enzyme that can convert N2 to NH3. Crystallographic structures have indicated that one of the sulfide ligands of the active‐site FeMo cluster, S2B, can be replaced by an inhibitor, like CO and OH−, and it has been suggested that it may be displaced also during the normal reaction. We have investigated possible proton transfer pathways within the FeMo cluster during the conversion of N2H2 to two molecules of NH3, assuming that the protons enter the cluster at the S3B, S4B or S5A sulfide ions and are then transferred to the substrate. We use combined quantum mechanical and molecular mechanical (QM/MM) calculations with the TPSS and B3LYP functionals. The calculations indicate that the barriers for these reactions are reasonable if the S2B ligand remains bound to the cluster, but they become prohibitively high if S2B has dissociated. This suggests that it is unlikely that S2B reversibly dissociates during the normal reaction cycle.
Keywords: nitrogenase, proton transfer, S2B dissociation, QM/MM, reaction mechanisms
Possible proton transfer pathways in the active‐site FeMo cluster of nitrogenase from the S5A, S4A or S3B sulfide ions to the substrate were studied through combined quantum and molecular mechanical calculations. The results show that the barriers are prohibitively high if the S2B sulfide ion has dissociated from the cluster, but are reasonable with S2B bound.

Introduction
Nitrogenase (EC 1.18/19.6.1) is the only group of enzymes that can cleave the strong triple bond in N2, making nitrogen available for biological lifeforms. It is found in some groups of bacteria and archaebacteria, but many higher plants live in symbiosis with such organisms. Crystal structures of the most active form of nitrogenase show that the active site consists of a MoFe7S9C(homocitrate) cluster (the FeMo cluster, shown in Figure 1).[ 1 , 2 , 3 , 4 , 5 ] It is connected to the protein by a cysteine ligand to one of the Fe ions and a histidine ligand to the Mo ion. There also exist alternative nitrogenases with the Mo ion replaced with either vanadium or iron, which have lower activities towards N2. [6]
Figure 1.
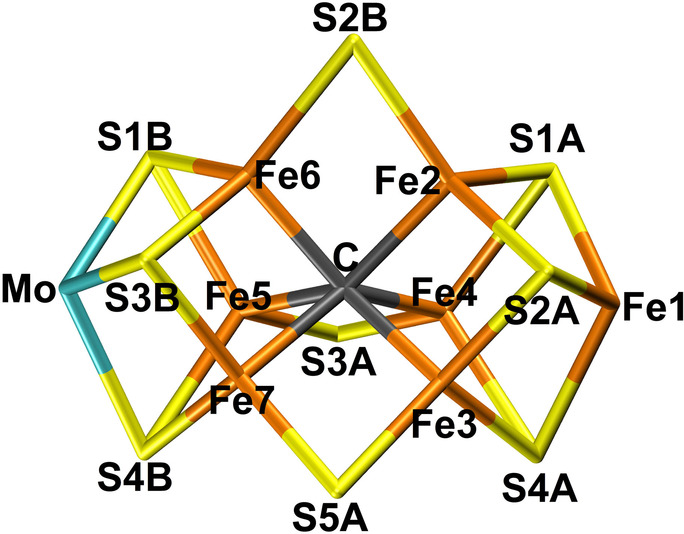
The FeMo cluster with atom names indicated. All structures in the figures use the same perspective.
