

Abstract
The Notch signaling pathway is a key regulator of skeletal muscle development and regeneration. Over the past decade, the discoveries of three new muscle disease genes have added a new dimension to the relationship between the Notch signaling pathway and skeletal muscle: MEGF10, POGLUT1, and JAG2. We review the clinical syndromes associated with pathogenic variants in each of these genes, known molecular and cellular functions of their protein products with a particular focus on the Notch signaling pathway, and potential novel therapeutic targets that may emerge from further investigations of these diseases. The phenotypes associated with two of these genes, POGLUT1 and JAG2, clearly fall within the realm of muscular dystrophy, whereas the third, MEGF10, is associated with a congenital myopathy/muscular dystrophy overlap syndrome classically known as early‐onset myopathy, areflexia, respiratory distress, and dysphagia. JAG2 is a canonical Notch ligand, POGLUT1 glycosylates the extracellular domain of Notch receptors, and MEGF10 interacts with the intracellular domain of NOTCH1. Additional genes and their encoded proteins relevant to muscle function and disease with links to the Notch signaling pathway include TRIM32, ATP2A1 (SERCA1), JAG1, PAX7, and NOTCH2NLC. There is enormous potential to identify convergent mechanisms of skeletal muscle disease and new therapeutic targets through further investigations of the Notch signaling pathway in the context of skeletal muscle development, maintenance, and disease.
Keywords: JAG2, MEGF10, muscular dystrophy, Notch signaling pathway, POGLUT1
Abbreviations
- ABCA1
adenosine triphosphate–binding cassette transporter 1
- ADAM10
A disintegrin and metalloproteinase domain–containing protein 10
- ADAMTS1
A disintegrin‐like and metalloproteinase with thrombospondin type 1 motif
- BMD
Becker muscular dystrophy
- C1q
complement component 1q
- CK
creatine kinase
- CMD
congenital muscular dystrophy
- CSL
CBF‐1/RBPJ‐κ, Suppressor of Hairless, Lag‐1
- DLL1
delta‐like canonical Notch ligand 1
- DLL4
delta‐like canonical Notch ligand 4
- DMD
Duchenne muscular dystrophy
- DOS
Delta and OS M‐11‐like protein
- DRPR
Draper
- DSL
Delta‐Serrate‐LAG2
- EC
endothelial cell
- EGF
epidermal growth factor–like domain
- EMARDD
early‐onset myopathy, areflexia, respiratory distress, and dysphagia
- EMG
electromyography
- EMI
elastin microfibril interfacer 1
- FDA
US Food and Drug Administration
- HEK293
human embryonic kidney
- hnRNPL
heterogeneous nuclear ribonucleoprotein L
- ICD
intracellular domain
- IL‐4
interleukin‐4
- ITAM
immunoreceptor tyrosine‐based activator motif
- ITIM
immunoreceptor tyrosine‐based inhibitory motif
- JAG1
Jagged1
- JAG2
Jagged2
- LGMD
limb‐girdle muscular dystrophy
- MAML1
mastermind‐like 1
- MEGF10
multiple epidermal growth factor–like domains protein 10
- MEGF11
multiple epidermal growth factor–like domains protein 11
- MEGF12
multiple epidermal growth factor–like domains protein 12
- MuSC
muscle stem cell
- NCS
nerve conduction studies
- NECD
Notch extracellular domain
- NFATc2
nuclear factor of activated T cells c2
- N1ICD
Notch1 intracellular domain
- NICD
Notch intracellular domain
- NIID
neuronal intranuclear inclusion disease
- NPxY
Asn‐Pro‐x‐Tyr
- OMIM
online Mendelian Inheritance in Man
- OPDM
oculopharyngodistal myopathy
- PAX7
paired box 7
- POFUT1
protein‐O‐fucosyltransferase 1
- POGLUT1
protein‐O‐glucosyltransferase 1
- RNAi
RNA interference
- SERCA
sarco/endoplasmic reticulum Ca ATPase
- SMARD1
spinal muscular atrophy with respiratory distress 1
- TM
transmembrane domain
- TRIM32
tripartite motif containing 32
1. INTRODUCTION
Since the landmark discovery in 1986 of DMD (dystrophin), 1 the causative gene for Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD), dozens of additional genes have been associated with various phenotypic subtypes of muscular dystrophy. Common disease mechanisms across multiple subtypes have, however, been more difficult to identify, with only a few major clusters such as the dystroglycanopathies identified to date. Given the common phenotypic features within muscular dystrophy categories, such as limb‐girdle muscular dystrophy (LGMD), there is a high likelihood that convergent disease mechanisms exist across more muscular dystrophy subtypes than is currently recognized.
There are therapeutic implications of identifying deeper biological ties between muscular dystrophy subtypes. In recent years, the US Food and Drug Administration (FDA) has approved several molecular and genetic therapies for neuromuscular disorders that target specific genes and even specific mutation types within those genes. These approaches are being applied to ever rarer forms of muscular dystrophies. However, proceeding through the preclinical and clinical research studies needed to attain FDA approval for a new therapy is lengthy and costly, and on the current trajectory it will be decades before molecular and genetic therapies are available for all known subtypes of muscular dystrophy.
The identification and characterization of disease mechanisms that are shared by multiple muscular dystrophy subtypes could pave the way for new pathway‐based treatments that have therapeutic effects for multiple disease subtypes. 2 This has the potential to accelerate the timeline for broader therapeutic coverage of patients with muscular dystrophy, with a greater impact on the entire muscular dystrophy population.
One disease mechanism that bears further analysis is the Notch signaling pathway, which is known to maintain muscle stem cell (MuSC, also known as satellite cell) quiescence. Recently, three different muscle disease genes that are known to interact with the Notch signaling pathway have been identified: MEGF10, POGLUT1, and most recently JAG2. In this review we examine the clinical, genetic, biochemical, and cellular knowledge of these genes and their protein products, as well as their interactions with each other and with the Notch signaling pathway.
1.1. The Notch signaling pathway in muscle development
The Notch signaling pathway has been a high‐profile subject of investigation since its discovery in the early 20th century, with intensified interest after the Drosophila Notch gene was first reported in 1983. 3 This pathway is well‐conserved across species and is regulated by a set of ligands and receptors that promote cell‐to‐cell communications. Among other activities, it plays a key role in determining specification and differentiation of cell fates during various aspects of invertebrate and vertebrate development and regeneration, including myogenesis. 4 , 5 , 6 , 7
In mammals, Notch1, Notch2, Notch3, and Notch4 are the core receptors in the Notch signaling pathway (Table 1). Each of these receptors consists of a Notch extracellular domain (NECD), a transmembrane domain (TM), and a Notch intracellular domain (NICD). 27 Both trans (intercellular) and cis (intracellular) interactions of ligands with the Notch receptors have been discovered 28 , 29 , 30 (Figure 1). In mammals, the canonical Notch ligands include Delta‐like1, Delta‐like3, Delta‐like4, Jagged1, and Jagged2 31 (Table 1). These proteins are encoded by the genes DLL1, DLL3, DLL4, JAG1, and JAG2, respectively. In Drosophila there are two canonical Notch ligands: Delta and Serrate (orthologous to the mammalian Delta‐like and Jagged proteins, respectively). 32 The typical trans interaction begins with binding of the extracellular domain of the ligand from the signaling cell to the NECD of the signal receiving cell, initiating two cleavage events in the Notch receptor of the receiving cell (Figure 1A). In the first cleavage event, members of the ADAM family of metalloproteinases separate the ligand‐bound NECD from the TM‐NICD. 27 The NECD undergoes endocytosis by the signaling cell, whereas the TM domain and NICD are separated from each other by γ‐secretase in the second cleavage event. 33 The NICD then enters the nucleus and binds to the DNA transcription factor CBF‐1/RBPJ‐κ, Suppressor of Hairless, Lag‐1 (also known as CSL), converting it from a transcription‐repressing state to an activating state by displacing co‐repressors and recruiting Mastermind‐like protein (MAML1) and other coactivators, initiating the transcription of downstream Notch signaling pathway genes. 34 , 35 Notch ligands also have an inhibitory effect on Notch within the same cells (cis‐inhibition) of Drosophila. 36 , 37 , 38 The extracellular Delta‐Serrate‐LAG2 (DSL) domain of human Jagged1 contributes to both trans‐activation and cis‐inhibition. 39 Most of the canonical Notch signaling pathway components have known functions in skeletal muscle development or function, yet aside from JAG2, the other canonical Notch signaling pathway genes are associated with diseases that do not primarily manifest in skeletal muscle (Table 1).
TABLE 1.
Expression and function of canonical Notch signaling pathway components in skeletal muscle
| Notch component | RNA expression | Protein expression | Muscle function | Disease association |
|---|---|---|---|---|
| NOTCH1 | 5.0 nTPM | Medium | Regulates MuSC fates 8 | Adams‐Oliver syndrome 5, 9 aortic valve disease 1 10 |
| NOTCH2 | 6.5 nTPM | Not detected | MuSC self‐renewal 11 | Alagille syndrome 2, 12 Hajdu‐Cheney syndrome 13 |
| NOTCH3 | 30.0 nTPM | Low | Inhibits Notch1 in MuSCs 14 | CADASIL 15 |
| NOTCH4 | 5.7 nTPM | No data | Unknown | None known |
| DLL1 | 5.4 nTPM | No data | Inhibits myoblast differentiation 16 | Neurodevelopmental disorder 17 |
| DLL3 | No data | No data | Unknown | Spondylocostal dysostosis 18 |
| DLL4 | 10.1 nTPM | Low | Regulates skeletal muscle mass 19 | Adams‐Oliver syndrome 6 20 |
| JAG1 | 9.8 nTPM | Medium | Rescues DMD in dogs 21 | Alagille syndrome 1, 22 Charcot‐Marie‐Tooth disease 2HH, 23 tetralogy of Fallot 24 |
| JAG2 | 9.7 nTPM | Medium | Canonical Notch ligand | JAG2‐related muscular dystrophy 25 |
Note: RNA and protein expression data from the Human Protein Atlas. 26 Protein expression levels were measured in myocytes.
Abbreviations: CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; DLL, delta‐like canonical Notch ligand; DMD, Duchenne muscular dystrophy; MuSC, muscle stem cell; nTPM, normalized transcripts per million.
FIGURE 1.
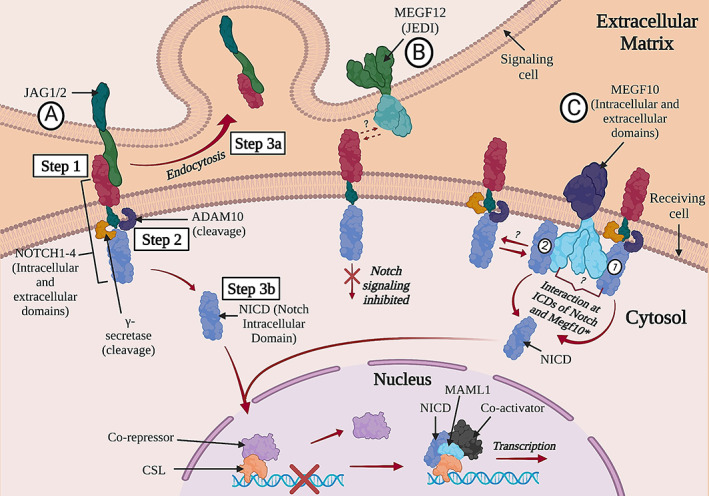
Diagram of the Notch signaling pathway showing putative interactions between Notch receptors and several key molecules of interest: JAG1/JAG2 (A); MEGF12, also known as JEDI (B); and MEGF10 (C). Several key steps in the interaction between a Notch receptor and a ligand are numbered in the diagram. Abbreviations: ADAM10, A Disintegrin and metalloproteinase domain–containing protein 10; CSL, CBF‐1/RBPJ‐κ, Suppressor of Hairless, LAG‐1; JAG1/2, Jagged1/2; MAML1, mastermind‐like1; MEGF10/12, multiple epidermal growth factor–like domains protein 10/12; NECD, notch extracellular domain; NICD, Notch intracellular domain; TM, transmembrane domain.
