

Abstract
Objective
γ‐Aminobutyric acid (GABA)A‐receptor subunit variants have recently been associated with neurodevelopmental disorders and/or epilepsy. The phenotype linked with each gene is becoming better known. Because of the common molecular structure and physiological role of these phenotypes, it seemed interesting to describe a putative phenotype associated with GABAA‐receptor–related disorders as a whole and seek possible genotype–phenotype correlations.
Methods
We collected clinical, electrophysiological, therapeutic, and molecular data from patients with GABAA‐receptor subunit variants (GABRA1, GABRB2, GABRB3, and GABRG2) through a national French collaboration using the EPIGENE network and compared these data to the one already described in the literature.
Results
We gathered the reported patients in three epileptic phenotypes: 15 patients with fever‐related epilepsy (40%), 11 with early developmental epileptic encephalopathy (30%), 10 with generalized epilepsy spectrum (27%), and 1 patient without seizures (3%). We did not find a specific phenotype for any gene, but we showed that the location of variants on the transmembrane (TM) segment was associated with a more severe phenotype, irrespective of the GABAA‐receptor subunit gene, whereas N‐terminal variants seemed to be related to milder phenotypes.
Significance
GABAA‐receptor subunit variants are associated with highly variable phenotypes despite their molecular and physiological proximity. None of the genes described here was associated with a specific phenotype. On the other hand, it appears that the location of the variant on the protein may be a marker of severity. Variant location may have important weight in the development of targeted therapeutics.
Keywords: channelopathy, developmental and epileptic encephalopathy, GABA A receptor, genetic generalized epilepsy
Key points.
γ‐Aminobutyric acid (GABA)A–receptor subunit variants lead to a fever‐related epilepsy, a generalized epilepsy, or a developmental and epileptic encephalopathy
Phenotype was not associated with a given gene, but with the location of variants within the protein
Variants of the transmembrane domain are significantly more severe than other variants in our cohort and in 402 published cases.
1. INTRODUCTION
GABA (γ‐aminobutyric acid) is the most abundant inhibitory neurotransmitter, 1 and its receptor is an important pharmacological target of many antiseizure medications. 2 , 3 Three major receptor classes are activated by GABA: the abundant GABA type A receptors, which are ligand‐dependent, postsynaptic anion channels that exert a rapid inhibitory action when activated 4 ; the metabotropic G protein–coupled GABA type B presynaptic receptors 5 ; and the less widespread GABA type C receptors. 6 GABAA‐receptor subunits are combined as heteropentamers from various combinations of proteins—most frequently two α, two β, and one γ subunit. 7 All GABAA‐receptor subunits exhibit a similar structure, with four transmembrane domains (TM1 to TM4) and a long extracellular N‐terminal domain (NT). The ion channel pore is formed by the second transmembrane domain (TM2) of each subunit. These receptors include several binding sites for ligands (agonist, antagonist, or benzodiazepine [BZD] 8 , 9 ).
GABAA‐receptor subunit heterozygous variants have recently been identified as an important cause of childhood epilepsy with or without intellectual disability (ID). 10 GABAA‐receptor subunit variants were first identified in GABRG2 and GABRA1 using classical linkage analysis, and then candidate gene sequencing in large families with autosomal dominant genetic generalized epilepsy. 4 , 11 With the availability of next‐generation sequencing, many different epileptic phenotypes have been linked to GABAA‐receptor subunit variants with or without neurodevelopmental disorder, 2 these genes being one of the most frequently implicated in epilepsy phenotypes. 12 Given the structural and functional proximity of GABAA‐receptor subunits, it seemed relevant to leave the gene‐by‐gene description and consider as a coherent whole the disorders associated with GABAA‐receptor subunit mutations. This approach has been proposed recently and guided us for this study at the clinical, electrophysiological, and developmental levels. 13 , 14 , 15 , 16
Here, we report a series of 37 patients with pathogenic variants in one of the GABAA‐receptor subunits—GABRA1, GABRB2, GABRB3, and GABRG2—and we analyzed 402 cases reported in the literature, identifying the clinical, electrophysiological, and molecular characteristics, with an emphasis on genotype–phenotype correlations and protein domain phenotype correlations to highlight the link between variant localization and neurodevelopmental impairment severity.
2. MATERIAL AND METHODS
We collected clinical, electrophysiological, therapeutic, and molecular data from patients affected with the most frequent GABAA‐receptor subunit variants (GABRA1, GABRB2, GABRB3, and GABRG2) through a national French collaboration using the EPIGENE network. We used a standardized survey completed by the clinician following these patients. Seizures were classified according to the International League Against Epilepsy (ILAE) classification. 17 This study has been declared to the health data access portal of Assistance Publique‐Hôpitaux de Marseille under the reference YNRSXY and registered under the number PADS22‐23.
We gathered epileptic phenotypes in three spectrums using the ILAE diagnosis criteria:
Epilepsy associated with fever sensibility: Genetic epilepsy with febrile seizures plus (GEFS+), Dravet syndrome (DS) spectrum, and epilepsies in which fever sensitivity is a preeminent sign.
Early developmental epileptic encephalopathy (EDEE) comprising epilepsy of infancy with migrating focal seizures (EIMFS), early infantile epileptic encephalopathy (EIEE), and early‐onset epilepsies that did not fit with any epileptic syndrome.
Genetic generalized epilepsy: epilepsy with myoclonic‐atonic seizure (MAE), juvenile myoclonic epilepsy (JME), atypical absences, and unclassified generalized epilepsy with normal background activity and generalized paroxysmal activities. MAE epilepsy was retained if seizures with myoclonic‐atonic falls were present, whether or not associated with other types of seizures (atypical absences, generalized tonic‐clonic seizures, myoclonia, and so on). Electroencephalography (EEG) could show focal or generalized anomalies.
Cognitive development assessment was based predominantly on psychomotor development assessed by the referring physicians, with occasional formal neuropsychological testing. Informed consent for study inclusion was obtained from patients and parents or their legal guardians, in compliance with the Declaration of Helsinki.
At the molecular level, we considered variants as pathogenic based on a combination of the following criteria as suggested by Richards et al. (2015) 18 : (1) the presence of nonsense, missense, nonsynonymous, frameshift, and splicing modifier variants; (2) the absence of variants from human polymorphism databases (such as gnomAD 19 ); (3) predicted as pathogenic by in silico prediction software (SIFT, 20 PolyPhen, 21 MutationTaster, 22 and UMD Predictor 23 ); (4) the results of segregation analyses with the presence of a variant in the affected individual or transmission by an affected parent. All variants are heterozygous except in three siblings with a homozygous variant in a consanguineous pedigree. All variants were identified using high‐throughput targeted gene‐sequencing panels designed for neurodevelopmental disorders (ID and epilepsy panels) except for patient 4, for whom whole‐exome sequencing was performed. All variants were confirmed by Sanger sequencing, as was familial segregation.
