

Abstract
Background
Transfusion‐Related Acute Lung Injury (TRALI) is a life‐threatening complication of blood transfusions characterized by pulmonary endothelial cell damage and edema, with a high incidence in critically ill patients. The pathophysiology of TRALI is unresolved, but can generally be hypothesized to follow a 2‐hit model in which the first hit is elicited by the underlying clinical condition of the patient (e.g., inflammation, which can be reflected by LPS in experimental models), and the second hit is delivered by the blood transfusion product (e.g., HLA class I antibodies). Here, we report a synergistic role for LPS and HLA class I antibody binding to pulmonary endothelium resulting in enhanced inflammatory responses.
Materials and Methods
Pulmonary endothelial cells were treated with PBS or low‐dose LPS, exclusively or in combination with anti‐HLA class I. Endothelial surface expression of HLA class I, TLR4, and inflammatory marker ICAM‐1 were measured, and trans‐endothelial migration (TEM) of neutrophils was investigated.
Results
LPS treatment of pulmonary endothelium enhanced HLA class I antibody binding, and combined LPS and HLA class I antibody binding enhanced TLR4 (LPS receptor) and ICAM‐1 expression on the endothelial cell surface. Low‐dose LPS and HLA antibody together also increased neutrophil TEM under physiological flow by on average 5‐fold.
Conclusion
We conclude that LPS and anti‐HLA class I antibody have the ability to activate the pulmonary endothelium into a spiral of increasing inflammation, opening the opportunity to potentially block TLR4 to prevent or reduce the severity of TRALI in vivo.
Keywords: HLA class I, pulmonary EC, TEM, TLR4, TRALI
1. INTRODUCTION
Transfusion‐Related Acute Lung Injury (TRALI) is a life‐threatening complication of blood transfusions with high incidence reported in critically ill patients. It is generally accepted that TRALI follows a two‐hit model, in which the first hit is the underlying condition of the patient (e.g., inflammation or sepsis, which can experimentally be induced by lipopolysaccharide (LPS)) and the second hit the blood transfusion, 1 generally including anti‐Human Leukocyte Antigen (HLA)‐ or Human Neutrophil Antigen (HNA) antibodies.
Neutrophils are considered the main effector cells of TRALI, 1 , 2 however, prominent roles for macrophages, dendritic‐ and endothelial cells (ECs) have also emerged. 1 , 3 , 4 , 5 Recently, it was reported that HLA expression on ECs may be an important determinant in experimental murine LPS and MHC class I antibody‐mediated TRALI. 6 However, the interaction between LPS and HLA‐antibody in the onset of endothelial damage in TRALI is unclear.
In this study, we investigated the effects of low‐dose LPS (first hit in TRALI) and HLA‐antibodies (second hit in TRALI) on lung ECs for their ability to increase inflammation by enhancing surface expression of inflammatory markers, TLR4 and HLA, and facilitate neutrophil trans‐endothelial migration (TEM), a key process occurring during the onset of TRALI.
2. METHODS
2.1. Antibodies
See Table 1.
TABLE 1.
List of antibodies
| Anti | Clone | Fluorophore | Company |
|---|---|---|---|
| PECAM‐1 | WM59 | BV‐510 | BD Biosciences |
| VE‐cadherin | 55‐7H1 | AF‐647 | BD Biosciences |
| ICAM‐1 | BBIG‐I1 | FITC | Biolegend |
| TLR4 | HTA125 | AF‐488 | Invitrogen |
| HLA‐ABC | W6/32 | PE | Biolegend |
| HLA‐ABC | W6/32 | BioConnect | |
| IgG2aκ | MOPC‐173 | PE | Biolegend |
| IgG2aκ | BioConnect | ||
| a‐IgG2a | AF568 | Invitrogen |
2.2. Cell culture
Human lung microvascular endothelial cells (HMVEC) from three different donors were purchased from PeloBiotech (Germany) and Lonza (Switzerland). Cells were cultured on fibronectin‐coated dishes in EGM‐2MV (5%FCS) (PeloBiotech, Germany) at 37°C:5% CO2 and used at passages 5–11. LPS from escherichia coli [O55:B5] (Sigma‐Aldrich, Germany) diluted in PBS was added at concentrations of 1–10 ng/ml for 5 h. Potency of LPS was 3,000,000 EU/mg. HLA‐ABC or isotype control IgG2a were added at 10 ug/ml.