The Notch1 intracellular domain (N1ICD) is an active regulator of cell fate choice for MuSCs, promoting their self‐renewal via Pax7 upregulation while inhibiting MuSC proliferation. 40 The N1ICD also has the capability of dedifferentiating myocytes into Pax7+ quiescent MuSCs, suggesting that the Notch signaling pathway has potential as a therapeutic target for muscle diseases. 41 The mechanisms of Notch signaling pathway regulation of MuSCs remains incompletely understood, but one key component is A disintegrin‐like and metalloproteinase with thrombospondin type 1 motif (ADAMTS1), which is secreted by macrophages, targets NOTCH1, stimulates MuSC activation, and promotes muscle regeneration. 42 Notch signaling and p53 activity diminish with aging, and stabilizing this Notch‐p53 axis improves the regenerative capacity of aged MuSCs. 43
The Notch signaling pathway regulates other components of developing and mature skeletal muscle. For example, delta‐like canonical Notch ligand 1 (DLL1) activation restrains fibroadipogenic progenitor (FAP) differentiation, yet dystrophin‐deficient FAPs are unresponsive to this regulatory mechanism. 44 There is emerging evidence that Delta‐like canonical Notch ligand 4 (DLL4) derived from muscle endothelial cells (ECs) induces quiescence in MuSCs. 45 With regard to potential immune system interactions, protein O‐fucosyltransferase 1 (POFUT1) modulates myogenesis via Notch signaling 46 and myoblast fusion via nuclear factor of activated T cells c2/interleukin‐4 (NFATc2/IL‐4) signaling. 47 POFUT1 deficiency in skeletal myofibers is also associated with reduced Notch signaling and degeneration of motor nerve innervation at the neuromuscular junction. 48
A growing list of noncanonical or less‐characterized Notch ligands that fine‐tune Notch signaling is becoming recognized. Among these is MEGF12 (also known as JEDI or PEAR1), a transmembrane protein that has an inhibitory role in the Notch signaling pathway. 49 Although the specific mechanism of this interaction is yet to be understood, it is hypothesized that MEGF12 competes with other Notch ligands, such as Jagged1/2, to exert its inhibitory effect by repressing Notch activation 49 (Figure 1B). Notably, MEGF12 is a paralog of the muscle disease gene MEGF10 (Figure 1C), which is discussed in greater depth in the following sections.
1.2. MEGF10 myopathy
Individuals with clinical features similar to spinal muscular atrophy with respiratory distress type 1 (SMARD1) but with muscle histological features indicating a primary myopathy, were described in 2007. 50 The affected individuals in the initial case series had infantile‐onset weakness with early respiratory failure, normal echocardiograms, normal to mildly elevated serum creatine kinase (CK) levels, normal velocities on nerve conduction studies (NCS), and myopathic findings on electromyography (EMG). 50 Some affected individuals achieve independent ambulation, typically with some limitations. The disease was named early‐onset myopathy, areflexia, respiratory distress, and dysphagia (EMARDD, OMIM 614399). Several years later, biallelic pathogenic variants in the gene MEGF10 were discovered in a set of individuals affected by EMARDD, 51 followed by additional reports substantiating the initial findings and expanding the phenotype to include some features of muscular dystrophy, cleft palate in some affected individuals, and in some individuals a milder clinical course. 52 , 53 , 54 , 55 , 56 , 57 , 58 Histological features on muscle biopsies range from mild fiber size variability to dystrophic findings to minicores. 50 , 52 These diseases are now collectively referred to as MEGF10 myopathy to reflect the phenotypic diversity. A range of pathogenic variants has been described for MEGF10 (Figure 2A,B), 51 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59 with a high degree of conservation of affected amino acid residues (Figure S1). MEGF10 myopathy could be classified as an ultrarare disease; aside from the first article linking the disease to pathogenic variants in MEGF10, most subsequent reports in the literature describe one or two kindreds.
FIGURE 2.
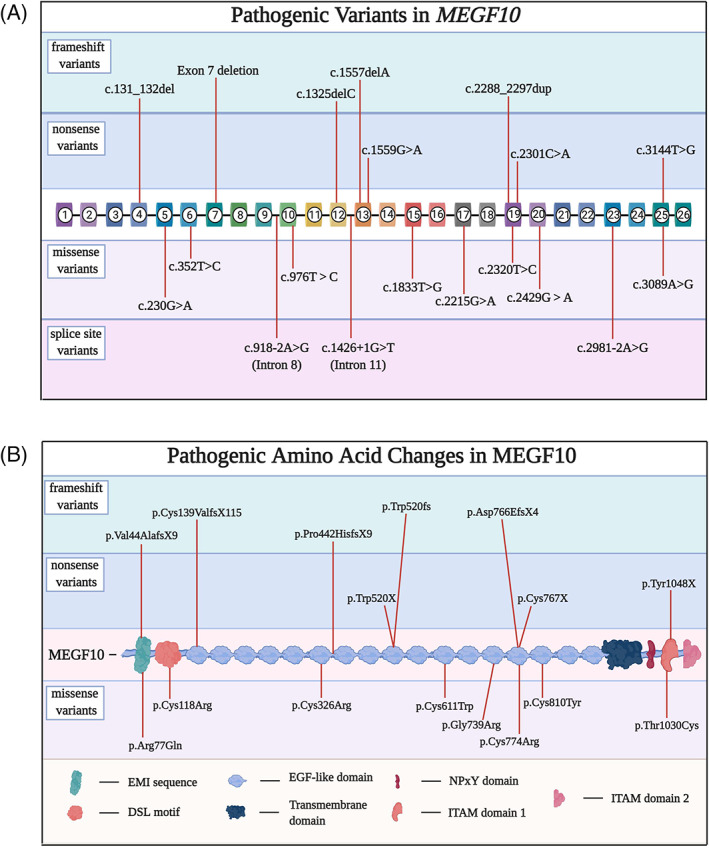
A, Diagram of pathogenic variants in MEGF10 mutations categorized by variant type: nonsense, frameshift, missense, and splice site. Variants’ positions were determined using the reference human MEGF10 transcript variant 1 (NM_032446.3). B, Diagram of amino acids in the human MEGF10 protein affected by pathogenic variants.
1.3. MEGF10 and its orthologs and paralogs
The human gene MEGF10 is located on chromosome 5q23.2 60 ; the longest known transcript contains 25 exons with a repetitive element at the 3′ end. The gene is highly expressed in the fetal and adult brain, adult spinal cord, and regenerating skeletal muscle. 61 , 62 The protein product MEGF10 is a single transmembrane 1147 amino acid protein with an N‐terminal extracellular signal sequence, an elastin microfibril interfacer 1 (EMI) domain, a DSL motif, 17 epidermal growth factor–like domain (EGF)‐like domains, a transmembrane domain, and a C‐terminal intracellular domain (ICD) with 13 tyrosine residues. 60 , 61 , 63 Homologous genes are descended from a common ancestral gene; orthologs are homologous genes found in different organisms, and paralogs are homologous genes that are found in the same genomes of the same organisms. The mouse and zebrafish orthologs also have an EMI domain, a DSL motif, 17 EGF‐like domains and a transmembrane domain. Several isoforms of the orthologous Drosophila protein Drpr have been identified, and three of them, named Drpr A, B, and C, are well‐characterized (these were previously known as Drpr II, I, and III, respectively). 64 , 65 Each has an EMI domain, a DSL motif, and a transmembrane domain as well. However, the Drpr proteins differ from their human, mouse, and zebrafish counterparts regarding the number of EGF‐like domains; Drpr B has 14 of these domains, whereas Drpr A and C each has 4. Drpr A has an immunoreceptor tyrosine‐based inhibitory motif (ITIM) domain that is not seen in the other isoforms (Figure 3). There is generally good conservation of key domains among these orthologs and isoforms (Figure 3 and Table S1).
FIGURE 3.
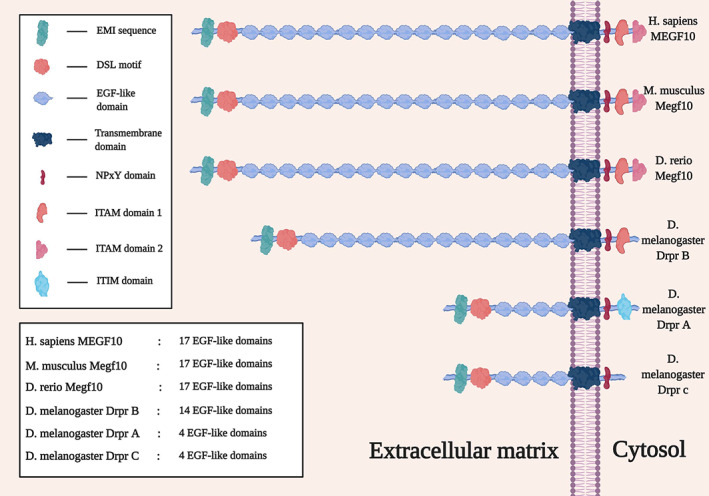
Isoforms of the Megf10 protein in human, mouse, zebrafish, and fruit fly. Each isoform has an EMI domain, a Delta and OS M‐11‐like (DSL) motif, epidermal growth factor–like domains, a transmembrane domain, and an Asn‐Pro‐x‐Tyr (NPxY) domain. Immunoreceptor tyrosine‐based activator motif (ITAM) domain 2 is only conserved in human, mouse, and zebrafish. ITAM domain 1 is very well conserved across species. Drpr A has an immunoreceptor tyrosine‐based inhibitory motif (ITIM) domain, which is not present in the other isoforms or species.
