

Abstract
Background & Aims
Experimental evidence indicates that systemic inflammation (SI) promotes liver fibrogenesis. This study investigated the potential link between SI and fibrogenesis in patients with advanced chronic liver disease (ACLD).
Methods
Serum biomarkers of SI (CRP, IL‐6, procalcitonin [PCT]) and extracellular matrix (ECM) turnover (i.e., fibrogenesis/fibrolysis) were analysed in 215 prospectively recruited patients with ACLD (hepatic venous pressure gradient [HVPG] ≥6 mm Hg) undergoing hepatic vein catheterization. Patients with non‐elective hospitalization or bacterial infection were excluded. Histological alpha‐smooth muscle actin (α‐SMA) area was quantified on full biopsy scans by automated morphometric quantification in a subset of 34 patients who underwent concomitant transjugular liver biopsy.
Results
Histological α‐SMA proportionate area correlated with enhanced liver fibrosis (ELF) score (Spearman's ρ = 0.660, p < .001), markers of collagen formation (PRO‐C3, ρ = 0.717, p < .001; PRO‐C6, ρ = 0.526, p = .002) and tissue inhibitor of metalloproteinases‐1 (TIMP1; ρ = 0.547, p < .001), indicating that these blood biomarkers are capable of reflecting the dynamic process of ECM turnover. CRP, IL‐6 and PCT levels correlated with ELF, biomarkers of collagen synthesis/degradation and TIMP1, both in compensated and decompensated patients. Multivariate linear regression models (adjusted for HVPG) confirmed that CRP, IL‐6 and PCT were independently linked to markers of liver fibrogenesis and ECM turnover.
Conclusion
Systemic inflammation is linked to both liver fibrogenesis and ECM turnover in ACLD and this association is not confounded by the severity of liver disease, as evaluated by HVPG. Our study confirms experimental data on the detrimental impact of SI on ECM deposition and fibrosis progression in a thoroughly characterized cohort of patients with ACLD.
Keywords: bacterial translocation, cirrhosis, extracellular matrix, gut‐liver‐axis, HSC, portal hypertension
Abbreviations
- ACLD
advanced chronic liver disease
- ACLF
acute‐on‐chronic liver failure
- ALD
alcohol‐related liver disease
- BT
bacterial translocation
- c/dACLD
compensated/decompensated ACLD
- C3M
neo‐epitope of MMP‐9 mediated degradation of type III collagen
- CRP
C‐reactive protein
- CSPH
clinically significant portal hypertension
- CTP
Child‐Turcotte‐Pugh
- DAMP
danger‐associated molecular pattern
- ECM
extracellular matrix
- ELF
enhanced liver fibrosis score
- HSC
hepatic stellate cell
- HVPG
hepatic venous pressure gradient
- IL‐6
interleukin‐6
- LBP
lipopolysaccharide binding protein
- LPS
lipopolysaccharide
- MELD
Model of End Stage Liver Disease
- NAFLD
non‐alcoholic fatty liver disease
- NSBB
non‐selective betablocker
- PAMP
pathogen‐associated molecular pattern
- PCT
procalcitonin
- PH
portal hypertension
- PRO‐C3
released N‐terminal pro‐peptide of type III collagen
- PRO‐C6
C‐terminal of released C5 domain of type VI collagen α3 chain
- SI
systemic inflammation
- TIMP1
tissue inhibitor of metalloproteinases‐1
- TIPS
transjugular intrahepatic portosystemic shunt
- TJBX
transjugular liver biopsy
- α‐SMA
alpha‐smooth muscle actin
Lay summary.
Cirrhosis is caused by chronic liver damage, a process characterized by development of fibrosis (‘fibrogenesis’), which describes the scarring of the liver. Systemic inflammation is common in patients with cirrhosis and has a detrimental impact on disease prognosis. Our study found a link between liver fibrogenesis and systemic inflammation in patients with cirrhosis, and thus, supports the concept that systemic inflammation promotes liver fibrogenesis in humans with cirrhosis.
1. INTRODUCTION
Fibrosis represents an important pathophysiological feature and key prognostic factor in advanced chronic liver disease (ACLD) and results from chronic liver injury and activation of hepatic stellate cells (HSCs). 1 , 2 HSCs play a central role in the structural remodelling of liver tissue and are the dominant cellular source of extracellular matrix (ECM), that is mainly different types of collagen. Liver fibrosis ultimately causes structural disruption and parenchymal loss‐of‐function. 3 In addition, HSC activation also promotes their vasoconstrictive phenotype. 4 , 5 Concordantly, these structural and functional changes contribute to the development of portal hypertension (PH). 6 ECM composition is considered to undergo constant remodelling and the balance may tip either towards liver fibrosis progression or regression. 7
Experimental studies have indicated that ACLD is accompanied by increased bacterial translocation (BT) from the gut into the portal circulation. Bacteria and/or their constituents are recognized as pathogen‐associated molecular patterns (PAMPs) by several cell types such as liver sinusoidal endothelial and innate immune cells, HSCs, and hepatocytes. 8 , 9 , 10 In turn, hepatocellular injury leads to the release of danger‐associated molecular patterns (DAMPs). PAMPs, DAMPs and cytokines disseminate into the systemic circulation where they may trigger systemic inflammation (SI). This hypothesis is strengthened by the observation that bacterial antigens have been linked to SI in humans with different aetiologies and severity of ACLD. 11 , 12 In vitro and in vivo experiments have found that a proinflammatory environment promotes the (in‐)direct activation of HSCs. 13 , 14 , 15 BT has been described to trigger SI and, moreover, SI emerged as an important factor contributing to the progression of ACLD. 16 However, the link between SI and the dynamic process of liver fibrogenesis in humans remains to be investigated.
Our study aimed to elucidate the association between SI and fibrogenesis in prospectively recruited patients with clinically stable ACLD. Therefore, we determined an advanced blood biomarker panel reflecting fibrogenesis and ECM turnover and also provide histological evidence that these biomarkers actually reflect HSC activation in patients with ACLD.
2. PATIENTS AND METHODS
2.1. Study design and clinical characterization
Patients with ACLD (defined by a hepatic venous pressure gradient [HVPG] ≥6 mm Hg) underwent liver vein catheterization between 01/2017 and 06/2020 at the Medical University of Vienna and were included in the Vienna Cirrhosis Study (VICIS). Patients with pre−/post‐hepatic/non‐cirrhotic PH, transjugular intrahepatic portosystemic shunt, non‐selective betablockers, hepatocellular carcinoma beyond Milan criteria, previous liver transplantation, bacterial infection or non‐elective hospitalization at the time of liver vein catheterization, or intake of non−/poorly absorbable antibiotics for treatment of hepatic encephalopathy or spontaneous bacterial peritonitis prophylaxis were excluded (Figure S1). Patient selection was performed by review of prospectively collected data. In the final study cohort of 215 patients, serum and plasma biomarkers of SI, fibrogenesis and matrix turnover were analysed. Among patients recruited after the first case of COVID‐19 reported in Austria (n = 27), no patient had tested positive for SARS‐CoV‐2 within 14 days prior to HVPG measurement, exhibited COVID‐19 symptoms or fever when presenting at our clinic. Furthermore, histological alpha‐smooth muscle actin (α‐SMA) stainings were performed in a subset of 34 (16%) patients undergoing concomitant transjugular liver biopsy (TJBX) within the same session (Figure 1). Previous hepatic decompensation events were determined according to national 17 and European guidelines for decompensated cirrhosis. 18
FIGURE 1.
