

Abstract
c‐Ros oncogene 1, receptor tyrosine kinase (ROS1) genomic rearrangements have been reported previously in rare cases of colorectal cancer (CRC), yet little is known about the frequency, molecular characteristics, and therapeutic vulnerabilities of ROS1‐driven CRC. We analyzed a clinical dataset of 40 589 patients with CRC for ROS1 genomic rearrangements and their associated genomic characteristics (Foundation Medicine, Inc [FMI]). We moreover report the disease course and treatment response of an index patient with ROS1‐rearranged metastatic CRC. ROS1 genomic rearrangements were identified in 34 (0.08%) CRC samples. GOPC‐ROS1 was the most common ROS1 fusion identified (11 samples), followed by TTC28‐ROS1 (3 samples). Four novel 5′ gene partners of ROS1 were identified (MCM9, SRPK1, EPHA6, P4HA1). Contrary to previous reports on fusion‐positive CRC, ROS1‐rearrangements were found exclusively in microsatellite stable (MSS) CRCs. KRAS mutations were significantly less abundant in ROS1‐rearranged vs ROS1 wild type cases. The index patient presented with chemotherapy‐refractory metastatic right‐sided colon cancer harboring GOPC‐ROS1. Molecularly targeted treatment with crizotinib induced a rapid and sustained partial response. After 15 months on crizotinib disseminated tumor progression occurred and KRAS Q61H emerged in tissue and liquid biopsies. ROS1 rearrangements define a small, yet therapeutically actionable molecular subgroup of MSS CRC. In summary, the high prevalence of GOPC‐ROS1 and noncanonical ROS1 fusions pose diagnostic challenges. We advocate NGS‐based comprehensive molecular profiling of MSS CRCs that are wild type for RAS and BRAF and patient enrollment in precision trials.
Keywords: acquired resistance, colorectal cancer, crizotinib, molecular subgroups, precision treatment, ROS1 rearrangement
What's new?
The frequency, molecular characteristics, and therapeutic vulnerabilities of ROS1‐driven colorectal cancer remain largely unknown. Using a clinical dataset of 40 589 colorectal cancer patients, here the authors characterize the rare but highly actionable molecular colorectal cancer subgroup harboring genomic rearrangements of the receptor tyrosine kinase ROS1. They show that GOPC‐ROS1 is the most common ROS1 fusion and that unlike previously‐described fusion oncogenes, ROS1 rearrangements occur exclusively in microsatellite‐stable colorectal cancers. Moreover, they report the treatment course of an index patient, underlining the diagnostic challenge and therapeutic potential of ROS1 targeting in colorectal cancer.
Abbreviations
- CAP
College of American Pathologists
- cfDNA
cell‐free DNA
- CGP
comprehensive genomic profiling
- CLIA
Clinical Laboratory Improvement Amendments
- CNV
copy‐number variations
- CRC
colorectal cancer
- FFPE
formalin‐fixed paraffin‐embedded
- FISH
fluorescence in situ hybridization
- FMI
Foundation Medicine, Inc
- gLOH
genome‐wide loss of heterozygosity
- IHC
immunohistochemistry
- mCRC
metastatic CRC
- MSI‐H
microsatellite instable
- MSS
microsatellite stable
- Muts/Mb
mutations per megabase
- NGS
next‐generation sequencing
- NSCLC
nonsmall cell lung cancer
- ROS1 RE
ROS1 rearrangement
- ROS1
c‐Ros oncogene 1 receptor tyrosine kinase
- SBRT
stereotactic body radiation therapy
- SNV
single nucleotide variants
- TMB
tumor mutational burden
- TMB‐H
TMB‐high
1. INTRODUCTION
ROS1 genomic rearrangements in colorectal cancer (CRC) have been reported previously. 1 , 2 , 3 , 4 , 5 , 6 However, for CRC, only limited information is available on the prevalence of ROS1 fusion oncogenes, their genomic characteristics and molecular context, as well as the clinicopathological features and therapeutic vulnerabilities of ROS1‐rearranged CRC toward ROS1 inhibitors. Globally, up to 2.5% of CRCs harbor fusion oncogenes, and genomic rearrangements have been identified for RET, ALK, NTRK, ROS1, BRAF and FGFR. 7 , 8 , 9 , 10 , 11 , 12 Of note, oncogenic fusions in CRC have been reported to be strongly enriched in hypermutated microsatellite instable (MSI‐H) tumors, 1 , 6 , 13 , 14 , 15 with yet to be defined consequences for the targeting strategy for these tumors. Clinical data on molecularly targeted treatment of ROS1‐positive CRC are very limited, 6 which stands in stark contrast to ROS1‐driven nonsmall cell lung cancer (NSCLC), for which exist elaborate testing and targeting algorithms based on prospective clinical trials. 16 , 17 , 18
The ROS1 gene is localized on chromosome 6 (region 6q22.1) and encodes a receptor tyrosine kinase with a large extracellular domain (exons 1‐32), a transmembrane (exon 33) and an intracellular kinase domain (exons 36‐41). 19 The pathogenic breakpoints usually occur within introns 31 to 35, 20 , 21 preserving ROS1 kinase domain while quenching the majority of the ROS1 extracellular domain, and, redirecting subcellular distribution of the fusion protein. 19 ROS1 fusions arise through either intrachromosomal (eg, GOPC‐ROS1) or interchromosomal (eg, CD74‐ROS1) translocations. Downstream signaling of ROS1 activates the MAPK, 7 PI3K‐AKT‐mTOR, JAK‐STAT3 22 , 23 and VAV3‐RHO pathways, as well as the tyrosine phosphatase SHP2. 19 While ROS1 mutations, amplifications and overexpression do not hold clear independent oncogenic potential, ROS1 rearrangements act as oncogenic drivers and have been reported across 22 distinct types of malignancies, recombining with >55 known 5′ fusion partners. 19 , 24 These fusions lead to ROS1 hyperfunction due to ligand‐independent activity of the tyrosine kinase domain. 19 ROS1 fusions previously reported in metastatic CRC (mCRC) are limited to a small number of cases with GOPC‐ROS1, 4 , 5 , 7 SLC34A2‐ROS1 3 and TTC28‐ROS1. 5 Reported clinical data for targeting ROS1 fusions in CRC is, to our knowledge, restricted to a single case 6 and no data on the molecular mechanisms of acquired resistance to ROS1‐targeted treatment have been reported for CRC.
Here, we investigated 40 589 patients with CRC receiving genomic profiling during routine care, for the presence of ROS1 rearrangements. We describe the prevalence and molecular characteristics of the identified ROS1 fusions, as well as the molecular landscape of ROS1‐positive CRC. Additionally, we present an index clinical case harboring GOPC‐ROS1, illustrating diagnostic challenges as well as the therapeutic potential of ROS1‐positive CRC.