2.3. Neutrophil (PMN) isolation and transmigration under flow assay
ECs seeded at 5 × 104 cells/channel in fibronectin‐coated Ibidi μ‐slides VI0.4 (Ibidi, Germany) were cultured for 2–3 days. Primary human neutrophils (PMN) isolation and transmigration assay was performed as previously described, 7 except activation of isolated PMN at 37°C were done for 30 min. Leukocyte‐endothelial interactions were recorded on a wide‐field microscope (Observer Z1/Axiovert; Zeiss, Germany). Transmigrated PMNs were distinguished by their transition from bright to phase‐dark morphology and manually quantified using ImageJ.
2.4. Volunteers
All volunteers signed an informed consent, under the rules and legislation in place within the Netherlands and Sanquin Medical Ethical Committee. The rules and legislations are based on the Declaration of Helsinki (informed consent for participation of human subjects in medical and scientific research) and guidelines for Good Clinical Practice.
2.5. Flow cytometry
Cells were detached with Accutase (GE‐Healthcare L11‐007) and harvested with FACS buffer (PBS++:0.5% BSA). 5 × 104 cells/well were added to a 96‐wells plate and blocked on ice. Cells were incubated with antibodies cold and dark for 1 h and measured on an LSR Fortessa (BD) cell analyzer using FACS Diva software. PECAM‐1 and VE‐cadherin were used as markers.
2.6. Statistical analysis
Statistical analysis between experimental groups was analyzed by methods indicated in figure legends. A two‐sided p ≤ .05 was considered significant.
3. RESULTS
3.1. LPS treatment of lung endothelium increases HLA class I binding and enhances ICAM‐1 and TLR4 expression upon additional treatment with HLA class I antibodies
Pulmonary human microvascular endothelial cells (pHMVEC) were treated with PBS as control or low‐dose LPS and analyzed with flow cytometry. LPS treatment resulted in 1,5‐fold increased fluorescence of anti‐HLA class I compared to PBS‐treated samples (Figure 1A) indicating an increased binding of anti‐HLA class I to pHMVEC upon LPS treatment.
FIGURE 1.
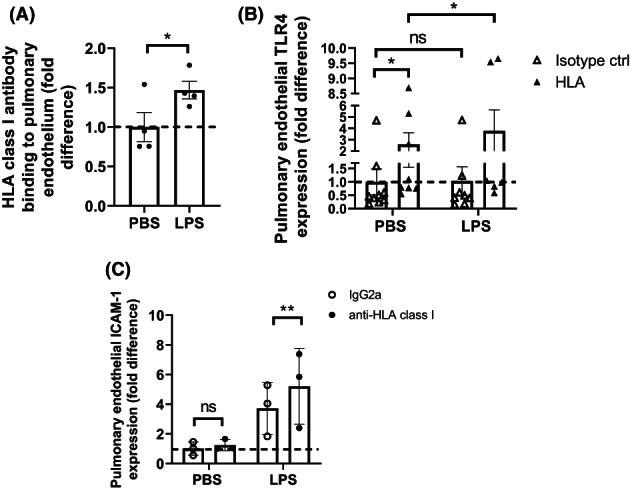
Surface expression of HLA on pulmonary ECs is enhanced by LPS, leading to enhanced TLR4 and ICAM‐1 expression upon HLA class I antibody binding (a) quantification of HLA‐ABC surface expression after treatment of pulmonary ECs with PBS or 10 ng/ml LPS as measured with FACS. All PBS‐treated data points were divided by their average to indicate variation. All LPS datapoints were divided by PBS‐treated data points for fold difference (1.5 ± 0.2). Dotted line indicates 1‐fold. A ratio‐paired T‐test was used to determine statistical significance. ns = non‐significant, * = p < .5, n = 4. (B) Quantification of TLR4 surface expression after treatment of pulmonary ECs with PBS or 10 ng/ml LPS and 10 μg/ml anti‐HLA class I or isotype control, as measured with FACS. All PBS + IgG2a‐treated data points were divided by their average to indicate variation. All anti‐HLA class I and LPS datapoints were divided by PBS + IgG2a‐treated data points for fold difference (fold increase PBS + anti‐HLA class I 2.6 ± 1.0, LPS + anti‐HLA class I 3.8 ± 1.8). Dotted line indicates 