Nitrogenase catalyses the reaction:
| (1) |
Thus, it requires eight electrons and protons, and consumes 16 molecules of ATP. H2 is a mandatory byproduct. The reaction is often described by the Lowe–Thorneley scheme, [7] which contains nine states E0 to E8, differing in the number of added electrons and protons. Thorough biochemical, kinetic, structural and spectroscopic studies have suggested that the enzyme typically needs to be loaded with four electrons and protons before the N2 substrate can bind, through reductive elimination of H2.[ 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 ] The substrate probably binds to the Fe2 or Fe6 ions (atom names are shown in Figure 1).[ 12 , 17 ]
Several crystallographic studies have shown that one of the sulfide ligands, S2B, which bridges the Fe2 and Fe6 ions, can be replaced by inhibitors, like CO and OH−.[ 4 , 18 ] The process is reversible and a putative storage site for the dissociated SH− ion has been identified. Therefore, it has been suggested that S2B may dissociate also during the normal reaction mechanism, opening up an obvious binding site for the substrate.[ 19 , 20 ] In fact, a recent crystal structure was suggested to show such replacement of S2B (and also the S3A and S5A sulfide ions) by N2, [21] although the interpretation has been disputed.[ 22 , 23 ] Nitrogenase has also been extensively investigated by computational methods, [24] but so far no consensus has been reached regarding the reaction mechanism or even of the structure and protonation of the crucial E4 state.[ 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 ]
Between each E n level, one electron and one proton are delivered to the FeMo cluster. The electrons are provided by the Fe protein via the P‐cluster,[ 11 , 12 ] and they can move freely within the FeMo cluster. However, movements of the protons are more restricted. They should ultimately come from the bulk solvent. His‐195 forms a hydrogen bond to the S2B sulfide ion and it has therefore often been assumed to provide protons to the substrate.[ 27 , 32 ] However, to donate more than one proton to the substrate, the sidechain of His‐195 must rotate and calculations by Dance have indicated that such rotations are unlikely in the protein.[ 33 , 34 ]
Therefore, he investigated alternative proton pathways from the solvent to the FeMo cluster and found that there is a conserved chain of water molecules from the surface that ends with a water molecule close to the S3B atom of the FeMo cluster.[ 33 , 35 , 36 , 37 ] He suggested that protons are delivered to the FeMo cluster on the S3B atom and that they are then transferred to the substrate via various Fe and sulfide ions in the cluster. He identified six local minima for the binding of a proton on S3B and showed that the proton may move between these with barriers of 10–60 kJ mol−1.[ 33 , 38 ] Based on this finding, he also studied possible proton transfers within the FeMo cluster, starting from the S3B atom and ending up at the substrate‐binding site at Fe6 and Fe2.[ 33 , 36 , 38 ] However, he studied mainly the first four steps of the reaction mechanism (E0 to E4) and partly with an outdated model of the FeMo cluster.
Recently, we have studied putative reaction mechanisms of nitrogenase, starting from bound and protonated N2H2 and going to two NH3 molecules, with either S2B bound to or dissociated from the cluster.[ 20 , 39 ] In both cases, reasonable pathways could be identified, following mainly alternating mechanisms (i.e. the two N atoms are protonated alternatively and the products do not dissociate before the E6 intermediate). Therefore, the calculations could not discriminate between the two scenarios. Moreover, between each E n state, protons were simply added to every possible site on the substrates, assuming that they can freely move around in the FeMo cluster. Therefore, the studies mainly determined the thermodynamically most stable protonation state and binding conformation of the substrate at each E n level.
In this study, we go one step further and study proton‐transfer reactions within the FeMo cluster, from the sulfide ions closest to the end of the water chain and to the substrate. In this case, we observe a significant effect of the S2B ligand.
Results and Discussion
We have studied possible proton transfers within the FeMo cluster for the E5–E8 states both when S2B has dissociated and when it remains bound to the cluster. Following the investigations by Dance,[ 33 , 36 , 38 ] we assume that the protons are delivered to the cluster via the proton wire ending close to S3B.[ 33 , 35 , 36 , 37 ] However, looking at the most accurate crystal structure of nitrogenase (3U7Q [3] ), the last water molecule in the chain (HOH‐519) is close to three sulfide ions, S3B (4.05 Å), S5A (4.03 Å) and S4B (3.77 Å). Therefore, we considered all three ions as possible entry sites for the proton.
First, we studied possible proton‐transfer paths at the E5 level and with S2B dissociated. We started from the best E4 structure in our previous investigation, [20] i.e. with NNH2 bridging Fe2 and Fe6. To this structure, we added an electron and a proton that initially was placed on either S5A, S3B or S4B. The results are shown in Figure 2. It turned out that protonation of S5A is 97 and 69 kJ mol−1 more favourable than protonation of S4B and S3B. The proton on each sulfide ion can point in several different directions that represent different local minima. For example, the proton on S5A can point either towards S2B or S3A (the two conformations are called S5A(2) or S5A(3) in Figure 2. The latter is 16 kJ mol−1 more stable.
Figure 2.