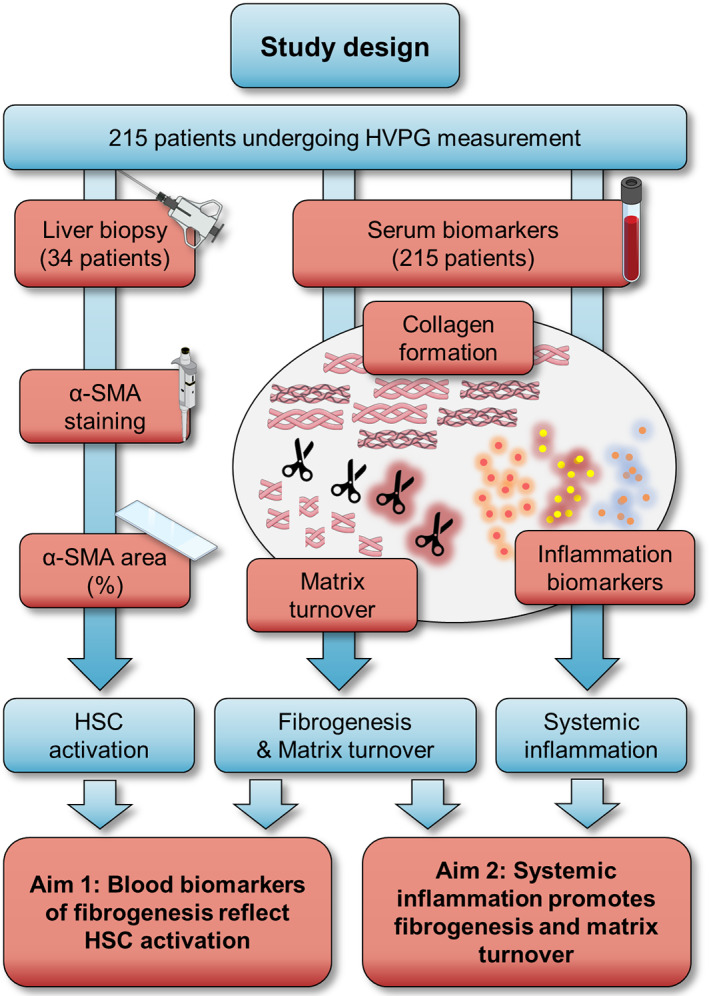
Graphical summary of the study design. Abbreviations: α‐SMA, alpha‐smooth muscle actin; HSC, hepatic stellate cell; HVPG, hepatic venous pressure gradient.
The study was conducted in accordance with the principles of the Declaration of Helsinki and its amendments, was approved by the local ethics committee of the Medical University of Vienna (EK1262/2017) and included patients of the prospective VICIS study (NCT03267615). All patients provided written informed consent for the invasive procedures of hepatic vein catheterization and TJBX.
2.2. Measurement of hepatic venous pressure gradient and transjugular biopsy
Liver vein catheterization was performed to assess HVPG in all patients and to undertake TJBX in a subset of patients in accordance with a standard operating procedure. 19 Detailed descriptions of the procedures are delineated in the Supplementary material.
2.3. Serum and plasma biomarkers
Blood biomarkers were analysed in samples obtained at the timepoint of HVPG measurement via the catheter introducer sheath. C‐reactive protein (CRP), interleukin‐6 (IL‐6), procalcitonin (PCT), lipopolysaccharide binding protein (LBP) and enhanced liver fibrosis score (ELF; including tissue inhibitor of metalloproteinases‐1 [TIMP1]) were assessed according to standardized protocols by the ISO‐certified Department of Laboratory Medicine, Medical University of Vienna, following the manufacturers' instructions, and as previously published. 20 , 21 The Protein Fingerprint™ biomarkers PRO‐C3 (released N‐terminal pro‐peptide of type III collagen), C3M (neo‐epitope of MMP‐9 mediated degradation of type III collagen) and PRO‐C6 (C‐terminal of released C5 domain of type VI collagen α3 chain) were assessed by competitive enzyme‐linked immunosorbent assay (ELISA). Further details on biomarker measurements are provided in the Supplementary material.
2.4. Histological α‐SMA staining
Liver tissue was formalin‐fixed, paraffin‐embedded, cut into 2‐μm‐thick slides and mounted on super frost glass slides. For assessment of α‐SMA proportionate area (%), liver tissue was stained using a standardized immunohistochemistry protocol according to the manufacturer's instructions (DAKO M851, Denmark). Stained proportionate areas of α‐SMA were assessed by automated quantification using the histomorphometry software Definiens TissueStudio® V4.3.1 (Definiens Inc.) on high‐resolution 40x slide scans of liver biopsy sections (Pannoramic MIDI Slidescanner, 3DHISTECH, Hungary) (Figure 2). Personnel performing histological stainings and proportionate area quantification were blinded to other patient characteristics.
FIGURE 2.
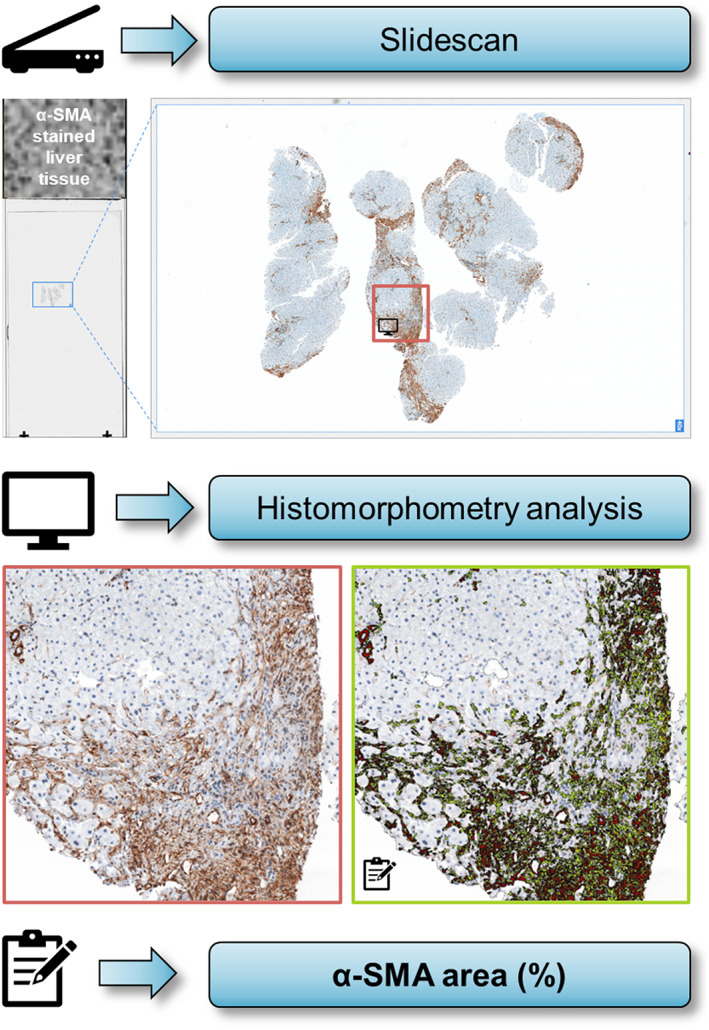
Assessment of α‐SMA proportionate area by slidescanner and automated quantification with histomorphometry analysis. Abbreviations: α‐SMA, alpha‐smooth muscle actin.
2.5. External validation cohort
External validation was performed in a patient cohort undergoing HVPG measurement at the Hvidovre Hospital, Denmark. Data on fibrogenesis and matrix turnover markers have previously been reported. 22 CRP levels were measured by ELISA as described previously 23 in patients without infection in whom stored (at −80°C) femoral artery plasma trazylol samples were available. The reported study was conducted in accordance with the principles of the Declaration of Helsinki and its amendments and was approved by the local ethics committee (J‐No.2008‐41‐2020). Patients gave written informed consent to participate in the study. 22
2.6. Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics 27 (IBM) or GraphPad Prism 9 (GraphPad Software). Categorical variables are reported as absolute (n) and relative (%) frequencies, while continuous variables are displayed as mean ± standard error of the mean or median with interquartile range (IQR), as appropriate. Normal distribution was determined by Kolmogorov–Smirnov and Shapiro–Wilk tests. Student's t test, Mann–Whitney U test, analysis of variance (ANOVA), or Kruskal–Wallis test were used for group comparisons of continuous variables, as applicable. Group comparisons of categorical variables were performed using Chi squared or Fisher's exact test. Correlations between parameters were assessed by calculating Pearson's or Spearman's correlation coefficients (95% confidence interval), as appropriate. Linear regression models were performed to identify associations between biomarkers of inflammation (predictor variables) and biomarkers of fibrogenesis and matrix turnover (dependent variables). For these analyses, some variables (indicated by footnotes) were log‐transformed based on the respective histogram, Q‐Q‐Plots, Kolmogorov–Smirnov and Shapiro–Wilk tests and diagnostic plots. The level of significance was set at a two‐sided p‐value < .05 for all analyses.