2.1. Literature review
We analyzed all cases reported to date (March 2022) with a pathogenic mutation of one of the following GABAA‐receptor subunits: GABRA1, GABRB2, GABRB3, and GABRG2 using the HGMD Pro database 24 and PubMed website. English‐language articles were selected.
For each case, we noted the clinical phenotype according to the classification previously described: (1) epilepsy associated with fever sensibility, (2) early developmental epileptic encephalopathy, and (3) genetic generalized epilepsy. The cases for which the classification was not possible due to missing data or for which the epileptic phenotype did not fit with any of these three phenotypes have nevertheless been reported. We also collected the age at the first seizure and the severity of ID, when available.
2.2. Statistical analysis
Descriptive statistics are reported as frequencies (sample size and percentages) and means for categorical and continuous variables, respectively. The groups were compared using χ2 or Fisher exact tests as appropriate. All analyses were performed using IBM SPSS Statistics 20.0 (IBM Inc.). For all two‐tailed tests used, a significance level of p < .05 was considered statistically significant.
3. RESULTS
We report 34 unpublished patients with a pathogenic variant in GABAA‐receptor subunits (3 of them have already been reported in Johannesen et al. 25 ): 6 in GABRA1, 5 in GABRB2, 16 in GABRB3, and 10 in GABRG2, representing 16 male and 21 female patients with an age at epilepsy onset from birth to 16 years. Thirty‐four index cases with heterozygous variants and three sibling cases with a homozygous variant are presented. All clinical and molecular results are summarized in Table 1.
TABLE 1.
Clinical characteristics of GABRA1, GABRB2, GABRB3, and GABRG3 patients from our national cohort
| Patient (gender) | Gene | Mutation inheritance | Age at onset | Seizure types | Epileptic phenotype | Diagnosis spectrum | ID | Clinical features | First EEG | Brain MRI | Effective drugs |
|---|---|---|---|---|---|---|---|---|---|---|---|
| N‐terminal domain | |||||||||||
| 1 (F) | GABRA1 |
c.274 T > C p.Phe92Leu de novo |
NP | GTCS | GEFS+ | Fever‐related epilepsy | Moderate | Limb tremor | Normal rhythm activity with localized fast rhythmic activity and GSW | Normal | LEV, LTG, PMP |
| 2 (F) |
c.335G > A p.Arg112Gln de novo |
26m | Atypical absence, GTCS | GEFS+ | Fever‐related epilepsy | Moderate | ASD, regression | Photo S, normal rhythm activity (34 m) | Ventriculomegaly, thin CC | VPA, TPM, LTG | |
| 3 (M) |
c.335G > A p.Arg112Gln de novo |
7m | GTCS | GEFS+ | Fever‐related epilepsy | No | Macrocephaly, macrosomia | Normal (2y) | Normal | VPA, TPM, CLB | |
| 4 (F) | GABRB3 |
c.229G > A p.Glu77Lys de novo |
16y | GTCS, myoclonia | MJE | Generalized epilepsy spectrum | Moderate | Trunk dystonia, spasticity, cerebellar syndrome, bilateral optic atrophy | Generalized SW (16y) | Normal | LTG, ZNS |
| 5 (M) |
c.238A > G p.Met80Val de novo |
8m | Focal, myoclonic | MAE | Generalized epilepsy spectrum | Moderate | Ataxia, stereotypies, ogival palate and dysmorphism | Angelman like trace (4y) | Normal | TPM, LTG, PIR | |
| 6 (M) |
c.343C > T p.Gln115* Inherited father |
36m | Atypical absences | unclassified | Generalized epilepsy spectrum | Mild | ASD, macrosomia | Regular generalized SW 3 Hz | NP | VPA, LTG | |
| 7 (F) | c.358G > A p.Asp120Asn de novo | 11m | Atypical absence, GTCS and atonic | MAE | Generalized epilepsy spectrum | No | Congenital macrocephaly | Slow bilateral SW (15 m) | Normal | LTG, LEV | |
| 8 (F) | c.674 T > C, p.Phe225Ser de novo | 13m | Nighttime focal secondarily generalized | GEFS+ | Fever‐related epilepsy | Moderate | Stereotypies, strabismus, severe GOR | Normal (<2y) | Arachnoid cyst | TPM | |
| 9 (F) | c.674 T > G p.Phe225Cys de novo | 2m | Spasms, GTCS | Unclassified | Generalized epilepsy spectrum | Mild | Ataxia, tremor, nystagmus, auto‐aggressivity, dyspraxia, neuro visual impairment, microcephaly |
Right occipital slow waves; Angelman like |
Normal | LTG, CLB | |
| 10 (M) | c.695G > A p.Arg232Gln de novo | 9m | GTCS, myoclonic atonic | Incomplete Dravet | Fever‐related epilepsy | Moderate | ADHD | GSW (5y) | Normal | VPA, ZNS | |
| 11 (F) |
c.695G > A p.Arg232Gln Inherited (mosaicism) |
18m | Focal, GTCS | Incomplete Dravet | Fever‐related epilepsy | Moderate | Behavior problems, ADHD, morbid obesity | Bifrontal SW asymptomatic | Hypersignal WM right temporal | VPA, CBZ, CLB | |
| 12 (M) | 18m | Focal, GTCS, atypical absences, atonic | GEFS+ | Fever‐related epilepsy | Moderate | Left foot and ankle deformation, macrocephaly | Asymptomatic GSW (2y) | Normal | LVT | ||
| 13 (F) | GABRG2 |
c.282_306dup, p. Pro103Argfs*6 Inherited |
12m | FS, GTCS | GEFS+ | Fever‐related epilepsy | No | NP | Normal (age NP) | Normal | LTG |
| Transmembrane localization | |||||||||||
| 14 (M) | GABRA1 |
c.787A > G p.Met263Val de novo |
1m | Myoclonic, tonic, atonic, GTCS | EIEE | EDEE | Severe | Postnatal microcephaly, hypotonia, choreoathetoid movement, nystagmus | Suppression burst (5m) | Normal | CBZ, PMP |
| 15 (F) | c.851 T > C p.Val284Ala de novo | 0.5m | Tonic, clonic | EIEE | EDEE | Severe (died at 8m) | Microcephaly, axial hypotonia, growth delay | Hypsarrhythmia (7m) | Cortical and CC atrophia | VGB | |
| 16 (M) |
c.888G > T p.Leu296Phe de novo |
No | No | NP | No | Mild | Drooling, bruxism, anxiety, growth delay | Normal (2y) | Normal | No | |
| 17 (M) | GABRB2 | c.815C > G p.Ala272Gly de novo | 4.5y | Night tonic clonic | GTCA | Generalized epilepsy spectrum | Moderate | Normal | Brief flashes of temporal ample bilateral spikes during sleep (4.5y) | NP | CLB, OXC |