Reaction and activation energies, as well as structures for proton transfers in the E5 state without S2B bound to the cluster, from S5A, S4B or S3B to the NNH2 intermediate bound to Fe6 and Fe2, leading to a HNNH2 product. Energies in black and red indicate that the BS10‐147 and BS7‐235 states are most stable, respectively. S4B(7) differs from S4B in that the proton points towards Fe7, rather than towards Mo. Fe7(2) differs from Fe7 in that the proton points towards Fe2, rather than towards S4B. In S3B(7) and S3B(6), the proton points towards Fe7 or Fe6, respectively. Fe6(Mo) differs from Fe6 in that the proton points towards Mo, rather than towards Fe2 (sometimes called the exo and endo positions, respectively).[ 33 , 35 , 36 , 37 ] HNNH2(3) differs from HNNH2(5) in that the added proton points towards S5A rather than S3A. A final prime in the name of the structures (e.g. S3B(6)′) indicates that the NNH2 substrate has changed its conformation so that the NH2 group binds to Fe6 (while the other N atom still bridges Fe2 and Fe6).
Then, we tried to find a proton‐transfer pathway from the three sulfide ions to the NNH2 ligand. The proton can first be transferred from S5A to Fe7 with a barrier of 48 kJ mol−1. During this reaction the most stable broken‐symmetry (BS) state changes from BS10‐147 to BS7‐235. Similar changes are found throughout the reaction mechanism and in the figures only the energy of the lowest BS state is indicated. A proton on S4B can easily be transferred to Fe7 with a barrier of only 3 kJ mol−1. Next, the proton on Fe7 changes its conformation to point towards Fe2 with a barrier of 68 kJ mol−1. Then, it is transferred to Fe6 (11 kJ mol−1 barrier) and to Fe2 (11 kJ mol−1 barrier), before it can be transferred to the substrate with a barrier of 40 kJ mol−1. This results in a HNNH2 state with only the NH group bridging Fe2 and Fe6. It is 16 kJ mol−1 more stable than the starting S5A structure, but it needs to be reorganised by a barrier of 30 kJ mol−1 before it reaches the most stable HNNH2 structure with also the NH2 group coordinating to Fe6, 55 kJ mol−1 more stable than the initial state with the proton on S5A.
Unfortunately, even if none of the individual barriers is prohibitively high, the early part of proton‐transfer path is uphill and the highest net barrier (calculated from S5A(3)) is actually 114 kJ mol−1. This is too high, compared to the net reaction rate of nitrogenase, ≈5 s−1, [12, 26] corresponding to a barrier of ≈70 kJ mol−1. Owing to this prohibitively large barrier, we looked for alternative paths. Several of these are included in Figure 2 and paths with net barriers of 107 or 109 kJ mol−1 can be found, leading to an alternative HNNH2 product with a slightly different orientation, 4 kJ mol−1 less stable than the one of the other path. We also tested different variants of the method and the model (e.g. adding an extra proton on S5A). The results of these calculations are described in the Supporting Information. For the E5 intermediates, it turned out that more reasonable barriers could be obtained for calculations with the B3LYP functional, giving a maximum barrier of 80 kJ mol−1.
Next, we added an electron and a proton to the HNNH2 structure to study proton transfers for the E6 state. In this case, protonation of S5A was 67 and 78 kJ mol−1 more favourable than protonation of S3B and S4B. As can be seen in Figure 3, the proton can be transferred from S5A to Fe7 and, after some changes in the conformation, to Fe6 and finally to the ligand, forming H2NNH2 (hydrazine). The individual barriers are 13–55 kJ mol−1. However, because all intermediate states are higher in energy than the initial protonation of S5A, the net barrier is 138 kJ mol−1, which is prohibitively large. An alternative path involving protonation of S3B has a slightly larger net barrier of 140 kJ mol−1. Unfortunately, the barrier becomes even higher with B3LYP, 162 kJ mol−1.
Figure 3.

Reaction and activation energies, as well as structures for proton transfers in the E6 state without S2B bound to the cluster. Energies in black and red indicate that BS10‐147 and BS7‐235 states are most stable, respectively. Fe7(5) differs from Fe7 in that the proton points towards S5A.
