

Abstract
Safe and effective use of drugs requires an understanding of metabolism and transport. We identified the 100 most prescribed drugs in six countries and conducted a literature search on in vitro data to assess contribution of Phase I and II enzymes and drug transporters to metabolism and transport.
Eighty‐nine of the 100 drugs undergo drug metabolism or are known substrates for drug transporters. Phase I enzymes are involved in metabolism of 67 drugs, while Phase II enzymes mediate metabolism of 18 drugs. CYP3A4/5 is the most important Phase I enzyme involved in metabolism of 43 drugs followed by CYP2D6 (23 drugs), CYP2C9 (23 drugs), CYP2C19 (22 drugs), CYP1A2 (14 drugs) and CYP2C8 (11 drugs). More than half of the drugs (54 drugs) are known substrates for drug transporters. P‐glycoprotein (P‐gp) is known to be involved in transport of 30 drugs, while breast cancer resistance protein (BCRP) facilitates transport of 11 drugs. A considerable proportion of drugs are subject to a combination of Phase I metabolism, Phase II metabolism and/or drug transport.
We conclude that the majority of the most frequently prescribed drugs depend on drug metabolism or drug transport. Thus, understanding variability of drug metabolism and transport remains a priority.
Keywords: ADME, CYP3A4, drug metabolism, drug transport, P‐gp
1. INTRODUCTION
The use of medicines is increasing worldwide. 1 A Danish study found that 51% of individuals ≥75 years are prescribed five or more different medications. 2 Enzymatic drug metabolism and drug transport are important for absorption, disposition, metabolism and elimination of a drug within the body. 3 Drug metabolism and drug transport are known to vary both between and within individuals, and this is a problem as it causes variable treatment efficacy and toxicity. Variation in drug metabolism and drug transport may be caused by drug–drug interactions, 3 epigenetics, 4 genetic polymorphisms in genes that encode drug‐metabolizing enzymes or drug transporters [e.g. cytochrome P450 (CYP)2D6, CYP2C9 and CYP2C19], 5 or intrinsic (e.g. sex and inflammation) and extrinsic factors (e.g. diet and chemical exposure from the environment). 6
A previous literature study described the Phase I enzymes involved in drug metabolism of the 200 most prescribed drugs in the United States. 7 In this paper, we provide an updated mapping of Phase I metabolism but also include Phase II metabolism and drug transporters relevant for the 100 most prescribed drugs in five European countries and Australia. We included data from six countries to make the list more international. Furthermore, we summarized the total enzyme and transporter contribution to understand the number of drugs that will be affected because of variation in metabolism or drug transport. Finally, we reviewed main aspects from the literature of the most important Phase I and II enzymes and drug transporters.
2. METHODS
We identified the 100 most prescribed drugs in five European countries and Australia and performed a literature search for each drug to understand its metabolism and transport.
2.1. Identifying the Top 100 most prescribed drugs
We obtained data on prescription drug use from six countries: Denmark, Sweden, Norway, England, Scotland and Australia. Data were available upon specific requests to collaborators in the six countries. For each country, we identified the most prescribed drugs. The ranking of drugs, on the individual lists, was based on different measures. Data from Denmark and Sweden were based on the number of prescriptions per 1000 citizens, data from Norway were based on the number of unique individuals buying a drug, data from England and Scotland were based on the number of dispensed items, and data from Australia were based on the number of prescriptions. Medications that contain two active substances were split into their individual components; for example, the drug combination of codeine and paracetamol was counted as the use of both drugs individually. We only included orally administered drugs for systemic treatment, thus excluding inhalation preparations, intravenous preparations, dermatological preparations, drugs with no absorption from the intestine and dietary supplements. Only prescription data from pharmacies were available, thus excluding prescription data from hospitals. As the Norwegian data contained the lowest number of individual medications (n = 179), we restricted all the remaining datasets to the 179 most used medications as well. For each country, we calculated the percentage the individual 179 drugs on the list constituted. Based on these percentages, we calculated the average ranking across all six countries with the following equation, where X was the percentage of a specific drug in the six countries:
A drug was assigned X = 0 if it was absent from the individual list of a country. Calculation of the Top 100 drug list was carried out using the Stata Statistical Software: Release 16 (StataCorp College Station, TX: StataCorp LLC, USA).
From the final list, we chose the 100 most prescribed drugs and conducted a literature search.
2.2. Search strategy
Two authors (DBI and NA) performed a literature search from January to October 2021 using the database PubMed (Medline). We performed separate literature searches for drug‐metabolizing enzymes and drug transporters.
The following search strings were applied for the individual drugs:
Drug metabolism: (drug name AND (hepatic OR Liver OR intestinal OR CYP OR cytochrome P450) AND (pharmacokinetic OR ADME OR metabolism OR metabolized) AND in vitro).
Drug transporter: (drug name AND substrate AND (transporter OR transport) AND (elimination OR excretion OR excreted OR efflux OR uptake)).
The search term ‘drug name’ was defined as the drug name specified on the Top 100 list. Synonyms for the drug name were automatically included by PubMed, for example, a search for paracetamol also included acetaminophen. Additionally, we identified and included relevant references from articles identified via the search string. We included articles regardless of publication year but strived to use the most recently published.
Furthermore, we conducted a literature search for each drug to determine if the drug was categorized as a prodrug. A prodrug was defined as an inactive substance that needs to be converted to a pharmacologically active substance through metabolism or physico‐chemical processes. 8
2.3. Study selection for Top 100 most prescribed drugs
We created three inclusion criteria before conducting the literature search. The inclusion criteria were that articles included in the analysis had to be specific for the drug in question and had to report (i) original data, (ii) human in vitro studies and (iii) data of Phase I or II metabolism or drug transport.
Documents from Food and Drug Administration (FDA) or European Medicines Agency (EMA) [e.g. Summary of Product Characteristics (SmPCs) or Highlights of Prescribing Information] were included in the analysis if they were found as references to articles in the literature search. Two authors (DBI and NA) were each responsible for the search of 50 drugs from the Top 100 list and independently screened relevant articles found in the literature search. We did not screen all articles obtained in the literature search, but only until the metabolism and transport of a drug was confirmed through an acceptable article that was deemed valid by meeting the inclusion criteria. After completion of the literature search, one author (TBS) double‐checked the results to uncover any mistakes or errors concerning the matching of drugs with enzymes and drug transporters known to be involved in drug metabolism or drug transport.
2.4. Data analysis
For the drug in question, we flagged an enzyme or transporter as major if one of two conditions were achieved, (i) if the article from the literature search stated that the enzyme or transporter were responsible for more than 50% of the fraction metabolized or transported or (ii) if an original paper defined an enzyme or transporter as ‘major’. We did not define a lower cut‐off to categorize enzymes or transporters as minor based on contribution to drug metabolism or drug transport. Furthermore, the status of the drug (prodrug or not) was recorded. We divided Phase I enzymes into subcategories of isoforms, for example, CYP2C9 and CYP2D6, and calculated the number of the 100 drugs metabolized by each isoform. All isoforms from the Top 100 list were included equally in the calculation regardless of if they were flagged as major enzyme or transporter. Isoforms metabolizing ≤5 of the 100 drugs were categorized as ‘Others’. CYP3A4 and CYP3A5 isoforms were grouped as one isoform, CYP3A4/5, as they share 80% structural similarity and have overlapping substrate specificity making it difficult to distinguish the isoforms. 9
For Phase II enzymes, we only divided the uridine 5′‐diphospho‐glucuronosyltransferases (UGTs) into isoforms, for example, UGT2B7 and UGT1A9, as UGTs are the largest Phase II superfamily. 10 The remaining Phase II enzymes were grouped to the superfamily sulfotransferases (SULT). For drug transporters, we described P‐glycoprotein (P‐gp), breast cancer resistance protein (BCRP) and organic anion transporting polypeptide (OATP) in isoforms and grouped organic anion transporter (OAT), organic cation transporter (OCT), multidrug resistance‐associated protein 1–4 (MRP1–4) and multidrug and toxic compound extrusion (MATE) into superfamilies. Isoforms and superfamilies, of both Phase II enzymes and drug transporters, were grouped as ‘Others’ if they metabolized or transported ≤2 of the 100 drugs.
For both Phase I and Phase II enzymes and drug transporters, we calculated the substrate overlap within each group (Phase I, Phase II and drug transporters). We grouped substrate overlap in the following groups for Phase I enzymes: 1–5 drugs, 6–10 drugs and more than 10 drugs. For Phase II enzymes and drug transporters, we changed the range of substrate overlap to better reflect substrate overlap within these groups. We grouped substrate overlap as 1–2 drugs, 3–4 drugs and more than 4 drugs. We also calculated the substrate overlap between the drug transporters (P‐gp, BCRP and OATP1B1/3) and four CYP enzymes (CYP3A4/5, CYP2D6, CYP2C9 and CYP2C19). We grouped substrate overlap between drug transporters and CYP enzymes as 1–5 drugs, 6–10 drugs and more than 10 drugs. Data visualization was carried out using the statistical software RStudio Team (2021) (RStudio: Integrated Development Environment for R. RStudio, PBC, Boston, MA; http://www.rstudio.com/).