| 18 (M) | c.847C > A p.Leu283IleI de novo | 3d | Clonic, automatism (chewing), migrating | EIEE | EDEE | Severe (died at 2m) | Axial hypotonia, peripheral hypertonia, no ocular interaction | Suppression burst | Diffuse hypersignal of the WM | Pharmacoresistant | |
| 19 (F) | GABRB3 | c.851 T > C p.Leu284Pro de novo | 7m | Arm twitching, myoclonic, absence | MAE | Generalized epilepsy spectrum |
Severe |
Microcephaly (NP); Hypotonia; spasticity, regression (9y) | GSW (NP) | Normal | Pharmacoresistant |
| 20 (M) |
c. 851 T > C p.Leu284Pro de novo |
6m | Absence, GTCS | EIEE | EDEE | Severe | Normal, prognathism | GSW (6m) | Normal | ETX | |
| 21 (F) | c.913G > A p.Ala305Thr de novo | NP | Atypical focal, migrating; GTCS | EIEE | EDEE | Severe | Congenital microcephaly; hypotonia, regression after 3m | Migrating PSW (3m) | Normal then atrophia | TPM, VPA, LEV | |
| 22 (M) | GABRG2 | c.844C > G p.Pro282Ala de novo | Birth | Trembling, focal, myoclonic | EIEE | EDEE | Severe (died at 18y) | Hypotonia, dystonia, strabismus, nystagmus, dysmorphism | suppression burst (2m) | Atrophia, WM abnormality | Pharmacoresistant |
| Loop localization | |||||||||||
| 23 (F) | GABRB2 |
c.892A > G p.Lys298Glu de novo |
3m | Atonic, GTCS, absence, alternating hemiplegia | GEFS+ | Fever‐related epilepsy | Mild | Ataxia | Focal SW, non‐clinical frontal SW sleeping | Normal | BZD, VPA, ZNS |
| 24 (F) |
c.902A > G p.Tyr301Cys de novo |
9m | Myoclonic, atonic, GTCS, atypical absence, photo S and sonosensibility | MAE | Generalized epilepsy spectrum | Moderate | Ataxia, hypotonia, macrosomia | Generalized ample PSW | Normal | LEV | |
| 25 (M) |
c.908A > G p.Lys303Arg de novo |
3d | Myoclonic, GTCS, tonic | EIEE | EDEE | Severe | Postnatal microcephaly, axial hypotonia, spasticity, strabismus, global growth delay at 7m | Suppression burst (3d) | WM hypersignal, cortical and CC atrophia | Vit. B6, PHB, TPM, ZNS, VPA | |
| 26 (F) | GABRB3 |
c.911A > G p.Lys304Arg de novo |
21d | Spasm, clonic, | EIEE | EDEE | Severe (died at 2m) | Macrocephaly, axial hypotonia, choreoathetoid movement, micro retrognathism | Hypsarrhythmia (21d) | Normal | Pharmacoresistant |
| 27 (F) |
c.1347_1350 delCAGA p.Arg450Glyfs*15 homozygous inherited |
6d | GTCS, spasms | EIEE | EDEE | Severe | Axial hypotonia, aggressively, stereotypies | Suppression bust (6d) | Hypersignal WM | CLB, PHB, VPA | |
| 28 (F) | 1d | GTCS, absence | EIEE | EDEE | Severe | Axial hypotonia, postnatal microcephaly, cleft palate | Suppression bust (2d) | NP | VGB, PHB | ||
| 29 (F) | 2d | Myoclonic, GTCS | EIEE | EDEE | Severe | Postnatal microcephaly, micro retrognathism | Normal (2d) | Hypersignal WM | PHB | ||
| 30 (M) | GABRG2 |
c.967C > T p.Arg323Trp de novo |
15m | Tonic clonic, atonic, atypical absence | MAE | Generalized epilepsy spectrum | Moderate | Hyperactivity, aggressivity, attention disorder | Multifocal SW (anterior and temporal) asymptomatic | Normal | LTG, ETX |
| 31 (M) |
c.986G > A p.Arg323Gln Inherited |
9m | Focal hemicorporal, GTCS | Dravet syndrome | Fever‐related epilepsy | Severe | No social interaction, stereotypies, ASD | Normal (9m) | Normal | CLB, VPA | |
| 32 (F) | 8m | Atonic, absence, myoclonic; focal | Dravet syndrome | Fever‐related epilepsy | Severe | Postnatal macrocephaly, stereotypies, ASD, dysmorphism, sleep trouble | Normal (8m) | Hydrocephalus | CLB, LTG | ||
| 33 (M) |
c.968G > A p.Arg323Gln de novo |
10m | Focal, GTCS clonic, myoclonic‐atonic | Incomplete Dravet | Fever‐related epilepsy | Mild | Comportment disorders, attention disorders | Focal SW right parietal | Hypersignal WM |
VPA, CLB KD |
|
| 34 (F) |
c.992A > G p.Tyr331Cys de novo |
12m | GTCS | GEFS+ | Fever‐related epilepsy | Moderate | Postnatal macrocephaly, ataxia, visuomotor apraxia, choreoathetoid movement, obesity | Normal (1y) | Normal | VPA | |
| 35 (F) |
c.1128 + 2 T > C Inherited |
10m | Absences +/− clonic, ocular revulsions | GEFS+ | Fever‐related epilepsy | No | Comportment disorder | Multifocal SW (NP) | Normal | VPA, LEV | |
| 36 (M) | 3y | Infantile spasms, tonic clonic, atonic, atypical absences | MAE | Generalized epilepsy spectrum | Mild to moderate | Concentration difficulties, ADHD | Multifocal SW, asymptomatic generalized PSW during sleep, normal (11y) | Normal | VPA, ETX, LTG, CLB, LEV | ||
| 37 (M) | 12m | Absences, GTCS | GEFS+ | Fever‐related epilepsy | Mild | Writing difficulties, aggressiveness | GSW | GSW(8y) normal after | VPA | ||
Note: c.DNA newly reported mutation highlighted in gray. GABRA1: NM_000806.5; GABRG2: NM_000816.3; GABRB3: NM_000814.4; GABRB2: NM_021911.2.
Abbreviations: Diagnosis and clinical features: ADHD: attention‐deficit/hyperactivity disorder, ASD: autism spectrum disorder, CC: corpus callosum, d: days, DS: Dravet syndrome, E: epilepsy, EDEE: early developmental epileptic encephalopathy, EOEE: early‐onset epileptic encephalopathy, EEG: electroencephalography, EOEE: early‐onset epileptic encephalopathy, FeS: febrile seizure, F: female, FS: focal seizure, GE: generalized epilepsy, GEFS+: generalized epilepsy with febrile seizure plus, GOR: gastroesophageal reflux, GSW: general spike waves, GTCS: generalized tonic‐clonic seizure, GTCA: generalized tonic‐clonic seizure alone, HC: head circumference, ID: intellectual disability, m: months, M: male, MAE: myoclonic‐atonic epilepsy, NP: not precise, PSW: polyspike waves, photo S: photosensibility, sens.: sensitive, Sz: seizure, SD: standard deviation, SW: spike waves, TM: transmembrane domain, y: years, WM: white matter.Treatment: BZD: benzodiazepine, CBZ: carbamazepine, CLB: clobazam, ETX: ethosuximide, KD: ketogenic diet, LEV: levetiracetam, LTG: lamotrigine, OXC: oxcarbazepine, PHB: phenobarbital, PIR: piracetam, PMP: perampanel, TPM: topiramate, VGB: vigabatrin, Vit. B6: vitamin B6, VPA: valproic acid, ZNS: zonisamide.