1‐fold. A Wilcoxon‐rank paired T‐test was used to determine statistical significance. ns = non‐significant, * = p < .5, n = 5. (C) Quantification of ICAM‐1 surface expression after treatment of pulmonary ECs with PBS or 10 ng/ml LPS and 10 μg/ml HLA‐ABC or isotype IgG2a antibodies as indicated and measured with FACS. All PBS + IgG2a‐treated data points were divided by their average to indicate variation. All LPS datapoints were divided by PBS + IgG2a‐treated data points for fold difference. LPS + IgG2a (3.7 ± 1.0) and LPS + anti‐HLA class I (5.2 ± 1.5) increase compared to PBS + IgG2a. Dotted line indicates 1‐fold. The ratio‐paired T‐test was used to determine statistical significance. ns = non‐significant, ** = p < .01, n = 3
Next, TLR4 and endothelial inflammatory marker Intra‐Cellular Adhesion Molecule (ICAM‐1; CD54) were measured on pHMVEC pre‐treated with PBS or LPS and anti‐HLA class I or isotype control antibodies. TLR4 expression was enhanced on the surface of both PBS + HLA class I (roughly 3‐fold) and LPS + HLA class I‐treated (roughly 4‐fold) pHMVEC compared to PBS + IgG2a‐treated cells, whereas no significant difference was observed from cells treated with LPS and IgG2a (Figure 1B). Surface ICAM‐1 expression was not altered by anti‐HLA treatment alone (PBS + HLA compared to PBS + IgG2a) but was almost 4‐fold enhanced in LPS‐treated pHMVEC compared to PBS + IgG2a‐treated samples, and more than 5‐fold enhanced when pHMVEC were treated with LPS and anti‐HLA Ab in combination (figure 1C).
3.2. HLA class I antibody exacerbates neutrophil TEM in LPS‐treated pHMVECs
pHMVEC treated with PBS or LPS in combination with isotype IgG2a or anti‐HLA class I antibody were exposed to flow with freshly isolated PMN. pHMVEC treated with PBS and IgG2a did not facilitate PMN TEM. The other treatments did lead to an increase in PMN TEM, but none to the same extent as combined LPS and anti‐HLA class I treatment, which facilitated up to 6‐fold more PMN TEM than LPS and isotype treatment (Figure 2).
FIGURE 2.
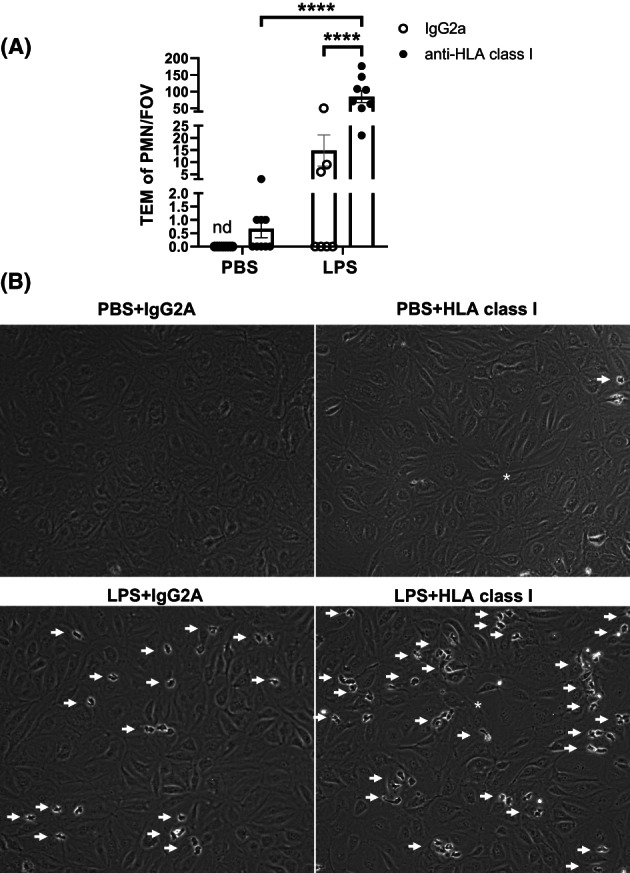
Pulmonary ECs facilitate more neutrophil transmigration upon addition of HLA class I antibodies compared to IgG2a treated cells (A) number of transmigrated neutrophils during flow assay after 5 h treatment with PBS or 1 ng/ml LPS and 2 h with IgG2a or HLA‐ABC antibody as indicated. Data points represent neutrophil (PMN) trans‐endothelial migration (TEM) per field of view (FOV). Three fields of view per channel were obtained during time‐lapse imaging, followed by tile scans with 25 additional fields of view per condition and replicate at endpoint to determine the distribution of neutrophils. Bars represent average TEM of PMN/FOV for all experiments. Two‐way ANOVA was used to determine statistical significance. **** = p < .0001, error bars are presented as standard error of mean (SEM), n = 3. (b) Representative images from time‐lapse videos as indicated. White arrows indicate neutrophils (individual or clusters), white star indicates EC disturbance
4. DISCUSSION
Here, using an ex‐vivo model of antibody‐mediated TRALI, we identify a novel mechanism by which TRALI may develop.