3. RESULTS
3.1. Patient characteristics
In this study, 215 patients with a median age of 69 (51–67) years, 67% male sex and predominantly alcohol‐related liver disease (ALD; 45%) and viral hepatitis (18%) were included (Table 1). Clinically significant PH (CSPH) was present in most (n = 188, 87%) patients, and 57% had decompensated cirrhosis. Regarding PH/disease severity, median HVPG was 17 (12–21) mm Hg, median Model for End‐Stage Liver Disease (MELD) score was 11 (9–13) points and 62%/31%/7% were classified as Child‐Turcotte‐Pugh (CTP) stage A/B/C respectively.
TABLE 1.
Patient characteristics in the overall cohort and patients stratified by HVPG
| Parameter | Overall cohort (n = 215) | HVPG 6–9 mm Hg (n = 27) | HVPG 10–19 mm Hg (n = 112) | HVPG ≥20 mm Hg (n = 76) | p‐value |
|---|---|---|---|---|---|
| Age (years) | 59 (51–67) | 59 (51–67) | 59 (51–67) | 58 (51–67) | .938 |
| Sex (M, %) | 144 (67) | 22 (82) | 73 (65) | 49 (65) | .229 |
| Aetiology (n, %) | |||||
| ALD | 97 (45) | 8 (30) | 44 (39) | 45 (59) | .009 |
| Viral | 38 (18) | 8 (30) | 21 (19) | 9 (12) | |
| ALD + Viral | 14 (6) | 2 (7) | 8 (7) | 4 (5) | |
| NASH | 23 (11) | 3 (11) | 19 (17) | 1 (1) | |
| Cholestatic | 8 (4) | 0 (0) | 3 (3) | 5 (7) | |
| Other | 35 (16) | 6 (22) | 17 (15) | 12 (16) | |
| cACLD (n, %) | 93 (43) | 24 (89) | 53 (47) | 16 (21) | <.001 |
| CTP score (points) | 6 (5–7) | 5 (5–5) | 6 (5–7) | 7 (6–8) | <.001 |
| MELD score (points) | 11 (9–13) | 8 (7–9) | 10 (9–13) | 12 (10–14) | <.001 |
| Varices (n, %) | |||||
| None | 79 (37) | 20 (74) | 43 (38) | 16 (21) | <.001 |
| Small | 54 (25) | 3 (11) | 28 (25) | 23 (30) | |
| Large | 76 (35) | 1 (4) | 38 (34) | 37 (49) | |
| (Unknown) | 6 (3) | 3 (11) | 3 (3) | 0 (0) | |
| Ascites (n, %) | |||||
| None | 120 (56) | 26 (96) | 70 (63) | 24 (32) | <.001 |
| Mild | 82 (38) | 1 (4) | 39 (35) | 42 (55) | |
| Severe | 13 (6) | 0 (0) | 3 (3) | 10 (13) | |
| HE (n, %) | |||||
| None | 185 (86) | 26 (96) | 97 (87) | 62 (82) | .288 |
| Mild | 29 (13) | 1 (4) | 15 (13) | 13 (17) | |
| Severe | 1 (1) | 0 (0) | 0 (0) | 1 (1) | |
| BMI | 26.7 (23.5–30.7) | 27.2 (25.3–31.7) | 26.7 (23.5–31.0) | 26.2 (22.9–29.9) | .353 |
| WBC (G/L) | 4.70 (3.30–6.14) | 5.60 (4.41–7.00) | 4.54 (3.30–6.22) | 4.54 (3.17–5.85) | .021 |
| CRP (mg/dl) | 0.25 (0.11–0.53) | 0.14 (0.06–0.29) | 0.23 (0.10–0.45) | 0.40 (0.18–0.69) | <.001 |
| IL‐6 (pg/ml) | 7.17 (4.57–12.2) | 4.68 (3.01–8.46) | 6.46 (3.95–12.2) | 8.71 (5.88–18.1) | <.001 |
| PCT (ng/ml) | 0.07 (0.05–0.13) | 0.06 (0.03–0.07) | 0.08 (0.05–0.13) | 0.09 (0.06–0.15) | <.001 |
| LBP (μg/ml) | 6.89 (5.40–8.59) | 7.31 (5.71–9.48) | 6.92 (5.41–8.60) | 6.54 (5.10–8.01) | .341 |
| ELF score | 11.2 (10.3–12.2) | 9.69 (9.07–10.1) | 11.0 (10.4–12.2) | 11.8 (11.2–12.6) | <.001 |
| PRO‐C3 (ng/ml) | 16.4 (11.4–25.7) | 10.0 (7.40–13.8) | 17.4 (11.7–28.2) | 18.2 (14.2–33.1) | <.001 |
| PRO‐C6 (ng/ml) | 13.9 (10.6–19.3) | 10.9 (8.91–12.4) | 14.3 (10.7–19.0) | 15.4 (12.0–21.2) | <.001 |
| TIMP1 (ng/ml) | 316 (246–435) | 252 (174–325) | 299 (243–421) | 376 (277–481) | <.001 |
| C3M (ng/ml) | 13.6 (11.2–18.0) | 11.6 (10.4–13.2) | 13.4 (11.2–16.7) | 16.2 (12.0–20.8) | <.001 |
Note: Statistical Analysis: Kruskal–Wallis and one‐way ANOVA were used to compare continuous variables across HVPG strata. Group comparisons of categorical variables were performed using Chi squared or Fisher's Exact test. p‐values < .05 are indicated in bold.
Abbreviations: ALD, alcohol‐related liver disease; BMI, body‐mass index; C3M, neo‐epitope of MMP‐9 mediated degradation of type III collagen; cACLD, compensated advanced chronic liver disease; CRP, C‐reactive protein; CSPH, clinically significant portal hypertension; CTP, Child‐Turcotte‐Pugh; ELF, enhanced liver fibrosis; HE, hepatic encephalopathy; HVPG, hepatic venous pressure gradient; IL‐6, interleukin‐6; LBP, lipopolysaccharide binding protein; M, male sex; MELD, Model for End‐Stage Liver Disease; NASH, non‐alcoholic steatohepatitis; PCT, procalcitonin; PRO‐C3, released N‐terminal pro‐peptide of type III collagen; PRO‐C6, C‐terminal of released C5 domain of type VI collagen α3 chain; TIMP1, tissue inhibitor of metalloproteinases‐1; WBC, white blood cell.
Increasing severity of PH was paralleled by an increasing prevalence and severity of ascites, presence and size of oesophageal varices and higher CTP and MELD scores (all p < .001) (Table 1). White blood cell (WBC) count decreased, while serum levels of systemic inflammation continuously increased across HVPG strata (6–9 mm Hg, 10–19 mm Hg, ≥20 mm Hg): median CRP was 0.14 vs. 0.23 vs. 0.40 mg/dl (p = .001), IL‐6 was 4.68 vs. 6.46 vs. 8.71 pg/ml (p < .001) and PCT was 0.06 vs. 0.08 vs. 0.09 ng/ml (p < .001) respectively. Similarly, serum biomarkers of liver fibrogenesis and matrix turnover exhibited stepwise increases with more severe PH (all p < .001; Table 1).
3.2. Histological validation of fibrogenesis biomarkers
Alpha‐SMA proportionate area in liver biopsies was compared to serum markers of fibrogenesis and matrix turnover to assess whether these parameters represent HSC activation. Importantly, patient characteristics in the subset of 34 patients undergoing concomitant liver biopsies were not profoundly different, as compared to patients without biopsy, thus, excluding major selection bias (Table 2). However, a trend towards a discrepancy in the distribution of aetiologies between groups was observed and patients undergoing liver biopsy had lower TIMP1 concentrations. Serum biomarkers displayed an excellent correlation with α‐SMA area (%) on liver histology: Spearman's ρ = 0.660 (0.41–0.82; p < .001) for ELF score, ρ = 0.717 (0.49–0.85; p < .001) for PRO‐C3, ρ = 0.526 (0.21–0.74, p = .002) for PRO‐C6, ρ = 0.547 (0.25–0.75, p < .001) for TIMP1 (Figure 3) and ρ = 0.422 (0.08–0.67; p = .014) for C3M. Notably, SI levels exhibited a significant correlation with α‐SMA proportionate area, with ρ = 0.444 (0.11–0.69; p = .009) for CRP, ρ = 0.355 (0.01–0.63; p = .040) for IL‐6 and ρ = 0.345 (0.00–0.62; p = .046) for PCT, while LBP showed no relevant association with α‐SMA (p = .515) (Figure S2).
TABLE 2.