3.1. Clinical results
3.1.1. Developmental and electroclinical phenotypes
The analysis of the electroclinical phenotype of the patients led us to describe three groups:
3.1.1.1. Group 1: Fever‐related epilepsy (40%—n = 15)
This group includes six patients with a GABRG2 variant, four with a GABRB3 variant, three with a GABRA1 variant, and two with a GABRB2 variant. The epilepsy began on average at 10.9 months (range 3–26 months) with fever‐related generalized tonic‐clonic seizures (n = 12) and/or focal seizures (n = 7). Some patients also had other types of seizures at first evaluation: absence seizures (n = 6), atonic seizures (n = 5), myoclonic seizures (n = 3), and clonic seizures (n = 2). One patient also had an alternating hemiplegia (Patient 25, Figure 1A). Initial EEG abnormalities were generalized (n = 4); two patients had focal anomalies and three have multifocal anomalies. Seven of these patients had a normal EEG at first evaluation. Most of the patients from this group had a moderate intellectual disability (or ID) (n = 7), three with mild ID and two with severe ID; it should be noted that three patients had no ID (Figure 1B). The most effective antiseizure medications (ASMs) were valproic acid (n = 10), clobazam and other benzodiazepines alone or in association (n = 6), and lamotrigine (n = 4). One patient became seizure‐free under a ketogenic diet. All the patients of this group had good control of their epilepsy under ASMs at the end of the follow‐up. Five patients had electroclinical features of Dravet syndrome (Table 1).
FIGURE 1.
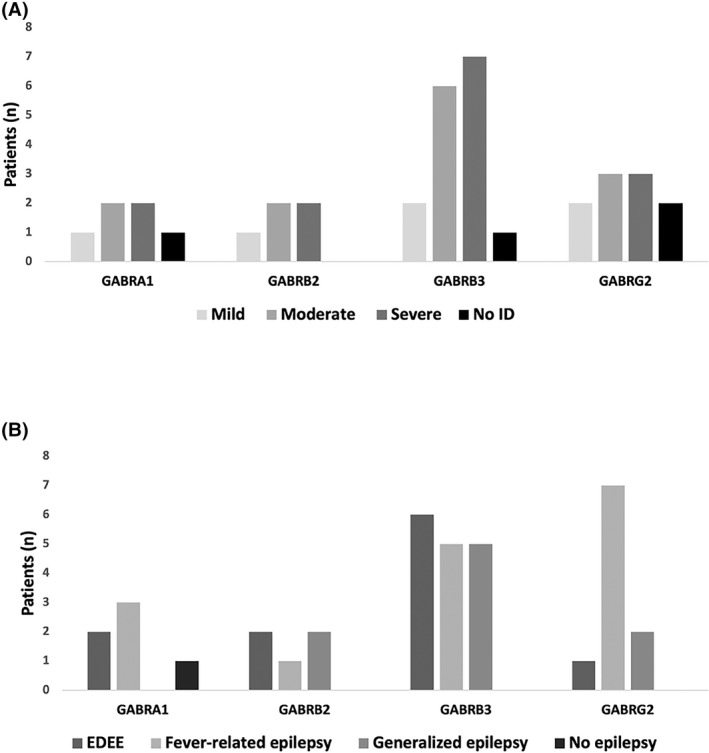
(A) Epileptic phenotype and (B) cognitive status related to the γ‐aminobutyric acid (GABA) gene variant. EDEE: early developmental epileptic encephalopathy, ID: Intellectual disability
3.1.1.2. Group 2: Early developmental epileptic encephalopathy (EDEE) (29%—n = 11)
Six patients had a variant in GABRB3, including three siblings; two had a variant in GABRB2, two had a variant in GABRA1, and one had a variant in GABRG2. The epilepsy began before 6 months of age, with motor symptoms in all cases: clonic and tonic seizures (n = 8) or clonic/myoclonic seizures (n = 4). One patient had focal migrating clonic seizures (Patient 21), and two patients also had infantile spasms (Patients 28 and 29) (Table 1 and Figure 1A). Of interest, we found a recurrent EEG pattern in those patients; most of them (6/11) had a suppression burst on the initial EEG, 2 of 11 patients had hypsarrhythmia, 1 had generalized spike waves, and 1 had migrating polyspike waves. One patient had a normal initial EEG. Ongoing EEGs showed asymptomatic burst of slow waves, predominantly anterior (Figure 2). This abnormal activity was not symptomatic, lasted for 5 to 20 seconds, and occurred preferentially in the quiet vigil and sleep states. All the patients had a severe ID (four patients died between 2 months and 18 years) (Figure 1B). Development was abnormal before or at epilepsy onset in all cases. The most effective ASMs were phenobarbital (n = 4) and valproic acid (n = 3), but most of the patients in this group had drug‐resistant epilepsy.
FIGURE 2.
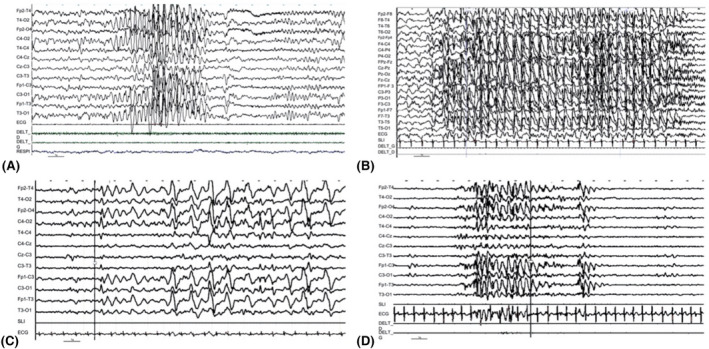
Interictal electroencephalography (EEG) recordings of patients with GABRB3 variants. (A) Patient 12, wakefulness. Asymptomatic episode of theta rhythmic pattern. (B) Patient 9, sleep. (C) Patient 11, wakefulness, asymptomatic. (D) Patient 10, wakefulness, asymptomatic bursts of slow wave, predominantly anterior
3.1.1.3. Group 3: Generalized epilepsy spectrum (27%—n = 10)
Six patients had a GABRB3 variant, two had a GABRB2 variant, and two had a GABRG2 variant (Figure 1A). Epilepsy began at a mean age of 37 months (range 2 months to 16 years) with generalized seizures, including generalized tonic–clonic seizures (n = 7), absences (n = 6), myoclonic (n = 4), atonic (n = 4), and spasms (n = 2). One patient had infantile spasms and then generalized tonic–clonic seizures (patient 9). In this group, the patients endured various types of epilepsy, including myoclonic‐atonic epilepsy (n = 6), myoclonic juvenile epilepsy (n = 1), and generalized tonic–clonic seizure alone (n = 1). Two patients had an unclassified epilepsy (n = 2). Most patients (n = 6) presented with generalized anomalies on the EEG, and two patients had an Angelman‐like pattern on the EEG (Patients 5 and 9, Table 1). Five patients had moderate ID, three had mild ID, one had severe ID, and one had standard intelligence (Figure 1B). Lamotrigine was the most effective drug in seven patients and levetiracetam in three patients. Valproic acid and ethosuximide were rarely effective.