Next, we added an electron and a proton to the H2NNH2 structure, to study possible proton transfers for the E7 state. As for the E5 and E6 states, protonation of S5A was 67 and 83 kJ mol−1 more stable than the S3B and S4B states. Figure 4 shows that the proton on S5A can be transferred to Fe7 and, after some reorientations, to Fe6 and finally to H2NNH2. However, the barriers for latter step is prohibitive, 153 or 213 kJ mol−1 relative to S5A(3) and the resulting H2NNH3 states (bound to Fe2 or Fe6) are 135 or 166 kJ mol−1 less stable than the starting S5A(3) state. However, if the N−N bond is cleaved, a state with NH2 bridging between Fe2 and Fe6 and with NH3 coordinated to Fe6 is 188 kJ mol−1 more stable than the best starting state. Yet, to reach that state, barriers of 182–222 kJ mol−1 need to be passed. The B3LYP method also gives high barriers, at least 215 kJ mol−1.
Figure 4.

Reaction and activation energies, as well as structures for proton transfers in the E7 state without S2B bound to the cluster. Energies in black and red indicate that BS10‐147 and BS7‐235 states are most stable, respectively.
Finally, we studied proton transfers within the E8 state, starting from NH2 bridging Fe2 and Fe6 (i.e. after dissociation of one NH3 molecule). As usual, protonation of S5A(3) was 72 and 60 kJ mol−1 more favourable than protonation of S4B and S3B. The results in Figure 5 show that the proton on S5A can be transferred to Fe7 and, after some rotations, further to Fe6, before it reaches NH2, forming the NH3 product. The individual barriers are rather small, 3–51 kJ mol−1, but most of the steps are upwards, leading to a net barrier of 77 kJ mol−1 for the transfer of the proton from Fe7 to Fe6. An alternative path involving S3B has a net barrier of 102 kJ mol−1. At the B3LYP level, the net barriers are higher, at least 128 kJ mol−1, because Fe‐bound hydride ions are strongly disfavoured, compared to sulfur‐bound protons.
Figure 5.

Reaction and activation energies, as well as structures for proton transfers in the E8 state without S2B bound to the cluster. Energies in black and red indicate that BS10‐147 and BS7‐235 states are most stable, respectively.
We have also performed similar calculations for structures with S2B remaining bound to the FeMo cluster. In that case, the binding site of the substrate is less clear. In our previous studies, we found that binding to Fe2 or Fe6 is thermodynamically most favourable for the E4 state,[ 20 , 40 ] in agreement with mutation studies. [12] For the E5 state, binding of H2NNH2 to Fe6 is at least 54 kJ mol−1 more stable than any state binding to Fe2. [39] When binding to Fe6, the substrate can form hydrogen bonds to His‐195, Gln‐191 and the homocitrate ligand. Moreover, homocitrate may constitute a proton buffer, by donating a proton to the substrate, thereby stabilising certain protonation states, in particular H2NNH2 and NH3. Consequently, we considered only states with the substrate bound to Fe6.
First, we studied the E5 state. We started from the Fe6‐HNNH2 structure, which was our most favourable E4 structure [39] and aimed for the Fe6‐H2NNH2 structure, which was the most stable E5 structure. In both cases, the substrate has abstracted a proton from homocitrate. The suggested proton‐transfer path is shown in Figure 6. As usual, protonation of S5A is more favourable than protonation of S4B or S3B by 70 and 32 kJ mol−1, respectively.
Figure 6.

Reaction and activation energies, as well as structures for proton transfers in the E5 state with S2B remaining bound to the cluster.
The proton on S5A or S4B can be transferred to Fe7 (with barriers of 2 or 43 kJ mol−1), then to S3B (35 kJ mol−1 barrier) and after a change in conformation (28 kJ mol−1 barrier), the proton can be transferred to the substrate (30 kJ mol−1 barrier). The resulting H2NNH2 intermediate is 123 kJ mol−1 more stable than the starting S5A(3) structure. Figure 6 also shows some alternative paths, involving Fe6 and other product states. The highest barrier along the entire proton‐transfer reaction path is 83 kJ mol−1 (for the rotation of the proton at S3B). This is slightly higher than the turnover rate of the enzyme. However, considering the approximations involved in the calculations (approximate transition states, no entropies, no tunnelling) and the known DFT‐functional sensitivity of energetic result for nitrogenase, [30] the result is acceptable.
Next, we studied the transfer of a proton in the E6 state. Since the most stable intermediate of both the E5 and E6 states have H2NNH2 bound to Fe6, we expected the proton to end up on the alcohol oxygen of homocitrate. The results are shown in Figure 7. As usual, protonation of S5A was more favourable than protonation of S4B or S3B (by 59 and 43 kJ mol−1, respectively). From S5A or S4B, the proton can be transferred to Fe7 (barriers of 12 or 52 kJ mol−1), then to S3B (34 kJ mol−1) and it many then change its conformation to point towards Fe6 (26 kJ mol−1 barrier).