3. RESULTS
Of the 100 most prescribed drugs (Table S1), 89 drugs are known to be subject to drug metabolism or drug transport, while the remaining 11 drugs are not metabolized or known to be substrates for drug transporters. Phase I enzymes metabolize 67 drugs, 18 drugs are metabolized by Phase II enzymes, and 54 drugs are known to be transported by drug transporters (Figure 1). A total of 27 drugs are known to be substrates for both Phase I metabolism and drug transport, while only seven drugs are known to be substrates for Phase I and Phase II metabolism and drug transport (Figure 1). We included a supplementary table (Table S2) with a ranked list of the 179 drugs from each country combined with the percentage each drug constituted from the list.
FIGURE 1.
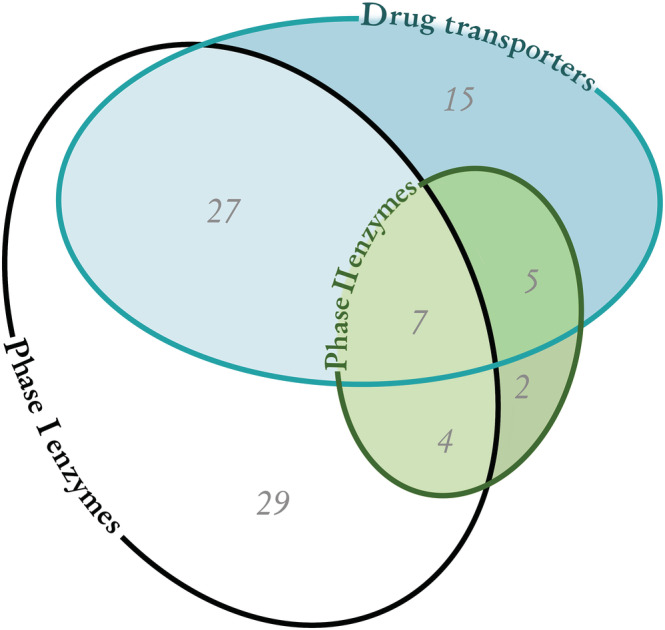
Number of the 100 most prescribed drugs metabolized by Phase I enzymes or Phase II enzymes or transported by drug transporters. Overlapping areas reflect a combination of pathways. Of the 100 most prescribed drugs, 11 drugs are not metabolized or transported.
3.1. Phase I metabolism
CYP3A4/5 is the most important Phase I enzyme as it is involved in the metabolism of 43 of the 100 drugs, followed by CYP2D6 (23 drugs), CYP2C9 (23 drugs), CYP2C19 (22 drugs), CYP1A2 (14 drugs) and CYP2C8 (11 drugs) (Figure 2).
FIGURE 2.
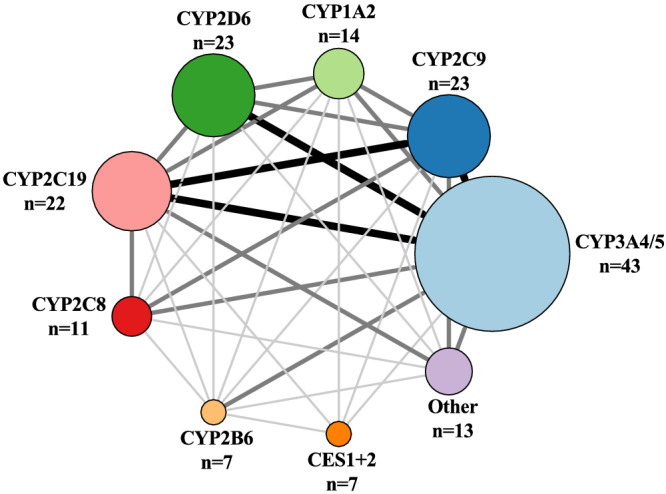
Number of drugs metabolized by Phase I enzymes is illustrated with increasing circle size. The size and darkness of the lines between enzymes illustrate the substrate overlap between enzymes. Thin/light‐grey line corresponds to 1–5 drugs as substrate overlap. Medium/grey line corresponds to 6–10 drugs as substrate overlap. Thick/black line corresponds to >10 drugs as substrate overlap. Overlap within a group is not illustrated in the figure, but we refer to Table S1 for further information. The group Other includes CYP1A1, CYP2E1, CP2C18, CYP2J2, CYP3A3, CYP3A7, CYP2A6, CYP1B1, AO, FMO and MAO‐A + B. AO, aldehyde oxidase; CES, carboxylesterase; CYP, cytochrome P450 enzyme; FMO, flavin‐containing monooxygenase; MAO, monoamine oxidase.
3.1.1. CYP3A4/5
We find that CYP3A4/5 metabolizes 43 of the 100 drugs and has substrate overlap with three CYP isoforms, CYP2D6, CYP2C9, and CYP2C19 (>10 drugs for all enzymes; Figure 2).
CYP3A4/5 is predominantly expressed in the liver and intestine and is the most dominant drug‐metabolizing enzyme in the body. 11 , 12 Top 3 drugs from our list with CYP3A4/5 as the major enzyme include bisoprolol, ethinylestradiol and zopiclone.
CYP3A4/5 is transcriptionally regulated by pregnane X receptor (PXR) and constitutive androstane receptor (CAR). PXR and CAR are members of the nuclear receptor (NR) superfamily, and PXR is one of the most important receptors in the regulation of metabolism and drug transport. Flucloxacillin (number 61 of the 100 drugs), dicloxacillin (number 77) and rifampicin (not on the list) are examples of PXR agonists that cause upregulation of CYP3A4 by PXR. 13 , 14 Contrary, ciprofloxacin (number 92) is an example of a moderate CYP3A4 inhibitor. 15 The time it takes for induction or inhibition to occur and the time it takes for an enzyme to recover from this is important when optimizing pharmacotherapy. One clinical trial investigated the recovery time for CYP3A4 after use of rifampicin for 7 days and discovered that 8 days was needed to recover from rifampicin‐mediated induction. 16 Another clinical trial investigated CYP3A4 induction after 28 days of rifampicin treatment. Discontinuation for 28 days was needed to completely recover from rifampicin‐mediated induction. 17 Duration of competitive inhibition depends on the elimination half‐life of the inhibitor, whereas duration of non‐competitive inhibition and induction depends on different biological factors such as de‐induction of PXR‐driven transcription (e.g. for CYP3A4), degradation of induced mRNAs and their encoded proteins in the liver and gut, CYP protein synthesis and cell turnover. 17
Food, drinks, herbal drugs and inflammation can also regulate the expression of CYP3A4/5 and lead to drug concentrations out of the therapeutic range. Both Seville orange and grapefruit inhibit CYP3A4. 18 St. John's wort, a herbal drug used against depression, is a potent ligand to PXR and an inducer of CYP3A4. 19 CYP3A4/5 can also be downregulated by proinflammatory cytokines during inflammation. 20
Despite multiple studies on genetic polymorphism in CYP3A4, there is currently no evidence of common and clinically relevant polymorphisms in CYP3A4. Individuals with CYP3A5 genetic polymorphism can be characterized as CYP3A5 expressors or as CYP3A5 non‐expressors. CYP3A5 non‐expressors make up 80%–85% of Caucasians, leading CYP3A5 expressors to be the minority in Europe. 21 This is in contrast to the Asian and African American population where 60%–73% and 32% are CYP3A5 non‐expressors, respectively. 22 The Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for treatment with tacrolimus (CYP3A4/5 substrate) (not on the list) recommends a standard starting dose for CYP3A5 non‐expressor and a higher starting dose for CYP3A5 expressors. 21
3.1.2. CYP2D6
Our findings show that CYP2D6 metabolizes 23 of the 100 drugs and has the largest substrate overlap with CYP3A4/5 (10 drugs; Figure 2). CYP2D6 is mainly expressed in the liver. 11 , 12 It is the second most important CYP enzyme in our review, and it accounts for 5% of the total CYP protein content in the human liver. 23 Top 3 drugs from our list with CYP2D6 as the major enzyme include metoprolol, venlafaxine and fluoxetine.
