

Abstract
Aim
To determine whether treatment with empagliflozin was able to affect the myocardial glucose metabolic rate, as assessed by cardiac dynamic 18F‐fluorodeoxyglucose‐positron emission tomography (18F‐FDG‐PET) combined with euglycaemic‐hyperinsulinaemic clamp compared with glimepiride in patients with type 2 diabetes.
Materials and Methods
To further investigate the cardioprotective mechanism of sodium‐glucose co‐transporter‐2 inhibitors, we performed a 26‐week, randomized, open‐label, crossover, active‐comparator study to determine the effects of empagliflozin 10 mg versus glimepiride 2 mg daily on the myocardial glucose metabolic rate assessed by cardiac dynamic 18F‐FDG‐PET combined with euglycaemic‐hyperinsulinaemic clamp in 23 patients with type 2 diabetes. We also measured cardiac geometry and myocardial mechano‐energetic efficiency, as well as systolic and diastolic function by echocardiography.
Results
Compared with glimepiride, treatment with empagliflozin resulted in a greater reduction in the myocardial glucose metabolic rate from baseline to 26 weeks (adjusted difference −6.07 [−8.59, −3.55] μmol/min/100 g; P < .0001). Moreover, compared with glimepiride, empagliflozin led to significant reductions in left atrial diameter, left ventricular end‐systolic and end‐diastolic volumes, N‐terminal pro b‐type natriuretic peptide levels, blood pressure, heart rate, stroke work, and myocardial oxygen consumption estimated by the rate pressure product, and increases in ejection fraction, myocardial mechano‐energetic efficiency, red blood cells, and haematocrit and haemoglobin levels.
Conclusions
The present study provides evidence that empagliflozin treatment in subjects with type 2 diabetes without coronary artery disease leads to a significant reduction in the myocardial glucose metabolic rate.
Keywords: cardiovascular disease, empagliflozin, inhibitor, insulin resistance, randomized trial, SGLT2 inhibitor, type 2 diabetes
1. INTRODUCTION
There is clear evidence from cardiovascular outcome trials (CVOTs) that treatment with sodium‐glucose co‐transporter‐2 inhibitors (SGLT2is) reduces major cardiovascular (CV) events in patients with type 2 diabetes and established atherosclerotic CV disease, and decreases the risk of hospitalization for heart failure (HHF) and CV mortality. 1 , 2 , 3 , 4 , 5 Results from the DAPA‐HF, 6 the EMPEROR‐Reduced 7 and the EMPEROR‐Preserved 8 have extended the findings of reduced HHF in patients with heart failure (HF), regardless of the presence or absence of type 2 diabetes. The putative mechanisms explaining the beneficial CV effects of SGLT2is have been the subject of intense debate. 9 It has been suggested that these beneficial effects in subjects with HF might be, at least in part, a result of improved cardiac energetics and efficiency. Indeed, it has been shown that treatment with SGLT2is is associated with increasing ketogenesis, leading to an elevation in circulation ketone bodies, which serve as alternative cardiac fuel substrate. 10 Because ketone bodies may act as a ‘super fuel’, requiring less oxygen to generate ATP, it has been proposed that SGLT2i treatment induces a metabolic shift, characterized by a decrease in glucose uptake and glucose oxidation to an increase in oxidation of ketone bodies. This metabolic shift towards a more energy‐efficient fuel like ketone bodies may improve myocardial energetics and cardiac efficiency. Recently, it was reported that, compared with placebo, 4 weeks of treatment with empagliflozin in patients with type 2 diabetes resulted in a 57% reduction in myocardial glucose uptake, as measured by 18F‐fluorodeoxyglucose (18F‐FDG) positron emission tomography (PET)/computed tomography (CT) after an overnight fast. 11 Treatment with empagliflozin did not affect myocardial free fatty acids (FFAs) uptake, a finding similar to that observed in another placebo‐controlled study evaluating the effects of 6 weeks of treatment with dapagliflozin in patients with type 2 diabetes. 12 On the other hand, it has been reported that, in patients with chronic HF, a short‐term reduction in serum FFA concentration induced an impairment of left ventricular energy metabolism and function. 13 Nevertheless, whether the observed reduction in myocardial glucose uptake oxidation is only a transient phenomenon rather than a long‐term adaptive metabolic process, remains unsettled. To date, no head‐to‐head trial has compared the effect of SGLT2is on the myocardial glucose metabolic rate (MrGlu) in patients with type 2 diabetes. The primary aim of this study was to determine, in patients with type 2 diabetes without coronary artery disease, whether treatment with empagliflozin was able to affect myocardial MrGlu, as assessed by cardiac dynamic 18F‐FDG‐PET combined with euglycaemic‐hyperinsulinaemic clamp, compared with glimepiride, a common antidiabetic drug with neutral CV effects. 14 Additional aims were assessments of the effects of empagliflozin versus glimepiride on cardiac geometry, and systolic and diastolic function assessed by echocardiography, as well as myocardial mechano‐energetic efficiency (MEE).