There are two mammalian paralogs of MEGF10: MEGF11 and MEGF12. 49 , 60 Despite expression of MEGF10 in the brain and retina, patients with EMARDD do not have structural or functional brain abnormalities, or visual defects. 51 , 52 However, deficiencies in Drpr, the Drosophila homolog of MEGF10, MEGF11, and MEGF12, lead to muscle and brain defects in fruit flies. 66 These observations suggest that functional redundancy by MEGF11 and/or MEGF12 may compensate for MEGF10 deficiency in mammalian brains. MEGF12, which encodes a transmembrane protein, contains 14 EGF‐like repeats and a DSL domain. MEGF10 and MEGF11 are known to have a small but significant protein structure homology with MEGF12 at their extracellular domains. 60
1.4. Animal models of MEGF10 deficiency (Mus musculus, Danio rerio, and Drosophila melangogaster)
Expression of Megf10 at neuromuscular junctions in Mus musculus suggested a role of Megf10 in neuromuscular transmission, a process that allows for communication between the central nervous system and skeletal muscle. 51 Repetitive nerve stimulation studies in the first reported cases of EMARDD were normal and trials of cholinesterase inhibitors were not therapeutic, suggesting that there is not a dramatic physiological defect in the neuromuscular junction in the setting of human MEGF10 myopathy. 50
A Cre‐mediated Megf10 knockout (Megf10 −/− ) mouse model was originally created for the study of retinal neurons. 67 Our laboratory reported a neuromuscular phenotype in skeletal muscle tissue and MuSCs from these Megf10 −/− mice. 68 Megf10 −/− mice showed reduced motor activity, and their skeletal muscles displayed mild endomysial fibrosis and intracellular infiltration upon intraperitoneal injections of Evans blue dye. 68 Intramuscular barium chloride injections led to impaired muscle regeneration in Megf10 −/− mice compared with wild‐type mice, providing more evidence of Megf10 involvement in myofiber regeneration. 68 , 69
Morpholino knockdown of megf10 in Danio rerio (zebrafish) resulted in muscle phenotype abnormalities similar to those reported in humans with mutations in MEGF10. 52 Zebrafish with megf10 knockdown had curved tails, difficulty swimming, and disorganized muscle morphology. Subsequently, a zebrafish line with a germline nonsense mutation demonstrated muscle defects and delayed somite formations. 54 Zebrafish models are thus useful for elucidating disease mechanisms for MEGF10 myopathy.
Drosophila melanogaster, commonly known as the fruit fly, is a useful system for modeling multiple human diseases, including neurological and neuromuscular diseases. Drosophila can recapitulate structural and functional features of human neuromuscular diseases. Key cellular and molecular processes are shared between Drosophila and humans, including the neuromuscular unit. 70 , 71 Two Drosophila genetic models of Drpr (ortholog of MEGF10) deficiency in skeletal muscle have been studied. One is an amorphic allele containing a deletion in the promoter and first exon of the drpr gene, and the other is a knockdown of drpr mediated by RNA interference (RNAi) in muscle. 66 , 72 In both genetically modified Drosophila models, the fruit flies showed abnormal position of legs, decreased locomotor activity, and pathological alterations in the thoracic striated muscle. In contrast, overexpression of Drpr in fly muscle resulted in pre‐adult lethality (toxic) when targeted at specific stages of myogenesis. 65 Escaper flies that survived presented evident muscle abnormalities. The gain‐of‐function of Drpr in Serrate‐positive wing cells caused extra branching at the wing margin, which phenocopies wing vein defects observed with Notch loss of function. 73 Further investigations of the spatiotemporal expression of MEGF10 and its orthologs will be crucial to understanding its function and provide insight into potential therapies.
1.5. Megf10 in the central nervous system
In the eye, MEGF10 contributes to the formation of retinal mosaics. 67 Elsewhere in the central nervous system, MEGF10 binds to dead neurons, contributing to the engulfment activities of glial cells 74 and phagocytic activities of neurons, 75 indicating a key role in neuronal apoptosis. Parallel functions have been identified for the Drosophila ortholog Drpr. 76 , 77 , 78 The ATP binding cassette transporter ABCA1 contributes to the engulfment activity of MEGF10. 79 MEGF10 is a receptor for C1Q, a signaling molecule that marks apoptotic cells. 80 This binding interaction is impaired when pathogenic variants in MEGF10 are expressed on human embryonic kidney‐293 (HEK‐293) cells. 80 A discovery with neurodevelopmental implications focuses on contributions of MEGF10 to the engulfment and phagocytosis of neuronal synapses, suggesting a role for MEGF10 in the synaptic pruning process. 81 At the other end of the lifespan, MEGF10 has been found to mediate uptake of amyloid‐β into neuroblastoma cells, suggesting that a deficiency of MEGF10 may contribute to the accumulation of senile plaques containing amyloid‐β in Alzheimer disease. 82
1.6. MEGF10 in skeletal muscle development and regeneration
During muscle development, Notch signaling regulates myoblast proliferation, migration, and differentiation 4 , 5 , 6 and cell adhesion activities. 7 Activation of the Notch signaling pathway helps maintain MuSCs in the quiescent state, 83 , 84 and contributes to their self‐renewal 40 and to their homing to target myofibers. 85 In particular, it is likely that MEGF10 contributes to the regulation of some of these processes in myoblasts and MuSCs.
Megf10 expression is higher during myoblast proliferation than differentiation 68 , 86 and Megf10 deficiency impairs myoblast proliferation. 68 In Drosophila, muscle and motor phenotypes were observed with knockdown of drpr (the fly homolog of Megf10) in adult muscle precursors. 66 Overexpression of Drpr, however, was deleterious at the differentiation and specification stages. 65 Drosophila thus provides an additional tool to further probe the role of Megf10 in regulating myoblast proliferation and myoblast differentiation.
Murine MuSCs that express Pax7 also express Megf10, 62 and expression of both Megf10 and myogenin spikes after cardiotoxin‐induced skeletal muscle injury in mice. 87 Myogenin is a transcription factor that binds sequences upstream of the Megf10 gene and activates its expression, 87 suggesting that it helps trigger a Megf10‐mediated response of MuSCs to muscle injury. In the setting of muscle injury induced by intramuscular barium chloride injections, Megf10 deficiency is associated with reduced regenerative potential. 69
In addition, MEGF10 promotes the adhesion of HEK‐293 cell membranes to substrates, 63 and Megf10 deficiency reduces murine myoblast adhesion capabilities. 68 It is not clear how adhesion may contribute to muscle development or repair, but myoblast 68 and MuSC 69 migration are both impaired in the setting of Megf10 deficiency, suggesting that movement and positioning of these muscle cells during these processes rely in part on Megf10 function.
1.7. Interactions of MEGF10/Megf10/Drpr with the Notch signaling pathway
Reports from several groups, 49 , 62 , 85 including ours, 68 demonstrate that MEGF10 and Drpr interact with the highly conserved Notch signaling pathway. The canonical DSL and Delta and OS M‐11‐like protein (DOS) domains, which are established Notch ligand motifs, are both found in the extracellular N termini of MEGF10, MEGF11, MEGF12, and Drpr, suggesting that these proteins may act as Notch ligands. 49 Our work has shown that MEGF10 and Notch interact at their intracellular domains, that pathogenic mutations impair these interactions, and that part of the MEGF10 interacting domain lies in the stretch from M1003 to E1140 68 (Figure 1C). Thus, MEGF10 does not appear to be a canonical Notch ligand. MEGF10 and MEGF12 are tyrosine phosphorylated and regulate phagocytosis of apoptotic neurons via the Src family kinase‐Syk pathway. 88 , 89 Our studies suggest that tyrosine phosphorylation of Megf10 regulates the binding of Megf10 with Notch1. 68 Further investigations are needed to elucidate the nature of the Megf10‐Notch interaction in greater depth.
1.8. POGLUT1‐related muscular dystrophy
The first human disease associated with pathogenic variants in POGLUT1 was Dowling‐Degos disease, an autosomal dominant dermatologic condition characterized by progressive reticulate hyperpigmentation. 90 Subsequently, biallelic pathogenic variants in POGLUT1 were associated with muscular dystrophy. 91 , 92 , 93 , 94 The first reported family was a large consanguineous kindred with multiple individuals affected by a progressive limb‐girdle muscular dystrophy (LGMD) phenotype that included scapular winging and loss of ambulation, with evidence for reduced PAX7+ cells in muscle samples from affected individuals. 91 A more recent cohort of nine unrelated families with muscular dystrophy showed biallelic pathogenic variants in POGLUT1 (Figure 4A,B) affecting conserved amino acids (Figure S2), firmly establishing the disease association. 93 This cohort included affected individuals with congenital muscular dystrophy (CMD) as well as those with LGMD phenotypes. Serum CK levels ranged from normal to a high of 10 times the upper limit of normal. A distinct radiological finding of “inside‐to‐outside” fatty degeneration was found on magnetic resonance images of skeletal muscle in both reports. Based on the sparse reports from the literature to date, POGLUT1‐related muscular dystrophy could be classified as an ultrarare disease. The original designation for POGLUT1‐related muscular dystrophy was LGMD type 2Z (LGMD2Z). However, the designation “Z” meant that this classification system had been exhausted; thus, as part of a reclassification effort, the disease phenotype is now recognized as LGMDR21. 95
FIGURE 4.
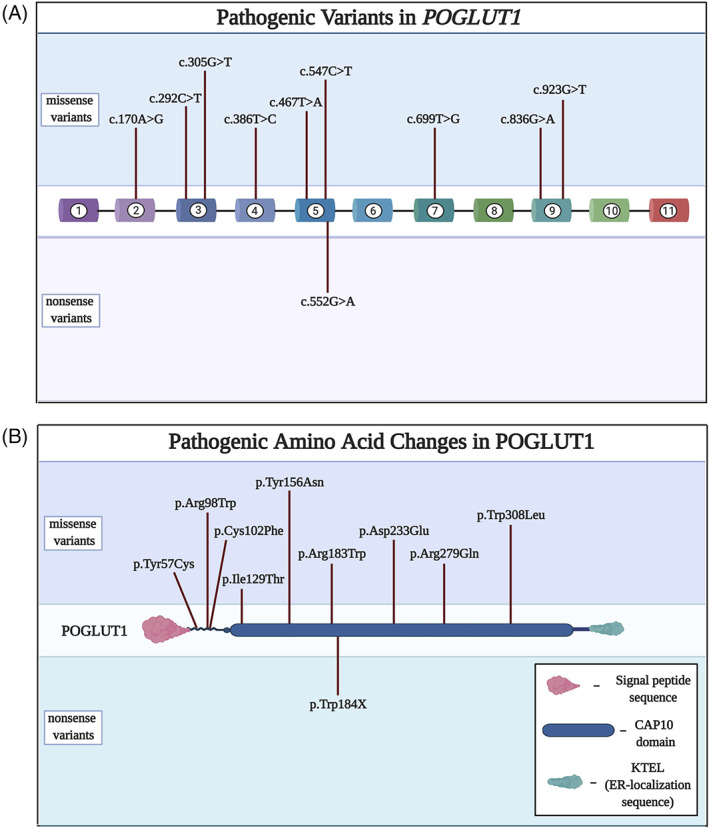
A, Pathogenic variants in POGLUT1 are predominantly missense changes distributed widely throughout the gene. B, Amino acid changes corresponding to the nucleotide changes in A. The predominance of missense variants indicates that the protein is sensitive to an array of conformational changes.
The gene POGLUT1, previously known as hCLP46, contains 11 exons and encodes a protein with 392 amino acids 96 that has orthologs across multiple species (Figure 5 and Table S2). 97 POGLUT1's ortholog in Drosophila, Rumi, is a protein O‐glycosyltransferase that glycosylates serine residues in the extracellular domain of Notch 98 and regulates Notch signaling. 99 Studies in mammalian cell culture systems, including C2C12 myoblasts, identified corresponding regulatory activities of POGLUT1 on the Notch signaling pathway. 100 , 101 However, POGLUT1 has two known enzymatic functions, serving as a glucosyltransferase and xylosyltransferase, with variable effects on cellular proliferation under different circumstances. 97 Deficiency of POGLUT1 and its orthologs also has divergent effects on Notch receptor expression in different contexts, leading to accumulation in one context 98 and depletion in another. 100 This may explain in part why POGLUT1 deficiency has been associated with two different human disease phenotypes.