CYP2D6 is not susceptible to enzyme induction by drugs, 24 but CYP2D6 expression is increased during pregnancy. 25 In a clinical trial, the plasma metabolic ratio of the CYP2D6 probe dextromethorphan (not on the list) decreased by 53% during pregnancy compared to after pregnancy. 25 , 26 The antidepressants sertraline (number 19) and fluoxetine (number 52) are inhibitors of CYP2D6. 27 Inhibition lasts 5 days for sertraline and 42 days for fluoxetine after discontinuation. 27 Clinicians should be cautious when prescribing CYP2D6 substrates following initiation and discontinuation of these antidepressants. 27
Polymorphism in CYP enzymes plays a critical role, especially in CYP2D6. A meta‐analysis showed that the efficacy of metoprolol (number 16) is higher in poor metabolizers compared to non‐poor metabolizers. 28 CPIC guideline recommends alternative analgesics to the two prodrugs, codeine (number 7) and tramadol (number 21), in poor metabolizers or ultrarapid metabolizers, 29 as they are more likely to experience poor pain relief or more adverse effects compared to intermediate metabolizers and extensive metabolizers. 30 The nuclear factor 1B (NFIB) was recently discovered to regulate CYP2D6 gene expression in vitro. 31 The same study showed that NFIB rs28379954 T>C carriers that were also CYP2D6 extensive metabolizers had comparable CYP2D6 activity to ultrarapid metabolizers. This highlights that NFIB polymorphisms are important to consider in CYP2D6 drug metabolism. 31
3.1.3. CYP2C9
We show that CYP2C9 metabolizes 23 of the 100 drugs and has substrate overlap with CYP3A4/5 and CYP2C19 (>10 drugs for each enzyme; Figure 2). CYP2C9 is predominantly expressed in the liver and gastrointestinal tract. 11 , 12 The CYP2C family contains CYP2C8, CYP2C9 and CYP2C19, which in total comprise 33% of the total CYP protein content in the liver, with CYP2C9 accounting for 24%. 23 Top 3 drugs from our list with CYP2C9 as the major enzyme include naproxen, diclofenac and warfarin. CYP2C9 is regulated by three nuclear receptors: PXR, CAR and glucocorticoid receptor (GR). 32
S‐warfarin (number 45) is a CYP2C9 substrate with a narrow therapeutic index. 33 Fluconazole (number 87) inhibits CYP2C9, and an epidemiological study found that coadministration of fluconazole with warfarin leads to an increase in mean international normalized ratio (INR) of 0.83. 34 This clinically relevant increase in INR can result in adverse effects. 34 Dicloxacillin (number 77) is an inducer of CYP2C9 through PXR activation. 13 Two epidemiological studies showed that coadministration of dicloxacillin and warfarin leads to a decrease in mean INR at 0.62 after 2–4 weeks of dicloxacillin exposure 35 and an increased risk of ischaemic stroke and systemic embolism (hazard ratio 2.19). 36
Genetic polymorphisms may affect CYP2C9 activity. CYP2C9*2 and CYP2C9*3 are the most studied genotypes and carriers of either have decreased CYP2C9 activity. The plasma clearance of S‐warfarin is decreased by 56% (CYP2C9*1/3), 70% (CYP2C9*2/3) and 75% (CYP2C9*3/3) compared to wild type (CYP2C9*1/1). 37 Three large randomized clinical trials investigated if genotyping before initiating anticoagulant therapy improves the percentage of time in the therapeutic INR range. The studies reached contradicting conclusions, which complicated implementation of CYP2C9‐guided treatment with warfarin. 38 , 39 , 40 The CPIC guideline for warfarin therapy only recommends dosing based on genotype if it is known before initiating treatment. 41
3.1.4. CYP2C19
CYP2C19 metabolizes 22 of the 100 drugs and has considerable substrate overlap with CYP2C9 and CYP3A4/5 (>10 drugs for each enzyme Figure 2). CYP2C19 is expressed in the liver and gastrointestinal tract. 11 , 12 Top 3 drugs from our list with CYP2C19 as the major enzyme include omeprazole, pantoprazole and esomeprazole. CYP2C19 is regulated by the same three nuclear receptors as CYP2C9 (PXR, CAR and GR). 42 Rifampicin (not on the list) is an inducer of this enzyme through both CAR and PXR, and dexamethasone (number 60) induces CYP2C19 through GR. 42
CYP2C19 polymorphisms have been widely investigated, and some variants might be clinically relevant. Carriers of CYP2C19*17 are characterized as ultrarapid metabolizers, while carriers of CYP2C19*2/3 are characterized as poor metabolizers. 43 Patients that carry CYP2C19*2 who are treated with citalopram (number 30), a substrate of CYP2C19, had lower odds of tolerance to the drug. 44 A clinical study showed that individuals carrying CYP2C19*2 and CYP2C19*3 had lower omeprazole (number 4) metabolism compared to individuals carrying CYP2C9*1. 45 The same study showed that administration of the CYP2C19 inhibitor fluvoxamine (not on the list) reduced omeprazole metabolism in individuals carrying CYP2C19*1, but it had no impact on the metabolism of omeprazole in CYP2C19*2 and CYP2C19*3 carriers. 45 CYP2C19 intermediate and poor metabolizers who receive the prodrug clopidogrel (number 36) experience reduced platelet inhibition and increased risk of major adverse cardiovascular and cerebrovascular events. 46 CPIC guideline recommends considering an alternative to clopidogrel in intermediate metabolizers and to avoid clopidogrel in poor metabolizers. 46
3.1.5. CYP1A2
Our review shows that CYP1A2 metabolizes 14 of the 100 drugs and has substrate overlap with CYP3A4/5, CYP2C9, CYP2C19 and CYP2D6 (6–10 drugs for each enzyme; Figure 2). CYP1A2 is predominantly expressed in the liver. 11 , 12 From our list, CYP1A2 is not categorized as the major enzyme in drug metabolism, but the Top 3 CYP1A2 substrates from our list are naproxen, ethinylestradiol and mirtazapine. CYP1A2 is transcriptionally regulated by aryl hydrocarbon receptor (AhR), which is a ligand‐activated transcription factor. 47
Several drugs induce CYP1A2. 48 Omeprazole (number 4) is an inducer of CYP1A2 in vitro. 49 The most potent CYP1A2 inhibitors are planar molecules with a small volume that easily fit into the active site of CYP1A2. They often contain methyl, chloro or fluoro substitutions, for example, ciprofloxacin (number 92). 50 Ciprofloxacin and oral contraceptives containing ethinylestradiol (number 27) and gestodene were investigated for inhibition of tizanidine metabolism (CYP1A2 substrate) (not on the list). 51 , 52 , 53 Ciprofloxacin is a stronger inhibitor than oral contraceptives; however, care should be taken when tizanidine is administered to oral contraceptive users, as tizanidine has a narrow therapeutic range. 51
The activity of CYP1A2 is subject to individual differences from genetic factors 54 and environmental factors such as smoking. 48 Clozapine (not on the list) is a substrate of CYP1A2 and is an antipsychotic drug where therapeutic drug monitoring is used. 55 A meta‐analysis recommended decreasing the dosage of clozapine by 30% for patients who smoke and suddenly stop smoking and to analyse clozapine blood levels. 55
3.1.6. CYP2C8
We find that CYP2C8 metabolizes 11 of the 100 drugs and has substrate overlap with CYP3A4/5, CYP2C9 and CYP2C19 (6–10 drugs for each enzyme; Figure 2). CYP2C8 is highly expressed in the liver. 11 , 12 From our list, CYP2C8 is not categorized as the major enzyme in the metabolism of the 100 most used drugs, but the Top 3 CYP2C8 substrates from our list are ibuprofen, ethinylestradiol and zopiclone. The transcriptional regulation of CYP2C8 is the same as for CYP2C9 and CYP2C19 (PXR, CAR and GR). 56
Felodipine (number 76) is an example of a potent inhibitor in vitro, 57 and trimethoprim (number 55) is a weak inhibitor of CYP2C8 both in vitro and in vivo. 58 Clopidogrel (number 36) is a CYP2C8 inhibitor via its metabolite, clopidogrel acyl‐β‐D‐glucuronide. 59 A retrospective study showed that patients treated with paclitaxel (CYP2C8 substrate) (not on the list) had a ~2‐fold increased risk of developing neuropathy Grade 2 or higher when co‐treated with clopidogrel. 60 Only a few CYP2C8 inducers have been discovered. Dexamethasone (number 60), a corticosteroid, induces CYP2C8 through binding to GR. Rifampicin (not on the list) induces CYP2C8 and increases the expression of the enzyme through PXR activation. 56
The genetic polymorphism CYP2C8*3 is the most investigated polymorphism in CYP2C8. The allele is common in Caucasians but rare in African and Asian populations. 61 Many studies have investigated this allele, but data are conflicting regarding the effect on metabolism, and the activity of CYP2C8*3 might be substrate dependent. 62 , 63
3.2. Phase II metabolism
The UGT superfamily is the most important Phase II superfamily and is involved in the metabolism of 17 of the most used drugs, followed by sulfotransferase (SULT) that metabolizes three of the 100 drugs (Table S1).
3.2.1. UGTs
UGT superfamily can be divided into four families, UGT1, UGT2, UGT3 and UGT8; however, UGT3 and UGT8 are not significant in drug metabolism. 64 We highlight UGT1A and UGT2B as the most important family members for drug metabolism. We show that UGT2B7 is responsible for metabolism of nine of the 100 drugs (Figure 3). UGT1A3 and UGT1A9 are the second most important Phase II enzymes, and they metabolize eight of the 100 drugs (Figure 3). UGT2B7 and UGT1A9 share the largest substrate overlap with five drugs. UGTs are mainly expressed in the liver and intestine. 64
FIGURE 3.
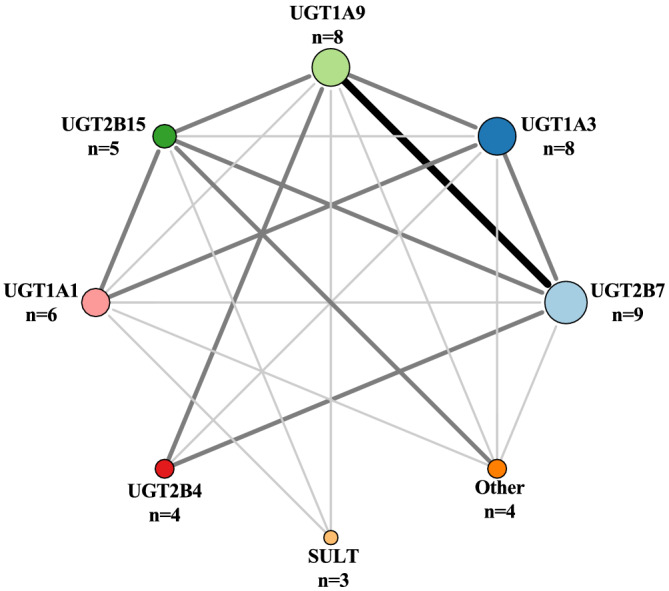
Number of drugs metabolized by Phase II enzymes is illustrated with increasing circle size. The size and darkness of the lines between enzymes illustrate the substrate overlap. Thin/light‐grey line corresponds to 1–2 drugs as substrate overlap. Medium/grey line corresponds to 3–4 drugs as substrate overlap. Thick/black line corresponds to >4 drugs as substrate overlap. Overlap within a group is not illustrated in the figure, but we refer to Table S1 for further information. The group Other contains UGT1A4, UGT1A6 and UGT2B10. SULT, sulfotransferases; UGT, uridine 5′‐diphospho‐glucuronosyltransferases.