2. MATERIALS AND METHODS
2.1. Study design
The effects of empagliFlozin on myocardIal metabOlic Rate of glucosE estimated through 18FDG PET (FIORE Study) was a 26‐week, single‐centre, prospective, randomized, open‐label, crossover, active‐comparator study aimed at investigating the effects of empagliflozin 10 mg versus glimepiride 2 mg daily on myocardial MrGlu and cardiac geometry and function in patients with type 2 diabetes. The study was conducted at the Department of Medical and Surgical Sciences of the University ‘Magna Graecia’ of Catanzaro (Italy). The inclusion criteria were subjects with type 2 diabetes in monotherapy treatment with a stable dose of metformin (daily dose of ≥1.500 mg), which remained unchanged throughout the study, age 40‐69 years and HbA1c 6.5%‐9% (48‐75 mmol/mol). The exclusion criteria were type 1 diabetes, a history of coronary artery disease or HF, prior cardio‐cerebral‐vascular events, pregnant women, childbearing women without an adequate birth control method, a history of malignant diseases, impaired kidney and liver function, valvular heart disease, anaemia, recurrent genital infections, and previous treatment with insulin, SGLT2is, glucagon‐like peptide‐1 receptor agonists or dipeptidyl peptidase‐4 inhibitors.
The primary outcome was the between‐groups (empagliflozin vs. glimepiride) change in the myocardial MrGlu from baseline to 26 weeks. Secondary outcomes included the between‐groups changes in cardio‐metabolic variables, peripheral insulin sensitivity, indexed MEE (MEEi) and cardiac function.
Eligible patients were randomly assigned in a 1:1 ratio to receive empagliflozin 10 mg or glimepiride 2 mg daily for 26 weeks. After the first 26 weeks, patients crossed over to the opposite treatment for another 26 weeks (Figure S1 ). Patients randomized to receive glimepiride were allowed to uptitrate to a maximum dose of 6 mg/day if they experienced fasting plasma glucose (FPG) levels of more than 112 mg/dl (6.2 mmol/L) at the scheduled visit at the sixth week or at any later scheduled visit.
At baseline and at the end of each 26‐week treatment period, after 12 hours of fasting, all subjects underwent an anthropometrical evaluation, including measurements of body mass index (BMI), waist circumference and body composition by bioelectrical impedance (BIA 101 ASE, Akern, Pontassieve, Florence, Italy), blood pressure and laboratory determinations, echocardiography and a 18F‐FDG‐PET scan combined with euglycaemic‐hyperinsulinaemic clamp in the morning, at the same time of the day, without taking the study drug. The last dose of the study drugs was taken by the patients during the previous dinner, 12 hours before the studies. A washout period between treatments was not performed, to avoid exposing patients to undesirable high amounts of CT radiation over a few days. The study was approved by the Hospital Ethical Committee (Comitato Etico Azienda Ospedaliera ‘Mater Domini’, approval code: 2016.43) and all participants provided written informed consent to be enrolled in this investigation in accordance with the principles of the Helsinki Declaration. The study was registered at https://www.clinicaltrials.gov (NCT04183868).
2.2. 18F‐FDG‐PET scan combined with euglycaemic‐hyperinsulinaemic clamp
Myocardial MRGlu was measured by 18F‐FDG‐PET acquired during euglycaemic‐hyperinsulinaemic clamp, as previously described. 15 Subjects received a priming dose of insulin (100 UI/ml) (Humulin R; Eli Lilly) during the initial 10 minutes to raise the serum insulin concentration acutely (80 mU/m2 × min), which was then maintained by continuous insulin infusion fixed at 40 mU/m2 × min. 16 The blood glucose level was kept constant at 100 mg/dl, with a coefficient of variation of less than 5% for the next 120 minutes, by infusing 20% glucose at varying rates according to blood glucose measurements performed at 5‐minute intervals. Glucose metabolized by the whole body (M) was calculated as the mean rate of glucose infusion measured during the last 60 minutes of the clamp examination (steady state) and was expressed in milligrams per minute per kilogram fat‐free mass assessed by bioelectrical impedance (MFFM). Urinary glucose concentrations were measured by collecting urine immediately after the end of the 18F‐FDG‐PET combined with euglycaemic‐hyperinsulinaemic clamp. Urinary glucose loss was subtracted from the total rate of glucose disposal to determine whole‐body insulin‐stimulated glucose disposal. The 18F‐FDG‐PET imaging procedure was performed on a hybrid PET/CT scanner (GE Discovery ST8‐ 2D PET scanner), starting 60 minutes after the insulin infusion. A 60‐minute dynamic acquisition was started simultaneously with the intravenous injection of 370 MBq of 18F‐FDG, according to the following time frame sampling: eight × 15 seconds, two × 30 seconds, two × 120 seconds, one × 180 seconds, six × 300 seconds and two × 600 seconds. 17 PET images were reconstructed in a 128 × 128 matrix using a Ordered subset expectation maximization algorithm, and were corrected for decay and attenuation based on co‐registered CT. The insulin‐glucose infusion continued during the entire PET acquisition. The estimation of myocardial MRGlu was performed by Patlak compartmental modelling, 15 , 18 using the graphical tool specific for cardiac images analysis (PCARD) implemented in PMOD Software platform (version 3.806). 17 In PCARD, the full dynamic study is used for myocardial MRGlu calculation, and the arterial input function is extracted from a volume of interest semiautomatically placed in the left ventricular cavity. 18 Both of the physicians who performed the PET scans, as well as the investigators who analysed the PET scan data, were blinded to the treatment of the participants.