First, we show that LPS treatment of pHMVEC increases expression of HLA class I. Similar results have previously been reported in HUVEC stimulated with LPS, 8 and enhanced HLA class II expression has been reported in response to IFN‐gamma, LPS, and mixtures of cytokines in a variety of cells. 9 , 10 These data strongly suggest that inflammatory stimuli can prime for TRALI by enhancing HLA molecules, which may explain why critically ill patients are at increased risk for TRALI upon transfusion. This may also explain the results of look back investigations in which transfusion products with anti‐HLA antibodies did not induce TRALI in each recipient even in the presence of cognate antigen. 11 Pro‐inflammatory activation of the endothelium with an inflammatory condition may be needed to sufficiently increase HLA expression for induction of TRALI by anti‐HLA abs, shedding light on the two‐hit mechanism of the TRALI pathophysiology.
Second, we show that binding of anti‐HLA class I antibodies to pHMVEC HLA class I molecules increases TLR4 expression on the endothelial surface. TLR4 is mainly known as a receptor for LPS, however, physiologically expressed oxidized lipids have also been shown to facilitate ALI via TLR4. 12 Thus, even in the absence of additional LPS insults, increased expression of the TLR4 receptor could be detrimental. Although Imai et al. reported that the contribution of endothelial TLR4 was negligible for acid‐induced ALI, LPS‐induced TRALI was not explored in their model. The additional increase of lung endothelial TLR4 upon treatment with LPS and anti‐HLA class I antibodies may indicate an ongoing amplification loop of LPS reactivity towards the endothelium during TRALI. These hypotheses could be tested by inhibiting TLR4 signaling, using specific inhibitor TAK‐242. 13
Thirdly, we show that treatment with both LPS and anti‐HLA class I antibodies together further enhances ICAM‐1 expression compared to LPS alone and that the combined treatment results in increased PMN TEM. It has been suggested that incubation of ECs with anti‐HLA class I antibodies leads to an additional increase of adhesion molecules, particularly ICAM‐1, on ECs. 14 , 15 Our study shows that anti‐HLA class I antibodies are only able to upregulate adhesion molecules in the additional presence of LPS in pHMVEC, again supporting a two‐hit requirement, with LPS as first hit, in the pathophysiology of TRALI.
We propose a mechanism that sheds light on the two‐hit mechanism in TRALI, with a crucial role for LPS, anti‐HLA, and the pulmonary endothelium: LPS primes the pulmonary endothelium to increase the expression of HLA class I molecules on the luminal cell surface, facilitating increased binding of anti‐HLA. This in turn increases pulmonary endothelial TLR4 and ICAM‐1 expression, creating an aggravated immune response culminating in increased PMN‐TEM. This may explain the high incidence of TRALI in the critically ill. Of note, future studies should focus on in vivo validation of the mechanism.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Video S1 Time‐lapse avi of pHMVEC treated for 5 h with 1 ng/ml of LPS and 2 h IgG2a antibody, subjected to flow with activated neutrophils.
Video S2 Time‐lapse avi of pHMVEC treated for 5 h with 1 ng/ml of LPS and 2 h HLA class I antibody, subjected to flow with activated neutrophils.
ACKNOWLEDGMENTS
This work was supported by ZonMW NWO Vici grant #91819632 (JDvB). Prof Dr Alexander P. J Vlaaris supported by ZonMW NWO VIDI grant #09150172010047.
Morsing SKH, Zeeuw van der Laan E, van Stalborch AD, van Buul JD, Kapur R, Vlaar AP. A pulmonary endothelial amplification loop aggravates ex‐vivo transfusion‐related acute lung injury via increased toll‐like receptor 4 and intra‐cellular adhesion molecule‐1 expression. Transfusion. 2022;62(10):1961–1966. 10.1111/trf.17076
Funding information ZonMw, Grant/Award Numbers: 09150172010047, 91819632
Contributor Information
Rick Kapur, Email: r.kapur@sanquin.nl.