UGTs are marked as the major enzyme for two of the top 100 drugs; this includes diclofenac and telmisartan. Different nuclear receptors are involved in regulation of different isoforms in the UGT superfamily. This includes AhR, CAR, PXR, farnesoid X receptor (FXR), liver X receptor (LXR) and peroxisome proliferator‐activated receptor (PPAR). 65 Studies investigating the regulation of UGTs are sparse compared to CYP enzymes. However, some drugs have been identified as inhibitors of UGTs in vitro though few are confirmed in in vivo studies. UGT1A1 is involved in glucuronidation of bilirubin, and a study found that tyrosine kinase inhibitors inhibit UGT1A1, which increases the risk of hyperbilirubinaemia in patients. 66 Atazanavir (not on the list) is used to treat HIV and inhibits UGT1A1. If UGT1A1 genotype is known before treatment start, CPIC guideline recommends considering an alternative agent to atazanavir in poor metabolizers as there is an increased risk of jaundice. 67 In vitro studies have also shown that UGTs are subject to induction. Many UGT inducers also induce other enzymes such as CYP enzymes and include rifampicin (not on the list), phenobarbital (not on the list) and carbamazepine (not on the list). 68 In vivo studies investigating induction or inhibition are difficult to conduct since there are few good and specific probe drugs for UGT isoforms. 69
3.3. Drug transporters
P‐gp is the most important drug transporter and known to transport 30 of the 100 drugs. Breast cancer resistance protein (BCRP) is known to transport 11 drugs, and organic anion transporting polypeptide 1B1 (OATP1B1) and OATP1B3 are both known to transport nine drugs (Figure 4).
FIGURE 4.
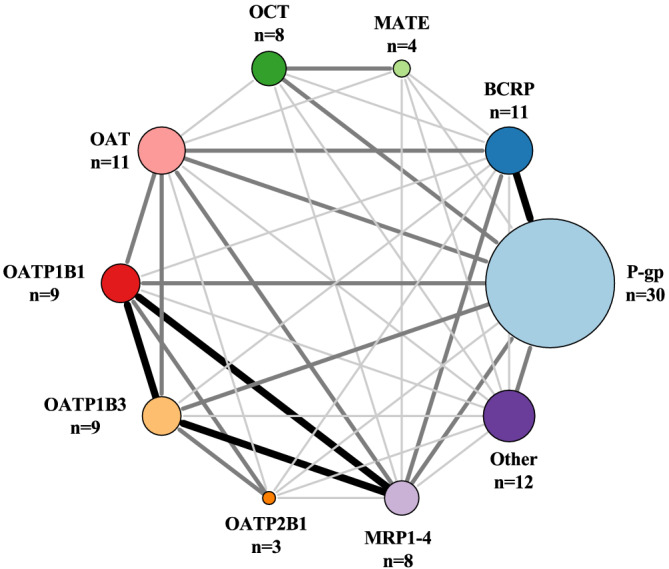
Number of drugs transported by drug transporters is illustrated with increasing circle size. The size and darkness of the lines between enzymes illustrate the substrate overlap. Thin/light‐grey line corresponds to 1–2 drugs as substrate overlap. Medium/grey line corresponds to 3–4 drugs as substrate overlap. Thick/black line corresponds to >4 drugs as substrate overlap. Overlap within a group is not illustrated in the figure, but we refer to Table S1 for further information. The group Other contains MCT, OATP1A2, OATP4C1, OCTN, LAT, SERT, PMAT, THTR, CHT, NTCP, PePT and AE. AE, anion exchange protein; BCRP, breast cancer resistance protein; CHT, choline transporter; LAT, L‐amino acid transporter; MATE, multidrug and toxic compound extrusion; MCT, monocarboxylate transporter; MRP, multidrug resistance‐associated protein; NTC, sodium‐taurocholate co‐transporting polypeptide; OAT, organic anion transporter; OATP, organic anion transporting polypeptide; OCT, organic cation transporter; OCTN, organic cation transporter novel; PePT, peptide transporter; PMAT, plasma membrane monoamine transporter; P‐gp, P‐glycoprotein; SERT, serotonin transporter; THTR, thiamine transporter protein.
3.3.1. P‐gp
We find that P‐gp is known to transports 30 drugs and has the largest substrate overlap with BCRP (five drugs; Figure 4). Substrate overlap of 3‐4 drugs is shared with OATP1B1 and OATP1B3, organic cation transporter (OCT), organic anion transporter (OAT) and multi‐drug resistance protein 1–4 (MRP1–4) (Figure 4). The overall largest substrate overlap for P‐gp is with CYP3A4/5 (>10 drugs; Figure 5A), and a minor substrate overlap is seen with CYP2D6, CYP2C9 and CYP2C19 (6–10 drugs; Figure 5A).
FIGURE 5.
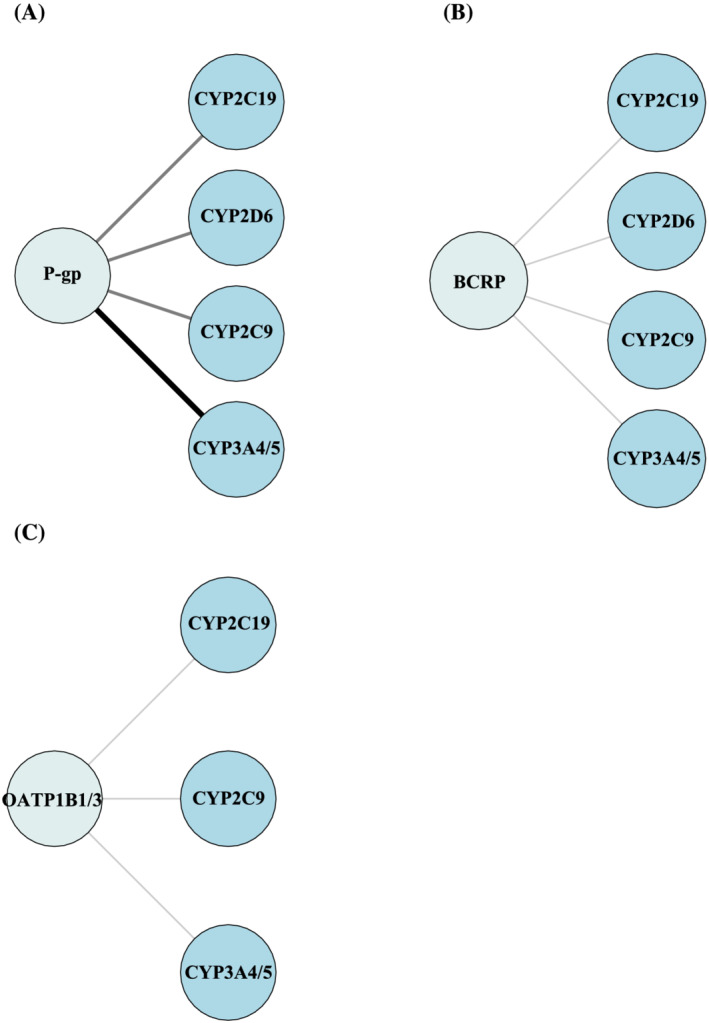
(A) Substrate overlap between P‐gp and CYP3A4/5, CYP2C9, CYP2C19 and CYP2D6. (B) Substrate overlap between BCRP and CYP3A4/5, CYP2C9, CYP2C19 and CYP2D6. (C) Substrate overlap between OATP1B1/3 and CYP3A4/5, CYP2C9 and CYP2C19. The size and darkness of the lines between drug transporter and enzymes illustrate the substrate overlap. Thin/light‐grey line corresponds to 1–5 drugs as substrate overlap. Medium/grey line corresponds to 6–10 drugs as substrate overlap. Thick/black line corresponds to >10 drugs as substrate overlap. BCRP, breast cancer resistance protein; CYP, cytochrome P450 enzyme; OATP, organic anion transporting polypeptide; P‐gp, P‐glycoprotein.
P‐gp has wide tissues distribution, for example, brain, endocrine tissues, gastrointestinal tract, liver and kidney. 11 , 12 It belongs to the ATP binding cassette (ABC) transporter superfamily, and the function of P‐gp is to limit cellular accumulation of endogenous metabolites and xenobiotics. 70
From our list, P‐gp is not categorized as the major transporter in drug transport of the 100 most used drugs, but the Top 3 P‐gp substrates from our list are atorvastatin, omeprazole and losartan. P‐gp is encoded by the human multidrug‐resistance (MDR1) gene, 71 which is regulated by two receptors, PXR and CAR. 72
Rifampicin (not on the list) is a well‐known inducer of P‐gp, and its impact on P‐gp is widely studied with different substrates, for example, fexofenadine (number 84). 73 A clinical study showed that rifampicin decreased area under the curve (AUC) of fexofenadine by 51%. 73 Verapamil (not on the list) is a well‐known P‐gp inhibitor, and coadministration with fexofenadine leads to a 2.5‐fold increase in AUC of fexofenadine in male volunteers. 74
Genetic polymorphisms in MDR1 can potentially alter the functional expression and activity, but despite extensive research within this field, there is still no consensus regarding the significance of P‐gp genetic polymorphism. 75 , 76 , 77
3.3.2. Breast cancer resistance protein (BCRP)
Our review shows that BCRP is known to transport 11 drugs, and besides a substrate overlap of five drugs with P‐gp, BCRP shares substrates of 3–4 drugs with OAT and MRP1–4 (Figure 4). Further, BCRP has substrate overlap with CYP2D6, CYP2C9 and CYP2C19 (1–5 drugs; Figure 5B). From our list, BCRP is not categorized as the major transporter in drug transport of the 100 most used drugs, but The top 3 BCRP substrates from our list are pantoprazole, furosemide and rosuvastatin.