Clinical characteristics and histological readouts of patients undergoing transjugular liver biopsy and comparison to patients without biopsy
| Parameter | Patients with biopsy (n = 34) | Patients without biopsy (n = 181) | p‐value |
|---|---|---|---|
| Age (years) | 60 (45–64) | 59 (51–68) | .542 |
| Sex (M, %) | 21 (62) | 123 (68) | .481 |
| Aetiology (n, %) | |||
| ALD | 14 (41) | 83 (46) | .068 |
| Viral | 2 (6) | 36 (20) | |
| ALD + Viral | 1 (3) | 13 (7) | |
| NASH | 6 (18) | 17 (9) | |
| Cholestatic | 1 (3) | 7 (4) | |
| Other | 10 (29) | 25 (14) | |
| cACLD (n, %) | 14 (41) | 79 (44) | .790 |
| CTP score (points) | 6 (5–7) | 6 (5–7) | .659 |
| MELD score (points) | 10 (8–13) | 11 (9–14) | .888 |
| CSPH (n, %) | 33 (97) | 155 (86) | .088 |
| HVPG (mm Hg) | 17 (13–19) | 18 (12–21) | .711 |
| BMI | 27.8 (23.2–31.6) | 26.5 (23.7–30.6) | .439 |
| WBC (G/L) | 4.04 (2.74–5.96) | 4.74 (3.38–6.21) | .145 |
| CRP (mg/dl) | 0.26 (0.10–0.53) | 0.25 (0.11–0.54) | .837 |
| IL‐6 (pg/ml) | 5.65 (3.47–9.79) | 7.29 (4.76–12.6) | .138 |
| PCT (ng/ml) | 0.07 (0.05–0.11) | 0.08 (0.05–0.13) | .632 |
| LBP (μg/ml) | 6.61 (4.78–9.52) | 6.91 (5.43–8.37) | .892 |
| ELF score | 10.9 (10.0–12.0) | 11.3 (10.4–12.3) | .247 |
| PRO‐C3 (ng/ml) | 14.0 (10.1–18.6) | 17.2 (11.8–26.0) | .115 |
| PRO‐C6 (ng/ml) | 12.2 (9.19–16.6) | 14.3 (10.8–19.5) | .065 |
| TIMP1 (ng/ml) | 268 (196–438) | 328 (252–434) | .049 |
| C3M (ng/ml) | 13.2 (11.2–19.0) | 13.6 (11.2–18.0) | .892 |
| Total liver tissue area (μm2) | 68.55 × 106 (18.46 × 106–18.99 × 07) | — | — |
| α‐SMA proportionate area (%) | 11.2 (6.73–24.62) | — | — |
Note: Statistical Analysis: Student's t test or Mann–Whitney U test were used to compare continuous variables. Group comparisons of categorical variables were performed using Chi squared or Fisher's exact test.
Abbreviations: ALD, alcohol‐related liver disease; BMI, body‐mass index; C3M, neo‐epitope of MMP‐9 mediated degradation of type III collagen; cACLD, compensated advanced chronic liver disease; CRP, C‐reactive protein; CSPH, clinically significant portal hypertension; CTP, Child‐Turcotte‐Pugh; ELF, enhanced liver fibrosis; HVPG, hepatic venous pressure gradient; IL‐6, interleukin‐6; LBP, lipopolysaccharide binding protein; M, male sex; MELD, Model for End‐Stage Liver Disease; NASH, non‐alcoholic steatohepatitis; PCT, procalcitonin; PRO‐C3, released N‐terminal pro‐peptide of type III collagen; PRO‐C6, C‐terminal of released C5 domain of type VI collagen α3 chain; TIMP1, tissue inhibitor of metalloproteinases‐1; WBC, white blood cell.
FIGURE 3.
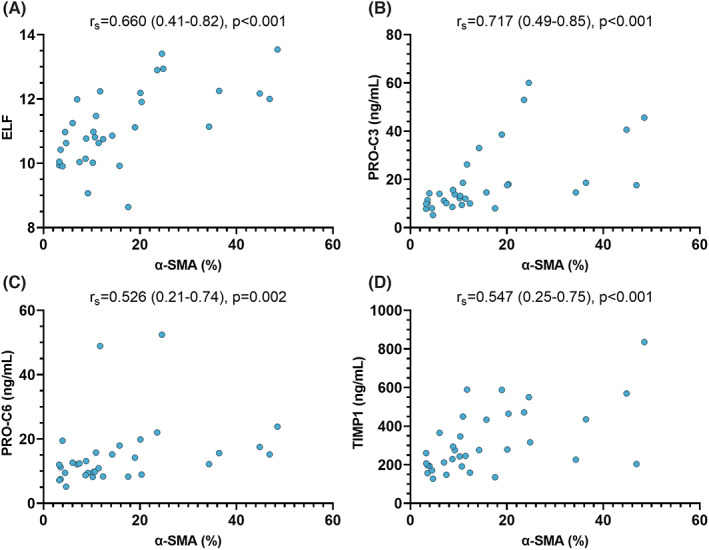
Correlation between α‐SMA proportionate area and biomarkers of fibrogenesis and matrix turnover. Statistical analysis: Correlation analyses were performed by calculating Spearman's ρ with 95% confidence intervals (in brackets). Abbreviations: α‐SMA, alpha‐smooth muscle actin proportionate area; ELF, enhanced liver fibrosis; TIMP1, tissue inhibitor of metalloproteinases‐1; PRO‐C3, released N‐terminal pro‐peptide of type III collagen; PRO‐C6, C‐terminal of released C5 domain of type VI collagen α3 chain.
3.3. Relation between fibrogenesis, matrix turnover and systemic inflammation
Serum biomarkers of collagen synthesis, degradation and inhibition of metalloproteinases were correlated with CRP, IL‐6, PCT and LBP to investigate whether increase in SI is linked to increased fibrogenesis. Results of correlation analyses are summarized in Table 3 and correlation plots are depicted in the Supplementary material (Figures S3–S6). Interestingly, IL‐6 and PCT exhibited the strongest and most consistent associations with the biomarker panel, followed by CRP. TIMP1 exhibited the strongest link to these SI markers: ρ = 0.446 (0.33–0.55, p < .001) for CRP, ρ = 0.554 (0.45–0.64, p < .001) for IL‐6 and ρ = 0.500 (0.39–0.60, p < .001) for PCT. Interestingly, LBP levels exhibited no meaningful correlation with markers of collagen formation and degradation, but a statistically significant positive correlation with TIMP1.
TABLE 3.
Correlations between systemic inflammation and liver fibrogenesis/matrix turnover
| CRP (mg/dl) | p‐value | IL‐6 (pg/ml) | p‐value | PCT (ng/ml) | p‐value | LBP (μg/ml) | p‐value | |
|---|---|---|---|---|---|---|---|---|
| ELF score | 0.356 (0.23–0.47) | <.001 | 0.424 (0.30–0.53) | <.001 | 0.505 (0.39–0.60) | <.001 | −0.148 (−0.28‐[−]0.01) | .030 |
| PRO‐C3 (ng/ml) | 0.342 (0.21–0.46) | <.001 | 0.322 (0.19–0.44) | <.001 | 0.445 (0.33–0.55) | <.001 | 0.000 (−0.13–0.13) | .999 |
| PRO‐C6 (ng/ml) | 0.289 (0.16–0.41) | <.001 | 0.420 (0.30–0.53) | <.001 | 0.412 (0.29–0.52) | <.001 | 0.096 (−0.04–0.23) | .161 |
| TIMP1 (ng/ml) | 0.446 (0.33–0.55) | <.001 | 0.554 (0.45–0.64) | <.001 | 0.500 (0.39–0.60) | <.001 | 0.265 (0.13–0.39) | <.001 |
| C3M (ng/ml) | 0.416 (0.29–0.52) | <.001 | 0.354 (0.23–0.47) | <.001 | 0.240 (0.11–0.37) | <.001 | 0.140 (0.00–0.27) | .041 |
Note: Statistical Analysis: Correlation analyses were performed by calculating Spearman's ρ with 95% confidence intervals (in brackets). p‐values <.05 are indicated in bold.
Abbreviations: C3M, neo‐epitope of MMP‐9 mediated degradation of type III collagen; CRP, C‐reactive protein; ELF, enhanced liver fibrosis; IL‐6, interleukin‐6; LBP, lipopolysaccharide binding protein; PCT, procalcitonin; PRO‐C3, released N‐terminal pro‐peptide of type III collagen; PRO‐C6, C‐terminal of released C5 domain of type VI collagen α3 chain; TIMP1, tissue inhibitor of metalloproteinases‐1.