2. MATERIALS AND METHODS
2.1. Patient cohort
A review of the Foundation Medicine (FMI) CRC dataset comprising 40 589 patients was performed to extract all patients with ROS1 rearrangements identified by FoundationOne or FoundationOneCDx assays between 2014 and 2021. Approval for our study, including a waiver of informed consent and a Health Insurance Portability and Accountability Act (HIPAA) waiver of authorization, was obtained from the Western Institutional Review Board Protocol No. 20152817. All patient cases were sent to Foundation Medicine, Inc for comprehensive genomic profiling (CGP) testing during routine clinical care. Site of specimen was extracted from accompanying pathology reports. We further report an index patient undergoing treatment for ROS1‐positive mCRC at our institution. The patient provided written consent to publication of her clinical and molecular data.
2.2. Statistical analysis
Comparative frequency of genomic alterations between patient subgroups was assessed with the Fisher's exact test. Adjustment for multiple comparisons was performed using the Benjamini‐Hochberg procedure, with P values ≤.05 considered to be statistically significant.
2.3. ROS1 immunohistochemistry and fluorescence in situ hybridization
Immunohistochemistry (IHC) staining for ROS1 was performed on 2 μm thick formalin‐fixed paraffin‐embedded (FFPE) tumor tissue sections using two distinct antibodies for ROS1: the rabbit monoclonal antibody clone D4D6 (Cell Signaling Technology) at 1:100 dilution, and the rabbit monoclonal antibody clone SP384 (ready to use antibody, Ventana ). The lung adenocarcinoma cell line HCC‐78 with known SLC34A2‐ROS1 rearrangement was used as positive control. Two experienced board‐certified pathologists assessed ROS1 IHC expression. The IHC cytoplasmic staining was qualified as 0 (negative), 1+ (weak), 2+ (moderate), or 3+ (strong). As there are no standardized cut off values, Score 1+ in any percentage of tumor cells was considered positive (requiring further testing).
Fluorescence in situ hybridization (FISH) assays were performed using a ROS1 break‐apart probe set (Abbott, Illinois), which hybridizes with the 5′ telomeric (labeled with Spectrum Orange) a 3′ centromeric (labeled with Spectrum Green) sequence of the ROS1 gene. A minimum of 50 tumor nuclei were evaluated with a Zeiss Axio Imager Z2 fluorescent microscope. The assay was considered positive when ≥15% of the tumor cells exhibited break‐apart or separate green signals.
2.4. DNA sequencing
FoundationOneCDx and an earlier version of the assay, FoundationOne, are CGP assays performed in a Clinical Laboratory Improvement Amendments (CLIA)‐certified and College of American Pathologists (CAP)‐accredited laboratory (Foundation Medicine, Inc). Hybrid capture was performed for at least 324 genes to detect base substitutions, insertion and deletion alterations, copy number alterations and gene rearrangements 25 (Table S1). An anatomic pathology board certified pathologist reviewed each sample's hematoxylin and eosin stained slide under light microscopy to determine suitability for CGP testing (at least 20% tumor nuclei present) and the diagnosis of the sample (the accompanying pathology report is also used to help determine diagnosis). The tumor mutational burden (TMB) was determined on 0.8 to 1.1 Mb of sequenced DNA, as previously described. 26 A TMB of 10 or more mutations per megabase (Muts/Mb) was considered TMB‐high (TMB‐H). Assessment of microsatellite instability was performed from DNA sequencing using a principal component analysis from compiling intronic homopolymer repeat loci. 26 , 27 Genome‐wide loss of heterozygosity (gLOH) was calculated as previously described, with samples with a gLOH of at least 16% of the genome considered as LOH high. 28 , 29 The sequencing coverage and quality statistics for each sample are summarized in Table S1. The list of genes/regions included in the FoundationOne CDx targeted NGS assay is summarized in Table S2.
2.5. RNA panel sequencing
The Ion Torrent Oncomine Focus Assay (RNA part) was performed (Thermo Fisher Scientific). Following deparaffinization, RNA was extracted from FFPE tumor samples using Maxwell 16 System Purification Kit (Promega) and concentration measured with Qubit 2.0 fluorometer (Life Technologies). Ten nanograms of RNA was reverse transcribed to cDNA (Superscript VILO, Invitrogen). cDNA was used for library preparation following manufacturer's instructions. Next‐generation sequencing (NGS) libraries were templated using Ion Chef, sequenced on an Ion S5 System (Thermo Fisher Scientific) and analyzed using Ion Reporter Software 5.14 (Oncomine Focus w2.6—Fusions—Single Sample, default settings). The sequencing coverage and quality statistics for the analyzed sample are summarized in Table S3.
2.6. Cell‐free DNA assay
OncomineTM Pan‐Cancer Cell‐free assay interrogates 52 genes for the presence of mutations, small insertion/deletions, copy number alterations and fusions. Shortly, circulating cell‐free DNA (cfDNA)/RNA was isolated using Qiagen Circulating Nucleic Acid Kit (Qiagen). The cfDNA/RNA concentration was measured with Qubit and up to 50 ng (cfNT) were used for NGS library preparation. Emulsion‐PCR, enrichment and chip loading was carried out on the Ion Chef, followed by sequencing on the Ion S5 System. The protocols from Thermo Fisher Scientific (Massachusetts) were followed in all steps. Alignment, variant calling, and annotation was performed with the Ion Reporter software 5.14 workflow from Thermo Fisher Scientific (Oncomine TagSeq Pan‐Cancer Liquid Biopsy w2.3—Single Sample, default settings).
3. RESULTS
3.1. Prevalence and molecular characteristics of ROS1 genomic rearrangements
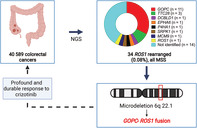
We analyzed sequencing data of 40 589 primary and metastatic CRC samples from the FMI CRC cohort for the presence of ROS1 genomic rearrangements. The following histological subtypes were included: colon adenocarcinoma (n = 33 440), rectal adenocarcinoma (n = 6993), rectal squamous carcinoma (n = 147), colon adenosquamous carcinoma (n = 9). All samples within the cohort had been submitted for testing during routine clinical care and underwent targeted DNA sequencing with FoundationOne or FoundationOneCDx assays. ROS1 rearrangements (ROS1 RE+) were detected in 34/40589 (0.084%) of cases (Figure 1A). GOPC was the most frequently detected 5′ ROS1 fusion partner in our cohort (11/34 of ROS1 RE+ samples) followed by TTC28 (3/34 samples). Other identified fusion partners were noncanonical (DCBLD1) or previously undescribed (EPHA6, P4HA1, SRPK1, MCM9) (Figure 1B). For 14/34 samples, breakpoints were detected between introns 30 and 35 of ROS1, suggesting a potentially pathogenic event, however, partner breakpoints were not uniquely mapped or were intergenic, such that no fusion partner could be definitively identified (Figure 1B). One case was annotated as an intragenic ROS1 rearrangement. The prevalence of 0.084% for CRC was significantly below the 0.23% (740/324103 samples) of ROS1 rearranged tumors identified within the overall pan‐solid tumor cohort (P < 10−5), with NSCLC and glioma showing the highest abundance of ROS1 rearrangements of all tumor types with 0.68% (431/63608) and 0.40% (42/10398), respectively (Figure 1C, Table S4).