BCRP is located in, for example, the brain, gastrointestinal tract, reproductive organs and muscle tissues. 11 , 12 A prominent BCRP substrate from our list includes allopurinol (number 51). BCRP belongs to the ABC transporter superfamily, and the BCRP transporter is encoded by the ABCG2 gene. 78 , 79 BCRP is regulated by AhR, CAR, PXR, GR, oestrogen receptor β (ER‐β), PPAR‐γ and nuclear factor erythroid 2‐related factor 2 (Nrf2). 80
Febuxostat (not on the list) is a newer xanthine oxidase inhibitor that inhibits BCRP‐mediated transport of rosuvastatin (number 24) both in vitro and in vivo. 81 The ABCG2 gene is polymorphic, and the ABCG2 c.421C>A variant is well studied. The minor A allele results in decreases of 30%–40% BCRP protein expression compared to the reference allele. 82 A study showed that ABCG2 c.421C>A variant resulted in poor response to the BCRP substrate allopurinol. 83 Pharmacokinetic data show that patients carrying the ABCG2 c.421C>A variant have increased rosuvastatin exposure (144% increased AUC), which leads to higher risk of myopathy. Furthermore, genome‐wide association studies showed that carriers of the variant have improved cholesterol‐lowering response of rosuvastatin. 82 Based on these results, the CPIC guideline recommends that patients with poor function ABCG2 reduce the starting dose of the BCRP substrate rosuvastatin to ≤20 mg or consider alternative statins if more than 20 mg is needed. 82
3.3.3. Organic anion transporting polypeptide 1B1 (OATP1B1) and OATP1B3
OATP1B1 and OATP1B3 are both known to transport nine drugs. The largest substrate overlap for the two isoforms are with each other (eight drugs; Figure 4) and with MRP1–4 (five drugs; Figure 4). OATP1B1 and OATP1B3 has also substrate overlap of three drugs with OAT (Figure 4) and substrate overlap with CYP3A4/5, CYP2C9 and CYP2C19 (1–5 drugs; Figure 5c). OATP1B1 and OATP1B3 are primarily expressed in the liver, 11 , 12 and they have 80% amino acid homology. 84 From our list, OATP1B1 and OATP1B3 are not categorized as the major transporter in drug transport of the 100 most used drugs, but the Top 3 OATP1B1 and OATP1B3 substrates from our list are atorvastatin, simvastatin and furosemide. OATP1B1 and OATP1B3 are members of the solute carrier (SLC) family that regulates cellular uptake and is encoded by SLCO1B1 (OATP1B1) and SLCO1B3 (OATP1B3). 85 , 86 Both transporters function as active uptake transporters. 86 The transcriptional regulation of OATP1B1 and OATP1B3 differ. The major transcriptional regulators for OATP1B1 are FXR and LXRα, but only FXR is known to be involved in regulation of OATP1B3. 87
Rifampicin (not on the list) is an inhibitor of OATP1B1 and OATP1B3. 88 A study with healthy volunteers gave the trial subjects a single dose of rifampicin and showed a sevenfold increase in AUC of atorvastatin (number 2), a substrate to OATP1B1 and OATP1B3. 88
The c.521T>C genotype in SLCO1B1 is the most widely studied polymorphism. This variant leads to reduced transport activity in vitro. 89 Several studies have shown that carriers of the c.521T>C genotype are at increased risk of simvastatin‐related myotoxicity (number 5). 90 CPIC guideline states that patients with decreased or poor function of OATP1B1 are at increased risk of myopathy upon treatment with atorvastatin and simvastatin, and dose should be adjusted accordingly. 82 There is currently no well‐validated polymorphism in OATP1B3 that require altered pharmacotherapy.
4. DISCUSSION
In this review, we identified the 100 most prescribed drugs in five European countries and Australia. We found that 89 of the 100 drugs are metabolized either by Phase I metabolism or Phase II metabolism or are known to be substrates for drug transporters, whereas 11 drugs are not subject to drug metabolism or drug transport. In total, 67 of the 100 drugs undergo Phase I metabolism where CYP3A4/5 is the predominant enzyme followed by CYP2D6, CYP2C9, CYP2C19, CYP1A2 and CYP2C8. UGTs are the most dominant Phase II enzymes responsible for metabolism of 17 of the 100 drugs. Lastly, P‐gp is the dominant transporter and is known to be responsible for the transport of 30 of the 100 drugs. CYP3A4/5 and P‐gp share a large substrate overlap of 15 drugs, and if expression or activity is altered for CYP3A4/5 or P‐gp, it will potentially affect 73 drugs, and this is of substantial clinical relevance.
A previous study investigated how the 200 most widely used drugs in the United States are metabolized through CYP enzymes and found the same five CYP enzymes to be the major contributors to drug metabolism. 7 A newer and updated version by the same authors looked at 248 clinically relevant drugs and found that the contribution of CYP3A4/5 to drug metabolism was 30% and therefore lower compared to their previous study (37%) and ours (43%). 7 , 91 Another literature review described that UGTs are involved in the metabolism of almost 8% of the 200 most prescribed drugs in the United States in 2002, 92 which is a smaller contribution compared to ours at 17%. They also found that UGT2B7 is the most dominant UGT isoform followed by UGT1A4 and UGT1A1. 92 Our update highlights that the latter two have a minor role today, while UGT2B7 retains its role as the most important UGT. The methods used to collect data on drug metabolism from the three previous articles differ from ours and may impact the results. Two articles 7 , 92 retrieved information on elimination route for the 200 most prescribed drugs through Rxlist.com. The third article 91 did not describe how the 248 drugs were chosen but found literature on drug metabolism. Contribution of drug transport to the most widely prescribed drugs has not previously been investigated. Drug transporters are known to be involved in the transport of more than half of the most prescribed drugs and are therefore important to consider in pharmacotherapy.
In our literature search, we assessed drug metabolism for each drug. For prodrugs activated through metabolism, drug metabolism covers both the activation route and the elimination route of the ingested drug. Different enzymes are responsible for either one of these routes, and this is important to consider as changes in one of these enzymes will affect the efficacy differently.
Our updated list is a snapshot of the enzymes and transporters known to be involved in drug metabolism and drug transport at the time of data extraction. It should be noted that pharmacotherapy is constantly evolving especially regarding Phase II enzymes and drug transporters, and thus, new knowledge is continuously obtained. Additionally, new drugs are approved, while others become obsolete. It is therefore important to update this list in the future.
The strengths of this review are that we obtained data on prescription medicine from five countries in Europe and from Australia, which makes the list applicable for multiple countries. Furthermore, we used original literature based on human in vitro studies. We chose human in vitro studies as these studies are the most accurate to explain underlying mechanisms for drug metabolism and drug transport. Additionally, a third author double‐checked our results from the literature search to reduce the risk of errors. The first limitation to our study is that we included prescription data from Scotland that were obtained 6 years ago and thus slightly outdated. Secondly, two authors each screened articles for 50 drugs until metabolism and drug transport were confirmed by a valid article. This could result in slight under‐representation of minor enzyme contributions though we believe this will be of minor relevance. Thirdly, we excluded animal studies in our literature search. If the assessment is performed in animal models or with murine drug transporters without utilization of human cell models, it is not included in our literature search. This might underestimate the contribution of especially drug transporters as knockout rodent models are widely used to establish involvement of drug transporters. One such example is the involvement of OAT3 in ciprofloxacin transport, which was studied in a mouse model 93 but not caught by our literature search. As this limitation is only relevant for drugs that are exclusively studied in animal models, we suspect that there is a relatively low risk of missing major metabolism and transport pathways. Fourthly, we were not able to confirm the proportional contribution (major or minor contribution) of enzymes and drug transporters to metabolism or drug transport as conclusive data are not available for most drugs. Fifthly, we only assessed metabolism and transport of the parent drug. Thus, we did not cover downstream metabolism and transport of metabolites. Sixthly, in some countries, drugs are available both as over‐the‐counter drugs and as prescriptions, whereas a prescription is required in others. This could result in slight under‐representation of specific drugs as we only included prescription data. Finally, we only had prescription data from pharmacies, while data on medicine used in hospitals were not available. Thus, drugs primarily prescribed in hospitals were not included. These may include chemotherapeutic drugs, biologics, etc.
5. CONCLUSION
In conclusion, we found that 89 of the 100 most prescribed drugs are metabolized and/or known to be transported. Only 11 drugs are not subject to either drug metabolism or drug transport. As involvement and overlap of enzyme and transporters are high, this study highlights the risk of drug–drug interactions in patients taking multiple medications. Thus, understanding variability of drug metabolism and drug transport remains a priority.