FIGURE 5.
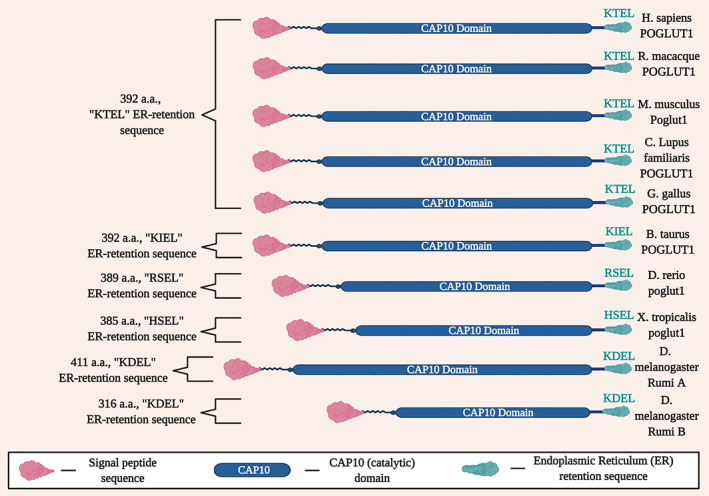
Conservation of key domains of POGLUT1 across species, including the signal peptide sequence, CAP10 domain, and the endoplasmic reticulum (ER) retention sequence.
The most sophisticated animal model of POGLUT1‐related muscular dystrophy to date has been developed in Drosophila. Rumi deficiency impairs muscle development in Drosophila, with more prominent rescue of the phenotype shown with overexpression of wild‐type POGLUT1 compared with POGLUT1 that harbors a pathogenic variant (c.699 T > G, p.D233E). 91
1.9. JAG2‐related muscular dystrophy
Biallelic pathogenic variants in JAG2 were found in a cohort of 13 unrelated families with muscular dystrophy, with severity ranging from an early‐onset congenital muscular dystrophy (CMD) phenotype to a later‐onset LGMD phenotype. 25 Serum CK levels ranged from normal to four times the upper limit of normal. Some individuals remained ambulatory into adulthood, whereas others lost ambulation in childhood or adolescence. Neck weakness was a prominent feature in a number of affected individuals, and EMG patterns were myopathic in all cases where this test was performed. Muscle biopsy findings included dystrophic patterns and increased fiber size variability.
Affected amino acids are conserved across species (Figure S3), 25 as well as key domains of the protein in general (Figure 6 and Table S3). Several of the affected individuals were found to have a distinct “outside‐to‐inside” pattern of fatty degeneration on magnetic resonance images of skeletal muscle, similar to the pattern seen in collagen VI–related muscular dystrophy 102 but the reverse of the pattern observed in POGLUT1‐related muscular dystrophy. 93 One additional report of JAG2‐related muscular dystrophy has since been published, 103 and the milder form of the disease is now classified as LGMD R27. Given the recent discovery of this genetic association, its epidemiology is currently unclear; it may be somewhat rare among muscular dystrophy subtypes.
FIGURE 6.
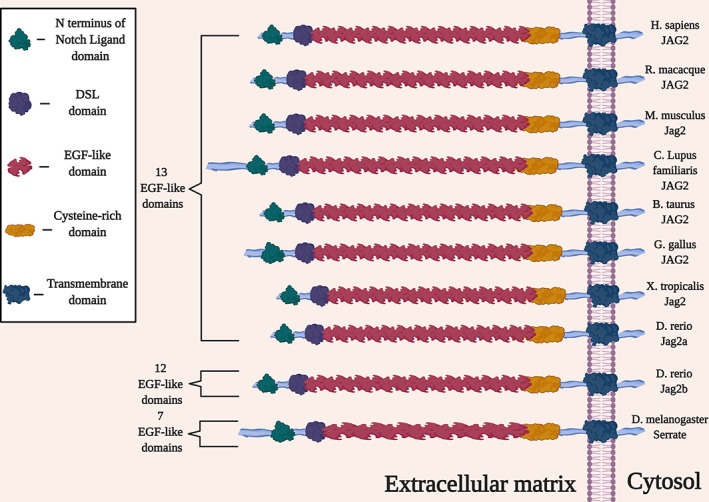
Key domains of the JAG2 (Jagged2) protein are conserved across multiple species.
In contrast to MEGF10, JAG2's protein product is an established ligand of the Notch receptors Notch1, 104 Notch2, 105 and Notch3, 104 and furthermore cleaves Notch2. 105 The association between JAG2 and muscle disease may come as a surprise in view of the complex literature that has accumulated on this gene and its orthologs over the past three decades. The Serrate gene was described for Drosophila in 1990, 106 followed by the orthologous rat gene Jagged2, 107 the murine Jag2, 108 and the human JAG2. 108 , 109 , 110 The protein products (in humans known as JAG2 or Jagged2) have EGF‐like domains 111 and are conserved canonical Notch ligands. In oncology, Jagged2 is known to promote metastasis 112 , 113 and tumorigenicity 114 in certain types of cancer, potentially due at least in part to pro‐angiogenic activity 115 ; thus, investigators are examining the means by which to inhibit Notch signaling in cancer biology. 116 , 117 , 118 JAG2 regulation is a potential target for this strategy for selected cancer subtypes.
Even in view of those findings, there are hints suggesting JAG2's link to skeletal muscle disease. JAG2 is expressed in mammalian skeletal muscle, 119 along with several other organs (including the brain, 120 , 121 gut 122 /enteric nervous system, 123 immune system, 124 , 125 and ovarian follicles 126 , 127 ). Expression patterns in zebrafish mirror those in mammals. 128 , 129 , 130 At the tissue level, JAG2 is also expressed in mammalian endothelial cells, 131 particularly in arterial vessels, 132 as well as MuSCs. 45 This raises the possibility that JAG2 may mediate signaling between MSCs and endothelial cells and trigger angiogenesis in response to muscle injury.
The regulatory environment upstream of JAG2 is not well characterized in skeletal muscle. However, hnRNP L is an intriguing molecule that is a known splice regulator, 133 and hnRNP L binding sites on the JAG2 transcript have been identified. 134 Previous reports in the literature have linked hnRNP L to the Notch signaling pathway. Overexpression of hnRNP I (also known as PTBP1), which is a partner of hnRNP L 135 in zebrafish, destabilizes the NICD and inhibits Notch signaling. 136 Studies in mice demonstrated that loss of the Notch inhibitory ligand DLL3 leads to increased levels in two proteins, including hnRNP L. 137 In addition, a screen carried out in Drosophila using Notch mutants identified smooth, the fly homolog of mammalian hnRNP L, as a genetic modifier of Notch. 138 Notably, hnRNP L downstream RNA targets also include Notch2, 139 Notch3, 134 Poglut1 ,140 and the Notch inhibitor Numb. 134 , 139 Although these studies point to hnRNP L as a major regulator of several Notch signaling pathway partners, 45 the muscle‐specific aspects of this relationship remain to be investigated. Further research is needed, but the studies to date strongly suggest that MEGF10, POGLUT1, and JAG2 interactions synergistically influence Notch signaling (Figure 7).
FIGURE 7.
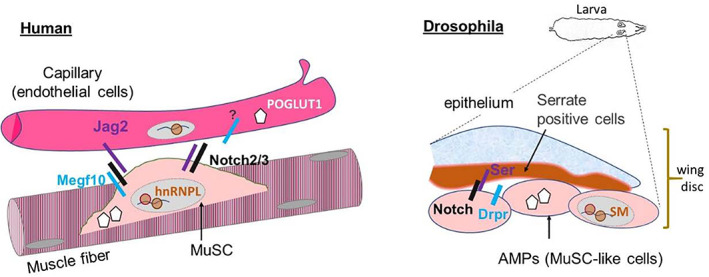
Schematic diagram of Notch signaling pathway proteins directly related to human skeletal muscle disease in the muscle fiber vs capillary (left) accompanied by a diagram showing the localization of orthologous proteins in Drosophila (right). Human and Drosophila orthologous pairs include the proteins JAG2 (Jagged2) and Ser (Serrate), MEGF10 and Drpr (Draper), and HNRNP L and Sm (Smooth). The question mark denotes the potential presence of Megf10 in endothelial cells.
1.10. Other links between the Notch signaling pathway and skeletal muscle disease
There are two other muscle disease genes with protein products that may have links to the Notch signaling pathway, with hints of such an association seen in the literature to date: TRIM32 141 and ATP2A1 (SERCA1). 142 , 143 In addition, JAG1 is not directly associated with a skeletal muscle disease but a specific variant in JAG1 appears to have modifying effects on muscular dystrophy. 21
Biallelic pathogenic variants in TRIM32 are associated with limb‐girdle muscular dystrophy type R8 (LGMDR8, formerly known as LGMD2H). 144 The protein product is a ubiquitin ligase that localizes to the Z disk of myofibers, regulates dysbindin, 145 and ubiquitylates thin filament and Z‐band proteins. 146 Expression of specific pathogenic variants in tn (ortholog of TRIM32) leads to myofibrillar abnormalities in Drosophila. 147 The link to the Notch signaling pathway arises in the mouse hippocampus, where Trim32 deficiency was found to be associated with upregulation of several relevant genes, including Notch1 and Hes1. 141 This relationship bears exploration for potential relevance to skeletal muscle function and disease.
Brody myopathy is a recessive muscle disorder characterized by childhood‐onset muscle stiffness and delayed muscle relaxation 148 , 149 that is associated with biallelic pathogenic variants in ATP2A1, 150 which encodes sarcoendoplasmic reticulum calcium ATPase 1 (SERCA1). The phenotype includes individuals with clinical paramyotonia but without electrical myotonia. 151 The association between SERCA1 and the Notch signaling pathway has been explored primarily in the context of cancer research, with SERCA1 having been identified as a potential therapeutic target. 142 , 143 Further study of potential interactions between SERCA1 and the Notch signaling pathway in skeletal muscle models may yield novel insights on disease mechanisms and therapeutic targets.