FIGURE 1.
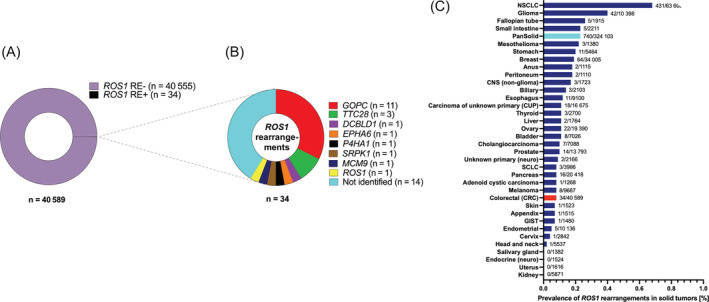
Frequency of ROS1 rearrangements and fusion partners in CRC. (A) Prevalence of ROS1 fusions in CRCs; 0.08% of CRC samples harbored ROS1 genomic rearrangements. (B) 5′ fusion partners of ROS1 across 34 identified CRC cases with ROS1 genomic rearrangements. (C) Prevalence of genomic ROS1 rearrangements across different tumor entities within the FMI solid tumor cohort. The overall frequency of ROS1 rearrangements is analyzed solid tumors is 0.23%. CRC, colorectal cancer; FMI, Foundation Medicine, Inc; RE, rearranged
The predicted genomic structures of the identified ROS1 rearrangements are illustrated in Figure 2. For 17/34 ROS1 RE+ CRC samples, the breakpoint in ROS1 was located within intron 34, for 6/34 in intron 32, 4/34 in intron 31 and 3/34 in intron 33; single cases with breakpoint in introns 30 and 35 were also identified (Figure 2A). Two cases had ROS1 breakpoints within exonic regions (one each in exon 31 and exon 35), both translocated onto intergenic fusion partners (Figure 2A). The resulting fusion genes are depicted in Figure 2B. The canonical shorter variant of GOPC‐ROS1 (breakpoint in intron 35) is the most commonly identified ROS1 fusion in our CRC cohort (10/34 ROS1 RE+ samples). TTC28 with variable breakpoints is the other previously described recurrent ROS1 fusion partner. 5 Several other singleton 5′ fusion partners were identified (Figure 2B). DCBLD1‐ROS1 is the result of a microdeletion on chromosome 6q22.1, previously found to express a GOPC‐ROS1 fusion transcript in glioma. 30 Of the newly identified ROS1 rearrangements, fusions with MCM9 and SRPK1 are intrachromosomal while rearrangements with EPHA6 and P4HA1 are interchromosomal. MCM9 is located on 6q22.31 and MCM9‐ROS1 is the result of a microdeletion of less than 2 megabases, while SRPK1 is localized at 6p21.3.1. Based on their structure, all fusions with identified 5′ fusion partners are predicted to be activating, leaving the kinase domain of ROS1 intact (Figure 2B).
FIGURE 2.
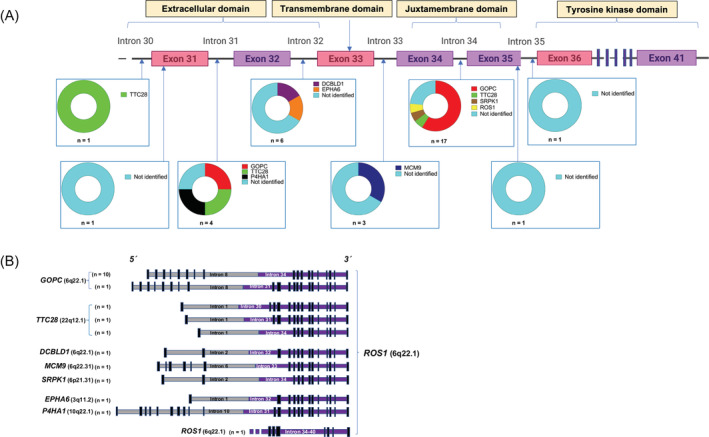
ROS1 fusion structure in CRC. (A) Schematic representation of ROS1 breakpoints located in introns 30 to 34, and exons 31 and 35 of the extracellular and transmembrane ROS1 domain. (B) Schematic of predicted intron‐exon structure of identified ROS1 fusions GOPC‐ROS1, DCBLD1‐ROS1 and MCM9‐ROS1 fusions occur caused by microdeletions within the chromosomal region 6q21
3.2. Molecular landscape of ROS1 ‐rearranged CRC
We next analyzed concomitant molecular alterations found in ROS1 RE+ CRCs. Importantly, within this large cohort, no ROS1 rearrangements were detected in MSI‐H tumors (0/2003 MSI‐H tumors) while 33/34 ROS1 rearrangements occurred in confirmed microsatellite‐stable (MSS) tumors (Table 1 and Figure 3A). In agreement with this, median TMB and the proportion of TMB high tumors, as defined by a TMB ≥10 Muts/mb, was similar between ROS1 RE+ and ROS1 RE− CRCs. Similarly, no differences in median genomic loss of heterozygosity (gLOH) score or the proportion of tumors with LOH high (defined by an gLOH score of 16 or higher) were found between ROS1 RE+ and ROS1 RE− CRCs (Table 1 and Figure 3A). The most frequent concomitant genomic alterations in CRCs with ROS1 rearrangements were single nucleotide variants (SNVs) in APC (82.4%) and TP53 (79.4%) (Figure 3B, Table S5). Overall, the comutational landscape of ROS1 RE+ and ROS1 RE− CRCs was similar with respect to single nucleotide variants (SNVs), copy‐number variations (CNV) and structural rearrangements (Figure 3B, Figure S1), with the sole exception of KRAS mutations, which were significantly less abundant in ROS1 RE+ CRCs (6/34 (17.7%) of ROS1 RE+ vs 19 576/40555 (48.3%) ROS1 RE− CRCs, P < .01; Figure 3B,C; Tables S5 and S6). With respect to patient and tumor characteristics, there was no sex preference or differential distribution between colon and rectum for ROS1 RE+ vs ROS1 RE− CRCs (Table 1).
TABLE 1.