Figure 7.

Reaction and activation energies, as well as structures for proton transfers in the E6 state with S2B remaining bound to the cluster, from S5A, S4B or S3B to the H2NNH2 intermediate bound to Fe6, leading to H2NNH2 or NH2 product. In the H2NNH2 structure, the extra proton has been transferred to homocitrate.
From this conformation, there are several possible paths. First, it can be transferred directly to homocitrate, passing a barrier of 56 kJ mol−1 and giving the most stable product, which is 54 kJ mol−1 more stable than the starting S5A(3) state. However, the maximum net barrier (for the last proton transfer) is prohibitively high, 98 kJ mol−1. Second, the proton can be transferred to the substrate, giving H2NNH3 with the homocitrate still deprotonated. It has a lower barrier of 36 kJ mol−1, but the product is 18 kJ mol−1 less stable than the H2NNH2 isomer. Third, the proton may be transferred to Fe6 with a barrier of only 4 kJ mol−1 and then to S2B with a barrier of 7 kJ mol−1. Finally it can be transferred to the substrate (again forming H2NNH3) with a barrier of 71 kJ mol−1. Thus, this third pathway has a slightly lower barrier than the second path, and both are dominated by the net barrier of forming S3B(6) from S3B(7), which is 84 kJ mol−1 above S5A(3). However, Fe6 can also be reached via Fe7 with a slightly lower net barrier of 78 kJ mol−1.
Interestingly, the N−N bond in H2NNH3 can be cleaved with a barrier of 65 kJ mol−1, forming a product with NH3 dissociated from the FeMo cluster and NH2 coordinated to Fe6 (and homocitrate still deprotonated). This state is only 4 kJ mol−1 less stable than the H2NNH2 isomer (with homocitrate protonated). According to the activation barriers, this is actually the preferred path for the E6 state, since the barrier forming the H2NNH2 product is 14 kJ mol−1 higher than that forming the NH3+NH2 product. Thus, we can conclude that this reaction is possible with a maximum barrier of 78 kJ mol−1.
Next, we considered proton‐transfer reactions for the E7 state. We started from two different structures, viz. with either H2NNH2 or NH2 bound to Fe6. According to the previous paragraph, the latter should be the proper starting state, but since hydrazine (H2NNH2 is a known substrate of the enzyme, the former reaction should also be possible and was therefore also tested.
Possible proton‐transfer reactions from the sulfide ions to H2NNH2 are shown in Figure 8. Protonation of S5A is 57 and 29 kJ mol−1 more stable than protonation on S4B and S3B. The proton on S5A or S4B can first be transferred to Fe7 with barriers of 16 or 38 kJ mol−1. The proton can then be transferred to S3B with a barrier of 60 kJ mol−1. This proton can rotate to point towards Fe6 with a barrier of only 11 kJ mol−1, leading to a stabilisation by 46 kJ mol−1. Then, the proton can be transferred successively to Fe6, S2B and finally to the substrate, passing barriers of 20, 24 and 31 kJ mol−1, with reaction energies of −5, −24 and −19 kJ mol−1. The product is the H2NNH3 intermediate, but it does not directly coordinate to the cluster. Therefore, the barrier for the cleavage of the N−N is rather high, 91 kJ mol−1. The net barrier of the reaction (for the isomerisation of the S3B‐protonated state) is 86 kJ mol−1. However, there is also an alternative path, involving another conformation of the proton on Fe7 (Fe7(2), i.e. with the proton pointing towards Fe2) and with no proton transfer to S3B, giving a lower net barrier of 74 kJ mol−1 (also shown in Figure 8).
Figure 8.

Reaction and activation energies, as well as structures for proton transfers in the E7 state with S2B remaining bound to the cluster, from S5A, S4B or S3B to the H2NNH2 intermediate bound to Fe6, leading to a NH2 product.