Alexander P. Vlaar, Email: a.p.vlaar@amsterdamumc.nl.
REFERENCES
- 1. Semple JW, Rebetz J, Kapur R. Transfusion‐associated circulatory overload and transfusion‐related acute lung injury. Blood. 2019;133(17):1840–53. [DOI] [PubMed] [Google Scholar]
- 2. Rebetz J, Semple JW, Kapur R. The pathogenic involvement of neutrophils in acute respiratory distress syndrome and transfusion‐related acute lung injury. Transfus Med Hemother. 2018;45(5):290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zeeuw van der Laan EAN, van der Velden S, Porcelijn L, Semple JW, van der Schoot CE, Kapur R. Update on the pathophysiology of transfusion‐related acute lung injury. Curr Opin Hematol. 2020;27(6):386–91. [DOI] [PubMed] [Google Scholar]
- 4. Kapur R, Kasetty G, Rebetz J, Egesten A, Semple JW. Osteopontin mediates murine transfusion‐related acute lung injury via stimulation of pulmonary neutrophil accumulation. Blood. 2019;134(1):74–84. [DOI] [PubMed] [Google Scholar]
- 5. Kapur R, Kim M, Aslam R, McVey MJ, Tabuchi A, Luo A, et al. T regulatory cells and dendritic cells protect against transfusion‐related acute lung injury via IL‐10. Blood. 2017;129(18):2557–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cleary SJ, Kwaan N, Tian JJ, Calabrese DR, Mallavia B, Magnen M, et al. Complement activation on endothelium initiates antibody‐mediated acute lung injury. J Clin Investig. 2020;130(11):5909–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kroon J, Daniel AE, Hoogenboezem M, van Buul JD. Real‐time imaging of endothelial cell‐cell junctions during neutrophil transmigration under physiological flow. JoVE. 2014;90:51766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Otsuka A, Hanafusa T, Kono N, Tarui S. Lipopolysaccharide augments HLA‐A,B,C molecule expression but inhibits interferon‐gamma‐induced HLA‐DR molecule expression on cultured human endothelial cells. Immunology. 1991;73(4):428–32. [PMC free article] [PubMed] [Google Scholar]
- 9. Etienne S, Bourdoulous S, Strosberg AD, Couraud PO. MHC class II engagement in brain endothelial cells induces protein kinase A‐dependent IL‐6 secretion and phosphorylation of cAMP response element‐binding protein. J Immunol. 1999;163(7):3636–41. [PubMed] [Google Scholar]
- 10. Kelher MR, Banerjee A, Gamboni F, Anderson C, Silliman CC. Antibodies to major histocompatibility complex class II antigens directly prime neutrophils and cause acute lung injury in a two‐event in vivo rat model. Transfusion. 2016;56(12):3004–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kopko PM, Bux J, Toy P. Antibodies associated with TRALI: differences in clinical relevance. Transfusion. 2018;13:trf.15094. [DOI] [PubMed] [Google Scholar]
- 12. Imai Y, Kuba K, Neely GG, Yaghubian‐Malhami R, Perkmann T, van Loo G, et al. Identification of oxidative stress and toll‐like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ono Y, Maejima Y, Saito M, Sakamoto K, Horita S, Shimomura K, et al. TAK‐242, a specific inhibitor of toll‐like receptor 4 signalling, prevents endotoxemia‐induced skeletal muscle wasting in mice. Sci Rep. 2020;10(1):694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Valenzuela NM, Mulder A, Reed EF. HLA class I antibodies trigger increased adherence of monocytes to endothelial cells by eliciting an increase in endothelial P‐selectin and, depending on subclass, by engaging FcγRs. J Immunol. 2013;190(12):6635–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hosenpud JD, Shipley GD, Morris TE, Hefeneider SH, Wagner CR. The modulation of human aortic endothelial cell ICAM‐1 (CD‐54) expression by serum containing high titers of anti‐HLA antibodies. Transplantation. 1993;55(2):405–11. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1 Time‐lapse avi of pHMVEC treated for 5 h with 1 ng/ml of LPS and 2 h IgG2a antibody, subjected to flow with activated neutrophils.
Video S2 Time‐lapse avi of pHMVEC treated for 5 h with 1 ng/ml of LPS and 2 h HLA class I antibody, subjected to flow with activated neutrophils.