Patient 16 was not epileptic, had mild ID, and behavioral disorders, such as drooling, bruxism, and anxiety. At 2 years of age, the EEG was normal, whereas at 5 years, it was diffuse, high, and slow, with rhythmic spikes and waves. No correlation was observed between genetic pathogenic variants and the phenotype of the patient.
3.1.2. Imaging characteristics
Thirty four of the 37 patients had brain magnetic resonance imaging (MRI). The majority (21/34) were reported as normal. The other displayed nonspecific abnormalities, including white matter hypersignal (n = 7), cortical atrophy (n = 4), corpus callosum anomalies (n = 3), ventriculomegaly (n = 2), and unilateral temporal arachnoid cyst (n = 1).
3.1.3. Other neurological features
Seven patients had movement disorders, with stereotypies (n = 5), choreoathetosis movements (n = 3), tremor (n = 2), and dystonia (n = 2). Five patients had ataxic gait and three had spastic diplegia. No genotype–phenotype correlation was found in terms of motor or nonepileptic abnormalities.
Eleven patients had head circumference abnormalities: microcephaly (n = 7) and macrocephaly (n = 4). These abnormalities were observed particularly in patients carrying a mutation in GABRB3 (8/16 patients: 5 with microcephaly and 3 with macrocephaly).
3.1.4. Ophthalmological features
Two patients with variants in GABRB3 had visual impairment. Patient 4 had a nystagmus with bilateral optic atrophy (confirmed with optic coherence tomography). Patient 9 had GEFS+ and mild ID, a nystagmus, bilateral hyperopia, reduced corrected visual acuity (4/10 for the right eye and 2.5/10 for the left), and a heterogeneous aspect of the peripheral retina. Two patients with the GABRG2 variant had a visuospatial apraxia. In addition, three had nystagmus and three had strabismus; no specific gene variant association was found.
3.1.5. Dysmorphism
Eight patients were described with particular morphological facial features, particularly patients carrying a variant in GABRB3 (four patients). These morphological features affected the lower and middle parts of the face and included high‐arched palate, chin anomalies (microretrognathism, prognathism), and cleft palate.
3.2. Molecular results
3.2.1. Type of variants
In our cohort of 37 unreported patients, 27 different pathogenic GABAA‐receptor subunits were noted: 5 in GABRA1, 5 in GABRB2, 11 in GABRB3, and 6 in GABRG2; 17 variants were unpublished (4 in GABRA1, 4 in GABRB2, 5 in GABRB3, and 4 in GABRG2). Variant types included 23 missense, 2 frameshifts, 1 nonsense, and 1 intronic variant. Variants were heterozygous in 34 cases and homozygous in 3 siblings in a consanguineous pedigree.
3.2.2. Inheritance
The mutations observed were heterozygous de novo in 25 patients and inherited in 12 patients (Figure S1). Three patients from the same family (Patients 27, 28, and 29) had a homozygous GABRB3 pathogenic variant associated with a particularly severe phenotype of EDEE, with seizures beginning from 1 to 6 days of age. In this consanguineous pedigree, mutations were inherited from their heterozygous parents with milder phenotypes. Both parents displayed mild ID, and the mother exhibited medication‐sensitive epilepsy during childhood. Although this variant has never been reported, it has been considered pathogenic. The other inherited cases are detailed in the supplemental data.
3.2.3. Variant location
We analyzed the identified variants based on their localization within the GABA receptor protein: NT as N‐terminus, TM as transmembrane domain, and the loop domain. Ten variants were located in the extracellular NT of the protein, eight in TM domains (two in TM1, five in TM2, and one in TM3), and nine in loops (seven in loop TM2‐3 and two in loop TM3‐4) (Figure 3). The variants in the intracellular domain (C‐term) were rare in our cohort, as in the literature.
FIGURE 3.
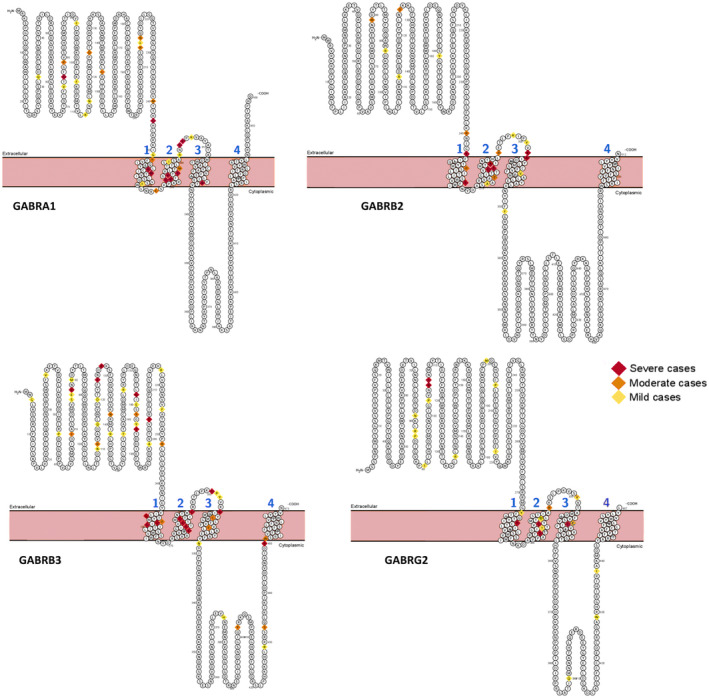
Location of pathogenic variants identified in GABRA1, GABRB2, GABRB3, and GABRG2 protein classified by the intellectual disability severity of the reported associated phenotype. Variants for which no information is available in the literature regarding the phenotype are not noted
3.3. Literature review and phenotype–genotype correlation
We reviewed 402 previously published cases: 71 patients with a mutation in GABRA1, 28 in GABRB2, 133 in GABRB3, and 170 in GABRG2. The distribution according to the phenotype was as follows: 68 reported mutations had an undetailed clinical description, and 77 reported mutations had a phenotype that did not fit into any of the groups we described (20%). Some mutations were reported without clinical description. Finally, the clinical and genetic data were available in 334 cases, and 257 patients were analyzed.
Most of the patients were epileptic, but eight had ID without epilepsy (except for one patient without ID or epilepsy, the mother of three affected children).
Fifty‐two mutations were recurrent (15 in GABRA1, 5 in GABRB2, 10 in GABRG2, and 22 in GABRB3) concerning 185 patients and 129 families (25 localized in the NT domain and 27 in TM or loop domains; Figure S2). Among these recurrent mutations, 18 were reported in more than three families (Figure S2); c.968G > A in GABRG2 was reported mostly in 13 different families associated with a milder phenotype.