Then, we studied the protonation of NH2 to NH3 in the E7 state (in this case, the substrate has abstracted the hydroxyl proton from homocitrate). The results in Figure 9 show that protonation of S5A is 68 and 34 kJ mol−1 more favourable than protonation of S4B and S3B. The proton can be transferred to Fe7 with a barrier of 57 kJ mol−1. After a rotation of the proton (15 kJ mol−1 barrier), it can be transferred to S3B with a barrier of 28 kJ mol−1. After another rotation (with a barrier of 18 kJ mol−1), the proton can be transferred to NH2, passing a barrier of only 6 kJ mol−1. This step is strongly exothermic (229 kJ mol−1). The net barrier of the full reaction (compared to the state protonated on S5A) is 73 kJ mol−1, for the rotation of the proton on S3B.
Figure 9.

Reaction and activation energies, as well as structures for proton transfers in the E7 state with S2B remaining bound to the cluster, from S5A, S4B or S3B to the NH2 intermediate bound to Fe6, leading to a NH3 product.
Finally, we studied the transfer of a proton from the sulfide ions to NH3 in the E8 state (again, with a proton abstracted from homocitrate). The results in Figure 10 show that it is 51 or 47 kJ mol−1 more favourable to have the proton on S5A than on S4B or S3B. The proton can be transferred to Fe7 with a barrier of 2–47 kJ mol−1. After a rotation (29 kJ mol−1 barrier), it can be transferred to Fe6 (34 kJ mol−1 barrier) and then to S2B (with a barrier of 11 kJ mol−1) before it can move to NH3 with a barrier of 49 kJ mol−1. The product is NH4 +, which dissociates from Fe6. The highest effective barrier of the reaction (relative to protonated S5A) is 69 kJ mol−1 for the transfer of the proton from Fe7(2) to Fe6. There are several other possible paths with slightly higher barriers shown in Figure 10.
Figure 10.

Reaction and activation energies, as well as structures for proton transfers in the E8 state with S2B remaining bound to the cluster. In the S3B(HCA) structure, the extra proton points towards homocitrate.
Conclusion
In this investigation, we have studied proton transfers within the FeMo cluster, assuming that the proton enters on either S3B, S4B or S5A, and is then transported to the substrate via the sulfide and Fe ions. Interestingly, we find that the net barriers for the proton transfers are in general higher when S2B has dissociated from the cluster than if S2B remains bound. In fact, in the former case, the maximum barriers are prohibitively large (107–213 kJ mol−1) for the E5–E7 levels. When S2B remains bound, the maximum barriers are lower, 69–83 kJ mol−1. We have checked that the barriers cannot be lowered by relaxing the surrounding of the QM system or by using another DFT method. Thus, our results provide a strong argument against the dissociation of S2B.
For all E n levels, protonation of S5A is always 29–98 kJ mol−1 more favourable than protonation of S4B and S3B. States with Fe7 and Fe2 are also 16–74 kJ mol−1 less stable. This shows that even if the proton initially is delivered to S3B, as Dance suggested, it would rapidly be transferred to S5A, which is thermodynamically more stable and the barriers for such a transfer is typically not higher than those involving a transfer towards the substrate.
However, a problem with the stable S5A protonation is that it becomes a thermodynamic sink for the reactions, increasing the effective barriers for the proton‐transfer reactions. The individual barriers for the proton‐transfer and proton‐rotation reactions are typically 6–67 kJ mol−1 for the seven paths in Figures 2–10. The highest individual barrier is typically observed for the first step of the path (i.e. moving the proton from S5A to Fe7) or the last step (moving the proton to the substrate). This corresponds to rates that are faster than the net reaction rate of nitrogenase. However, if the barriers are compared to the S5A state, the maximum effective barriers increase to 69–83 kJ mol−1 (with S2B bound), which are higher than the experimental reaction rate. The reason for this may be that proton tunnelling has not been considered, that the transition states are approximate, that no entropy and thermal effects are considered and that TPSS may not give accurate results for such reactions. However, an alternative explanation may be that S5A actually is always protonated throughout the reaction mechanism of nitrogenase. We have tested such reactions for one state and found that the barriers are indeed reduced by 16 kJ mol−1 (discussed in the Supporting Information).
Dance also studied proton‐transfer reactions within the FeMo cluster.[ 33 , 35 , 36 , 37 ] However, he never studied transfers to or from S5A and therefore did not observe its high stability. Consequently, he underestimated the barriers. S3B is in the middle of our reaction mechanisms, 29–69 kJ mol−1 above the S5A(3) state. The barriers backwards to S5A are always lower than those forwards towards the substrate.