2.3. Echocardiographic measurements
Tracings were taken using a VIVID‐ E95 ultrasound machine (GE Healthcare, Horten, Norway) with an annular phased array 2.5‐MHz transducer. To optimize the reproducibility, all the readings were performed by the same experienced investigator, who was blinded to the treatment of the participants. Measurement of interventricular septum thickness (IVS) was made at end‐diastole. LV end‐diastolic volume (LVEDV), end‐systolic volume (LVESV) and left ventricular ejection fraction (LVEF) were measured according to the Simpson method. 19 Left atrial diameter was measured from the parasternal long‐axis view. The left ventricular mass index (LVMI) was calculated using the Deveraux formula 20 normalized by body surface area. 19 , 21 Left ventricular diastolic function was evaluated according to diagnostic criteria proposed by the American Society of Echocardiography. 19 Systolic pulmonary arterial pressure (s‐PAP) was calculated with the Bernoulli equation 22 using the apical four chamber o short axis to obtain peak trans tricuspid regurgitation. 19 LV myocardial MEE was calculated using the method developed and validated by de Simone et al. as the ratio between external myocardial work and myocardial oxygen consumption (MVO2). 23 , 24 External myocardial work can be estimated as stroke work (SW), with SW being computed as systolic blood pressure (SBP) × echocardiographic stroke volume (SV). SV was calculated as the difference between LVEDV and LVESV by the z‐derived method. 25 , 26 , 27 , 28 MVO2 reflects the total amount of energy produced by the myocardium and can be estimated using the ‘double product’ of SBP × heart rate (HR). 29 LV myocardial MEE was calculated as SV × SBP/ SBP × HR = SV/ HR. Because of the close dependence of MEE on LV mass, MEE was normalized by LVM to obtain an estimate of MEE per gram of LVM (i.e. MEEi). 23 , 24 , 25 , 26
2.4. Laboratory determinations
Plasma glucose levels were measured by the enzymatic method (Roche Diagnostics, Mannheim, Germany). Serum creatinine was measured by a clinical chemistry analyser (Roche/Hitachi Modular Analytics System, P Module) using the Roche Creatinine Plus assay (Hoffman‐La Roche, Basel, Switzerland). Plasma insulin concentration was measured with a chemiluminescence‐based assay (Immulite, Siemens, Italy). HbA1c was measured with high performance liquid chromatography using an National Glycohemoglobin Standardization Program‐certified automated analyser (Adams HA‐8160 HbA1c analyser, Menarini, Italy). Serum N‐terminal pro b‐type natriuretic peptide (NT‐proBNP) was measured by chemiluminescent immunoassay (Roche Diagnostics). Blood β‐hydroxybutyrate were measured by a Glucomen LX ketone sensor (Menarini Diagnostics, Florence, Italy). Urinary glucose concentrations were measured by Aution Max (Menarini Diagnostics). Red blood cells, haemoglobin and haematocrit were determined using an automated particle counter (Siemens Healthcare Diagnostics ADVIA 120/2120 Haematology System, Milan, Italy). The estimated glomerular filtration rate (eGFR) was calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) equation 30 : eGFR = 141 × min (Scr/k, 1)α × max (Scr/k, 1)−1.209 × 0.993Age × 1.018 (if female) × 1.159 (if Black), where Scr is serum creatinine, k is 0.7 for females and 0.9 for males, α is −0.329 for females and −0.411 for males, min indicates the minimum of Scr/k or 1, and max indicates the maximum of Scr/k or 1.
2.5. Sample size estimation and statistical analyses
Sample size calculation was based on the results of a 4‐week study with empagliflozin showing a 57% reduction in myocardial glucose uptake measured by 18F‐FDG (PET)/CT in patients with type 2 diabetes under fasting state. 11 Considering an expected delta between the two groups on myocardial MrGlu of 6.0 μmol/min/100 g and a standard deviation (SD) of 9.0, 20 patients per treatment group are required to reject the null hypothesis that the population means of the two groups are equal with a power of 80% and an alpha of .05. With an addition of 15% to safeguard from potential missing values, a sample size of 23 was planned. With this sample size we were also able to reject the null hypothesis that the population means of the two groups are equal with a power of 80% and an alpha of .05, considering an expected delta between the two groups on LVEF of 2.5% and a SD of 4.1, on LVEDV of ‐8.1 ml and a SD of 13.4, and on LVESV of ‐5.6 ml and a SD of 9.2.
Triglyceride values showed a skewed distribution, and, therefore, were natural log‐transformed for the statistical analyses. Continuous variables are expressed as means ± SD. Categorical variables were compared by χ2 test. Treatment effects were analysed using ANCOVA, a linear model with treatment group as the categorical independent variable and the baseline value of the primary and secondary outcomes as a covariate. Changes in primary outcome during the intervention period, and effects of the treatment, were modelled by linear mixed‐effects models with a participant‐specific random intercept to account for the correlation of repeated measurements within participants and treatment, time and the interaction between treatment and time as fixed factors. Relationships between variables were determined by Pearson's correlation coefficient (r). For all the analyses, a P value of .05 or less was considered to be statistically significant. All analyses were performed using SPSS software version 25 for Windows.