Clinicopathological characteristics of ROS1 RE (+) vs ROS1 RE (−) CRCs
| n = 40 589 | ||
|---|---|---|
| Characteristics | ROS1 RE (+) (n = 34) | ROS1 RE (−) (n = 40 555) |
| Age—median (IQR) (y) | 62.0 (54.8‐69.8) | 60.0 (51‐69) |
| Histological subtype (%) | ||
| Colon adenocarcinoma (n = 33 440) | 27 (79.4) | 33 413 (82.4) |
| Rectum adenocarcinoma (n = 6993) | 6 (17.6) | 6987 (17.2) |
| Rectum squamous cell carcinoma (n = 147) | 1 (2.9) | 146 (0.4) |
| Colon adenosquamous carcinoma (n = 9) | 0 (0.00) | 9 (0.02) |
| Sex—n (%) | ||
| Female | 15 (44.1) | 18 356 (45.3) |
| Male | 19 (55.9) | 22 176 (54.7) |
| Unknown | 0 (0.0) | 23 (0.0) |
| Analyzed tumor‐sample—n (%) | ||
| Local tumor | 14 (41.2) | 20 844 (51.4) |
| Metastasis | 14 (41.2) | 15 125 (37.3) |
| Lymph node | 1 (2.9) | 1311 (3.2) |
| Unknown | 5 (14.7) | 3235 (8.0) |
| MSI status—n (%) | ||
| Available | 33 (97.1) | 39 359 (97.1) |
| MSI‐H | 0 (0.0) | 2003 (5.1) |
| Unknown | 1 (2.9) | 1196 (2.9) |
| LOH score—n (%) | 24 (70.6) | 24 931 (61.5) |
| Available | 5.62 (4.24‐9.50) | 6.01 (3.23‐9.44) |
| Median LOH score (IQR) | ||
| LOH‐high | 2 (8.3) | 1390 (5.6) |
| Unknown | 10 (29.4) | 15 624 (38.5) |
| TMB—n (%) | ||
| Available | 34 (100.0) | 40 554 (100.0) |
| Median (IQR) | 3.75 (1.74‐5.22) | 3.75 (1.74‐6.78) |
| TMB‐high | 2 (5.9) | 3492 (8.6) |
| Unknown | 0 (0.0) | 1 (0.0) |
Abbreviations: IQR, interquartile range; n, number of samples; RE, rearrangement.
FIGURE 3.
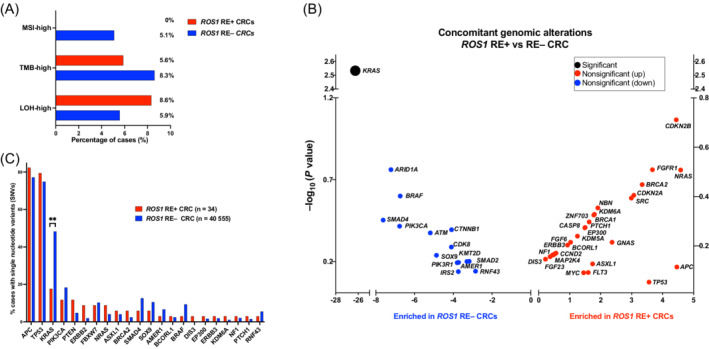
Molecular landscape of ROS1‐fusion‐positive CRCs. (A) Frequency of tumors classified as MSI‐H, genome‐wide loss of heterozygosity (gLOH) high and TMB‐high by FoundationOne test in the ROS1 rearranged vs nonrearranged cohort. (B) Most common concomitant mutations (SNVs) in ROS1 rearranged vs nonrearranged CRCs; **P = .0029; (C) Difference in relative mutational frequency of concomitant genomic alterations, enriched in the ROS1 rearranged vs nonrearranged cohort; p values (Fisher's exact test) are shown in the Y axis. CRC, metastatic colorectal cancer; RE+, rearranged; RE−, nonrearranged; SNV, single nucleotide variants
3.3. Clinical presentation of index case and identification of GOPC‐ROS1
A 48‐year‐old female was referred to our center with a hitherto chemotherapy‐refractory metastatic right‐sided colorectal cancer, following two lines of state‐of‐the‐art systemic treatment. Initial tumor stage had been pT4a, pN1b (3/31), cM1a (HEP). Initial molecular profiling (Sanger Sequencing and IHC) had shown KRAS, NRAS and BRAF wild type, HER2 score 0, and normal expression of mismatch repair proteins MLH1, MSH2, MSH6 and PMS2 (data not shown). The patient had experienced disease progression as best response during first line chemotherapy with FOLFOX (5‐FU, leucovorin, oxaliplatin), as well as second line treatment with FOLFOXIRI (5‐FU, leucovorin, oxaliplatin, irinotecan) plus bevacizumab. Following a 3‐month period of third line treatment at our institution with FOLFIRI (folinic acid, 5‐fluorouracil and irinotecan) plus cetuximab, combined with hepatic intraarterial floxuridine treatment, peritoneal and hepatic disease progression occurred (Figure 4A, Figure S2). Following our internal diagnostic algorithm for stage IV CRC (Figure S3), we performed comprehensive next‐generation sequencing (FoundationOne CDx) of a tumor biopsy, which detected GOPC‐ROS1 in a KRAS, NRAS, BRAF wild type, microsatellite‐stable tumor with a TMB of 4 (Figure 4B). We performed targeted RNA panel sequencing (Oncomine Focus Assay), which confirmed high read counts of GOPC‐ROS1 on transcript level (Figure 4B). ROS1 IHC staining showed negative tumor cell staining with ROS1 antibody clone D4D6 (Cell Signaling Technology, Figure 5A), however positive staining with clone SP384 (Ventana, Figure 5B). Additionally, ROS1 FISH was negative, as previously described for GOPC‐ROS1 31 , 32 (Figure 5C).
FIGURE 4.
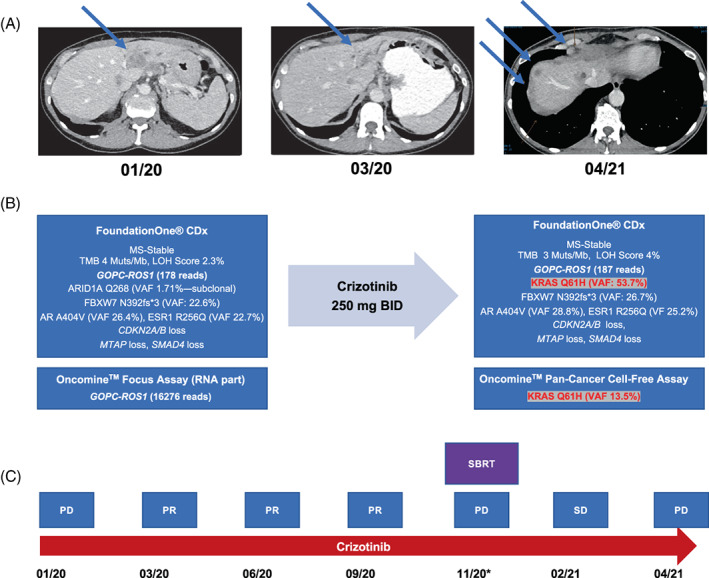
Disease course and treatment response to crizotinib in GOPC‐ROS1‐positive mCRC (index case). (A) CT scans (transversal) showing disease course along treatment with crizotinib. (B) Results of tumor tissue NGS with FoundationOne CDx assay, confirmation of ROS1 liquid biopsy NGS with Oncomine Pan‐Cancer Cell‐free DNA assay before and after acquired resistance. (C) Schematic representation of disease and therapeutic course. *Oligometastatic progression. amp, amplification; BID, twice daily; CNV, copy number variation; LOH, loss of heterozygosity; MS‐stable, microsatellite‐stable; PD, progressive disease; PR, partial response; SD, stable disease; SBRT, stereotactic body radiotherapy; TMB, tumor mutational burden; VAF, variant allele frequency
FIGURE 5.