We observed an overrepresentation of severe cognitive impairment in patients with variants located in the TM region. To analyze the potential genotype–phenotype correlation according to protein location, we compared the clinical characteristics of patients in the cohort (n = 37) and those reported in the literature (n = 402), depending on the location of variants (Table S1 and Appendix S1). We selected three relevant and available clinical data: age at epilepsy onset, epilepsy severity, and ID severity.
Severe ID was noted in 39% of patients in our cohort and 38% of cases in the literature (Table S1). The proportion of patients with severe ID was enriched among patients with variants in TM (87%), higher than in cases with variants localized to the loop (38%) or NT (22%) (χ2 (2)=16.9, p < .001, in our cohort and χ2 (2)=42.6, p < .001 in the literature) (Figures 4, 5).
FIGURE 4.
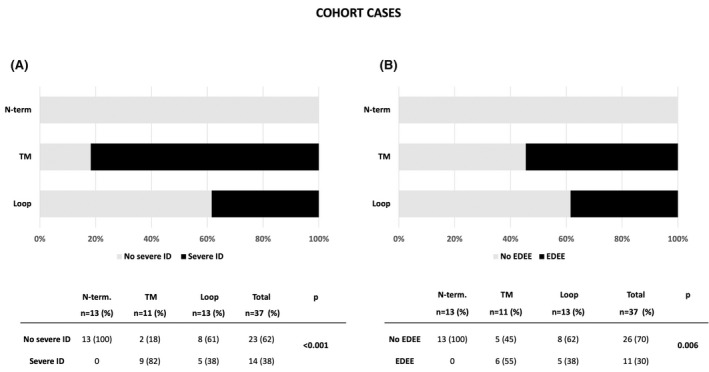
Correlation between phenotype (A. severe ID, B. EDEE) and location of mutated GABA‐R protein domains in our cohort (n = 37). ID: intellectual disability, EDEE: early developmental epileptic encephalopathy, pathogenic variant location: N‐term, loop, and TM (transmembrane).
FIGURE 5.
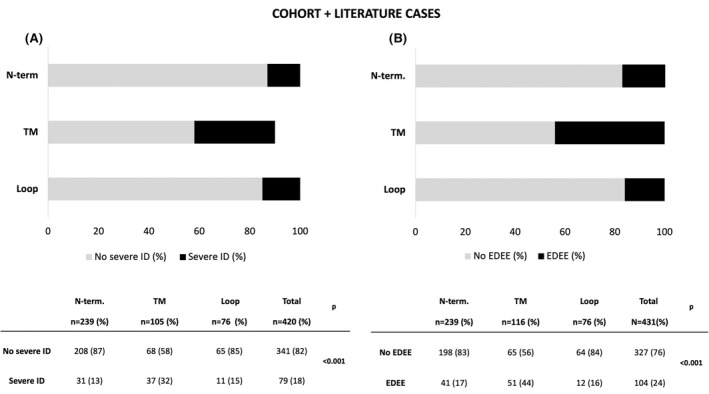
Correlation between phenotype (A. severe ID and B. EDEE) and location of mutated GABA‐R protein domains in our cohort and literature cases. ID: Intellectual disability, EDEE: Early developmental epileptic encephalopathy, pathogenic variant location: N‐term, loop, and TM (transmembrane)
In our cohort, more than half of patients with EDEE had TM variants (55%). Indeed, the TM variants were more often associated with EDEE; 30% in our cohort (χ2 (2)=10.2, p = .006) and 25% in the literature (χ2 (2)=27.8, p < .001) (Figures 4, 5).
However, no correlation was observed between variant location and abnormal movements, behavioral disorders, or abnormalities on brain MRI.
4. DISCUSSION
Here, we have described a cohort of 37 patients with epilepsy and/or ID, carrying pathogenic variants in several genes (GABRA1, GABRB2, GABRB3, and GABRG2) encoding most common GABAA‐receptor subunits.
We reported three main epilepsy phenotypes of almost equal quantitative importance: a fever‐relative epilepsy spectrum (GEFS+ and DS), an early‐onset DEE spectrum, and a genetic generalized epilepsy spectrum, represented primarily by myoclonic‐astatic epilepsy. None of these phenotypes was associated with a given gene (i.e., a selective subunit dysfunction), and in contrast, our study emphasizes the overlapping phenotypes of patients with GABAA‐receptor subunit variants with regard to epilepsy subtypes, ID severity, neurological findings, and brain imaging. Studies of recurrent variants highlight the complexity of a genotype–phenotype correlation approach, particularly regarding genes encoding receptors and channels. 27 Indeed, for example, patients carrying the mutation c.968G>A; p.Arg323Gln of GABRG2 had either DS (Patients 31 and 32), a GEFS+ with mild cognitive impairment (Patients 33 and 31–32’s mother), a GEFS+ with normal cognitive status, 28 childhood epilepsy with centrotemporal spikes, 29 or early‐onset epileptic encephalopathy with severe ID. 15 This mutation is highly recurrent and has been found in 13 different families from the literature (Figure S2). Other recurrent mutations were associated with a more homogeneous phenotype; for example, the p.Lys303Arg in GABRB2 was always associated with EDEE. 30 Most of the inherited variants reported here led to a homogeneous epileptic phenotype in a given family when they were heterozygous. This was not always the case regarding the degree of ID within a given family (Figure S1). Finally, we did not find any correlation between antiseizure medication effectiveness and a given gene or a specific subunit localization.
We found a homozygous frameshift variant of GABRB3 in three consanguineous siblings with an EDEE and a suppression‐burst EEG pattern. These patients had a highly severe phenotype, and their heterozygous parents had a mild one. In the present case, the variant was located in the last exon of the gene and was probably not subject to nonsense‐mediated decay. It is then likely that a transcript still exists in the patients' cells that is different from the normal transcript in the C‐terminal region. A functional study would be particularly interesting in this case. It is likely that biallelic mutations of GABA receptor subunits will be further described in the future because many mutations are not associated with a highly severe phenotype at the heterozygous state. Genes coding for the GABA receptor subunits join the long list of genes involved in both recessive and dominant diseases.
Disease‐causing variants were located in critical functional protein domains, including the extracellular NT domain (12 cases); the extracellular loop between TM2 and TM3 (9 cases); and TM2, which forms the GABAA channel pore (6 cases) (Figure 3, Table S1 and (Ref. 10)). We observed a significant correlation between ID degree, epilepsy severity, and variant localization through this cohort but also among the patients published to date (402 cases). Patients with NT variants presented with milder epilepsy and ID than patients with variants in other regions of the protein, regardless of the mutated subunit, whereas variants in TM domains had the most severe phenotype in terms of epilepsy and ID. This correlation has been reported recently for GABRB2 13 and GABRB3 cohorts. 25 In this recent report, milder phenotypes (generalized epilepsy associated with mild to moderate ID) were associated with a mutation in the extracellular domain, whereas patients with early‐onset epilepsy and severe ID had a mutation in TM or the extracellular domain. Our findings expand this correlation to three other GABAA‐receptor subunit genes.