3. RESULTS
Among 50 screened individuals with type 2 diabetes, 26 met the inclusion criteria and were randomized to receive either empagliflozin 10 mg (n = 13) or glimepiride 2 mg (n = 13). Among those randomized, three participants withdrew prematurely from the trial. One patient was withdrawn from the study because of genital infection. One patient was excluded from the evaluable dataset because he refused to undergo an 18F‐FDG‐PET scan, and one patient was lost to follow‐up. Therefore, the evaluable study population comprised 23 patients (Figure S2). The baseline characteristics of the participants who completed the trial are reported in Table 1.
TABLE 1.
Baseline anthropometric and clinical characteristics of the patients completing the trial
| Total study population (n = 23) | |
|---|---|
| Female/male (n) | 8/15 |
| Age (y) | 55.5 ± 8 |
| BMI (kg/m2) | 31.2 ± 4.2 |
| Waist circumference (cm) | 107.2 ± 9 |
| Systolic blood pressure (mmHg) | 127 ± 13 |
| Diastolic blood pressure (mmHg) | 80 ± 11 |
| Heart rate (bpm) | 74 ± 10 |
| Fasting glucose (mg/dl) | 149 ± 25 |
| Fasting plasma insulin (mU/ml) | 10.7 ± 6.2 |
| HbA1c (%) (mmol/mol) |
7.5 ± 1 (58 ± 7.6) |
| Total cholesterol (mg/dl) | 176 ± 36 |
| LDL cholesterol (mg/dl) | 112 ± 24 |
| HDL cholesterol (mg/dl) | 45 ± 9 |
| Triglycerides (mg/dl) | 143 ± 69 |
| Insulin‐stimulated glucose disposal corrected for fat‐free mass (mg/min × kg FFM) | 3.8 ± 2.4 |
| Myocardial MrGlu (μmol/min/100 g) | 14.1 ± 9 |
| Diabetes duration (y) | 5.6 ± 5 |
| Hypertension (%) | 65 |
| Lipid‐lowering therapy (%) | 61.5 |
| Antihypertensive therapy (%) | 65.4 |
Note: Data are means ± SD.
Abbreviations: BMI, body mass index; FFM, fat‐free mass; MrGlu, glucose metabolic rate.
At the end of the study, all the subjects randomized to glimepiride received a dose of 2 mg daily. None of the subjects in the study reported hypoglycaemic events.
3.1. Primary outcome
3.1.1. Change in myocardial MrGlu
Patients randomized to empagliflozin, compared with patients randomized to glimepiride, exhibited a greater reduction in myocardial MrGlu after 26 weeks (14.4 ± 9 vs. 7.9 ± 8 μmol/min/100 g, P < .0001 in the empagliflozin group, 10.3 ± 9 vs. 13.8 ± 10 μmol/min/100 g, P = .01 in the glimepiride group). The adjusted difference between the groups was −6.07 (−8.59, −3.55) μmol/min/100 g; P < .0001; treatment: P = .003; treatment × time interaction: P = .002) (Table 2, Figure 1). The differences in myocardial MrGlu remained significant after adjustment for changes in BMI (P = .001) (Table 2).
TABLE 2.
Changes in myocardial glucose metabolic rate, insulin sensitivity, glucose and insulin concentrations and myocardial oxygen consumption during 18F‐FDG‐PET combined with euglycaemic‐hyperinsulinaemic clamp after exposure to empagliflozin or glimepiride for 26 weeks
| Empagliflozin n = 23 | Glimepiride n = 23 | Adjusted difference between groups (95% CI) | P value | P value a | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Before treatment | After 26 weeks of treatment | Treatment difference | P value | Before treatment | After 26 weeks of treatment | Treatment difference | P value | |||||
| Myocardial MrGlu (μmol/min/100 g) | 14.4 ± 9 | 7.9 ± 8 | −6.55 ± 6.4 | <.0001 | 10.3 ± 9 | 13.8 ± 10 | 3.49 ± 5.8 | .01 | −6.07 | (−8.59, −3.55) |
<.0001 |
.001 |
| Insulin‐stimulated glucose disposal corrected for FFM and urinary glucose loss (mg/min × kg FFM) | 3.49 ± 2 | 5.52 ± 2.1 | 2.02 ± 2.2 | <.0001 | 5.5 ± 3.4 | 3.97 ± 2.4 | −1.53 ± 2.1 | .003 | 1.53 | (0.73, 2.33) |
<.0001 |
<.0001 |
| Fasting plasma glucose concentration at the beginning of the clamp (mg/dl) | 120 ± 24 | 111 ± 16 | −9.56 ± 21 | .04 | 112 ± 21 | 106 ± 16 | −6.52 ± 15 | .052 | −7.35 | (−13.1, −1.59) |
.7 |
– |
| Plasma glucose concentration at the end of the clamp (mg/dl) | 101 ± 6 | 99 ± 2 | −1.9 ± 4 | .055 | 101 ± 4 | 99 ± 1 | −1.69 ± 4 | .055 | ‐1.69 | (−4.32, −2.5) |
.9 |
– |
| Fasting plasma insulin levels at the beginning of the clamp (mU/ml) | 11.2 ± 5.9 | 8.4 ± 5.8 | −2.87 ± 2.5 | <.0001 | 10.8 ± 7 | 12 ± 6 | 1.75 ± 5.4 | .2 | −2.83 | (−4.56, −1.09) |
.001 |
.002 |
| Steady state plasma insulin levels during the clamp (mU/ml) | 50.9 ± 23 | 48.6 ± 26 | −2.3 ± 15 | .2 | 50.03 ± 26 | 47.4 ± 18 | −2.63 ± 9 | .2 | −1.7 | (−8.6, 5.2) |
.9 |
.8 |
| MVO2 during the clamp (mmHg*bpm) | 9777 ± 1915 | 8536 ± 1359 | −1240 ± 1678 | .002 | 9001 ± 1694 | 9685 ± 2140 | 684 ± 793 | <.0001 | −1666 | (−2283, −948) | <.0001 | – |
Note: Data are means ± SD, unless otherwise indicated, and were analysed using ANCOVA adjusting for baseline values. Changes in individual outcomes during the intervention period and effects of the treatment were modelled by linear mixed‐effects models.