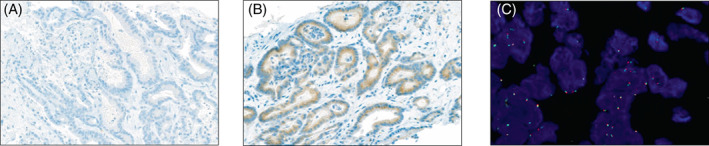
ROS1 immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH) of a GOPC‐ROS1‐positive mCRC (index case). (A) Negative IHC with monoclonal anti‐rabbit antibody D4D6 (Cell Signaling Technology). (B) Positive IHC with monoclonal anti‐rabbit antibody SP384 (Ventana). (C) Negative FISH in GOPC‐ROS1 rearranged mCRC. Green dots: 6q22 ROS1 centromeric probe; orange dots: 6q22 ROS1 telomeric probe; overlapping or adjacent green and orange signals: negative result (no break‐apart). FFPE, formalin‐fixed paraffin embedded; FISH, fluorescence in situ hybridization; IHC, immunohistochemistry; mCRC, metastatic colorectal cancer
3.4. Molecularly targeted treatment with crizotinib
The patient was switched to fourth line off‐label treatment with crizotinib and experienced a rapid and sustained clinical and radiologic response (Figure 4A,C). Symptomatic oligo‐progression with two peritoneal lesions occurred after 11 months on crizotinib, and stereotactic body radiation therapy (SBRT) with 5 × 9 Gy (45 Gy) was performed. After 15 months on crizotinib, disseminated peritoneal and hepatic tumor progression occurred (Figure 4C). We performed tumor tissue and liquid biopsies, both uncovering the emergence of KRAS Q61H at high allele frequencies within an otherwise unchanged tumor molecular profile (Figure 4B). The patient was switched fifth line treatment with trifluridine/tipiracil (TAS‐102), however her overall clinical condition deteriorated rapidly.
4. DISCUSSION
Triggered by the reported index case, we analyzed a large cohort of CRC cases for the presence of ROS1 genomic rearrangements. We find that ROS1 rearrangements are rare genomic events in CRC, with a prevalence of less than 1 in 1000 patients. Individual cases of ROS1 rearrangements in colorectal cancer have been reported previously. 1 , 2 , 3 , 4 , 5 , 6 However, to our knowledge, our analysis queried the hitherto largest CRC cohort and reports the most comprehensive analysis of ROS1 rearrangements in this tumor entity, uncovering several further aspects with respect to fusion partners, concomitant molecular landscape, and clinical strategy.
The analyzed FMI CRC cohort can be considered a large real‐world molecular data base, since all cases had been submitted for molecular profiling during routine treatment. Despite susceptibility for bias, the distribution of key molecular features of CRC across the cohort (50% mutations in KRAS, 4% NRAS, 10% BRAF, 5% microsatellite instability), is in excellent agreement with other CRC cohorts and trial populations 33 , 34 suggesting that the various molecular subgroups of CRC are well‐represented. Another strength of our cohort is analysis of all samples with the very same NGS assays (FoundationOne and FDA‐approved FoundationOneCDx), with rigorous quality controls. 35 A relevant limitation of the cohort in the context of fusion oncogenes is that expression of the identified rearrangements cannot be confirmed and further characterized on genomic, transcript and protein level, as we undertook for the reported index case. Another limitation of the cohort is the lack of more detailed clinical data, which precluded the analysis of relevant clinical characteristics for the cohort including tumor sidedness, metastatic pattern, histopathologic features, response to state‐of‐the‐art chemotherapy and survival endpoints, which in the context of CRC fusion oncogenes have been reported by others for smaller cohorts. 6 The hitherto largest cohort with detailed clinical data analyzed 27 fusion‐driven CRC including 3 patients with ROS1 rearrangement and reported a significant association with higher age, right‐sided primary tumors, lymphatic spread and poor overall survival independent of microsatellite instability. 1 In our cohort, we observed no significant age difference between ROS1 RE+ and ROS1 RE− patients (median 62 vs 60 years, Table 1). However, our index patient's disease course strongly echoed the overall poor outcome of fusion‐driven mCRC. Clinical features specifically of ROS1‐positive tumors have best been studied for NSCLC, where ROS1 fusions are enriched in younger nonsmoker patients, tend to metastasize to the brain, however, appear to be associated with a more favorable outcome than ROS1‐negative tumors. 36 , 37
Our data indicate that GOPC‐ROS1 is the most common ROS1 fusion in CRC, which is in agreement with previously reported individual cases. 1 , 4 GOPC‐ROS1 was the first identified ROS1 fusion 38 and appears to be a predominant alteration in glioma 30 and other non‐NSCLC tumors. 5 Interestingly, DCBLD1‐ROS1 (n = 1) induces expression of a GOPC‐ROS1 fusion transcript. 30 None of the canonical ROS1 fusions found in NSCLC 19 (eg, CD74‐ROS1, EZR‐ROS1, SDC4‐ROS1) were present in our CRC cohort, indicating significant site‐ and entity‐specific differences in the molecular landscape of ROS1 fusion oncogenes and the underlying pathogenic mechanisms. It is also notable that 14/20 translocations with identified fusion partners in CRC are intrachromosomal rearrangements, with only 5/20 samples showing interchromosomal rearrangements. Four identified 5′ fusions partners of ROS1 have not been previously described. 5 MCM9‐ROS1 is the result of a microdeletion on chromosome 6q22, making a strong case for a pathogenic event. SRPK1‐ROS1 is an intrachromosomal rearrangement, while EPHA6‐ROS1 and P4HA1‐ROS1 are interchromosomal events. To this date, more than 55 fusion partners of ROS1 have been described illustrating profound molecular heterogeneity of ROS1‐driven tumors. 19
Fourteen of the identified ROS1 rearrangements in CRC are translocations onto intergenic regions, one rearrangement was described as an intragenic event within ROS1. These cases are difficult to assess with a DNA‐based targeted assay alone. FoundationOne uses a hybrid capture methodology and detects gene rearrangements on the genomic level. 25 Obtaining additional resolution on the expression of a functional fusion protein resulting from these complex rearrangements is challenging. 39 Therefore, such cases require careful individual assessment through complementary approaches including targeted RNA sequencing and IHC or FISH. No RNA sequencing data or histopathological analyses are available for these samples to positively confirm expression of an in‐frame functional fusion construct. However, based on their breakpoints within introns or exons 31 to 35 of ROS1, we speculate that a significant subset of those alterations might be pathogenic leading to expression of a functional fusion protein.