Dominant pathogenic variants in JAG1 have been associated with Alagille syndrome, a multiorgan system disease that does not have prominent skeletal muscle manifestations, 22 , 152 familial tetralogy of Fallot, 24 and an axonal form of Charcot‐Marie‐Tooth disease, known as type 2HH. 23 The protein product JAG1 (also known as Jagged1) is a canonical Notch ligand, along with JAG2 (Jagged2). There have been hints of JAG1 activity in skeletal muscle, including a reduction of JAG1 muscle expression in older humans 153 and in mdx mice. 154 JAG1 expression also appears to be induced in activated MuSCs. 155 Intriguingly, a recent study identified a variant in the promoter region of Jag1 that creates a novel myogenin binding site, increasing Jag1 expression in skeletal muscle and rescuing Duchenne muscular dystrophy in Golden Retriever dogs. 21
Recessive pathogenic variants in PAX7 are associated with a congenital myopathy phenotype, as described in five affected individuals from four unrelated consanguineous kindreds. 156 The disease manifestations include hypotonia, ptosis, and scoliosis, with atrophic fibers and fibroadipose replacement on muscle biopsy. N1ICD expression restores the proliferative potential of Pax7‐deficient MuSCs, linking Pax7 to the Notch signaling pathway. 157 Elevated Notch signaling activity has also been associated with an undifferentiated myogenic cell population, characterized by high levels of Pax7 expression. 158
NOTCH2NLC bears some resemblance to NOTCH2 and its protein product is also involved in the regulation of Notch signaling. Pathogenic CGG repeat expansions in the 5′ untranslated region of NOTCH2NLC have been associated with oculopharyngodistal myopathy (OPDM), 159 neuronal intranuclear inclusion disease (NIID), 160 , 161 and hereditary essential tremor. 162 Clinical features of this form of OPDM include ptosis, ophthalmoplegia, dysarthria, and muscle weakness. 159
2. POTENTIAL THERAPEUTIC TARGETS
Components of the Notch signaling pathway are drawing increasing attention as therapeutic targets. For example, some investigational compounds target γ‐secretase, which cleaves the TM from the NICD in the Notch receptor. 163 These and other compounds targeting the Notch signaling pathway have primarily been assessed in nonmuscle diseases 164 ; given the emerging body of work linking the Notch signaling pathway to muscle disease, examinations of the effects of some of these compounds in muscle contexts may be warranted. There is also emerging evidence that the Notch signaling pathway is a promising target for therapy in skeletal muscle. Specific small molecule candidate drugs have been found to promote human myotube formation, 165 and sertraline has been found to ameliorate MEGF10 myopathy in cellular, Drosophila, and zebrafish model systems. 54
2.1. Future directions
Numerous insights into the key contributions of the Notch signaling pathway in skeletal muscle development, maintenance, and repair have been described. The clinical relevance of the Notch signaling pathway for muscle disease has become apparent over the past decade with the discoveries of three Notch signaling pathway‐related muscle disease genes: MEGF10, POGLUT1, and JAG2. Further study of these genes and their encoded proteins, along with exploration of TRIM32, ATP2A1 (SERCA1), and JAG1 in the same context, are likely to yield a better understanding of skeletal muscle disease. It is likely that additional genes and proteins related to the Notch signaling pathway will be linked to muscle diseases in the future. Sertraline has shown therapeutic effects in models of MEGF10 myopathy, with evidence indicating that it acts via the Notch signaling pathway in this context. 54 This finding, coupled with the potential therapeutic effects of JAG1 augmentation in muscular dystrophy, indicate that the Notch signaling pathway promises to be a robust area of investigation for new therapeutic targets in muscular dystrophy and other skeletal muscle diseases.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose.
Supporting information
APPENDIX S1 Supporting information
ACKNOWLEDGMENT
Figures 1, 2, 3, 4, 5, 6 were created with BioRender (biorender.com) under a paid academic subscription that includes a license to publish diagrams in journal publications.
Vargas‐Franco D, Kalra R, Draper I, Pacak CA, Asakura A, Kang PB. The Notch signaling pathway in skeletal muscle health and disease. Muscle & Nerve. 2022;66(5):530‐544. doi: 10.1002/mus.27684
Answer questions and earn CME https://education.aanem.org/URL/JR98.
The objectives of this activity are to: 1) Recognize the phenotypes of muscle disease produced by pathogenic variants in 3 different muscle disease genes involved in the Notch signaling pathway; 2) Be able to order appropriate genetic testing in these patients.
The AANEM is accredited by the American Council for Continuing Medical Education (ACCME) to providing continuing education for physicians. AANEM designates this Journal‐based CME activity for a maximum of 1.0 AMA PRA Category 1 Credit™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
DATA AVAILABILITY STATEMENT
Data sharing not applicable ‐ no new data generated
REFERENCES
- 1. Monaco AP, Neve RL, Colletti‐Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986;323:646‐650. doi: 10.1038/323646a0. [DOI] [PubMed] [Google Scholar]
- 2. Barton ER, Pacak CA, Stoppel WL, Kang PB. The ties that bind: functional clusters in limb‐girdle muscular dystrophy. Skelet Muscle. 2020;10:22. doi: 10.1186/s13395-020-00240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Artavanis‐Tsakonas S, Muskavitch MA, Yedvobnick B. Molecular cloning of Notch, a locus affecting neurogenesis in Drosophila melanogaster . Proc Natl Acad Sci USA. 1983;80:1977‐1981. doi: 10.1073/pnas.80.7.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trenerry MK, Della Gatta PA, Cameron‐Smith D. JAK/STAT signaling and human in vitro myogenesis. BMC Physiol. 2011;11:6. doi: 10.1186/1472-6793-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pézeron G, Millen K, Boukhatmi H, Bray S. Notch directly regulates the cell morphogenesis genes Reck, talin and trio in adult muscle progenitors. J Cell Sci. 2014;127:4634‐4644. doi: 10.1242/jcs.151787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Price FD, von Maltzahn J, Bentzinger CF, et al. Inhibition of JAK‐STAT signaling stimulates adult satellite cell function. Nat Med. 2014;20:1174‐1181. doi: 10.1038/nm.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bradley RS, Cowin P, Brown AM. Expression of Wnt‐1 in PC12 cells results in modulation of plakoglobin and E‐cadherin and increased cellular adhesion. J Cell Biol. 1993;123:1857‐1865. doi: 10.1083/jcb.123.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shan T, Xu Z, Wu W, Liu J, Wang Y. Roles of Notch1 SIGNALING in regulating satellite cell fates choices and postnatal skeletal myogenesis: roles of Notch1 signaling during myogenesis. J Cell Physiol. 2017;232:2964‐2967. doi: 10.1002/jcp.25730. [DOI] [PubMed] [Google Scholar]
- 9. Stittrich A‐B, Lehman A, Bodian DL, et al. Mutations in NOTCH1 cause Adams‐Oliver syndrome. Am J Hum Genet. 2014;95:275‐284. doi: 10.1016/j.ajhg.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270‐274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 11. Yartseva V, Goldstein LD, Rodman J, et al. Heterogeneity of satellite cells implicates DELTA1/NOTCH2 signaling in self‐renewal. Cell Rep. 2020;30:1491‐1503.e6. doi: 10.1016/j.celrep.2019.12.100. [DOI] [PubMed] [Google Scholar]
- 12. McDaniell R, Warthen DM, Sanchez‐Lara PA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the Notch signaling pathway. Am J Hum Genet. 2006;79:169‐173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Simpson MA, Irving MD, Asilmaz E, et al. Mutations in NOTCH2 cause Hajdu‐Cheney syndrome, a disorder of severe and progressive bone loss. Nat Genet. 2011;43:303‐305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- 14. Kitamoto T, Hanaoka K. Notch3 null mutation in mice causes muscle hyperplasia by repetitive muscle regeneration. Stem Cells. 2010;28:2205‐2216. doi: 10.1002/stem.547. [DOI] [PubMed] [Google Scholar]
- 15. Joutel A, Corpechot C, Ducros A, et al. Notch3 mutations in CADASIL, a hereditary adult‐onset condition causing stroke and dementia. Nature. 1996;383:707‐710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- 16. Zhang H, Shang R, Bi P. Feedback regulation of Notch signaling and myogenesis connected by MyoD‐Dll1 axis. PLoS Genet. 2021;17:e1009729. doi: 10.1371/journal.pgen.1009729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fischer‐Zirnsak B, Segebrecht L, Schubach M, et al. Haploinsufficiency of the Notch ligand DLL1 causes variable neurodevelopmental disorders. Am J Hum Genet. 2019;105:631‐639. doi: 10.1016/j.ajhg.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bulman MP, Kusumi K, Frayling TM, et al. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat Genet. 2000;24:438‐441. doi: 10.1038/74307. [DOI] [PubMed] [Google Scholar]
- 19. Fujimaki S, Matsumoto T, Muramatsu M, et al. The endothelial Dll4‐muscular Notch2 axis regulates skeletal muscle mass. Nat Metab. 2022;4:180‐189. doi: 10.1038/s42255-022-00533-9. [DOI] [PubMed] [Google Scholar]
- 20. Meester JAN, Southgate L, Stittrich A‐B, et al. Heterozygous loss‐of‐function mutations in DLL4 cause Adams‐Oliver syndrome. Am J Hum Genet. 2015;97:475‐482. doi: 10.1016/j.ajhg.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vieira NM, Elvers I, Alexander MS, et al. Jagged 1 rescues the Duchenne muscular dystrophy phenotype. Cell. 2015;163:1204‐1213. doi: 10.1016/j.cell.2015.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oda T, Elkahloun AG, Pike BL, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235‐242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- 23. Sullivan JM, Motley WW, Johnson JO, et al. Dominant mutations of the Notch ligand Jagged1 cause peripheral neuropathy. J Clin Invest. 2020;130:1506‐1512. doi: 10.1172/JCI128152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eldadah ZA, Hamosh A, Biery NJ, et al. Familial tetralogy of Fallot caused by mutation in the jagged1 gene. Hum Mol Genet. 2001;10:163‐169. doi: 10.1093/hmg/10.2.163. [DOI] [PubMed] [Google Scholar]