3.4. Fibrogenesis in compensated and decompensated ACLD
Patients were stratified by compensated (cACLD) and decompensated ACLD (dACLD) to explore whether the observed link between SI and fibrogenesis was restricted to one of these stages. As expected, disease and PH severity, as well as fibrogenesis and SI biomarkers were significantly higher in patients with dACLD (Table S1). Interestingly, SI displayed a consistent significant correlation with fibrogenesis and matrix turnover in both cACLD and dACLD (Table S2). PCT exhibited the strongest association with these biomarkers in cACLD, particularly with ELF score (ρ = 0.443, 0.26–0.60, p < .001) and TIMP1 (ρ = 0.440, 0.25–0.59, p < .001). In dACLD, PCT displayed the strongest correlation with ELF score (ρ = 0.509, 0.36–0.63, p < .001) and PRO‐C3 (ρ = 0.467, 0.31–0.60, p < .001), and PRO‐C6 (ρ = 0.426, 0.26–0.57, p < .001), while IL‐6 was most firmly linked TIMP1 (ρ = 0.605, 0.48–0.71, p < .001). Again, LBP displayed a significant correlation with TIMP1 in both cACLD (ρ = 0.241, 0.03–0.43, p = .020) and dACLD (ρ = 0.339, 0.17–0.49, p < .001), but was not significantly associated with other fibrogenesis markers in these patient strata.
3.5. Systemic inflammation is linked to fibrogenesis independent from portal hypertension severity
Due to the observation that markers of SI and fibrogenesis/matrix turnover increase with portal hypertension/disease severity (Table 1), multivariate linear regression models were performed to rule‐out that the link between SI and fibrogenesis/matrix turnover was confounded by the severity of underlying liver disease. Results of univariate and multivariate linear regression models are depicted in Table 4. After adjustment for HVPG, the multivariate analysis confirmed that CRP levels were independently linked to ELF score (p = .013), PRO‐C3, PRO‐C6, TIMP1 and C3M (all p < .001). Similarly, IL‐6 was independently associated with ELF score, PRO‐C3, PRO‐C6, TIMP1 and C3M (all p < .001). PCT was an independent predictor variable for ELF, the collagen formation markers PRO‐C3 and PRO‐C6 and the matrix turnover biomarker TIMP1 (all p < .001), as well as the fibrolysis marker C3M (p = .031). Finally, LBP levels were independently associated with PRO‐C6 (p = .012), TIMP1 (p = .003) and C3M (p = .002) but exhibited no association with other biomarkers.
TABLE 4.
Univariate and multivariate (adjusted for HVPG) linear regression models to assess the link between systemic inflammation (predictor variables) and liver fibrogenesis/matrix turnover (dependent variables)
| Parameters | Univariate analysis | Multivariate analysis a | |||||
|---|---|---|---|---|---|---|---|
| Dependent variable | Predictor variable | Estimate | 95% CI | p‐value | Estimate | 95% CI | p‐value |
| ELF score | CRP b | 0.879 | 0.51–1.25 | <.001 | 0.447 | 0.10–0.80 | .013 |
| IL‐6 c | 0.063 | 0.05–0.08 | <.001 | 0.042 | 0.03–0.06 | <.001 | |
| PCT b | 3.500 | 1.92–5.08 | <.001 | 7.825 | 5.21–10.4 | <.001 | |
| LBP b | −0.062 | −0.13–0.01 | .068 | −0.059 | −0.14 − 0.02 | .150 | |
| PRO‐C3 | CRP c | 0.207 | 0.14–0.28 | <.001 | 0.173 | 0.10–0.25 | <.001 |
| IL‐6 c | 0.222 | 0.13–0.31 | <.001 | 0.170 | 0.07–0.27 | <.001 | |
| PCT d | 1.380 | 0.66–2.10 | <.001 | 1.242 | 0.54–1.94 | <.001 | |
| LBP d | 0.007 | −0.02 to 0.04 | 0.656 | 0.014 | ‐0.02 to 0.04 | .344 | |
| PRO‐C6 | CRP c | 0.129 | 0.07–0.19 | <.001 | 0.105 | 0.04–0.17 | <.001 |
| IL‐6 c | 0.224 | 0.15–0.30 | <.001 | 0.201 | 0.12–0.28 | <.001 | |
| PCT c | 0.295 | 0.21–0.38 | <.001 | 0.270 | 0.18–0.36 | <.001 | |
| LBP c | 0.188 | 0.00–0.37 | .045 | 0.230 | 0.05–0.41 | .012 | |
| TIMP1 | CRP c | 0.173 | 0.12–0.23 | <.001 | 0.139 | 0.08–0.20 | <.001 |
| IL‐6 c | 0.286 | 0.22–0.36 | <.001 | 0.248 | 0.17–0.32 | <.001 | |
| PCT c | 0.263 | 0.17–0.35 | <.001 | 0.212 | 0.12–0.31 | <.001 | |
| LBP d | 0.029 | 0.00–0.05 | .025 | 0.036 | 0.01–0.06 | .003 | |
| C3M | CRP c | 0.152 | 0.11–0.20 | <.001 | 0.125 | 0.08–0.17 | <.001 |
| IL‐6 c | 0.178 | 0.12–0.24 | <.001 | 0.140 | 0.08–0.20 | <.001 | |
| PCT d | 0.605 | 0.13–1.08 | .013 | 0.497 | 0.05–0.95 | .031 | |
| LBP c | 0.163 | 0.02–0.31 | .027 | 0.211 | 0.08–0.35 | .002 | |
Note: Statistical Analysis: Linear regression models were performed using non‐transformed or log‐transformed variables as appropriate based on the respective histogram, Q‐Q‐Plot as well as Kolmogorov–Smirnov and Shapiro–Wilk tests (indicated by footnotes). Multivariate models were calculated by adjusting for HVPG. p‐values <.05 are indicated in bold.
Abbreviations: C3M, neo‐epitope of MMP‐9 mediated degradation of type III collagen; CRP, C‐reactive protein; ELF, enhanced liver fibrosis; HVPG, hepatic venous pressure gradient; IL‐6, interleukin‐6; LBP, lipopolysaccharide binding protein; PCT, procalcitonin; PRO‐C3, released N‐terminal pro‐peptide of type III collagen; PRO‐C6, C‐terminal of released C5 domain of type VI collagen α3 chain; TIMP1, tissue inhibitor of metalloproteinases‐1.
Adjusted by hepatic HVPG.
Without log‐transformation of included variables.
Log‐transformation of all included variables (except HVPG).
Log‐transformation of the dependent variable.
The link between inflammation and fibrogenesis markers was also observed in HVPG‐adjusted subgroup analyses restricted to patients with cACLD or dACLD. Notably, IL‐6 displayed the most consistent association with fibrogenesis and ECM turnover markers in patients with both cACLD and dACLD (Tables S3/S4).
Finally, all inflammation parameters as well as HVPG were entered into multivariate regression models to evaluate which parameters displayed an independent link with fibrogenesis and ECM turnover markers (Table S5). All inflammation parameters except CRP exhibited an independent link with ELF (all p < .001). IL‐6 and/or PCT remained independently associated with other markers of fibrogenesis as well as ECM turnover. Again, subgroup analyses restricted to patients with cACLD and dACLD were performed. Interestingly, HVPG was independently associated with ELF test, PRO‐C3 and TIMP1 in patients with cACLD, however, only remained independently associated with the fibrolysis marker C3M (in contrast to fibrogenesis markers) in patients with dACLD.
3.6. Confirmation of the link between systemic inflammation and fibrogenesis as well as ECM turnover in an external cohort
Seventy‐three patients served as an external validation cohort. The median age was 56 (49–63) years, the patients were predominantly male (n = 57, 78%) and had a median HVPG of 15 (11–18) mm Hg and a median MELD of 15 (11–19) points (Table S6). CRP levels tended to correlate with PRO‐C3 (r s = 0.221, −0.02 to 0.43, p = .061) and C3M (r s = 0.486, 0.28–0.65, p < .001) levels in this patient cohort (Figure S7), confirming the link between systemic inflammation and fibrogenesis as well as ECM turnover.