Abbreviations: 18F‐FDG‐PET, 18F‐fluorodeoxyglucose‐positron emission tomography; BMI, body mass index; FFM, fat‐free mass; MrGlu, glucose metabolic rate; MVO2, myocardial oxygen consumption.
Adjusted for changes in BMI.
FIGURE 1.
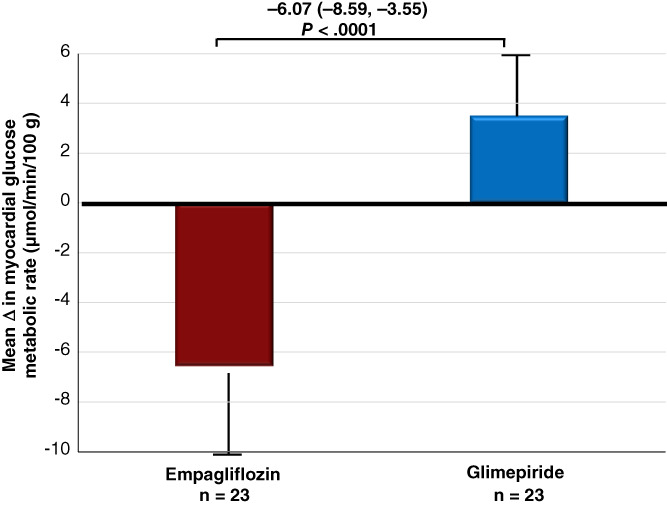
Changes in myocardial glucose metabolic rate after exposure to empagliflozin or glimepiride for 26 weeks. Data are means ± SD, and analysed using ANCOVA adjusting for baseline values. The mean change in myocardial glucose metabolic rate is presented as mean (95% CI), and the adjusted difference between the groups is shown with 95% CI
To address the issue of a potential risk of carry‐over effect between treatment periods, we analysed primary and secondary outcomes, also after the first treatment period. As shown in Table S1 , changes in myocardial MrGlu, whole body insulin‐stimulated glucose disposal, SBP, HR and fasting ketone bodies from baseline to 26 weeks after the first treatment period were consistent with the results obtained after the crossover (Table 2). These findings do not support a carry‐over effect between the treatment periods.
3.2. Secondary outcomes
3.2.1. Change in cardio‐metabolic variables
After 26 weeks of treatment, patients randomized to empagliflozin, compared with patients randomized to glimepiride, exhibited a greater reduction in BMI (adjusted difference −0.93 [−1.64, −0.22] kg/m2; P = .01), waist circumference (adjusted difference −1.94 [−3.07, −0.81] cm; P = .001), SBP (adjusted difference −11.94 [−17.5, −6.36] mmHg; P < .0001), diastolic blood pressure (DBP) (adjusted difference −5.33 [−9.08, −1.57] mmHg; P = .007) and resting HR (adjusted difference −9.55 [−13.79, −5.3] bpm; P < .0001) (Table 3). Changes in FPG and HbA1c were not significantly different between the two treatment arms (Table 3). Patients randomized to empagliflozin exhibited a significant increase in fasting β‐hydroxybutyrate (P < .0001), red blood cells (P < .0001), haematocrit (P = .004) and haemoglobin (P < .0001) (Table 3).
TABLE 3.