- 25. Coppens S, Barnard AM, Puusepp S, et al. A form of muscular dystrophy associated with pathogenic variants in JAG2. Am J Hum Genet. 2021;108:840‐856. doi: 10.1016/j.ajhg.2021.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue‐based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 27. Groot AJ, Vooijs MA. The role of Adams in Notch signaling. Adv Exp Med Biol. 2012;727:15‐36. doi: 10.1007/978-1-4614-0899-4_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nandagopal N, Santat LA, Elowitz MB. Cis‐activation in the Notch signaling pathway. Elife. 2019;8:e37880. doi: 10.7554/eLife.37880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fleming RJ, Hori K, Sen A, et al. An extracellular region of Serrate is essential for ligand‐induced cis‐inhibition of Notch signaling. Development. 2013;140:2039‐2049. doi: 10.1242/dev.087916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. LeBon L, Lee TV, Sprinzak D, Jafar‐Nejad H, Elowitz MB. Fringe proteins modulate Notch‐ligand cis and trans interactions to specify signaling states. Elife. 2014;3:e02950. doi: 10.7554/eLife.02950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. D'Souza B, Meloty‐Kapella L, Weinmaster G. Canonical and non‐canonical Notch ligands. Curr Top Dev Biol. 2010;92:73‐129. doi: 10.1016/S0070-2153(10)92003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zacharioudaki E, Bray SJ. Tools and methods for studying Notch signaling in Drosophila melanogaster . Methods. 2014;68:173‐182. doi: 10.1016/j.ymeth.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Strooper B, Annaert W, Cupers P, et al. A presenilin‐1‐dependent gamma‐secretase‐like protease mediates release of Notch intracellular domain. Nature. 1999;398:518‐522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 34. Wilson JJ, Kovall RA. Crystal structure of the CSL‐Notch‐mastermind ternary complex bound to DNA. Cell. 2006;124:985‐996. doi: 10.1016/j.cell.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 35. Wu L, Sun T, Kobayashi K, Gao P, Griffin JD. Identification of a family of mastermind‐like transcriptional coactivators for mammalian notch receptors. Mol Cell Biol. 2002;22:7688‐7700. doi: 10.1128/MCB.22.21.7688-7700.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. de Celis JF, Bray S. Feed‐back mechanisms affecting Notch activation at the dorsoventral boundary in the Drosophila wing. Development. 1997;124:3241‐3251. [DOI] [PubMed] [Google Scholar]
- 37. Klein T, Brennan K, Arias AM. An intrinsic dominant negative activity of serrate that is modulated during wing development in Drosophila . Dev Biol. 1997;189:123‐134. doi: 10.1006/dbio.1997.8564. [DOI] [PubMed] [Google Scholar]
- 38. Micchelli CA, Rulifson EJ, Blair SS. The function and regulation of cut expression on the wing margin of Drosophila: Notch, Wingless and a dominant negative role for Delta and Serrate. Development. 1997;124:1485‐1495. [DOI] [PubMed] [Google Scholar]
- 39. Cordle J, Johnson S, Tay JZY, et al. A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans‐activation and cis‐inhibition. Nat Struct Mol Biol. 2008;15:849‐857. doi: 10.1038/nsmb.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wen Y, Bi P, Liu W, Asakura A, Keller C, Kuang S. Constitutive Notch activation upregulates Pax7 and promotes the self‐renewal of skeletal muscle satellite cells. Mol Cell Biol. 2012;32:2300‐2311. doi: 10.1128/MCB.06753-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bi P, Yue F, Sato Y, et al. Stage‐specific effects of Notch activation during skeletal myogenesis. Elife. 2016;5:e17355. doi: 10.7554/eLife.17355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Du H, Shih C‐H, Wosczyna MN, et al. Macrophage‐released ADAMTS1 promotes muscle stem cell activation. Nat Commun. 2017;8:669. doi: 10.1038/s41467-017-00522-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu L, Charville GW, Cheung TH, et al. Impaired Notch signaling leads to a decrease in p53 activity and mitotic catastrophe in aged muscle stem cells. Cell Stem Cell. 2018;23:544‐556.e4. doi: 10.1016/j.stem.2018.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marinkovic M, Fuoco C, Sacco F, et al. Fibro‐adipogenic progenitors of dystrophic mice are insensitive to NOTCH regulation of adipogenesis. Life Sci Alliance. 2019;2:e201900437. doi: 10.26508/lsa.201900437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verma M, Asakura Y, Murakonda BSR, et al. Muscle satellite cell cross‐talk with a vascular niche maintains quiescence via VEGF and Notch signaling. Cell Stem Cell. 2018;23:530‐543.e9. doi: 10.1016/j.stem.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Der Vartanian A, Audfray A, Al Jaam B, et al. Protein O‐fucosyltransferase 1 expression impacts myogenic C2C12 cell commitment via the Notch signaling pathway. Mol Cell Biol. 2015;35:391‐405. doi: 10.1128/MCB.00890-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Der Vartanian A, Chabanais J, Carrion C, Maftah A, Germot A. Downregulation of POFUT1 impairs secondary myogenic fusion through a reduced NFATc2/IL‐4 signaling pathway. Int J Mol Sci. 2019;20:E4396. doi: 10.3390/ijms20184396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zygmunt DA, Singhal N, Kim M‐L, et al. Deletion of Pofut1 in mouse skeletal myofibers induces muscle aging‐related phenotypes in cis and in trans. Mol Cell Biol. 2017;37:e00426‐e00416. doi: 10.1128/MCB.00426-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krivtsov AV, Rozov FN, Zinovyeva MV, et al. Jedi‐‐‐a novel transmembrane protein expressed in early hematopoietic cells. J Cell Biochem. 2007;101:767‐784. doi: 10.1002/jcb.21232. [DOI] [PubMed] [Google Scholar]
- 50. Hartley L, Kinali M, Knight R, et al. A congenital myopathy with diaphragmatic weakness not linked to the SMARD1 locus. Neuromuscul Disord. 2007;17:174‐179. doi: 10.1016/j.nmd.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 51. Logan CV, Lucke B, Pottinger C, et al. Mutations in MEGF10, a regulator of satellite cell myogenesis, cause early onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD). Nat Genet. 2011;43:1189‐1192. doi: 10.1038/ng.995. [DOI] [PubMed] [Google Scholar]
- 52. Boyden SE, Mahoney LJ, Kawahara G, et al. Mutations in the satellite cell gene MEGF10 cause a recessive congenital myopathy with minicores. Neurogenetics. 2012;13:115‐124. doi: 10.1007/s10048-012-0315-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pierson TM, Markello T, Accardi J, et al. Novel SNP array analysis and exome sequencing detect a homozygous exon 7 deletion of MEGF10 causing early onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD). Neuromuscul Disord. 2013;23:483‐488. doi: 10.1016/j.nmd.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Saha M, Rizzo SA, Ramanathan M, et al. Selective serotonin reuptake inhibitors ameliorate MEGF10 myopathy. Hum Mol Genet. 2019;28:2365‐2377. doi: 10.1093/hmg/ddz064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liewluck T, Milone M, Tian X, Engel AG, Staff NP, Wong L‐J. Adult‐onset respiratory insufficiency, scoliosis, and distal joint hyperlaxity in patients with multiminicore disease due to novel Megf10 mutations. Muscle Nerve. 2016;53:984‐988. doi: 10.1002/mus.25054. [DOI] [PubMed] [Google Scholar]
- 56. Takayama K, Mitsuhashi S, Shin J‐Y, et al. Japanese multiple epidermal growth factor 10 (MEGF10) myopathy with novel mutations: a phenotype‐genotype correlation. Neuromuscul Disord. 2016;26:604‐609. doi: 10.1016/j.nmd.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 57. Fujii K, Hirano M, Terayama A, et al. Identification of a novel mutation and genotype‐phenotype relationship in MEGF10 myopathy. Neuromuscul Disord. 2022;32:P436‐P440. doi: 10.1016/j.nmd.2022.01.009. [DOI] [PubMed] [Google Scholar]
- 58. Harris E, Marini‐Bettolo C, Töpf A, et al. MEGF10 related myopathies: a new case with adult onset disease with prominent respiratory failure and review of reported phenotypes. Neuromuscul Disord. 2018;28:48‐53. doi: 10.1016/j.nmd.2017.09.017. [DOI] [PubMed] [Google Scholar]
- 59. Alabdullatif MA, Al Dhaibani MA, Khassawneh MY, El‐Hattab AW. Chromosomal microarray in a highly consanguineous population: diagnostic yield, utility of regions of homozygosity, and novel mutations. Clin Genet. 2017;91:616‐622. doi: 10.1111/cge.12872. [DOI] [PubMed] [Google Scholar]
- 60. Nagase T, Nakayama M, Nakajima D, Kikuno R, Ohara O. Prediction of the coding sequences of unidentified human genes. XX. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2001;8:85‐95. doi: 10.1093/dnares/8.2.85. [DOI] [PubMed] [Google Scholar]
- 61. Suzuki E, Nakayama M. MEGF10 is a mammalian ortholog of CED‐1 that interacts with clathrin assembly protein complex 2 medium chain and induces large vacuole formation. Exp Cell Res. 2007;313:3729‐3742. doi: 10.1016/j.yexcr.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 62. Holterman CE, Le Grand F, Kuang S, Seale P, Rudnicki MA. Megf10 regulates the progression of the satellite cell myogenic program. J Cell Biol. 2007;179:911‐922. doi: 10.1083/jcb.200709083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Suzuki E, Nakayama M. The mammalian Ced‐1 ortholog MEGF10/KIAA1780 displays a novel adhesion pattern. Exp Cell Res. 2007;313:2451‐2464. doi: 10.1016/j.yexcr.2007.03.041. [DOI] [PubMed] [Google Scholar]
- 64. Logan MA, Hackett R, Doherty J, Sheehan A, Speese SD, Freeman MR. Negative regulation of glial engulfment activity by Draper terminates glial responses to axon injury. Nat Neurosci. 2012;15:722‐730. doi: 10.1038/nn.3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Draper I, Saha M, Stonebreaker H, Salomon RN, Matin B, Kang PB. The impact of Megf10/Drpr gain‐of‐function on muscle development in Drosophila . FEBS Lett. 2019;593:680‐696. doi: 10.1002/1873-3468.13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Draper I, Mahoney LJ, Mitsuhashi S, Pacak CA, Salomon RN, Kang PB. Silencing of drpr leads to muscle and brain degeneration in adult Drosophila . Am J Pathol. 2014;184:2653‐2661. doi: 10.1016/j.ajpath.2014.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kay JN, Chu MW, Sanes JR. MEGF10 and MEGF11 mediate homotypic interactions required for mosaic spacing of retinal neurons. Nature. 2012;483:465‐469. doi: 10.1038/nature10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Saha M, Mitsuhashi S, Jones MD, et al. Consequences of MEGF10 deficiency on myoblast function and Notch1 interactions. Hum Mol Genet. 2017;26:2984‐3000. doi: 10.1093/hmg/ddx189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Li C, Vargas‐Franco D, Saha M, et al. Megf10 deficiency impairs skeletal muscle stem cell migration and muscle regeneration. FEBS Open Biol. 2021;11:114‐123. doi: 10.1002/2211-5463.13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lloyd TE, Taylor JP. Flightless flies: Drosophila models of neuromuscular disease. Ann NY Acad Sci. 2010;1184:e1‐e20. doi: 10.1111/j.1749-6632.2010.05432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Draper I. Model organisms offer new possibilities for discovery of therapeutics. Drug Discov Today Technol. 2013;10:e61‐e64. doi: 10.1016/j.ddtec.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 72. Freeman MR, Delrow J, Kim J, Johnson E, Doe CQ. Unwrapping glial biology: Gcm target genes regulating glial development, diversification, and function. Neuron. 2003;38:567‐580. doi: 10.1016/s0896-6273(03)00289-7. [DOI] [PubMed] [Google Scholar]
- 73. Cornell M, Evans DA, Mann R, et al. The Drosophila melanogaster suppressor of deltex gene, a regulator of the Notch receptor signaling pathway, is an E3 class ubiquitin ligase. Genetics. 1999;152:567‐576. doi: 10.1093/genetics/152.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wu H‐H, Bellmunt E, Scheib JL, et al. Glial precursors clear sensory neuron corpses during development via Jedi‐1, an engulfment receptor. Nat Neurosci. 2009;12:1534‐1541. doi: 10.1038/nn.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fujita Y, Maeda T, Kamaishi K, et al. Expression of MEGF10 in cholinergic and glutamatergic neurons. Neurosci Lett. 2017;653:25‐30. doi: 10.1016/j.neulet.2017.05.029. [DOI] [PubMed] [Google Scholar]