CONFLICT OF INTEREST
Ann‐Cathrine Dalgård Dunvald has given paid lectures for Astellas Pharma. Tore B. Stage has given paid lectures for Pfizer and Eisai and done consulting for Pfizer. Anton Pottegård has participated in research projects funded by Alcon, Almirall, Astellas, AstraZeneca, Boehringer‐Ingelheim, Novo Nordisk, Servier and LEO Pharma, all regulator‐mandated Phase IV studies. All of this is unrelated to the work done in this review. Ditte Bork Iversen and Nanna Elman Andersen declare that they have no conflict of interest.
Supporting information
Table S1. Drugs on top 100 list linked with enzymes and drug transporters involved in metabolism and transportation.
Table S2. Most prescribed drugs from the six countries combined with the percentage each drug constitute from the list. The countries included are Australia, Denmark, England, Norway, Scotland, and Sweden.
ACKNOWLEDGEMENTS
We would like to acknowledge the following for providing prescription data from Norway, Sweden, Denmark, Scotland, England and Australia: Øystein Karlstad, Peter Bjødstrup Jensen, Nicole Pratt and Daniel Morales. The authors would also like to acknowledge Morten Olsen for helping with conducting the Top 100 drug list. Further, we would like to acknowledge Sissel Mogensen for drawing Figure 1.
Iversen DB, Andersen NE, Dalgård Dunvald A‐C, Pottegård A, Stage TB. Drug metabolism and drug transport of the 100 most prescribed oral drugs. Basic Clin Pharmacol Toxicol. 2022;131(5):311‐324. doi: 10.1111/bcpt.13780
Funding information This work was funded by Novo Nordisk Foundation (NNF19OC0058275), Lundbeck Foundation Fellowship (R307‐2018‐2980) and Danish Cancer Society (R279‐A16411).
Funding information Danish Cancer Society, Grant/Award Number: R279‐A16411; Lundbeck Foundation, Grant/Award Number: R307‐2018‐2980; Novo Nordisk Foundation, Grant/Award Number: NNF19OC0058275
REFERENCES
- 1. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Human drug metabolism and the cytochromes P450: application and relevance of in vitro models. J Clin Pharmacol. 2001;41(11):1149‐1179. doi: 10.1177/00912700122012724 [DOI] [PubMed] [Google Scholar]
- 2. Kornholt J, Christensen MB. Prevalence of polypharmacy in Denmark. Dan Med J. 2020;67(6):A12190680. [PubMed] [Google Scholar]
- 3. Issa NT, Wathieu H, Ojo A, Byers SW, Dakshanamurthy S. Drug metabolism in preclinical drug development: a survey of the discovery process, toxicology, and computational tools. CDM. 2017;18(6):556‐565. doi: 10.2174/1389200218666170316093301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao M, Ma J, Li M, et al. Cytochrome P450 enzymes and drug metabolism in humans. Int J Mol Sci. 2021;22(23):12808. doi: 10.3390/ijms222312808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ahmed S, Zhou Z, Zhou J, Chen SQ. Pharmacogenomics of drug metabolizing enzymes and transporters: relevance to precision medicine. Genom Proteom Bioinf. 2016;14(5):298‐313. doi: 10.1016/j.gpb.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thummel KE, Lin YS. Sources of interindividual variability. Methods Mol Biol. 2014;1113:363‐415. doi: 10.1007/978-1-62703-758-7_17 [DOI] [PubMed] [Google Scholar]
- 7. Zanger UM, Turpeinen M, Klein K, Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem. 2008;392(6):1093‐1108. doi: 10.1007/s00216-008-2291-6 [DOI] [PubMed] [Google Scholar]
- 8. Wu KM. A new classification of prodrugs: regulatory perspectives. Pharmaceuticals. 2009;2(3):77‐81. doi: 10.3390/ph2030077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lolodi O, Wang YM, Wright WC, Chen T. Differential regulation of CYP3A4 and CYP3A5 and its implication in drug discovery. Curr Drug Metab. 2017;18(12):1095‐1105. doi: 10.2174/1389200218666170531112038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jancova P, Anzenbacher P, Anzenbacherova E. Phase II drug metabolizing enzymes. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2010;154(2):103‐116. doi: 10.5507/bp.2010.017 [DOI] [PubMed] [Google Scholar]
- 11. Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue‐based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 12. The human protein atlas. Accessed November 1, 2021. https://www.proteinatlas.org
- 13. Stage TB, Graff M, Wong S, et al. Dicloxacillin induces CYP2C19, CYP2C9 and CYP3A4 in vivo and in vitro. Br J Clin Pharmacol. 2018;84(3):510‐519. doi: 10.1111/bcp.13467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Backman JT, Kivistö KT, Olkkola KT, Neuvonen PJ. The area under the plasma concentration‐time curve for oral midazolam is 400‐fold larger during treatment with itraconazole than with rifampicin. Eur J Clin Pharmacol. 1998;54(1):53‐58. doi: 10.1007/s002280050420 [DOI] [PubMed] [Google Scholar]
- 15. Shahzadi A, Javed I, Aslam B, et al. Therapeutic effects of ciprofloxacin on the pharmacokinetics of carbamazepine in healthy adult male volunteers. Pak J Pharm Sci. 2011;24(1):63‐68. [PubMed] [Google Scholar]
- 16. Inui N, Akamatsu T, Uchida S, et al. Chronological effects of rifampicin discontinuation on cytochrome P450 activity in healthy Japanese volunteers, using the cocktail method. Clin Pharmacol Ther. 2013;94(6):702‐708. doi: 10.1038/clpt.2013.167 [DOI] [PubMed] [Google Scholar]
- 17. Reitman ML, Chu X, Cai X, et al. Rifampin's acute inhibitory and chronic inductive drug interactions: experimental and model‐based approaches to drug‐drug interaction trial design. Clin Pharmacol Ther. 2011;89(2):234‐242. doi: 10.1038/clpt.2010.271 [DOI] [PubMed] [Google Scholar]
- 18. Malhotra S, Bailey DG, Paine MF, Watkins PB. Seville orange juice‐felodipine interaction: comparison with dilute grapefruit juice and involvement of furocoumarins. Clin Pharmacol Ther. 2001;69(1):14‐23. doi: 10.1067/mcp.2001.113185 [DOI] [PubMed] [Google Scholar]
- 19. Moore LB, Goodwin B, Jones SA, et al. John's wort induces hepatic drug metabolism through activation of the pregnane X receptor. Proc Natl Acad Sci USA. 2000;97(13):7500‐7502. doi: 10.1073/pnas.130155097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dunvald ACD, Järvinen E, Mortensen C. Stage TB clinical and molecular perspectives on inflammation‐mediated regulation of drug metabolism and transport. Clin Pharmacol Ther. 2021;4(2):277‐290. doi: 10.1002/cpt.2432 [DOI] [PubMed] [Google Scholar]
- 21. Birdwell KA, Decker B, Barbarino JM, et al. Clinical pharmacogenetics implementation consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin Pharmacol Ther. 2015;98(1):19‐24. doi: 10.1002/cpt.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Andrews LM, De Winter BC, Van Gelder T, Hesselink DA. Consideration of the ethnic prevalence of genotypes in the clinical use of tacrolimus. Pharmacogenomics. 2016;17(16):1737‐1740. doi: 10.2217/pgs-2016-0136 [DOI] [PubMed] [Google Scholar]
- 23. Zhang HF, Wang HH, Gao N, et al. Physiological content and intrinsic activities of 10 cytochrome P450 isoforms in human normal liver microsomes. J Pharmacol Exp Ther. 2016;358(1):83‐93. doi: 10.1124/jpet.116.233635 [DOI] [PubMed] [Google Scholar]
- 24. Ingelman‐Sundberg M, Sim SC, Gomez A, Rodriguez‐Antona C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther. 2007;116(3):496‐526. doi: 10.1016/j.pharmthera.2007.09.004 [DOI] [PubMed] [Google Scholar]
- 25. Claessens AJ, Risler LJ, Eyal S, Shen DD, Easterling TR, Hebert MF. CYP2D6 mediates 4‐hydroxylation of clonidine in vitro: implication for pregnancy‐induced changes in clonidine clearance. Drug Metab Dispos. 2010;38(9):1393‐1396. doi: 10.1124/dmd.110.033878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wadelius M, Darj E, Frenne G, Rane A. Induction of CYP2D6 in pregnancy. Clin Pharmacol Ther. 1997;62(4):400‐407. doi: 10.1016/S0009-9236(97)90118-1 [DOI] [PubMed] [Google Scholar]
- 27. Liston HL, DeVane CL, Boulton DW, Risch SC, Markowitz JS, Goldman J. Differential time course of cytochrome P450 2D6 enzyme inhibition by fluoxetine, sertraline, and paroxetine in healthy volunteers. J Clin Psychopharmacol. 2002;22(2):169‐173. doi: 10.1097/00004714-200204000-00010 [DOI] [PubMed] [Google Scholar]