Changes in clinical and metabolic variables after exposure to empagliflozin or glimepiride for 26 weeks
| Empagliflozin n = 23 | Glimepiride n = 23 | Adjusted difference between groups (95% CI) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Before treatment | After 26 weeks of treatment | Treatment difference | P value | Before treatment | After 26 weeks of treatment | Treatment difference | P value | P value | |||
| BMI (kg/m2) | 31.1 ± 4.2 | 30.1 ± 4.7 | −1.02 ± 1.4 | .003 | 30.7 ± 4.8 | 30.6 ± 4.5 | −0.07 ± 0.7 | .6 | −0.93 | (−1.64, −0.22) | .01 |
| Waist circumference (cm) | 107 ± 9 | 104.8 ± 9 | −2.26 ± 2.2 | <.0001 | 106.2 ± 10 | 105.9 ± 9 | −0.3 ± 1.4 | .3 | −1.94 | (−3.07, −0.81) | .001 |
| Systolic blood pressure (mmHg) | 129.5 ± 13 | 120 ± 12 | −9.34 ± 12 | .002 | 123.5 ± 15 | 128 ± 14 | 4.56 ± 7.1 | .006 | −11.94 | (−17.5, −6.36) | <.0001 |
| Diastolic blood pressure (mmHg) | 81.8 ± 11 | 78.4 ± 7 | −3.39 ± 7.6 | .04 | 79 ± 10 | 82.3 ± 9 | 3.21 ± 8.2 | .07 | −5.33 | (−9.08, −1.57) | .007 |
| Heart rate (bpm) | 76.4 ± 11 | 70.1 ± 7 | −6.3 ± 10 | .009 | 72.3 ± 9 | 77.1 ± 11 | 4.73 ± 3.7 | <.0001 | −9.55 | (−13.79, −5.3) | <.0001 |
| Fasting glucose (mg/dl) | 140 ± 28 | 126.3 ± 18 | −16.5 ± 19 | <.0001 | 136 ± 22 | 120 ± 17 | −15.4 ± 18 | .001 | −15.5 | (−21.1, 9.8) | .81 |
| HbA1c (%/mmol/mol) | 7.3 ± 1 (56 ± 7.6) | 6.7 ± 0.8 (50 ± 5.9) | −0.52 ± 0.7 (−6 ± 1.7) | .002 | 7.1 ± 0.8 (54 ± 5.9) | 6.7 ± 0.6 (50 ± 4.4) | −0.46 ± 0.6 (−4 ± 1.5) | .001 | −0.13 | (−0.29, 0.31) | .9 |
| Fasting β‐hydroxybutyrate (mmol/l) | 0.10 ± 0 | 0.28 ± 0.09 | 0.18 ± 0.09 | <.0001 | 0.10 ± 0 | 0.104 ± 0.02 | 0.004 ± 0.02 | .3 | 0.18 | (0.15, 0.21) | <.0001 |
| Red blood cells (million/mm3) | 4.9 ± 0.4 | 5.1 ± 0.4 | 0.28 ± 0.44 | .005 | 5.1 ± 0.4 | 4.8 ± 0.4 | −0.22 ± 0.4 | .01 | 0.41 | (0.19, 0.63) | <.0001 |
| Haematocrit (%) | 42.2 ± 5 | 43.5 ± 3.7 | 1.30 ± 2.6 | .02 | 43 ± 4 | 42 ± 4.7 | −1.06 ± 2.65 | .06 | 2.17 | (0.74, 3.60) | .004 |
| Haemoglobin (g/dl) | 14.1 ± 1.8 | 14.6 ± 1.4 | 0.50 ± 0.74 | .003 | 14.4 ± 1.5 | 14.0 ± 1.6 | −0.49 ± 0.75 | .005 | 0.92 | (0.52, 1.33) | <.0001 |
| Serum creatinine (mg/dl) | 0.78 ± 0.1 | 0.80 ± 0.1 | 0.018 ± 0.07 | .2 | 0.78 ± 0.1 | 0.785 ± 0.1 | 0.003 ± 0.06 | .8 | 0.018 | (−0.01, 0.04) | .4 |
| eGFR (ml/min per 1.73m2) | 129.6 ± 33 | 127.1 ± 35 | −2.54 ± 19 | .5 | 137.9 ± 52 | 136 ± 53 | −1.87 ± 13 | .5 | −2.68 | (−9.75, 4.37) | .8 |
| Albuminuria (mg/L) | 18.3 ± 11 | 14.4 ± 8 | −4.67 ± 12 | .2 | 13.9 ± 7 | 13.6 ± 6 | −0.64 ± 5.9 | .2 | −3.71 | (−8.38, 0.95) | .8 |
Note: Data are means ± SD, unless otherwise indicated, and were analysed using ANCOVA adjusting for baseline values.
Abbreviations: BMI, body mass index; eGFR, estimated glomerular filtration rate.
In addition, treatment with empagliflozin was not associated with a significant reduction in eGFR and proteinuria compared with treatment with glimepiride (Table 3).
3.2.2. Change in insulin‐stimulated glucose disposal
Treatment with empagliflozin was associated with a significant increase in insulin‐stimulated glucose disposal corrected for fat‐free mass and urinary glucose loss (MFFM) (3.49 ± 2 vs. 5.52 ± 2.9 mg/min × kg FFM; P < .0001), whereas treatment with glimepiride was associated with a decrease in MFFM (5.5 ± 3.4 vs. 3.97 ± 2 mg/min × kg FFM; P = .003) (adjusted difference 1.53 [0.73, 2.33] mg/min × kg FFM; P < .0001) (Table 2). This difference remained significant after adjustment for changes in BMI (P < .0001) (Table 2).