4. DISCUSSION
The present study investigates the interplay between SI and liver fibrogenesis as well as ECM turnover in a large cohort of prospectively recruited and extensively characterized patients with clinically stable ACLD. Besides the characterization of PH severity by HVPG measurement and simultaneous blood sampling for assessment of SI levels, we performed an advanced biomarker panel designed to reflect key molecules of hepatic ECM formation and degradation. Importantly, this study also provides novel histological data in a subset of the patients, confirming that these blood biomarkers reflect the dynamic process of fibrogenesis within the liver—considering that these patients have already progressed to ACLD. In contrast to previous work, 24 our patient cohort does not include subjects with infections or acute decompensation (AD) but rather clinically stable outpatients with ACLD. Accordingly, it captures the steady state of liver disease progression.
The study hypothesis is strongly based on the pathophysiological ‘gut‐liver‐axis’ concept that the liver is the first gatekeeper of the portal venous circulation and responds to immunological triggers—presumably mostly due to BT—by promoting proinflammatory signals. 25 , 26 Experimental studies have indicated that increased BT in cirrhotic animals was linked to an impaired mucosal antimicrobial defence in the intestine, 9 reduced expression of tight junction proteins (i.e. important barrier proteins) in the intestinal mucosa of animals with cirrhosis, 27 , 28 as well as increased permeability of the basolateral gut‐vascular barrier. 29 Previous studies in humans have suggested that SI is increased in the distinct clinical settings of AD and acute‐on‐chronic liver failure (ACLF)—which are both conditions associated with a high prevalence of infections. 30 , 31 Similarly, we have demonstrated recently that SI levels also predict disease progression in compensated and clinically stable decompensated patients with ACLD. 32 Although we want to acknowledge that our study only presents blood markers of SI and does not provide direct markers of BT (LBP is discussed below), previous studies have described that BT promotes a systemic proinflammatory response: Bacterial DNA in blood and ascitic fluid of rats with CCl4‐induced cirrhosis were linked to the detection of bacteria in mesenteric lymphnodes (indicating BT from the intestines) as well as SI biomarkers in the blood (e.g. tumour necrosis factor[TNF]‐α and IL‐6). 10 Similarly, different circulating bacterial antigens reflecting both gram‐positive and ‐negative BT in patients with non‐alcoholic fatty liver disease (NAFLD) were linked to SI 11 and serum levels of bacterial DNA were strongly associated with serum markers of SI in patients with ascites. 12 Therefore, biomarkers of SI seem capable of reflecting the response to BT in patients with cirrhosis. Finally, the release of DAMPs, for example by damaged hepatocytes, may also induce SI independently from BT (‘sterile’ inflammation) and activate HSCs. 33
Direct or indirect activation of HSCs causes transdifferentiation to myofibroblasts, a process accompanied by upregulation of α‐SMA and induction of ECM formation and fibrogenesis. 14 , 15 Fibrogenesis is increasingly recognized as a highly dynamic process with bidirectional impact of pathological triggers (e.g. alcohol use) and therapeutic interventions (e.g. cure of viral hepatitis). 7 For example, the histological decrease in α‐SMA expression on liver biopsies after antiviral therapy was reported previously. 34 However, due to the invasive nature of this procedure, repeated liver biopsies have limited utility for assessing the dynamic process of ECM turnover. Thus, several blood biomarkers reflecting ECM remodelling have been developed: ELF score comprises three different ECM markers and is well‐validated for the staging of liver fibrosis, 35 , 36 , 37 , 38 similar to the collagen formation markers PRO‐C3 and PRO‐C6 and the collagen degradation marker C3M. 39 , 40 , 41 Furthermore, previous studies reported that PRO‐C3 and ELF score decreased upon therapeutic interventions such as antiviral therapy 42 , 43 and that PRO‐C3 as well as the ratio between PRO‐C3 and C3M were linked to AD, ACLF and survival. 24 To assess whether ELF score and markers of collagen formation not only reflect fibrosis, but also the dynamic process of fibrogenesis, we compared the area of α‐SMA on liver biopsies to serum markers of fibrogenesis, and found an excellent correlation between histological α‐SMA area (i.e. activated HSCs) and the ELF score, as well as PRO‐C3 and TIMP1 levels. Of note, histological data were obtained by scanning the complete liver biopsy slide and automated quantitative analysis of the α‐SMA proportionate area, which minimizes the risk of human bias. Therefore, the significance of ELF and other biomarkers of hepatic ECM formation and turnover extends beyond the staging of liver fibrosis, as these markers additionally provide valuable information on the activity of hepatic fibrogenesis in ACLD patients.
Experimental studies have indicated that activation of inflammatory responses (e.g. mediated by immune cells such as Kupffer cells) upon exposure to BT may lead to activation of HSCs and ultimately result in promotion of fibrogenesis. 26 For example besides bacterial toxins like lipopolysaccharide (LPS), 44 bacterial cell wall components may induce the activation of HSCs in cell culture. 45 Concordantly, in vivo data by Seki et al. showed that LPS‐induced activation of toll‐like receptor‐4 promotes transforming growth factor‐beta signalling and hepatic fibrosis in mice. 13 However, no large‐scale human study has specifically addressed whether SI is connected with liver fibrogenesis in patients with ACLD. The present study confirms that fibrogenesis and ECM turnover are linked to SI, in particular CRP, IL‐6 and PCT levels. Importantly, we performed multivariate linear regression models adjusted by HVPG to account for potential confounding effects by the underlying severity of liver disease, which solidified these findings. CRP, IL‐6 and PCT displayed a consistent HVPG‐independent link with markers of fibrogenesis in the overall cohort and moreover a significant correlation with α‐SMA on liver histology in the subgroup of patients undergoing concomitant liver biopsy. More specifically, besides ELF score which comprises three different ECM biomarkers, the collagen formation markers PRO‐C3 and PRO‐C6 were linked to SI levels. Thus, SI appears to be not only linked to liver disease severity (i.e. ‘stage’), but also liver fibrogenesis as a dynamic process. Multivariate regression models indicated that systemic inflammation and liver fibrogenesis as well as ECM turnover (even when adjusting for HVPG) seem to be linked irrespective of disease stage, i.e. both in patients with cACLD and dACLD. However, we found that the link between HVPG and fibrogenesis markers was more consistent in patients with cACLD. These data suggest that fibrogenesis (responsible for the static component of increased intrahepatic resistance) and portal hypertension are less tightly interrelated in dACLD, which seems in line with the concept that the impact of SI on the progression of ACLD is particularly strong in the decompensated stage. 16 Nonetheless, despite the broad body of experimental evidence discussed above, we have to acknowledge that our clinical data cannot proof that SI directly causes HSC activation, which represents a limitation of this study. Therefore, further studies are warranted. Since interventions modulating SI may not only prevent the development of complications that are directly related to SI (e.g. AD, bacterial infections and ACLF), but may also prevent liver fibrosis progression, the assessment of markers of liver fibrogenesis/ECM turnover should be considered when designing clinical trials investigating disease‐modifying treatments that target SI. Of note, the potential impact of SI on liver fibrogenesis may also contribute to the non‐haemodynamic benefits of NSBB treatment. 46 , 47
Interestingly, besides indicators of fibrogenesis, the two markers reflecting matrix turnover, TIMP1 (also part of ELF score) and C3M, were independently associated with CRP, IL‐6 and PCT levels. These biomarkers reflect opposing states of matrix turnover, since TIMP1 impedes the degradation of ECM, while C3M indicates fibrolysis, as it depicts collagen type III degradation by macrophages via matrix metalloproteinases. 7 We have to acknowledge that our study cannot answer whether high absolute serum levels of C3M or their balance with PRO‐C3 in an individual patient may indicate that ECM remodelling tips towards fibrolysis, however, it may be speculated that increased liver fibrogenesis is paralleled by increased matrix turnover and degradation of collagen by compensatory mechanisms, thereby explaining the concordant upregulation of both mechanisms.
LBP levels have been used as surrogate parameter for BT in previous studies in patients with ACLD. 48 , 49 Controversially, we found that LBP levels did not clearly associate with fibrogenesis biomarkers in our study: LBP only showed a weak negative correlation with ELF, as well as positive association with C3M on multivariate regression analysis, despite exhibiting significant positive association with TIMP1. We can only speculate how to interpret these results, however, as our study strictly excluded patients with AD and bacterial infection at the timepoint of study inclusion, LBP may not represent an accurate marker of BT in clinically stable patients with ACLD. Similarly, WBC continuously decreased across PH strata, which denotes a difference from studies conducted in patients with AD. 30 Again, the selection of a stable patient collective may explain this difference, as PH and associated hypersplenism may have a stronger impact on WBC counts in these patients as compared to SI.