When S2B is bound, the energies of the various intermediates and transition state vary by only 4–20 kJ mol−1 between the E5 to E8 states (cf. Table S3), except for the last step. Two competing pathways are observed, either via S3B(7) and S3B(6) or via Fe7(2). Most pathways involve the transfer of the proton from S2B to the substrate.
When S2B has dissociated, the variation in the energies is larger and the barriers are higher. The same two pathways are observed but the proton needs to be transferred directly from Fe6 to the substrate (because S2B has dissociated). Figure 11 compares the energies with and without S2B. They are similar in the early steps but differ at the end. When S2B is present, it is normally used for the final transfer of the proton to the substrate. When it is not present, the final proton transfer to the substrate is often problematic, in three cases leading to prohibitively large barriers. This explains why the proton transfers are significantly higher if S2B has dissociated than if S2B is still bound to the cluster.
Figure 11.

Relative energies for the proton‐transfer reactions at the various E n states with and without S2B bound. As shown in Figures 2–10, there are often multiple possible paths, but this figure shows only the most favourable ones (those with the lowest net barriers). Alternative paths, inspired by the results for the other E n states were also tested, but sometimes failed owing to subtle differences in the structures.
We have tried to gain further understanding how the surrounding protein affects the proton transfers by dividing the total QM/MM energy into components from MM and from the point‐charge model, indicating the importance of steric and electrostatic effects from the surrounding protein and solvent, outside the QM system. The MM energy correction is quite small (−11 to 15 kJ mol−1), with a distribution that is only slightly biased to positive values (average 2 kJ mol−1). It often increases slightly as the proton approaches the substrate.
On the other hand, the point‐charge model has a quite large influence on the relative reaction energies, by −18 to 84 kJ mol−1. It is positive for most intermediates and transition states (average 28 kJ mol−1), indicating that protonation of S5A is more favoured by the electrostatics of the surrounding than the other protonation states.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Acknowledgements
This investigation has been supported by grants from the Swedish research council (project 2018‐05003) and from China Scholarship Council. The computations were performed on computer resources provided by the Swedish National Infrastructure for Computing (SNIC) at Lunarc at Lund University, NSC at Linköping University and HPC2N at Umeå University, partially funded by the Swedish Research Council (grant 2018‐05973).
H. Jiang, O. K. G. Svensson, L. Cao, U. Ryde, Angew. Chem. Int. Ed. 2022, 61, e202208544; Angew. Chem. 2022, 134, e202208544.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1. Kim J., Rees D. C., Science 1992, 257, 1677–1682. [DOI] [PubMed] [Google Scholar]
- 2. Einsle O., Tezcan F. A., Andrade S. L. A., Schmid B., Yoshida M., Howard J. B., Rees D. C., Science 2002, 297, 1696. [DOI] [PubMed] [Google Scholar]
- 3. Spatzal T., Aksoyoglu M., Zhang L., Andrade S. L. A., Schleicher E., Weber S., Rees D. C., Einsle O., Science 2011, 334, 940–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Spatzal T., Perez K. A., Einsle O., Howard J. B., Rees D. C., Science 2014, 345, 1620–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Einsle O., J. Biol. Inorg. Chem. 2014, 19, 737–745. [DOI] [PubMed] [Google Scholar]
- 6. Eady R. R., Chem. Rev. 1996, 96, 3013–3030. [DOI] [PubMed] [Google Scholar]
- 7. Thorneley R. N. F., Lowe D. J. in Molybdenum Enzym (Ed.: Spiro T. G.), Wiley, New York, 1985, pp. 221–284. [Google Scholar]