As a major difference between our findings and previous reports on fusion oncogene‐driven CRC, 1 , 6 , 13 , 14 , 15 we find ROS1 rearrangements exclusively in MSS CRC. Previous reports had found a strong association between fusion oncogenes and tumors with sporadic microsatellite instability caused by MLH1 promoter hypermethylation. 6 , 13 However, when performing a case‐by‐case analysis of the published data, it is intriguing that ROS1 fusions had been strongly outnumbered by ALK, RET and NTRK rearrangements in these studies, and while NTRK and ALK fusions genes clearly showed enrichment in MSI‐H tumors, most if not all of the few reported ROS1 fusions had actually been found in MSS CRC or tumors with unreported microsatellite status. 3 , 6 , 13 We interpret our results as representative given that no ROS1 fusions were identified in more than 2000 MSI‐H tumors in the cohort, making enrichment in this group of tumors highly unlikely. These findings indicate that ROS1‐driven CRCs might in pathobiology and clinical characteristics differ from other rare fusion oncogenes in CRC.
The comutational landscape of ROS1 RE+ CRC in our cohort is largely indistinguishable from ROS1 RE− tumors, with the significant analytical limitation that the two cohorts for comparison differ markedly in size precluding the identification of any subtle differences. The only significant finding is that ROS1 rearrangements are enriched in KRAS wild type CRC, which is in agreement with previous reports on CRC fusion oncogenes. 6 However, in contrast to other fusions‐driven CRCs, which have almost exclusively been reported in RAS/RAF wild type CRC, we did find a number of cases of ROS1‐rearranged CRC harboring activating coalterations in KRAS, NRAS and BRAF. It remains unclear, whether the identified few cases with concomitant ROS1 alterations and KRAS, NRAS or BRAF mutations are the consequence of intratumor heterogeneity or represent examples of primary or acquired resistance to targeted treatment, since no treatment data for these cases is available. The index patient acquired KRAS Q61H at high allele frequency during treatment with crizotinib and we cannot rule out that other patients in the cohort had been treated with ROS1‐directed treatment elsewhere.
The high prevalence of intrachromosomal microdeletion within the identified cohort of ROS1 rearrangements including GOPC‐ROS1, DLBCD‐1‐ROS1 and MCM9‐ROS1 has implications for the diagnostic approach to screen for ROS1‐fusions in CRC. For the index case harboring GOPC‐ROS1, IHC with ROS1 antibody clone D4D6 stained negative while antibody clone SP384 stained positive. The short variant of GOPC‐ROS1 has previously been reported to generate heterogenous results on IHC probably because partial deletion of epitopes during the recombination events. 2 Also ROS1 antibody clone D4D6 is considered less sensitive than SP384. 40 Because of the small microdeletion on chromosome 6q22.1, FISH remains consistently false‐negative with GOPC‐ROS1. 31 We therefore advocate an upfront NGS‐based approach to identify rare cases of ROS1‐driven CRC rather than routine IHC screening for ROS1. In unclear cases, targeted RNA sequencing should complement DNA sequencing. We restrict comprehensive profiling to mCRCs without known driver alteration (pan‐wild type tumors, diagnostic algorithm in Figure S3).
Treatment data for ROS1‐rearranged CRC are sparse. To our knowledge, the reported index case is only the second reported case of molecularly targeted treatment for ROS1‐driven CRC.
A recently published manuscript reports a sustained response of a GOPC‐ROS1‐positive CRC to entrectinib, ongoing 9 months after treatment initiation. 6 Our patient showed a 15‐month response to crizotinib, before disseminated tumor progression occurred. Data on chemotherapy sensitivity for mCRCs harboring fusion oncogenes are limited, with a small number of reported cases showing rather poor responses. 1 Our index patient's tumor was completely refractory to several lines of standard chemotherapy, as well as treatment with the molecularly targeted agents bevacizumab and the anti‐EGFR antibody cetuximab. In NSCLC, acceptable response rates (45%‐60%) to standard chemotherapy in ROS1‐positive tumors have been reported. 19 , 41 , 42 However, in NSCLC, first‐line treatment with crizotinib showed superior responses over pemetrexed‐based chemotherapy, as well as prolonged progression‐free survival (PFS). 43 Current international guidelines recommend either crizotinb or entrectinib, as first‐line targeted treatment for ROS1‐positive NSCLC. 17
Acquired resistance to crizotinib in our case was likely driven by KRAS Q61H, which is, to our knowledge, the first report of acquired resistance to ROS1‐targeted treatment in mCRC. Secondary resistance to ROS1‐targeted treatment can develop through secondary resistance mutations within the kinase domain of ROS1, 44 as well as reactivation of ROS1‐ effector pathways, including de novo mutations in the downstream MAPK pathway (eg, KRAS, NRAS, BRAF), KIT or MET amplifications. 19 , 45 , 46 , 47 , 48 KRAS‐mutations are a very characteristic secondary events in mCRC, emerging during molecularly targeted treatment, both with anti‐EGFR‐ and BRAF‐V600E directed therapy. 49 Identification of molecular alterations driving acquired drug resistance can in a subset of cases guide subsequent targeted treatment lines, 19 , 50 which was not the case with our index patient. Due to the rarity of fusion oncogenes in CRC, any patient should be enrolled in basket trials such as the TAPISTRY (NCT04589845) whenever feasible.