Finally, we observed a broadly similar picture in regard to the link between systemic inflammation (i.e. CRP) and fibrogenesis as well as ECM turnover (i.e. PRO‐C3 and C3M) markers in a patient cohort from Denmark, 22 thus, serving as external confirmation of our results. Previously reported data from this cohort already indicated a link between HVPG and different biomarkers of fibrogenesis and fibrolysis. 22 Of note, the study setup and patient characteristics were slightly differed from our study, as evident from the exclusive focus on patients with ALD and higher MELD in the Danish cohort. Furthermore, CRP levels in this cohort were measured after substantially longer storage duration, as compared to PRO‐C3/C3M. All these circumstances may account for differences regarding correlation strength between CRP and PRO‐C3 levels as compared to our cohort. Nevertheless, the similarities in results as compared to our cohort serve as further evidence towards the connection between systemic inflammation and liver fibrogenesis in clinically stable patients with ACLD. Concordantly, a recent publication also reported a link between systemic inflammation, fibrogenesis markers and α‐SMA on liver histology in patients with ACLF. 50
In summary, our study demonstrates that SI is linked to the dynamic process of fibrogenesis and fibrolysis in patients with ACLD. Blood biomarkers of ECM formation accurately reflect the intrahepatic activation of HSCs, as demonstrated by the strong correlations of α‐SMA area on liver histology and the ELF score as well as markers of collagen formation. Research efforts investigating whether therapeutic modulation of SI ameliorates liver disease progression are warranted.
AUTHORSHIP
The following authors contributed either to study concept and design (BeSi, TR, and MM) and/or data acquisition (all authors), analysis (BeSi, DR, TR and MM) or interpretation (all authors). BeSi and MM drafted the manuscript, which was critically revised by all other authors. All authors approved the final version of the manuscript.
FUNDING INFORMATION
BeSi and TR were supported by an International Liver Scholar by Gilead Sciences.
CONFLICT OF INTEREST
BeSi has received travel support from AbbVie and Gilead. IFV is a full‐time employee at Nordic Bioscience. PK was co‐supported by the Christian‐Doppler Society and Boehringer Ingelheim, and supported by the Medical Scientific Fund of the Mayor of the City of Vienna (Project: 18070) and awarded to PS. BeSc received travel support from Abbvie and Gilead. DB has received travel support from AbbVie and Gilead, as well as speaker fees from AbbVie. PS received speaking honoraria from Bristol‐Myers Squibb and Boehringer‐Ingelheim, consulting fees from PharmaIN, and travel support from Falk and Phenex Pharmaceuticals. MP is an investigator for Bayer, BMS, Lilly and Roche; he received speaker honoraria from Bayer, BMS, Eisai, Lilly and MSD; he is a consultant for Bayer, BMS, Ipsen, Eisai, Lilly, MSD and Roche; he received travel support from Bayer and BMS. SM received a grant from the NovoNordisk Foundation. MT received grant support from Albireo, Cymabay, Falk, Gilead, Intercept, MSD and Takeda, honoraria for consulting from BiomX, Boehringer Ingelheim, Falk, Genfit, Gilead, Intercept, Janssen, MSD, Novartis, Phenex and Regulus, speaker fees from BMS, Falk, Gilead, Intercept and MSD, as well as travel support from Abbvie, Falk, Gilead and Intercept. MK is a full‐time employee at Nordic Bioscience and among the original inventors and patent holders of the Nordic Bioscience biomarkers. DJL is a full‐time employee at Nordic Bioscience and among the original inventors and patent holders of the Nordic Bioscience biomarkers. TR received grant support from Abbvie, Boehringer‐Ingelheim, Gilead, MSD, Philips Healthcare, Gore; speaking honoraria from Abbvie, Gilead, Gore, Intercept, Roche, MSD; consulting/advisory board fee from Abbvie, Bayer, Boehringer‐Ingelheim, Gilead, Intercept, MSD, Siemens and travel support from Abbvie, Boehringer‐Ingelheim, Gilead and Roche. MM served as a speaker and/or consultant and/or advisory board member for AbbVie, Collective Acumen and W. L. Gore & Associates and received travel support from AbbVie and Gilead. RP, AFS, GT, DR, JS and KW declare no conflict of interest.
CLINICAL TRIAL NUMBER
Supporting information
Appendix S1
ACKNOWLEDGEMENTS
All authors have disclosed potential conflicts of interest (if applicable) and have read the journal's authorship agreement and policy on disclosure of potential conflicts of interest.
We thank our laboratory technicians, Kerstin Zinober and Martha Seif, for their excellent work to ensure the maintenance and quality of the VICIS biobank, and our nurses for their magnificent support for performing hepatic vein catheterizations and transjugular liver biopsies at the Vienna Hepatic Hemodynamic Lab.
Simbrunner B, Villesen IF, Königshofer P, et al. Systemic inflammation is linked to liver fibrogenesis in patients with advanced chronic liver disease. Liver Int. 2022;42:2501‐2512. doi: 10.1111/liv.15365
Handling Editor: Alejandro Forner
REFERENCES
- 1. Israelsen M, Guerrero Misas M, Koutsoumourakis A, et al. Collagen proportionate area predicts clinical outcomes in patients with alcohol‐related liver disease. Aliment Pharmacol Ther. 2020;52(11–12):1728‐1739. [DOI] [PubMed] [Google Scholar]
- 2. Buzzetti E, Hall A, Ekstedt M, et al. Collagen proportionate area is an independent predictor of long‐term outcome in patients with non‐alcoholic fatty liver disease. Aliment Pharmacol Ther. 2019;49(9):1214‐1222. [DOI] [PubMed] [Google Scholar]
- 3. Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371(9615):838‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schwabl P, Hambruch E, Seeland BA, et al. The FXR agonist PX20606 ameliorates portal hypertension by targeting vascular remodelling and sinusoidal dysfunction. J Hepatol. 2017;66(4):724‐733. [DOI] [PubMed] [Google Scholar]
- 5. Schwabl P, Brusilovskaya K, Supper P, et al. The soluble guanylate cyclase stimulator riociguat reduces fibrogenesis and portal pressure in cirrhotic rats. Sci Rep. 2018;8(1):9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bosch J, Groszmann RJ, Shah VH. Evolution in the understanding of the pathophysiological basis of portal hypertension: how changes in paradigm are leading to successful new treatments. J Hepatol. 2015;62(1 Suppl):S121‐S130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karsdal MA, Daniels SJ, Holm Nielsen S, et al. Collagen biology and non‐invasive biomarkers of liver fibrosis. Liver Int. 2020;40(4):736‐750. [DOI] [PubMed] [Google Scholar]
- 8. Simbrunner B, Mandorfer M, Trauner M, Reiberger T. Gut‐liver axis signaling in portal hypertension. World J Gastroenterol. 2019;25(39):5897‐5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Teltschik Z, Wiest R, Beisner J, et al. Intestinal bacterial translocation in rats with cirrhosis is related to compromised Paneth cell antimicrobial host defense. Hepatology. 2012;55(4):1154‐1163. [DOI] [PubMed] [Google Scholar]
- 10. Guarner C, González‐Navajas JM, Sánchez E, et al. The detection of bacterial DNA in blood of rats with CCl4‐induced cirrhosis with ascites represents episodes of bacterial translocation. Hepatology. 2006;44(3):633‐639. [DOI] [PubMed] [Google Scholar]
- 11. Gómez‐Hurtado I, Gallego‐Durán R, Zapater P, et al. Bacterial antigen translocation and age as BMI‐independent contributing factors on systemic inflammation in NAFLD patients. Liver Int. 2020;40(9):2182‐2193. [DOI] [PubMed] [Google Scholar]
- 12. Caro E, Francés R, Zapater P, Pascual S, Bellot P, Such J. Grade of soluble inflammatory response is mainly affected by circulating bacterial DNA concentrations in cirrhosis. Liver Int. 2016;36(10):1473‐1480. [DOI] [PubMed] [Google Scholar]