- 76. Ziegenfuss JS, Biswas R, Avery MA, et al. Draper‐dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling. Nature. 2008;453:935‐939. doi: 10.1038/nature06901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Macdonald JM, Doherty J, Hackett R, Freeman MR. The c‐Jun kinase signaling cascade promotes glial engulfment activity through activation of draper and phagocytic function. Cell Death Differ. 2013;20:1140‐1148. doi: 10.1038/cdd.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869‐881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 79. Hamon Y, Trompier D, Ma Z, et al. Cooperation between engulfment receptors: the case of ABCA1 and MEGF10. PLoS One. 2006;1:e120. doi: 10.1371/journal.pone.0000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Iram T, Ramirez‐Ortiz Z, Byrne MH, et al. Megf10 is a receptor for C1Q that mediates clearance of apoptotic cells by astrocytes. J Neurosci. 2016;36:5185‐5192. doi: 10.1523/jneurosci.3850-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chung W‐S, Clarke LE, Wang GX, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504:394‐400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Singh TD, Park S‐Y, Bae J, et al. MEGF10 functions as a receptor for the uptake of amyloid‐β. FEBS Lett. 2010;584:3936‐3942. doi: 10.1016/j.febslet.2010.08.050. [DOI] [PubMed] [Google Scholar]
- 83. Mourikis P, Tajbakhsh S. Distinct contextual roles for Notch signalling in skeletal muscle stem cells. BMC Dev Biol. 2014;14:2. doi: 10.1186/1471-213X-14-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bjornson CRR, Cheung TH, Liu L, Tripathi PV, Steeper KM, Rando TA. Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells. 2012;30:232‐242. doi: 10.1002/stem.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bröhl D, Vasyutina E, Czajkowski MT, et al. Colonization of the satellite cell niche by skeletal muscle progenitor cells depends on Notch signals. Dev Cell. 2012;23:469‐481. doi: 10.1016/j.devcel.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 86. Seale P, Ishibashi J, Holterman C, Rudnicki MA. Muscle satellite cell‐specific genes identified by genetic profiling of MyoD‐deficient myogenic cell. Dev Biol. 2004;275:287‐300. doi: 10.1016/j.ydbio.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 87. Park S‐Y, Yun Y, Kim M‐J, Kim I‐S. Myogenin is a positive regulator of MEGF10 expression in skeletal muscle. Biochem Biophys Res Commun. 2014;450:1631‐1637. doi: 10.1016/j.bbrc.2014.07.061. [DOI] [PubMed] [Google Scholar]
- 88. Scheib JL, Sullivan CS, Carter BD. Jedi‐1 and MEGF10 signal engulfment of apoptotic neurons through the tyrosine kinase Syk. J Neurosci. 2012;32:13022‐13031. doi: 10.1523/jneurosci.6350-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mitsuhashi S, Mitsuhashi H, Alexander MS, Sugimoto H, Kang PB. Cysteine mutations cause defective tyrosine phosphorylation in MEGF10 myopathy. FEBS Lett. 2013;587:2952‐2957. doi: 10.1016/j.febslet.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Basmanav FB, Oprisoreanu A‐M, Pasternack SM, et al. Mutations in POGLUT1, encoding protein O‐glucosyltransferase 1, cause autosomal‐dominant Dowling‐Degos disease. Am J Hum Genet. 2014;94:135‐143. doi: 10.1016/j.ajhg.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Servián‐Morilla E, Takeuchi H, Lee TV, et al. A POGLUT1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO Mol Med. 2016;8:1289‐1309. doi: 10.15252/emmm.201505815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wu J, Hunt SD, Matthias N, et al. Generation of an induced pluripotent stem cell line (CSCRMi001‐a) from a patient with a new type of limb‐girdle muscular dystrophy (LGMD) due to a missense mutation in POGLUT1 (Rumi). Stem Cell Res. 2017;24:102‐105. doi: 10.1016/j.scr.2017.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Servián‐Morilla E, Cabrera‐Serrano M, Johnson K, et al. POGLUT1 biallelic mutations cause myopathy with reduced satellite cells, α‐dystroglycan hypoglycosylation and a distinctive radiological pattern. Acta Neuropathol. 2020;139:565‐582. doi: 10.1007/s00401-019-02117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Töpf A, Johnson K, Bates A, et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb‐girdle weakness. Genet Med. 2020;22:1478‐1488. doi: 10.1038/s41436-020-0840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Straub V, Murphy A, Udd B, LGMD Workshop Study Group . 229th ENMC international workshop: limb girdle muscular dystrophies‐‐‐nomenclature and reformed classification Naarden, The Netherlands, 17‐19 March 2017. Neuromuscul Disord. 2018;28:702‐710. doi: 10.1016/j.nmd.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 96. Teng Y, Liu Q, Ma J, et al. Cloning, expression and characterization of a novel human CAP10‐like gene hCLP46 from CD34(+) stem/progenitor cells. Gene. 2006;371:7‐15. doi: 10.1016/j.gene.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 97. Mehboob MZ, Lang M. Structure, function, and pathology of protein O‐glucosyltransferases. Cell Death Dis. 2021;12:71. doi: 10.1038/s41419-020-03314-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Acar M, Jafar‐Nejad H, Takeuchi H, et al. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell. 2008;132:247‐258. doi: 10.1016/j.cell.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Fernandez‐Valdivia R, Takeuchi H, Samarghandi A, et al. Regulation of mammalian Notch signaling and embryonic development by the protein O‐glucosyltransferase Rumi. Development. 2011;138:1925‐1934. doi: 10.1242/dev.060020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Takeuchi H, Yu H, Hao H, et al. O‐glycosylation modulates the stability of epidermal growth factor‐like repeats and thereby regulates Notch trafficking. J Biol Chem. 2017;292:15964‐15973. doi: 10.1074/jbc.M117.800102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pélisse M, Der Vartanian A, Germot A, Maftah A. Protein O‐glucosyltransferase 1 expression influences formation of differentiated myotubes in C2C12 cell line. DNA Cell Biol. 2018;37:359‐372. doi: 10.1089/dna.2017.4052. [DOI] [PubMed] [Google Scholar]
- 102. Batra A, Lott DJ, Willcocks R, et al. Lower extremity muscle involvement in the intermediate and Bethlem myopathy forms of COL6‐related dystrophy and Duchenne muscular dystrophy: a cross‐sectional study. J Neuromuscul Dis. 2020;7:407‐417. doi: 10.3233/JND-190457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Nur Villar‐Quiles R, Romero NB, Tanya S. JAG2‐related muscular dystrophy: when differential diagnosis matters [in French]. Med Sci (Paris). 2021;37(1):40‐43. doi: 10.1051/medsci/2021191. [DOI] [PubMed] [Google Scholar]
- 104. Shimizu K, Chiba S, Saito T, Kumano K, Hirai H. Physical interaction of Delta1, Jagged1, and Jagged2 with Notch1 and Notch3 receptors. Biochem Biophys Res Commun. 2000;276:385‐389. doi: 10.1006/bbrc.2000.3469. [DOI] [PubMed] [Google Scholar]
- 105. Shimizu K, Chiba S, Hosoya N, et al. Binding of Delta1, Jagged1, and Jagged2 to Notch2 rapidly induces cleavage, nuclear translocation, and hyperphosphorylation of Notch2. Mol Cell Biol. 2000;20:6913‐6922. doi: 10.1128/MCB.20.18.6913-6922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Fleming RJ, Scottgale TN, Diederich RJ, Artavanis‐Tsakonas S. The gene Serrate encodes a putative EGF‐like transmembrane protein essential for proper ectodermal development in Drosophila melanogaster . Genes Dev. 1990;4:2188‐2201. doi: 10.1101/gad.4.12a.2188. [DOI] [PubMed] [Google Scholar]
- 107. Shawber C, Boulter J, Lindsell CE, Weinmaster G. Jagged2: a serrate‐like gene expressed during rat embryogenesis. Dev Biol. 1996;180:370‐376. doi: 10.1006/dbio.1996.0310. [DOI] [PubMed] [Google Scholar]
- 108. Valsecchi C, Ghezzi C, Ballabio A, Rugarli EI. JAGGED2: a putative Notch ligand expressed in the apical ectodermal ridge and in sites of epithelial‐mesenchymal interactions. Mech Dev. 1997;69:203‐207. doi: 10.1016/s0925-4773(97)00146-9. [DOI] [PubMed] [Google Scholar]
- 109. Luo B, Aster JC, Hasserjian RP, Kuo F, Sklar J. Isolation and functional analysis of a cDNA for human Jagged2, a gene encoding a ligand for the Notch1 receptor. Mol Cell Biol. 1997;17:6057‐6067. doi: 10.1128/MCB.17.10.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Deng Y, Madan A, Banta AB, et al. Characterization, chromosomal localization, and the complete 30‐kb DNA sequence of the human Jagged2 (JAG2) gene. Genomics. 2000;63:133‐138. doi: 10.1006/geno.1999.6045. [DOI] [PubMed] [Google Scholar]
- 111. Appella E, Weber IT, Blasi F. Structure and function of epidermal growth factor‐like regions in proteins. FEBS Lett. 1988;231:1‐4. doi: 10.1016/0014-5793(88)80690-2. [DOI] [PubMed] [Google Scholar]
- 112. Li W, Liu M, Feng Y, et al. High expression of Notch ligand Jagged2 is associated with the metastasis and recurrence in urothelial carcinoma of bladder. Int J Clin Exp Pathol. 2013;6:2430‐2440. [PMC free article] [PubMed] [Google Scholar]
- 113. Hu Y, Su H, Li X, et al. The NOTCH ligand JAGGED2 promotes pancreatic cancer metastasis independent of NOTCH signaling activation. Mol Cancer Ther. 2015;14:289‐297. doi: 10.1158/1535-7163.MCT-14-0501. [DOI] [PubMed] [Google Scholar]
- 114. Vaish V, Kim J, Shim M. Jagged‐2 (JAG2) enhances tumorigenicity and chemoresistance of colorectal cancer cells. Oncotarget. 2017;8:53262‐53275. doi: 10.18632/oncotarget.18391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Palano MT, Giannandrea D, Platonova N, et al. Jagged ligands enhance the pro‐angiogenic activity of multiple myeloma cells. Cancers (Basel). 2020;12:E2600. doi: 10.3390/cancers12092600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Platonova N, Parravicini C, Sensi C, et al. Identification of small molecules uncoupling the Notch::Jagged interaction through an integrated high‐throughput screening. PLoS One. 2017;12:e0182640. doi: 10.1371/journal.pone.0182640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Takam Kamga P, Dal Collo G, Midolo M, et al. Inhibition of Notch signaling enhances chemosensitivity in B‐cell precursor acute lymphoblastic leukemia. Cancer Res. 2019;79:639‐649. doi: 10.1158/0008-5472.CAN-18-1617. [DOI] [PubMed] [Google Scholar]
- 118. Yang M, Zhang G, Wang Y, et al. Tumour‐associated neutrophils orchestrate intratumoural IL‐8‐driven immune evasion through Jagged2 activation in ovarian cancer. Br J Cancer. 2020;123:1404‐1416. doi: 10.1038/s41416-020-1026-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gray GE, Mann RS, Mitsiadis E, et al. Human ligands of the Notch receptor. Am J Pathol. 1999;154:785‐794. doi: 10.1016/S0002-9440(10)65325-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Irvin DK, Nakano I, Paucar A, Kornblum HI. Patterns of Jagged1, Jagged2, Delta‐like 1 and Delta‐like 3 expression during late embryonic and postnatal brain development suggest multiple functional roles in progenitors and differentiated cells. J Neurosci Res. 2004;75:330‐343. doi: 10.1002/jnr.10843. [DOI] [PubMed] [Google Scholar]