No differences in changes in plasma glucose and insulin levels during the clamp study were observed between the two treatment arms (Table 2).
Furthermore, treatment with empagliflozin led to a significant reduction in MVO2 measured during the euglycaemic‐hyperinsulinaemic clamp compared with glimepiride (P < .0001) (Table 2).
3.2.3. Changes in cardiac geometry, myocardial mechano‐energetic efficiency and cardiac function
Compared with glimepiride, patients randomized to empagliflozin exhibited a significant reduction from baseline to 26 weeks in left atrial diameter (P < .0001), LVESV (P < .0001), LVEDV (P = .02), IVS (P = 0.01) and s‐PAP (P < .0001) (Table 4). Treatment with empagliflozin was associated with an increase in LVEF that fell just short of statistical significance (P = .06). However, because treatment with glimepiride was associated with a decrease in LVEF, the 2.36% adjusted difference between the two treatments was statistically significant (P = .01) (Table 4). Accordingly, treatment with empagliflozin was associated with a significant reduction in NT‐proBNP levels compared with treatment with glimepiride (P = .001). No differences between the two groups were observed in changes in diastolic function and LVMI (Table 4). The differences in LVEF (P = .01), LVESV (P = .006), left atrial diameter (P = .001), s‐PAP (P = .009) and NT‐proBNP (P = .01) remained significant after adjustment for changes in SBP and DBP (Table 4). Moreover, treatment with empagliflozin led to a significant reduction in SW (P = .03) and MVO2 (P < .0001) and a significant increase in myocardial MEEi (P = .003) compared with glimepiride (Table 4).
TABLE 4.
Changes in echocardiographic and energetic efficiency variables after exposure to empagliflozin or glimepiride for 26 weeks
| Empagliflozin n = 23 | Glimepiride n = 23 | Adjusted difference between groups (95% CI) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Before treatment | After 26 weeks of treatment | Treatment difference | P value | Before treatment | After 26 weeks of treatment | Treatment difference | P value | P value | P value a | |||
| LVEF (%) | 60.9 ± 3 | 62.5 ± 3 | 1.56 ± 3.8 | .06 | 62 ± 3 | 60.3 ± 3.7 | −1.6 ± 3.8 | .051 | 2.36 | (0.49, 4.24) | .01 | .01 |
| LVESV (ml) | 44.2 ± 14 | 39.08 ± 13 | −5.21 ± 7.8 | .004 | 40.5 ± 12 | 45.03 ± 14 | 4.51 ± 8.09 | .01 | −4.98 | (−8.32 −1.64) | <.0001 | .006 |
| LVEDV (ml) | 117.8 ± 27 | 111.9 ± 25 | −5.89 ± 12.8 | .03 | 114.3 ± 23 | 117.6 ± 27 | 3.27 ± 12.6 | .2 | −5.74 | (−11.12, −0.37) | .02 | .09 |
| Left atrial diameter (cm) | 3.9 ± 0.3 | 3.65 ± 0.3 | −0.25 ± 0.33 | .001 | 3.66 ± 0.3 | 3.9 ± 0.3 | 0.24 ± 0.32 | .002 | −0.19 | (−0.31, −0.07) | <.0001 | .001 |
| LVMI (g/m2) | 86 ± 22 | 83.9 ± 19 | −2.64 ± 14 | .3 | 86.4 ± 21 | 87.1 ± 20 | 0.69 ± 9.73 | .7 | −3.45 | (−10.13, 3.23) | .3 | .5 |
| IVS (cm) | 1.11 ± 0.13 | 1.08 ± 0.12 | −0.03 ± 0.09 | .9 | 1.10 ± 0.12 | 1.13 ± 0.12 | 0.02 ± 0.06 | .09 | −0.05 | (−0.10, −0.01) | .01 | .2 |
| Early to atrial filling velocity ratio | 0.83 ± 0.22 | 0.81 ± 0.17 | −0.07 ± 0.33 | .6 | 0.81 ± 0.17 | 0.81 ± 0.18 | −0.002 ± 0.1 | .9 | −0.059 | (−0.14, 0.025) | .4 | .9 |
| s‐PAP (mm/Hg) | 29.6 ± 5.6 | 22.9 ± 6 | −6.69 ± 7.8 | .001 | 24.8 ± 6.4 | 27.4 ± 5.3 | 2.60 ± 3.88 | .004 | −6.58 | (−9.82, −3.17) | <.0001 | .009 |
| MVO2 (mmHg*bpm) | 9960 ± 2094 | 8452 ± 1419 | −1508 ± 1792 | .001 | 8985 ± 1872 | 9938 ± 2140 | 952 ± 664 | <.0001 | −2155 | (−2899, −1411) | <.0001 | – |
| Stroke work (mmHg*ml) | 9496 ± 2237 | 8736 ± 2074 | −760 ± 1447 | .02 | 9124 ± 2255 | 9323 ± 2365 | 199 ± 1441 | .5 | −720 | (−1300, −140) | .03 | – |
| Myocardial MEEi (ml/s*g−1) | 0.34 ± 0.08 | 0.37 ± 0.07 | 0.02 ± 0.06 | .04 | 0.35 ± 0.07 | 0.33 ± 0.08 | −0.02 ± 0.04 | .02 | 0.027 | (0.005, 0.048) | .003 | – |
| NT‐ProBNP (pg/ml) | 38.2 ± 42 | 29.5 ± 39 | −8.7 ± 14 | .009 | 30.7 ± 32 | 37.9 ± 40 | 7.2 ± 14 | .02 | −15.87 | (−24.67, −7.07) | .001 | .01 |
Note: Data are means ± SD, unless otherwise indicated, and were analysed using ANCOVA adjusting for baseline values.