- 28. Meloche M, Khazaka M, Kassem I, Barhdadi A, Dubé MP, de Denus S. CYP2D6 polymorphism and its impact on the clinical response to metoprolol: a systematic review and meta‐analysis. Br J Clin Pharmacol. 2020;86(6):1015‐1033. doi: 10.1111/bcp.14247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crews KR, Monte AA, Huddart R, et al. Clinical pharmacogenetics implementation consortium guideline for CYP2D6, OPRM1, and COMT genotypes and select opioid therapy. Clin Pharmacol Ther. 2021;110(4):888‐896. doi: 10.1002/cpt.2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. St Sauver JL, Olson JE, Roger VL, et al. CYP2D6 phenotypes are associated with adverse outcomes related to opioid medications. Pharmgenomics Pers Med. 2017;10:217‐227. doi: 10.2147/PGPM.S136341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lenk HÇ, Klöditz K, Johansson I, et al. The polymorphic nuclear factor NFIB regulates hepatic CYP2D6 expression and influences risperidone metabolism in psychiatric patients. Clin Pharmacol Ther. 2022;6(5):1165‐1174. doi: 10.1002/cpt.2571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gerbal‐Chaloin S, Daujat M, Pascussi JM, Pichard‐Garcia L, Vilarem MJ, Maurel P. Transcriptional regulation of CYP2C9 gene. Role of glucocorticoid receptor and constitutive androstane receptor. J Biol Chem. 2002;277(1):209‐217. doi: 10.1074/jbc.M107228200 [DOI] [PubMed] [Google Scholar]
- 33. Johnson JA. Warfarin pharmacogenetics: a rising tide for its clinical value. Circulation. 2012;125(16):1964‐1966. doi: 10.1161/circulationaha.112.100628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Iversen DB, Hellfritzsch M, Stage TB, Aabenhus RM, Lind BS, Pottegård A. Antimycotic treatment of oral candidiasis in warfarin users. Am J Med. 2021;134(5):e308‐e312. doi: 10.1016/j.amjmed.2020.10.018 [DOI] [PubMed] [Google Scholar]
- 35. Pottegård A, Henriksen DP, Madsen KG, Hellfritzsch M, Damkier P, Stage TB. Change in international normalized ratio among patients treated with dicloxacillin and vitamin K antagonists. Jama. 2015;314(3):296‐297. doi: 10.1001/jama.2015.6669 [DOI] [PubMed] [Google Scholar]
- 36. Hellfritzsch M, Lund LC, Ennis Z, et al. Ischemic stroke and systemic embolism in warfarin users with atrial fibrillation or heart valve replacement exposed to dicloxacillin or flucloxacillin. Clin Pharmacol Ther. 2020;107(3):607‐616. doi: 10.1002/cpt.1662 [DOI] [PubMed] [Google Scholar]
- 37. Flora DR, Rettie AE, Brundage RC, Tracy TS. CYP2C9 genotype‐dependent warfarin pharmacokinetics: impact of CYP2C9 genotype on R‐ and S‐warfarin and their oxidative metabolites. J Clin Pharmacol. 2017;57(3):382‐393. doi: 10.1002/jcph.813 [DOI] [PubMed] [Google Scholar]
- 38. Kimmel SE, French B, Kasner SE, et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N Engl J Med. 2013;369(24):2283‐2293. doi: 10.1056/NEJMoa1310669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pirmohamed M, Burnside G, Eriksson N, et al. A randomized trial of genotype‐guided dosing of warfarin. N Engl J Med. 2013;369(24):2294‐2303. doi: 10.1056/NEJMoa1311386 [DOI] [PubMed] [Google Scholar]
- 40. Verhoef TI, Ragia G, de Boer A, et al. A randomized trial of genotype‐guided dosing of acenocoumarol and phenprocoumon. N Engl J Med. 2013;369(24):2304‐2312. doi: 10.1056/NEJMoa1311388 [DOI] [PubMed] [Google Scholar]
- 41. Johnson JA, Caudle KE, Gong L, et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for pharmacogenetics‐guided warfarin dosing: 2017 update. Clin Pharmacol Ther. 2017;102(3):397‐404. doi: 10.1002/cpt.668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Y, Ferguson SS, Negishi M, Goldstein JA. Identification of constitutive androstane receptor and glucocorticoid receptor binding sites in the CYP2C19 promoter. Mol Pharmacol. 2003;64(2):316‐324. doi: 10.1124/mol.64.2.316 [DOI] [PubMed] [Google Scholar]
- 43. Sienkiewicz‐Oleszkiewicz B, Wiela‐Hojeńska A. CYP2C19 polymorphism in relation to the pharmacotherapy optimization of commonly used drugs. Pharmazie. 2018;73(11):619‐624. doi: 10.1691/ph.2018.8689 [DOI] [PubMed] [Google Scholar]
- 44. Mrazek DA, Biernacka JM, O'Kane DJ, et al. CYP2C19 variation and citalopram response. Pharmacogenet Genomics. 2011;21(1):1‐9. doi: 10.1097/fpc.0b013e328340bc5a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kamiya C, Inui N, Hakamata A, et al. Effect of co‐administered inducer or inhibitor on omeprazole pharmacokinetics based on CYP2C19 genotype. J Pharmacol Sci. 2019;139(4):361‐366. doi: 10.1016/j.jphs.2019.03.001 [DOI] [PubMed] [Google Scholar]
- 46. Lee CR, Luzum JA, Sangkuhl K, et al. Clinical pharmacogenetics implementation consortium guideline for CYP2C19 genotype and clopidogrel therapy: 2022 update. Clin Pharmacol Ther. 2022;16. doi: 10.1002/cpt.2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vogel CFA, Van Winkle LS, Esser C, Haarmann‐Stemmann T. The aryl hydrocarbon receptor as a target of environmental stressors ‐ implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020;34:101530. doi: 10.1016/j.redox.2020.101530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dobrinas M, Cornuz J, Oneda B, Kohler Serra M, Puhl M, Eap CB. Impact of smoking, smoking cessation, and genetic polymorphisms on CYP1A2 activity and inducibility. Clin Pharmacol Ther. 2011;90(1):117‐125. doi: 10.1038/clpt.2011.70 [DOI] [PubMed] [Google Scholar]
- 49. Roymans D, Annaert P, Van Houdt J, et al. Expression and induction potential of cytochromes P450 in human cryopreserved hepatocytes. Drug Metab Dispos. 2005;33(7):1004‐1016. doi: 10.1124/dmd.104.003046 [DOI] [PubMed] [Google Scholar]
- 50. Zhou SF, Yang LP, Zhou ZW, Liu YH, Chan E. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. AAPS J. 2009;11(3):481‐494. doi: 10.1208/s12248-009-9127-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Granfors MT, Backman JT, Laitila J, Neuvonen PJ. Oral contraceptives containing ethinyl estradiol and gestodene markedly increase plasma concentrations and effects of tizanidine by inhibiting cytochrome P450 1A2. Clin Pharmacol Ther. 2005;78(4):400‐411. doi: 10.1016/j.clpt.2005.06.009 [DOI] [PubMed] [Google Scholar]
- 52. Granfors MT, Backman JT, Neuvonen M, Ahonen J, Neuvonen PJ. Fluvoxamine drastically increases concentrations and effects of tizanidine: a potentially hazardous interaction. Clin Pharmacol Ther. 2004;75(4):331‐341. doi: 10.1016/j.clpt.2003.12.005 [DOI] [PubMed] [Google Scholar]
- 53. Granfors MT, Backman JT, Neuvonen M, Neuvonen PJ. Ciprofloxacin greatly increases concentrations and hypotensive effect of tizanidine by inhibiting its cytochrome P450 1A2‐mediated presystemic metabolism. Clin Pharmacol Ther. 2004;76(6):598‐606. doi: 10.1016/j.clpt.2004.08.018 [DOI] [PubMed] [Google Scholar]
- 54. Rasmussen BB, Brix TH, Kyvik KO, Brøsen K. The interindividual differences in the 3‐demthylation of caffeine alias CYP1A2 is determined by both genetic and environmental factors. Pharmacogenetics. 2002;12(6):473‐478. doi: 10.1097/00008571-200208000-00008 [DOI] [PubMed] [Google Scholar]
- 55. Wagner E, McMahon L, Falkai P, Hasan A, Siskind D. Impact of smoking behavior on clozapine blood levels ‐ a systematic review and meta‐analysis. Acta Psychiatr Scand. 2020;142(6):456‐466. doi: 10.1111/acps.13228 [DOI] [PubMed] [Google Scholar]
- 56. Ferguson SS, Chen Y, LeCluyse EL, Negishi M, Goldstein JA. Human CYP2C8 is transcriptionally regulated by the nuclear receptors constitutive androstane receptor, pregnane X receptor, glucocorticoid receptor, and hepatic nuclear factor 4alpha. Mol Pharmacol. 2005;68(3):747‐757. doi: 10.1124/mol.105.013169 [DOI] [PubMed] [Google Scholar]
- 57. Walsky RL, Gaman EA, Obach RS. Examination of 209 drugs for inhibition of cytochrome P450 2C8. J Clin Pharmacol. 2005;45(1):68‐78. doi: 10.1177/0091270004270642 [DOI] [PubMed] [Google Scholar]
- 58. Hruska MW, Amico JA, Langaee TY, Ferrell RE, Fitzgerald SM, Frye RF. The effect of trimethoprim on CYP2C8 mediated rosiglitazone metabolism in human liver microsomes and healthy subjects. Br J Clin Pharmacol. 2005;59(1):70‐79. doi: 10.1111/j.1365-2125.2005.02263.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tornio A, Filppula AM, Kailari O, et al. Glucuronidation converts clopidogrel to a strong time‐dependent inhibitor of CYP2C8: a phase II metabolite as a perpetrator of drug‐drug interactions. Clin Pharmacol Ther. 2014;96(4):498‐507. doi: 10.1038/clpt.2014.141 [DOI] [PubMed] [Google Scholar]
- 60. Agergaard K, Mau‐Sørensen M, Stage TB, et al. Clopidogrel‐paclitaxel drug‐drug interaction: a Pharmacoepidemiologic study. Clin Pharmacol Ther. 2017;102(3):547‐553. doi: 10.1002/cpt.674 [DOI] [PubMed] [Google Scholar]