We found missense variants in 29 patients and protein truncation variants in 7 patients. Protein truncation most likely leads to a loss of function and may reduce inhibitory phasic GABAergic transmission. 13 , 14 , 31 , 32 Missense variants may either be a loss or a gain of function. 26 In GABRB3, gain‐of‐function variants were associated with early‐onset epilepsy, more severe intellectual disability, hypotonia, and more pharmacoresistance, whereas loss of function mutations were most frequently related to a milder phenotype and febrile seizures. 26 Gain‐of‐function variants were mostly localized in the TM domain and associated with the most severe phenotype. It is then possible that the variants localized in the TM domain could be linked with gain of function, whatever the subunit. This finding also suggests that within the GABAA receptor gene, the location of the mutation is more important than the time of expression of the gene during development in terms of predicting the phenotype. Although it is relatively easy to make the link between loss of function of GABAergic receptors and epilepsy, the association is less clear for gain‐of‐function mutations. It is possible that epilepsy is not the direct consequence of the mutation at the time of onset but the result of abnormal early developmental brain activities (due to the mutation). As suspected for KCNQ2‐related DEEs, epilepsy and ID would be the late consequence of alterations in network activities induced earlier (and transiently) by the mutation. 33
Among DEE patients, we often found an EEG pattern similar to that observed in Angelman syndrome, made of irregular bursts of ample slow/acute activity in frontal regions (Figure 2). This pattern has also been found in Gabrb3 +/− and Gabrb3 −/− mice 34 and may be a potential biomarker of a GABAergic dysfunction. 16
A number of ASMs (vigabatrin, topiramate, valproic acid, cenobamate) are known to affect GABA receptors via different mechanisms (GABA receptor activation, reuptake inhibition, neurotransmitter breakdown inhibition, and so on). 35 The effect of these molecules has already been reported, with inconsistent consequences. 15 , 36 , 37 , 38 , 39 The existence of gain‐of‐function mutations may explain the lack of efficacy or the worsening effect of these ASMs and the potential efficacy of sodium channel blockers in some cases. 40 , 41 , 42 Although interpreted with substantial caution, the most effective ASMs were different within the three groups in our cohort: valproic acid in the fever‐sensitive group, phenobarbital in the DEE group, and lamotrigine in the GGE group. It is important to note that these treatments were not chosen according to the mutation and were not modified after the molecular results had been obtained. It would be of significant interest to test the potential relationship between functional consequences and the effect of a given ASM.
Here, we reported variants in the most common GABAA‐receptor subunits in the human brain: ~60% of all GABAA‐receptor subunits have the combination α1β2γ2, and 15%–20% have the combination α2β3γ2. However, several other genes code for other rare subunits of this receptor (19 in total). Mutations in some of these genes have been identified in patients with similar phenotypes, with epilepsy (including EDEE) and ID. However, the conclusions drawn here cannot yet be generalized for all GABAA‐receptor subunits, and further work is still required.
5. CONCLUSION
We have described the phenotypes associated with variants in the four most common subunits of GABAA receptors in humans—GABRA1, GABRB2, GABRB3, and GABRG2—so that we could determine the epileptic and developmental features and identify genotype–phenotype associations. From our observations, no genotype–phenotype associations could be evidenced among GABAA‐receptor subunits regarding epilepsy, response to treatment, ID severity, neurological findings, and brain imaging. However, a significant genotype–phenotype correlation was found regarding the variant localization within the GABAA‐receptor subunit protein domains. Patients with an NT pathogenic variant showed significantly milder phenotype (milder ID and less severe epilepsy) compared to variants in TM domains (more severe) or other locations (intermediate). These results have also been confirmed by a comprehensive literature analysis of 402 reported cases. Then, in the case of finding a mutation of a GABAA‐receptor subunit in a patient, a rapid assessment of the mutation's location may be helpful to better anticipate the disease burden.
AUTHOR CONTRIBUTIONS
Pierre‐Yves Maillard, Sarah Baer, Béatrice Desnous, Anne de Saint Martin, and Mathieu Milh contributed to study design and conceptualization, data collection, analysis, interpretation of data, and the original draft of the manuscript. Élise Schaefer, Caroline Lacoste, Salima El Chehadeh, Amélie Piton, Giulia Barcia, Gaëtan Lesca, Véronique Paquis‐Flucklinger, and Laurent Villard performed the genetic analysis and interpretation. Nathalie Villeneuve, Anne Lépine, Alexandre Fabre, Caroline Lacoste, Salima El Chehadeh, Amélie Piton, Louise Frances Porter, Caroline Perriard, Marie‐Thérèse Abi Wardé, Marie‐Aude Spitz, Vincent Laugel, Gaëtan Lesca, Audrey Putoux, Dorothée ville, Cyril Mignot, Delphine Héron, Rima Nabbout, Marlène Rio, Agathe Roubertie, Pierre Meyer, Olivier Patat, Jérémie Lefranc, Marion Gerard, and Julietta de Bellescize contributed to data collection and interpretation, and to revision of the manuscript for intellectual content. Statistical analysis was done by Béatrice Desnous.
CONFLICT OF INTEREST
None of the authors have any conflict of interest in line with this study to declare.
Supporting information
Appendix S1
Figure S1
Figure S2
Table S1
ACKNOWLEDGMENTS
We thank the patients and families who participated in the collection of clinical data for this work. We thank our colleagues who referred families to our collaboration.
Maillard P‐Y, Baer S, Schaefer É, Desnous B, Villeneuve N & Lépine A et al. Epigen Consortium Molecular and clinical descriptions of patients with GABAA receptor gene variants ( GABRA1, GABRB2, GABRB3, GABRG2 ): A cohort study, review of literature, and genotype–phenotype correlation. Epilepsia. 2022;63:2519–2533. 10.1111/epi.17336
Pierre‐Yves Maillard and Sarah Baer contributed equally.