- 13. Seki E, de Minicis S, Österreicher CH, et al. TLR4 enhances TGF‐beta signaling and hepatic fibrosis. Nat Med. 2007;13(11):1324‐1332. [DOI] [PubMed] [Google Scholar]
- 14. Rockey DC, Boyles JK, Gabbiani G, Friedman SL. Rat hepatic lipocytes express smooth muscle Actin upon activation in vivo and in culture. J Submicrosc Cytol Pathol. 1992;24(2):193‐203. [PubMed] [Google Scholar]
- 15. Seki E, Schnabl B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J Physiol. 2012;590(3):447‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arroyo V, Angeli P, Moreau R, et al. The systemic inflammation hypothesis: towards a new paradigm of acute decompensation and multiorgan failure in cirrhosis. J Hepatol. 2021;74(3):670‐685. [DOI] [PubMed] [Google Scholar]
- 17. Reiberger T, Püspök A, Schoder M, et al. Austrian consensus guidelines on the management and treatment of portal hypertension (Billroth III). Wien Klin Wochenschr. 2017;129(Suppl 3):135‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. EASL clinical practice guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69(2):406‐460. [DOI] [PubMed] [Google Scholar]
- 19. Reiberger T, Schwabl P, Trauner M, Peck‐Radosavljevic M, Mandorfer M. Measurement of the hepatic venous pressure gradient and Transjugular liver biopsy. J Vis Exp. 2020;160. [DOI] [PubMed] [Google Scholar]
- 20. Simbrunner B, Marculescu R, Scheiner B, et al. Non‐invasive detection of portal hypertension by enhanced liver fibrosis score in patients with different etiologies of advanced chronic liver disease. Liver Int. 2020;40:1713‐1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simbrunner B, Semmler G, Stadlmann A, et al. Vitamin a levels reflect disease severity and portal hypertension in patients with cirrhosis. Hepatol Int. 2020;14:1093‐1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leeming DJ, Karsdal MA, Byrjalsen I, et al. Novel serological neo‐epitope markers of extracellular matrix proteins for the detection of portal hypertension. Aliment Pharmacol Ther. 2013;38(9):1086‐1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mortensen C, Andersen O, Krag A, Bendtsen F, Møller S. High‐sensitivity C‐reactive protein levels predict survival and are related to haemodynamics in alcoholic cirrhosis. Eur J Gastroenterol Hepatol. 2012;24(6):619‐626. [DOI] [PubMed] [Google Scholar]
- 24. Praktiknjo M, Lehmann J, Nielsen MJ, et al. Acute decompensation boosts hepatic collagen type III deposition and deteriorates experimental and human cirrhosis. Hepatol Commun. 2018;2(2):211‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simbrunner B, Trauner M, Reiberger T. Therapeutic aspects of bile acid signalling in the gut‐liver axis. Aliment Pharmacol Ther. 2021;54:1243‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tranah TH, Edwards LA, Schnabl B, Shawcross DL. Targeting the gut‐liver‐immune axis to treat cirrhosis. Gut. 2020;70:982‐994. [DOI] [PubMed] [Google Scholar]
- 27. Ubeda M, Lario M, Muñoz L, et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol. 2016;64(5):1049‐1057. [DOI] [PubMed] [Google Scholar]
- 28. Verbeke L, Farre R, Verbinnen B, et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am J Pathol. 2015;185(2):409‐419. [DOI] [PubMed] [Google Scholar]
- 29. Sorribas M, Jakob MO, Yilmaz B, et al. FxR‐modulates the gut‐vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J Hepatol. 2019;71:1126‐1140. [DOI] [PubMed] [Google Scholar]
- 30. Trebicka J, Fernandez J, Papp M, et al. The PREDICT study uncovers three clinical courses of acutely decompensated cirrhosis that have distinct pathophysiology. J Hepatol. 2020;73:842‐854. [DOI] [PubMed] [Google Scholar]
- 31. Moreau R, Jalan R, Gines P, et al. Acute‐on‐chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology, 2013. 144(7): p. 1426–37. e1‐9, 1437.e9. [DOI] [PubMed] [Google Scholar]
- 32. Costa D, Simbrunner B, Jachs M, et al. Systemic inflammation increases across distinct stages of advanced chronic liver disease and correlates with decompensation and mortality. J Hepatol. 2021;74(4):819‐828. [DOI] [PubMed] [Google Scholar]
- 33. Ignat SR, Dinescu S, Hermenean A, Costache M. Cellular interplay as a consequence of inflammatory signals leading to liver fibrosis development. Cell. 2020;9(2):461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Parkes J, Guha IN, Roderick P, et al. Enhanced liver fibrosis (ELF) test accurately identifies liver fibrosis in patients with chronic hepatitis C. J Viral Hepat. 2011;18(1):23‐31. [DOI] [PubMed] [Google Scholar]
- 36. Thiele M, Madsen BS, Hansen JF, Detlefsen S, Antonsen S, Krag A. Accuracy of the enhanced liver fibrosis test vs FibroTest, elastography, and indirect markers in detection of advanced fibrosis in patients with alcoholic liver disease. Gastroenterology. 2018;154(5):1369‐1379. [DOI] [PubMed] [Google Scholar]
- 37. Lichtinghagen R, Pietsch D, Bantel H, Manns MP, Brand K, Bahr MJ. The enhanced liver fibrosis (ELF) score: normal values, influence factors and proposed cut‐off values. J Hepatol. 2013;59(2):236‐242. [DOI] [PubMed] [Google Scholar]
- 38. Stasi C, Tsochatzis EA, Hall A, et al. Comparison and correlation of fibrosis stage assessment by collagen proportionate area (CPA) and the ELF panel in patients with chronic liver disease. Dig Liver Dis. 2019;51(7):1001‐1007. [DOI] [PubMed] [Google Scholar]
- 39. Dold L, Nielsen MJ, Praktiknjo M, et al. Circulating levels of PRO‐C3 reflect liver fibrosis and liver function in HIV positive patients receiving modern cART. PLoS One. 2019;14(7):e0219526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Daniels SJ, Leeming DJ, Eslam M, et al. ADAPT: an algorithm incorporating PRO‐C3 accurately identifies patients with NAFLD and advanced fibrosis. Hepatology. 2019;69(3):1075‐1086. [DOI] [PubMed] [Google Scholar]
- 41. Luo Y, Oseini A, Gagnon R, et al. An evaluation of the collagen fragments related to fibrogenesis and Fibrolysis in nonalcoholic steatohepatitis. Sci Rep. 2018;8(1):12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laursen TL, Villesen IF, Leeming DJ, et al. Altered balance between collagen formation and degradation after successful direct‐acting antiviral therapy of chronic hepatitis C. J Viral Hepat. 2020;28:236‐244. [DOI] [PubMed] [Google Scholar]
- 43. Mauro E, Crespo G, Montironi C, et al. Portal pressure and liver stiffness measurements in the prediction of fibrosis regression after sustained virological response in recurrent hepatitis C. Hepatology. 2018;67(5):1683‐1694. [DOI] [PubMed] [Google Scholar]
- 44. Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll‐like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37(5):1043‐1055. [DOI] [PubMed] [Google Scholar]
- 45. Brun P, Castagliuolo I, Pinzani M, Palù G, Martines D. Exposure to bacterial cell wall products triggers an inflammatory phenotype in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2005;289(3):G571‐G578. [DOI] [PubMed] [Google Scholar]
- 46. Jachs M, Hartl L, Schaufler D, et al. Amelioration of systemic inflammation in advanced chronic liver disease upon beta‐blocker therapy translates into improved clinical outcomes. Gut. 2020;70:1758‐1767. [DOI] [PubMed] [Google Scholar]
- 47. Reiberger T, Mandorfer M. Beta adrenergic blockade and decompensated cirrhosis. J Hepatol. 2017;66(4):849‐859. [DOI] [PubMed] [Google Scholar]
- 48. Albillos A, de la Hera A, González M, et al. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology. 2003;37(1):208‐217. [DOI] [PubMed] [Google Scholar]
- 49. Reiberger T, Ferlitsch A, Payer BA, et al. Non‐selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL‐6 in patients with cirrhosis. J Hepatol. 2013;58(5):911‐921. [DOI] [PubMed] [Google Scholar]
- 50. Kerbert AJC, Gupta S, Alabsawy E, et al. Biomarkers of extracellular matrix formation are associated with acute‐on‐chronic liver failure. JHEP Rep. 2021;3(6):100355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1