- 121. Stump G, Durrer A, Klein A‐L, Lütolf S, Suter U, Taylor V. Notch1 and its ligands Delta‐like and Jagged are expressed and active in distinct cell populations in the postnatal mouse brain. Mech Dev. 2002;114:153‐159. doi: 10.1016/s0925-4773(02)00043-6. [DOI] [PubMed] [Google Scholar]
- 122. Sander GR, Powell BC. Expression of notch receptors and ligands in the adult gut. J Histochem Cytochem. 2004;52:509‐516. doi: 10.1177/002215540405200409. [DOI] [PubMed] [Google Scholar]
- 123. Sander GR, Brookes SJH, Powell BC. Expression of Notch1 and Jagged2 in the enteric nervous system. J Histochem Cytochem. 2003;51:969‐972. doi: 10.1177/002215540305100712. [DOI] [PubMed] [Google Scholar]
- 124. DeHart SL, Heikens MJ, Tsai S. Jagged2 promotes the development of natural killer cells and the establishment of functional natural killer cell lines. Blood. 2005;105:3521‐3527. doi: 10.1182/blood-2004-11-4237. [DOI] [PubMed] [Google Scholar]
- 125. Kared H, Adle‐Biassette H, Foïs E, et al. Jagged2‐expressing hematopoietic progenitors promote regulatory T cell expansion in the periphery through notch signaling. Immunity. 2006;25:823‐834. doi: 10.1016/j.immuni.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 126. Johnson J, Espinoza T, McGaughey RW, Rawls A, Wilson‐Rawls J. Notch pathway genes are expressed in mammalian ovarian follicles. Mech Dev. 2001;109:355‐361. doi: 10.1016/s0925-4773(01)00523-8. [DOI] [PubMed] [Google Scholar]
- 127. Vanorny DA, Prasasya RD, Chalpe AJ, Kilen SM, Mayo KE. Notch signaling regulates ovarian follicle formation and coordinates follicular growth. Mol Endocrinol. 2014;28:499‐511. doi: 10.1210/me.2013-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Zecchin E, Conigliaro A, Tiso N, Argenton F, Bortolussi M. Expression analysis of jagged genes in zebrafish embryos. Dev Dyn. 2005;233:638‐645. doi: 10.1002/dvdy.20366. [DOI] [PubMed] [Google Scholar]
- 129. Yeo S‐Y, Chitnis AB. Jagged‐mediated Notch signaling maintains proliferating neural progenitors and regulates cell diversity in the ventral spinal cord. Proc Natl Acad Sci USA. 2007;104:5913‐5918. doi: 10.1073/pnas.0607062104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gwak J‐W, Kong HJ, Bae Y‐K, et al. Proliferating neural progenitors in the developing CNS of zebrafish require Jagged2 and Jagged1b. Mol Cells. 2010;30:155‐159. doi: 10.1007/s10059-010-0101-4. [DOI] [PubMed] [Google Scholar]
- 131. Tsai S, Fero J, Bartelmez S. Mouse Jagged2 is differentially expressed in hematopoietic progenitors and endothelial cells and promotes the survival and proliferation of hematopoietic progenitors by direct cell‐to‐cell contact. Blood. 2000;96:950‐957. [PubMed] [Google Scholar]
- 132. Villa N, Walker L, Lindsell CE, Gasson J, Iruela‐Arispe ML, Weinmaster G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech Dev. 2001;108:161‐164. doi: 10.1016/s0925-4773(01)00469-5. [DOI] [PubMed] [Google Scholar]
- 133. Griffith BN, Walsh CM, Szeszel‐Fedorowicz W, Timperman AT, Salati LM. Identification of hnRNPs K, L and A2/B1 as candidate proteins involved in the nutritional regulation of mRNA splicing. Biochim Biophys Acta. 2006;1759:552‐561. doi: 10.1016/j.bbaexp.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Fei T, Chen Y, Xiao T, et al. Genome‐wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc Natl Acad Sci USA. 2017;114:E5207‐E5215. doi: 10.1073/pnas.1617467114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Blatter M, Dunin‐Horkawicz S, Grishina I, et al. The signature of the five‐stranded vRRM fold defined by functional, structural and computational analysis of the hnRNP L protein. J Mol Biol. 2015;427:3001‐3022. doi: 10.1016/j.jmb.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 136. Yang J, Chan CY, Jiang B, et al. hnRNP I inhibits Notch signaling and regulates intestinal epithelial homeostasis in the zebrafish. PLoS Genet. 2009;5:e1000363. doi: 10.1371/journal.pgen.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sewell W, Sparrow DB, Smith AJ, et al. Cyclical expression of the Notch/Wnt regulator Nrarp requires modulation by Dll3 in somitogenesis. Dev Biol. 2009;329:400‐409. doi: 10.1016/j.ydbio.2009.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Kankel MW, Hurlbut GD, Upadhyay G, Yajnik V, Yedvobnick B, Artavanis‐Tsakonas S. Investigating the genetic circuitry of mastermind in Drosophila, a Notch signal effector. Genetics. 2007;177:2493‐2505. doi: 10.1534/genetics.107.080994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Shankarling G, Cole BS, Mallory MJ, Lynch KW. Transcriptome‐wide RNA interaction profiling reveals physical and functional targets of hnRNP L in human T cells. Mol Cell Biol. 2014;34:71‐83. doi: 10.1128/MCB.00740-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Bhattacharya S, Levy MJ, Zhang N, et al. The methyltransferase SETD2 couples transcription and splicing by engaging mRNA processing factors through its SHI domain. Nat Commun. 2021;12:1443. doi: 10.1038/s41467-021-21663-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Ntim M, Li Q‐F, Zhang Y, et al. TRIM32 deficiency impairs synaptic plasticity by excitatory‐inhibitory imbalance via Notch pathway. Cereb Cortex. 2020;30:4617‐4632. doi: 10.1093/cercor/bhaa064. [DOI] [PubMed] [Google Scholar]
- 142. Roti G, Carlton A, Ross KN, et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell. 2013;23:390‐405. doi: 10.1016/j.ccr.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Pagliaro L, Marchesini M, Roti G. Targeting oncogenic Notch signaling with SERCA inhibitors. J Hematol Oncol. 2021;14:8. doi: 10.1186/s13045-020-01015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Frosk P, Weiler T, Nylen E, et al. Limb‐girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3‐ubiquitin‐ligase gene. Am J Hum Genet. 2002;70:663‐672. doi: 10.1086/339083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Locke M, Tinsley CL, Benson MA, Blake DJ. TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum Mol Genet. 2009;18:2344‐2358. doi: 10.1093/hmg/ddp167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Cohen S, Zhai B, Gygi SP, Goldberg AL. Ubiquitylation by Trim32 causes coupled loss of desmin, Z‐bands, and thin filaments in muscle atrophy. J Cell Biol. 2012;198:575‐589. doi: 10.1083/jcb.201110067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Bawa S, Gameros S, Baumann K, et al. Costameric integrin and sarcoglycan protein levels are altered in a Drosophila model for limb‐girdle muscular dystrophy type 2H. Mol Biol Cell. 2021;32:260‐273. doi: 10.1091/mbc.E20-07-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Voermans NC, Laan AE, Oosterhof A, et al. Brody syndrome: a clinically heterogeneous entity distinct from Brody disease: a review of literature and a cross‐sectional clinical study in 17 patients. Neuromuscul Disord. 2012;22:944‐954. doi: 10.1016/j.nmd.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 149. Molenaar JP, Verhoeven JI, Rodenburg RJ, et al. Clinical, morphological and genetic characterization of Brody disease: an international study of 40 patients. Brain. 2020;143:452‐466. doi: 10.1093/brain/awz410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Odermatt A, Taschner PE, Khanna VK, et al. Mutations in the gene‐encoding SERCA1, the fast‐twitch skeletal muscle sarcoplasmic reticulum Ca2+ ATPase, are associated with Brody disease. Nat Genet. 1996;14:191‐194. doi: 10.1038/ng1096-191. [DOI] [PubMed] [Google Scholar]
- 151. Bruels CC, Li C, Mendoza T, et al. Identification of a pathogenic mutation in ATP2A1 via in silico analysis of exome data for cryptic aberrant splice sites. Mol Genet Genomic Med. 2019;7:e552. doi: 10.1002/mgg3.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243‐251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 153. Carey KA, Farnfield MM, Tarquinio SD, Cameron‐Smith D. Impaired expression of Notch signaling genes in aged human skeletal muscle. J Gerontol A Biol Sci Med Sci. 2007;62:9‐17. doi: 10.1093/gerona/62.1.9. [DOI] [PubMed] [Google Scholar]
- 154. Jiang C, Wen Y, Kuroda K, Hannon K, Rudnicki MA, Kuang S. Notch signaling deficiency underlies age‐dependent depletion of satellite cells in muscular dystrophy. Dis Model Mech. 2014;7:997‐1004. doi: 10.1242/dmm.015917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Gnocchi VF, White RB, Ono Y, Ellis JA, Zammit PS. Further characterisation of the molecular signature of quiescent and activated mouse muscle satellite cells. PLoS One. 2009;4:e5205. doi: 10.1371/journal.pone.0005205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Feichtinger RG, Mucha BE, Hengel H, et al. Biallelic variants in the transcription factor PAX7 are a new genetic cause of myopathy. Genet Med. 2019;21:2521‐2531. doi: 10.1038/s41436-019-0532-z. [DOI] [PubMed] [Google Scholar]
- 157. Pasut A, Chang NC, Gurriaran‐Rodriguez U, et al. Notch signaling rescues loss of satellite cells lacking Pax7 and promotes brown adipogenic differentiation. Cell Rep. 2016;16:333‐343. doi: 10.1016/j.celrep.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Mourikis P, Gopalakrishnan S, Sambasivan R, Tajbakhsh S. Cell‐autonomous Notch activity maintains the temporal specification potential of skeletal muscle stem cells. Development. 2012;139:4536‐4548. doi: 10.1242/dev.084756. [DOI] [PubMed] [Google Scholar]
- 159. Ogasawara M, Iida A, Kumutpongpanich T, et al. CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations. Acta Neuropathol Commun. 2020;8:204. doi: 10.1186/s40478-020-01084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Sone J, Mitsuhashi S, Fujita A, et al. Long‐read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet. 2019;51:1215‐1221. doi: 10.1038/s41588-019-0459-y. [DOI] [PubMed] [Google Scholar]
- 161. Ishiura H, Shibata S, Yoshimura J, et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet. 2019;51:1222‐1232. doi: 10.1038/s41588-019-0458-z. [DOI] [PubMed] [Google Scholar]
- 162. Sun Q‐Y, Xu Q, Tian Y, et al. Expansion of GGC repeat in the human‐specific NOTCH2NLC gene is associated with essential tremor. Brain. 2020;143:222‐233. doi: 10.1093/brain/awz372. [DOI] [PubMed] [Google Scholar]
- 163. Wen J, Liu D, Zhao L. Small molecules targeting γ‐secretase and their potential biological applications. Eur J Med Chem. 2022;232:114169. doi: 10.1016/j.ejmech.2022.114169. [DOI] [PubMed] [Google Scholar]
- 164. Jaiswal A, Murakami K, Elia A, et al. Therapeutic inhibition of USP9x‐mediated Notch signaling in triple‐negative breast cancer. Proc Natl Acad Sci USA. 2021;118:e2101592118. doi: 10.1073/pnas.2101592118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Choi IY, Lim HT, Che YH, Lee G, Kim YJ. Inhibition of the combinatorial signaling of transforming growth factor‐beta and NOTCH promotes myotube formation of human pluripotent stem cell‐derived skeletal muscle progenitor cells. Cells. 2021;10:1649. doi: 10.3390/cells10071649 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1 Supporting information
Data Availability Statement
Data sharing not applicable ‐ no new data generated