REFERENCES
- 1. Rudolph U, Knoflach F. Beyond classical benzodiazepines: novel therapeutic potential of GABA a receptor subtypes. Nat Rev Drug Discov. 2011;10(9):685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, Reid CA. Ion channels in genetic epilepsy: from genes and mechanisms to disease‐targeted therapies. Pharmacol Rev. 2018;70(1):142–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davies MF, Onaivi ES, Chen SW, Maguire PA, Tsai NF, Loew GH. Evidence for central benzodiazepine receptor heterogeneity from behavior tests. Pharmacol Biochem Behav. 1994;49(1):47–56. [DOI] [PubMed] [Google Scholar]
- 4. Baulac S, Huberfeld G, Gourfinkel‐An I, Mitropoulou G, Beranger A, Prud'homme JF, et al. First genetic evidence of GABAA receptor dysfunction in epilepsy: a mutation in the γ2‐subunit gene. Nat Genet. 2001;28(1):46–8. [DOI] [PubMed] [Google Scholar]
- 5. Couve A, Moss SJ, Pangalos MN. GABA(B) receptors: a new paradigm in G protein signaling. Mol Cell Neurosci. 2000;16(4):296–312. [DOI] [PubMed] [Google Scholar]
- 6. Bormann J. The “ABC” of GABA receptors. Trends Pharmacol Sci. 2000;21(1):16–9. [DOI] [PubMed] [Google Scholar]
- 7. Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. [DOI] [PubMed] [Google Scholar]
- 8. Laverty D, Desai R, Uchański T, Masiulis S, Stec WJ, Malinauskas T, et al. Cryo‐EM structure of the human α1β3γ2 GABAA receptor in a lipid bilayer. Nature. 2019;565(7740):516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Masiulis S, Desai R, Uchański T, Serna Martin I, Laverty D, Karia D, et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature. 2019;565(7740):454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maljevic S, Møller RS, Reid CA, Pérez‐Palma E, Lal D, May P, et al. Spectrum of GABAA receptor variants in epilepsy. Curr Opin Neurol. 2019;32(2):183–90. [DOI] [PubMed] [Google Scholar]
- 11. Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, et al. Mutant GABA a receptor γ2‐subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28(1):49–52. [DOI] [PubMed] [Google Scholar]
- 12. Feng YCA, Howrigan DP, Abbott LE, Tashman K, Cerrato F, Singh T, et al. Ultra‐rare genetic variation in the epilepsies: a whole‐exome sequencing study of 17,606 individuals. Am J Hum Genet. 2019;105(2):267–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. El Achkar CM, Harrer M, Smith L, Kelly M, Iqbal S, Maljevic S, et al. Characterization of the GABRB2‐associated neurodevelopmental disorders. Ann Neurol. 2021;89(3):573–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Møller RS, Wuttke TV, Helbig I, Marini C, Johannesen KM, Brilstra EH, et al. Mutations in GABRB3 from febrile seizures to epileptic encephalopathies. Neurology. 2017;88(5):483–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shen D, Hernandez CC, Shen W, Hu N, Poduri A, Shiedley B, et al. De novo GABRG2 mutations associated with epileptic encephalopathies. Brain. 2017;140(1):49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ahring PK, Liao VWY, Gardella E, Johannesen KM, Krey I, Selmer KK, et al. Gain‐of‐function variants in GABRD reveal a novel pathway for neurodevelopmental disorders and epilepsy. Brain. 2021;2021:2–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11(1):1–9. [DOI] [PubMed] [Google Scholar]
- 21. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwarz JM, Cooper DN, Schuelke M, Seelow D. Mutationtaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014;11(4):361–2. [DOI] [PubMed] [Google Scholar]
- 23. Salgado D, Desvignes JP, Rai G, Blanchard A, Miltgen M, Pinard A, et al. UMD‐predictor: a high‐throughput sequencing compliant system for pathogenicity prediction of any human cDNA substitution. Hum Mutat. 2016;37(5):439–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Hum Genet. 2017;136(6):665–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Johannesen KM, Iqbal S, Guazzi M, Mohammadi NA, Pérez‐Palma E, Schaefer E, et al. Structural mapping of GABRB3 variants reveals genotype‐phenotype correlations. Genet Med. 2022;24(3):681–93. 10.1016/j.gim.2021.11.004 [DOI] [PubMed] [Google Scholar]
- 26. Absalom NL, Liao VWY, Johannesen KMH, Gardella E, Jacobs J, Lesca G, et al. Gain‐of‐function and loss‐of‐function GABRB3 variants lead to distinct clinical phenotypes in patients with developmental and epileptic encephalopathies. Nat Commun. 2022;13(1):1822. 10.1038/s41467-022-29280-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hernandez CC, Macdonald RL. A structural look at GABA a receptor mutations linked to epilepsy syndromes. Brain Res. 2019;1714:234–47. [DOI] [PubMed] [Google Scholar]
- 28. Carvill GL, Weckhuysen S, McMahon JM, Hartmann C, Møller RS, Hjalgrim H, et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. 2014;82(14):1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reinthaler EM, Dejanovic B, Lal D, Semtner M, Merkler Y, Reinhold A, et al. Rare variants in γ‐aminobutyric acid type a receptor genes in rolandic epilepsy and related syndromes. Ann Neurol. 2015;77(6):972–86. [DOI] [PubMed] [Google Scholar]
- 30. Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent De novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet. 2017;101(5):664–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lachance‐Touchette P, Brown P, Meloche C, Kinirons P, Lapointe L, Lacasse H, et al. Novel α1 and γ2 GABA a receptor subunit mutations in families with idiopathic generalized epilepsy. Eur J Neurosci. 2011;34(2):237–49. [DOI] [PubMed] [Google Scholar]
- 32. Janve VS, Hernandez CC, Verdier KM, Hu N, Macdonald RL. Epileptic encephalopathy de novo GABRB mutations impair γ‐aminobutyric acid type a receptor function. Ann Neurol. 2016;79(5):806–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Biba‐Maazou N, Becq H, Pallesi‐Pocachard E, Sarno S, Granjeaud S, Montheil A, et al. Time‐limited alterations in cortical activity of a knock‐in mouse model of KCNQ2‐related developmental and epileptic encephalopathy. J Physiol. 2022;600:2429–60. [DOI] [PubMed] [Google Scholar]
- 34. DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, et al. Mice lacking the β3 subunit of the GABA(a) receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci. 1998;18(20):8505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rogawski MA, Löscher W, Rho JM. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb Perspect Med. 2016;6(5):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kodera H, Ohba C, Kato M, Maeda T, Araki K, Tajima D, et al. De novo GABRA1 mutations in Ohtahara and west syndromes. Epilepsia. 2016;57(4):566–73. [DOI] [PubMed] [Google Scholar]
- 37. Hamdan FF, Srour M, Capo‐Chichi JM, Daoud H, Nassif C, Patry L, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014;10(10):e1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dan B, Boyd SG. Angelman syndrome reviewed from a neurophysiological perspective. The UBE3A‐GABRB3 hypothesis. Neuropediatrics. 2003;34(4):169–76. [DOI] [PubMed] [Google Scholar]
- 39. Wang JF, Sun X, Chen B, Young LT. Lamotrigine increases gene expression of GABA‐A receptor β3 subunit in primary cultured rat hippocampus cells. Neuropsychopharmacology. 2002;26(4):415–21. [DOI] [PubMed] [Google Scholar]
- 40. David SP, Murthy NV, Rabiner EA, Munafó MR, Johnstone EC, Jacob R, et al. Novel GABRG2 mutation. Cancer Res. 2007;25(10):2586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boillot M, Morin‐Brureau M, Picard F, Weckhuysen S, Lambrecq V, Minetti C, et al. Novel GABRG2 mutations cause familial febrile seizures. Neurol Genet. 2015;1(4):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Absalom NL, Liao VWY, Kothur K, Indurthi DC, Bennetts B, Troedson C, et al. Gain‐of‐function GABRB3 variants identified in vigabatrin‐hypersensitive epileptic encephalopathies. Brain Commun. 2020;2(2):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Figure S1
Figure S2
Table S1