AUTHOR CONTRIBUTIONS
The work reported in the paper has been performed by the authors, unless clearly specified in the text. Conception and design: Dilara Akhoundova, Saskia Hussung, Ethan Sokol, Ralph M. Fritsch. Data curation: Smruthy Sivakumar, Antonia Töpfer, Markus Rechsteiner, Florian Angst, Ethan Sokol, Ralph M. Fritsch. Analysis and interpretation of data: Dilara Akhoundova, Saskia Hussung, Smruthy Sivakumar, Markus Rechsteiner, Antonia Töpfer, Abdullah Kahraman, Fabian Arnold, Ethan Sokol, Ralph M. Fritsch. Investigation: Dilara Akhoundova, Saskia Hussung, Smruthy Sivakumar, Antonia Töpfer, Markus Rechsteiner, Abdullah Kahraman, Fabian Arnold, Florian Angst, Christian Britschgi, Achim Weber, Ethan Sokol, Ralph M. Fritsch. Writing—original draft: Dilara Akhoundova, Saskia Hussung, Ralph M. Fritsch. Writing—manuscript review & editing: Smruthy Sivakumar, Antonia Töpfer, Christian Britschgi, Achim Weber, Ethan Sokol. Validation: Smruthy Sivakumar, Ralph M. Fritsch. Resources: Martin Zoche, Holger Moch, Supervision: Martin Zoche, Holger Moch, Achim Weber, Ethan Sokol, Ralph M. Fritsch. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST
Saskia Hussung reports fees from Pierre Fabre and Incyte outside the submitted work. Smruthy Sivakumar is an employee at Foundation Medicine, Inc, a wholly owned subsidiary of Roche Holdings, Inc and Roche Finance Ltd, and has equity interest in an affiliate of these Roche entities (shareholder at Roche). Christian Britschgi reports Consulting or Advisory Role for AstraZeneca, Pfizer, Roche, Takeda, Janssen Cilag, and Boehringer‐Ingelheim, and Travel, Accommodations, Expenses from AstraZeneca and Takeda, all outside the submitted work. Ethan Sokol is an employee at Foundation Medicine, Inc, a wholly owned subsidiary of Roche Holdings, Inc and Roche Finance Ltd, and has equity interest in an affiliate of these Roche entities (shareholder at Roche). The institution of Holger Moch received research funding from Roche. Martin Zoche received research grants from F. Hoffmann‐La Roche AG and consultancy fees from Astra Zeneca and Bayer AG. Ralph M. Fritsch reports grants and personal fees from Pierre Fabre, Bayer, Merck, MSD, Astra Zeneca, Novartis and BMS outside the submitted work. No disclosures were reported from the other authors.
ETHICS STATEMENT
Approval for our study, including a waiver of informed consent and a Health Insurance Portability and Accountability Act (HIPAA) waiver of authorization, was obtained from the Western Institutional Review Board Protocol No. 20152817. The index patient provided written consent to publication of her clinical and molecular data.
Supporting information
Appendix S1. Supporting information.
Appendix S2. Supporting information.
ACKNOWLEDGEMENTS
The graphical abstract was created with BioRender.com. Open access funding provided by Universitat Zurich.
Akhoundova D, Hussung S, Sivakumar S, et al. ROS1 genomic rearrangements are rare actionable drivers in microsatellite stable colorectal cancer. Int J Cancer. 2022;151(12):2161‐2171. doi: 10.1002/ijc.34257
Dilara Akhoundova and Saskia Hussung contributed equally to our study.
DATA AVAILABILITY STATEMENT
The results underlying Figures 1, 2, 3, Figure S1, Table 1, and Tables S1, S4, S5, and S6 are based on genomic sequencing data from Foundation Medicine Inc. Further data that support the findings of our study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Pietrantonio F, Di Nicolantonio F, Schrock AB, et al. ALK, ROS1, and NTRK rearrangements in metastatic colorectal cancer. J Natl Cancer Inst. 2017;109:djx098. https://academic.oup.com/jnci/article/109/12/djx089/3860155 [DOI] [PubMed] [Google Scholar]
- 2. Huang RSP, Gottberg‐Williams A, Vang P, et al. Correlating ROS1 protein expression with ROS1 fusions, amplifications, and mutations. JTO Clin Res Rep. 2021;2:100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aisner DL, Nguyen TT, Paskulin DD, et al. ROS1 and ALK fusions in colorectal cancer, with evidence of intratumoral heterogeneity for molecular drivers. Mol Cancer Res. 2014;12:111‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang M, Zhu L, Huang D, Shen X, Zhang H, She X. Abstract 2179: identification of ROS1 alterations in ctDNA from colorectal cancer patients in China. Cancer Res. 2021;81:2179. [Google Scholar]
- 5. Huang RSP, Haberberger J, Sokol E, et al. Clinicopathologic, genomic and protein expression characterization of 356 ROS1 fusion driven solid tumors cases. Int J Cancer. 2021;148:1778‐1788. [DOI] [PubMed] [Google Scholar]
- 6. Singh H, Li YY, Spurr LF, et al. Molecular characterization and therapeutic targeting of colorectal cancers harboring receptor tyrosine kinase fusions. Clin Cancer Res. 2021;27:1695‐1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sato H, Schoenfeld AJ, Siau E, et al. MAPK pathway alterations correlate with poor survival and drive resistance to therapy in patients with lung cancers driven by ROS1 fusions. Clin Cancer Res. 2020;26:2932‐2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choi Y, Kwon CH, Lee SJ, Park J, Shin J‐Y, Park DY. Integrative analysis of oncogenic fusion genes and their functional impact in colorectal cancer. Br J Cancer. 2018;119:230‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Svrcek M, Colle R, Cayre A, et al. Prevalence of NTRK1/3 fusions in mismatch repair‐deficient (dMMR)/microsatellite instable (MSI) tumors of patients with metastatic colorectal cancer (mCRC). J Clin Oncol. 2021;39:e15537. [Google Scholar]
- 10. Pietrantonio F, Di Nicolantonio F, Schrock AB, et al. RET fusions in a small subset of advanced colorectal cancers at risk of being neglected. Ann Oncol. 2018;29:1394‐1401. [DOI] [PubMed] [Google Scholar]
- 11. Lasota J, Chłopek M, Lamoureux J, et al. Colonic adenocarcinomas harboring NTRK fusion genes: a clinicopathologic and molecular genetic study of 16 cases and review of the literature. Am J Surg Pathol. 2020;44:162‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kloosterman WP, Coebergh van den Braak RRJ, Pieterse M, et al. A systematic analysis of oncogenic gene fusions in primary colon cancer. Cancer Res. 2017;77:3814‐3822. [DOI] [PubMed] [Google Scholar]
- 13. Cocco E, Benhamida J, Middha S, et al. Colorectal carcinomas containing hypermethylated MLH1 promoter and wild‐type BRAF/KRAS are enriched for targetable kinase fusions. Cancer Res. 2019;79:1047‐1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sato K, Kawazu M, Yamamoto Y, et al. Fusion kinases identified by genomic analyses of sporadic microsatellite instability‐high colorectal cancers. Clin Cancer Res. 2019;25:378‐389. [DOI] [PubMed] [Google Scholar]
- 15. Vaňková B, Vaněček T, Ptáková N, et al. Targeted next generation sequencing of MLH1‐deficient, MLH1 promoter hypermethylated, and BRAF/RAS‐wild‐type colorectal adenocarcinomas is effective in detecting tumors with actionable oncogenic gene fusions. Genes Chromosomes Cancer. 2020;59:562‐568. [DOI] [PubMed] [Google Scholar]