- 8. Lancaster K. M., Roemelt M., Ettenhuber P., Hu Y., Ribbe M. W., Neese F., Bergmann U., DeBeer S., Science 2011, 334, 974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bjornsson R., Lima F. A., Spatzal T., Weyhermüller T., Glatzel P., Bill E., Einsle O., Neese F., DeBeer S., Chem. Sci. 2014, 5, 3096–3103. [Google Scholar]
- 10. Burgess B. K., Lowe D. J., Chem. Rev. 1996, 96, 2983–3012. [DOI] [PubMed] [Google Scholar]
- 11. Schmid B., Chiu H.-J., Ramakrishnan V., Howard J. B., Rees D. C., Handbook of Metalloproteins, Wiley, Hoboken, 2006, pp. 1025–1036. [Google Scholar]
- 12. Hoffman B. M., Lukoyanov D., Yang Z.-Y., Dean D. R., Seefeldt L. C., Chem. Rev. 2014, 114, 4041–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Igarashi R. Y., Laryukhin M., Dos Santos P. C., Lee H.-I., Dean D. R., Seefeldt L. C., Hoffman B. M., J. Am. Chem. Soc. 2005, 127, 6231–6241. [DOI] [PubMed] [Google Scholar]
- 14. Hoeke V., Tociu L., Case D. A., Seefeldt L. C., Raugei S., Hoffman B. M., J. Am. Chem. Soc. 2019, 141, 11984–11996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roth L. E., Tezcan F. A., Methods Mol. Biol. 2011, 766, 147–164. [DOI] [PubMed] [Google Scholar]
- 16. Van Stappen C., Decamps L., Cutsail G. E., Bjornsson R., Henthorn J. T., Birrell J. A., DeBeer S., Chem. Rev. 2020, 120, 5005–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lukoyanov D., Khadka N., Yang Z.-Y., Dean D. R., Seefeldt L. C., Hoffman B. M., J. Am. Chem. Soc. 2016, 138, 10674–10683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sippel D., Einsle O., Nat. Chem. Biol. 2017, 13, 956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Varley J. B., Wang Y., Chan K., Studt F., Nørskov J. K., Phys. Chem. Chem. Phys. 2015, 17, 29541–29547. [DOI] [PubMed] [Google Scholar]
- 20. Cao L., Ryde U., J. Catal. 2020, 391, 247–259. [Google Scholar]
- 21. Kang W., Lee C. C., Jasniewski A. J., Ribbe M. W., Hu Y., Science 2020, 368, 1381–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bergmann J., Oksanen E., Ryde U., J. Biol. Inorg. Chem. 2021, 26, 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peters J. W., Einsle O., Dean D. R., DeBeer S., Hoffman B. M., Holland P. L., Seefeldt L. C., Science 2021, 371, eabe5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dance I., ChemBioChem 2020, 21, 1671–1709. [DOI] [PubMed] [Google Scholar]
- 25. Rohde M., Sippel D., Trncik C., Andrade S. L. A., Einsle O., Biochemistry 2018, 57, 5497–5504. [DOI] [PubMed] [Google Scholar]
- 26. Seefeldt L. C., Yang Z.-Y., Lukoyanov D. A., Harris D. F., Dean D. R., Raugei S., Hoffman B. M., Chem. Rev. 2020, 120, 5082–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Siegbahn P. E. M., J. Am. Chem. Soc. 2016, 138, 10485–10495. [DOI] [PubMed] [Google Scholar]
- 28. Siegbahn P. E. M., J. Comput. Chem. 2018, 39, 743–747. [DOI] [PubMed] [Google Scholar]
- 29. Cao L., Caldararu O., Ryde U., J. Phys. Chem. B 2017, 121, 8242–8262. [DOI] [PubMed] [Google Scholar]
- 30. Cao L., Ryde U., Phys. Chem. Chem. Phys. 2019, 21, 2480–2488. [DOI] [PubMed] [Google Scholar]
- 31. Cao L., Ryde U., J. Chem. Theory Comput. 2020, 16, 1936–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Siegbahn P. E. M., Phys. Chem. Chem. Phys. 2019, 21, 15747–15759. [DOI] [PubMed] [Google Scholar]
- 33. Dance I., J. Am. Chem. Soc. 2005, 127, 10925–10942. [DOI] [PubMed] [Google Scholar]
- 34. Dance I., J. Inorg. Biochem. 2017, 169, 32–43. [DOI] [PubMed] [Google Scholar]
- 35. Dance I., Dalton Trans. 2012, 41, 7647–7659. [DOI] [PubMed] [Google Scholar]
- 36. Dance I., Inorg. Chem. 2013, 52, 13068–13077. [DOI] [PubMed] [Google Scholar]
- 37. Dance I., Dalton Trans. 2015, 44, 18167–18186. [DOI] [PubMed] [Google Scholar]
- 38. Dance I., Biochemistry 2006, 45, 6328–6340. [DOI] [PubMed] [Google Scholar]
- 39. Jiang H., Ryde U., Chem. Eur. J. 2022, 28, e202103933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cao L., Ryde U., J. Biol. Inorg. Chem. 2020, 25, 521–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.