- 61. Daily EB, Aquilante CL. Cytochrome P450 2C8 pharmacogenetics: a review of clinical studies. Pharmacogenomics. 2009;10(9):1489‐1510. doi: 10.2217/pgs.09.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marcath LA, Kidwell KM, Robinson AC, et al. Patients carrying CYP2C8*3 have shorter systemic paclitaxel exposure. Pharmacogenomics. 2019;20(2):95‐104. doi: 10.2217/pgs-2018-0162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aquilante CL, Kosmiski LA, Bourne DWA, et al. Impact of the CYP2C8 *3 polymorphism on the drug‐drug interaction between gemfibrozil and pioglitazone. Br J Clin Pharmacol. 2013;75(1):217‐226. doi: 10.1111/j.1365-2125.2012.04343.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meech R, Hu DG, McKinnon RA, et al. The UDP‐glycosyltransferase (UGT) superfamily: new members, new functions, and novel paradigms. Physiol Rev. 2019;99(2):1153‐1222. doi: 10.1152/physrev.00058.2017 [DOI] [PubMed] [Google Scholar]
- 65. Mackenzie PI, Hu DG, Gardner‐Stephen DA. The regulation of UDP‐glucuronosyltransferase genes by tissue‐specific and ligand‐activated transcription factors. Drug Metab Rev. 2010;42(1):99‐109. doi: 10.3109/03602530903209544 [DOI] [PubMed] [Google Scholar]
- 66. Qosa H, Avaritt BR, Hartman NR, Volpe DA. In vitro UGT1A1 inhibition by tyrosine kinase inhibitors and association with drug‐induced hyperbilirubinemia. Cancer Chemother Pharmacol. 2018;82(5):795‐802. doi: 10.1007/s00280-018-3665-x [DOI] [PubMed] [Google Scholar]
- 67. Gammal RS, Court MH, Haidar CE, et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for UGT1A1 and Atazanavir prescribing. Clin Pharmacol Ther. 2016;99(4):363‐369. doi: 10.1002/cpt.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Soars MG, Petullo DM, Eckstein JA, Kasper SC, Wrighton SA. An assessment of udp‐glucuronosyltransferase induction using primary human hepatocytes. Drug Metab Dispos. 2004;32(1):140‐148. doi: 10.1124/dmd.32.1.140 [DOI] [PubMed] [Google Scholar]
- 69. Lv X, Zhang JB, Hou J, et al. Chemical probes for human UDP‐glucuronosyltransferases: a comprehensive review. Biotechnol J. 2019;14(1):e1800002. doi: 10.1002/biot.201800002 [DOI] [PubMed] [Google Scholar]
- 70. Silva R, Vilas‐Boas V, Carmo H, et al. Modulation of P‐glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol Ther. 2015;149:1‐123. doi: 10.1016/j.pharmthera.2014.11.013 [DOI] [PubMed] [Google Scholar]
- 71. Ueda K, Cornwell MM, Gottesman MM, et al. The mdr1 gene, responsible for multidrug‐resistance, codes for P‐glycoprotein. Biochem Biophys Res Commun. 1986;141(3):956‐962. doi: 10.1016/s0006-291x(86)80136-x [DOI] [PubMed] [Google Scholar]
- 72. Chan GNY, Hoque MT, Cummins CL, Bendayan R. Regulation of P‐glycoprotein by orphan nuclear receptors in human brain microvessel endothelial cells. J Neurochem. 2011;118(2):163‐175. doi: 10.1111/j.1471-4159.2011.07288.x [DOI] [PubMed] [Google Scholar]
- 73. Bosilkovska M, Samer CF, Déglon J, et al. Geneva cocktail for cytochrome p450 and P‐glycoprotein activity assessment using dried blood spots. Clin Pharmacol Ther. 2014;96(3):349‐359. doi: 10.1038/clpt.2014.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yasui‐Furukori N, Uno T, Sugawara K, Tateishi T. Different effects of three transporting inhibitors, verapamil, cimetidine, and probenecid, on fexofenadine pharmacokinetics. Clin Pharmacol Ther. 2005;77(1):17‐23. doi: 10.1016/j.clpt.2004.08.026 [DOI] [PubMed] [Google Scholar]
- 75. Kim RB, Leake BF, Choo EF, et al. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther. 2001;70(2):189‐199. doi: 10.1067/mcp.2001.117412 [DOI] [PubMed] [Google Scholar]
- 76. Peng R, Zhang H, Zhang Y, Wei DY. Impacts of ABCB1 (G1199A) polymorphism on resistance, uptake, and efflux to steroid drugs. Xenobiotica. 2016;46(10):948‐952. doi: 10.3109/00498254.2016.1138249 [DOI] [PubMed] [Google Scholar]
- 77. Öztaş E, Parejo Garcia‐Saavedra A, Yanar F, et al. P‐glycoprotein polymorphism and levothyroxine bioavailability in hypothyroid patients. Saudi Pharm J. 2018;26(2):274‐278. doi: 10.1016/j.jsps.2017.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nigam SK. What do drug transporters really do? Nat Rev Drug Discov. 2015;14(1):29‐44. doi: 10.1038/nrd4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bircsak KM, Moscovitz JE, Wen X, et al. Interindividual regulation of the breast cancer resistance protein/ABCG2 transporter in term human placentas. Drug Metab Dispos. 2018;46(5):619‐627. doi: 10.1124/dmd.117.079228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fohner AE, Brackman DJ, Giacomini KM, Altman RB, Klein TE. PharmGKB summary: very important pharmacogene information for ABCG2. Pharmacogenet Genomics. 2017;27(11):420‐427. doi: 10.1097/FPC.0000000000000305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lehtisalo M, Keskitalo JE, Tornio A, et al. Febuxostat, but not allopurinol, markedly raises the plasma concentrations of the breast cancer resistance protein substrate rosuvastatin. Clin Transl Sci. 2020;13(6):1236‐1243. doi: 10.1111/cts.12809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cooper‐DeHoff RM, Niemi M, Ramsey LB, et al. The clinical pharmacogenetics implementation consortium (CPIC) guideline for SLCO1B1, ABCG2, and CYP2C9 and statin‐associated musculoskeletal symptoms. Clin Pharmacol Ther. 2022;12(5):1007‐1021. doi: 10.1002/cpt.2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Brackman DJ, Yee SW, Enogieru OJ, et al. Genome‐wide association and functional studies reveal novel pharmacological mechanisms for allopurinol. Clin Pharmacol Ther. 2019;106(3):623‐631. doi: 10.1002/cpt.1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. König J, Cui Y, Nies AT, Keppler D. Localization and genomic organization of a new hepatocellular organic anion transporting polypeptide. J Biol Chem. 2000;275(30):23161‐23168. doi: 10.1074/jbc.M001448200 [DOI] [PubMed] [Google Scholar]
- 85. Alam K, Crowe A, Wang X, et al. Regulation of organic anion transporting polypeptides (OATP) 1B1‐ and OATP1B3‐mediated transport: an updated review in the context of OATP‐mediated drug‐drug interactions. Int J Mol Sci. 2018;19(3):E855. doi: 10.3390/ijms19030855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158(3):693‐705. doi: 10.1111/j.1476-5381.2009.00430.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Meyer Zu Schwabedissen HE, Böttcher K, Chaudhry A, Kroemer HK, Schuetz EG, Kim RB. Liver X receptor α and farnesoid X receptor are major transcriptional regulators of OATP1B1. Hepatology. 2010;52(5):1797‐1807. doi: 10.1002/hep.23876 [DOI] [PubMed] [Google Scholar]
- 88. Lau YY, Huang Y, Frassetto L, Benet LZ. Effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther. 2007;81(2):194‐204. doi: 10.1038/sj.clpt.6100038 [DOI] [PubMed] [Google Scholar]
- 89. Tirona RG, Leake BF, Merino G, Kim RB. Polymorphisms in OATP‐C: identification of multiple allelic variants associated with altered transport activity among European‐ and African‐Americans. J Biol Chem. 2001;276(38):35669‐35675. doi: 10.1074/jbc.M103792200 [DOI] [PubMed] [Google Scholar]
- 90. Lu B, Sun L, Seraydarian M, et al. Effect of SLCO1B1 T521C on statin‐related myotoxicity with use of lovastatin and atorvastatin. Clin Pharmacol Ther. 2021;110(3):733‐740. doi: 10.1002/cpt.2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138(1):103‐141. doi: 10.1016/j.pharmthera.2012.12.007 [DOI] [PubMed] [Google Scholar]
- 92. Williams JA, Hyland R, Jones BC, et al. Drug‐drug interactions for UDP‐glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32(11):1201‐1208. doi: 10.1124/dmd.104.000794 [DOI] [PubMed] [Google Scholar]
- 93. Vanwert AL, Srimaroeng C, Sweet DH. Organic anion transporter 3 (oat3/slc22a8) interacts with carboxyfluoroquinolones, and deletion increases systemic exposure to ciprofloxacin. Mol Pharmacol. 2008;74(1):122‐131. doi: 10.1124/mol.107.042853 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Drugs on top 100 list linked with enzymes and drug transporters involved in metabolism and transportation.
Table S2. Most prescribed drugs from the six countries combined with the percentage each drug constitute from the list. The countries included are Australia, Denmark, England, Norway, Scotland, and Sweden.