- 16. Lindeman NI, Cagle PT, Aisner DL, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch Pathol Lab Med. 2018;142:321‐346. [DOI] [PubMed] [Google Scholar]
- 17. Ettinger DS, Wood DE, Aggarwal C, et al. NCCN guidelines insights: non‐small cell lung cancer, version 1.2020. J Natl Compr Cancer Netw. 2019;17:1464‐1472. [DOI] [PubMed] [Google Scholar]
- 18. Shaw AT, Ou S‐HI, Bang Y‐J, et al. Crizotinib in ROS1‐rearranged non‐small‐cell lung cancer. N Engl J Med. 2014;371:1963‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Drilon A, Jenkins C, Iyer S, Schoenfeld A, Keddy C, Davare MA. ROS1‐dependent cancers—biology, diagnostics and therapeutics. Nat Rev Clin Oncol. 2021;18:35‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bubendorf L, Büttner R, Al‐Dayel F, et al. Testing for ROS1 in non‐small cell lung cancer: a review with recommendations. Virchows Arch. 2016;469:489‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li B, Qu H, Zhang J, et al. Genomic characterization and outcome evaluation of kinome fusions in lung cancer revealed novel druggable fusions. npj Precis Oncol. 2021;5:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crescenzo R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015;27:516‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bayard Q, Caruso S, Couchy G, et al. Recurrent chromosomal rearrangements of ROS1, FRK and IL6 activating JAK/STAT pathway in inflammatory hepatocellular adenomas. Gut. 2020;69:1667‐1676. [DOI] [PubMed] [Google Scholar]
- 24. Uguen A, De Braekeleer M. ROS1 fusions in cancer: a review. Future Oncol. 2016;12:1911‐1928. [DOI] [PubMed] [Google Scholar]
- 25. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trabucco SE, Gowen K, Maund SL, et al. A novel next‐generation sequencing approach to detecting microsatellite instability and Pan‐tumor characterization of 1000 microsatellite instability‐high cases in 67,000 patient samples. J Mol Diagn. 2019;21:1053‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Smyth EC, Cafferkey C, Loehr A, et al. Genomic loss of heterozygosity and survival in the REAL3 trial. Oncotarget. 2018;9:36654‐36665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pawlyn C, Loehr A, Ashby C, et al. Loss of heterozygosity as a marker of homologous repair deficiency in multiple myeloma: a role for PARP inhibition? Leukemia. 2018;32:1561‐1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davare MA, Henderson JJ, Agarwal A, et al. Rare but recurrent ROS1 fusions resulting from chromosome 6q22 microdeletions are targetable oncogenes in glioma. Clin Cancer Res. 2018;24:6471‐6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davies KD, Le AT, Sheren J, et al. Comparison of molecular testing modalities for detection of ROS1 rearrangements in a cohort of positive patient samples. J Thorac Oncol. 2018;13:1474‐1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Conde E, Hernandez S, Martinez R, et al. Assessment of a new ROS1 immunohistochemistry clone (SP384) for the identification of ROS1 rearrangements in patients with non‐small cell lung carcinoma: the ROSING study. J Thorac Oncol. 2019;14:2120‐2132. [DOI] [PubMed] [Google Scholar]
- 33. Lieu CH, Golemis EA, Serebriiskii IG, et al. Comprehensive genomic landscapes in early and later onset colorectal cancer. Clin Cancer Res. 2019;25:5852‐5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yaeger R, Chatila WK, Lipsyc MD, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33:125‐36.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. https://www.foundationmedicine.com/press-releases/f2b20698-10bd-4ac9-a5e5c80c398a57b5. Accessed March 1, 2022.
- 36. Zinsky R. Metaanalysis of ROS1‐positive lung cancer cases. Eur Respir J. 2016;48:PA2867. [Google Scholar]
- 37. Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Birchmeier C, Sharma S, Wigler M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc Natl Acad Sci U S A. 1987;84:9270‐9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li W, Liu Y, Li W, Chen L, Ying J. Intergenic breakpoints identified by DNA sequencing confound targetable kinase fusion detection in NSCLC. J Thorac Oncol. 2020;15:1223‐1231. [DOI] [PubMed] [Google Scholar]
- 40. Conde E, Hernandez S, Benito A, Caminoa A, Garrido P, Lopez‐Rios F. Screening for ROS1 fusions in patients with advanced non‐small cell lung carcinomas using the VENTANA ROS1 (SP384) rabbit monoclonal primary antibody. Expert Rev Mol Diagn. 2021;21:437‐444. [DOI] [PubMed] [Google Scholar]
- 41. Park S, Ahn B‐C, Lim SW, et al. Characteristics and outcome of ROS1‐positive non–small cell lung cancer patients in routine clinical practice. J Thorac Oncol. 2018;13:1373‐1382. [DOI] [PubMed] [Google Scholar]
- 42. Chen YF, Hsieh MS, Wu SG, et al. Clinical and the prognostic characteristics of lung adenocarcinoma patients with ROS1 fusion in comparison with other driver mutations in east Asian populations. J Thorac Oncol. 2014;9:1171‐1179. [DOI] [PubMed] [Google Scholar]
- 43. Shen L, Qiang T, Li Z, Ding D, Yu Y, Lu S. First‐line crizotinib versus platinum‐pemetrexed chemotherapy in patients with advanced ROS1‐rearranged non‐small‐cell lung cancer. Comp Study. 2020;9:3310‐3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Drilon A, Zhai D, Deng W, et al. Abstract 442: repotrectinib, a next generation TRK inhibitor, overcomes TRK resistance mutations including solvent front, gatekeeper and compound mutations. Cancer Res. 2019;79:442. [Google Scholar]
- 45. McCoach CE, Le AT, Gowan K, et al. Resistance mechanisms to targeted therapies in ROS1(+) and ALK(+) non‐small cell lung cancer. Clin Cancer Res. 2018;24:3334‐3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Drilon A, Somwar R, Wagner JP, et al. A novel crizotinib‐resistant solvent‐front mutation responsive to Cabozantinib therapy in a patient with ROS1‐rearranged lung cancer. Clin Cancer Res. 2016;22:2351‐2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin JJ, Johnson T, Lennerz JK, et al. Resistance to lorlatinib in ROS1 fusion‐positive non‐small cell lung cancer. J Clin Oncol. 2020;38:9611. [Google Scholar]
- 48. Cargnelutti M, Corso S, Pergolizzi M, et al. Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget. 2015;6:5182‐5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Misale S, Yaeger R, Hobor S, et al. Emergence of KRAS mutations and acquired resistance to anti‐EGFR therapy in colorectal cancer. Nature. 2012;486:532‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Akhoundova D, Pietge H, Hussung S, et al. Targeting secondary and tertiary resistance to BRAF inhibition in BRAF V600E‐mutated metastatic colorectal cancer. JCO Precis Oncol. 2021;5:1082‐1087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting information.
Appendix S2. Supporting information.
Data Availability Statement
The results underlying Figures 1, 2, 3, Figure S1, Table 1, and Tables S1, S4, S5, and S6 are based on genomic sequencing data from Foundation Medicine Inc. Further data that support the findings of our study are available from the corresponding author upon reasonable request.