Abbreviations: DBP, diastolic blood pressure; IVS, interventricular septal thickness; LVEDV, left ventricular end‐diastolic volume; LVEF, left ventricular ejection fraction; LVESV, left ventricular end‐systolic volume; LVMI, left ventricular mass index; MEEi, indexed myocardial mechano‐energetic efficiency; MVO2, myocardial oxygen consumption; NT‐ProBNP, N‐terminal pro b‐type natriuretic peptide; SBP, systolic blood pressure; s‐PAP, systolic pulmonary arterial pressure.
Adjusted for changes in SBP and DBP.
Univariate analysis showed that changes in myocardial MrGlu were directly correlated with changes in LVESV (r = 0.545; P < .0001), LVEDV (r = 0.462; P = .001) and MVO2, both under fasting state (r = 0.302; P = .04) and during the clamp study (r = 0.305; P = .03) and were inversely correlated with changes in LVEF (r = −0.399; P = .006), while changes in fasting β‐hydroxybutyrate levels were inversely correlated with changes in MVO2 (r = −0.619; P < .0001) and SW (r = −0.308; P = .03) and directly correlated with changes in myocardial MEEi (r = 0.422; P = .003).
4. DISCUSSION
4.1. Myocardial energetic metabolism
The present study was designed to explore the cardioprotective mechanism induced by empagliflozin. We found that 26 weeks of treatment with empagliflozin reduced the myocardial MrGlu by 45.1%. This effect appears specific for SGLT2 inhibition because treatment with glimepiride increased myocardial MrGlu by 33.9%, despite no differences in metabolic control being observed between the two treatments. The differences between the two treatments remained significant even after adjusting for changes in BMI, thus confirming that the reduction in myocardial MrGlu induced by empagliflozin occurred regardless of weight loss. The present findings are in agreement with those of a prior study showing that 4 weeks of treatment with empagliflozin reduced myocardial glucose uptake by 57% under fasting postabsorptive state, 11 but not with those of a study investigating the effect of 8 weeks of dapagliflozin treatment on insulin‐stimulated glucose uptake in myocardium, as measured by PET and 18F‐FDG during hyperinsulinaemic‐euglycaemic clamp in patients with type 2 diabetes. 31 Some differences in clinical characteristics might plausibly explain the discrepancies between the present results and those reported by Latva‐Rasku et al. In the latter study, the participants were older, had longer diabetes duration, were predominantly men (86%), and were treated with dapagliflozin for a shorter period of time (8 weeks) compared with the present study. Moreover, in the dapagliflozin group, nine out of 15 patients were treated with metformin and sitagliptin, and three subjects had coronary artery disease.
It has been hypothesized that treatment with SGLT2is by increasing concentrations of circulating ketone bodies might promote a shift in cardiac substrate utilization from glucose towards ketone bodies requiring less oxygen to generate ATP compared with glucose. 10 As observed in most studies with SGLT2is, 11 , 12 we found that treatment with empagliflozin resulted in a significant increase in fasting ketone bodies compared with glimepiride. Regrettably, we did not measure FFA and β‐hydroxybutyrate levels during the clamp study. Nevertheless, as shown by Ferrannini et al., SGLT2i treatment is associated with a 3‐fold increase in circulating postprandial β‐hydroxybutyrate levels, and a significant increase in circulating postprandial FFA concentrations. 32 The present and prior observations showing that treatment with SGLT2is is associated with a reduction in myocardial glucose uptake, 11 coupled with those of two previous studies showing that SGLT2 inhibition did not affect myocardial FFA uptake, 11 , 12 support the hypothesis that SGLT2is may improve myocardial energetics by using an alternative cardiac fuel substrate such as the ketone bodies.
Treatment with glimepiride was associated with worsening of peripheral insulin sensitivity without any changes in β‐hydroxybutyrate levels. Thus it is probable that glimepiride treatment may exacerbate the cardiac metabolic inflexibility, impairing the cardiac ability to switch from FFAs and glucose as a fuel source towards an energy‐efficient super fuel like ketone bodies, which improves myocardial work efficiency and function. 33
4.2. Insulin‐stimulated glucose disposal
We found that empagliflozin treatment increased insulin‐stimulated whole‐body glucose disposal by 58%, a greater increase than that reported in previous studies with dapagliflozin. 34 , 35 , 36 , 37 A possible explanation for this discrepancy may be the longer duration of treatment in the present study compared with the shorter duration of treatment with dapagliflozin observed in the earlier studies (2‐12 weeks). 34 , 35 , 36 , 37
We observed that glimepiride treatment reduces insulin‐stimulated whole‐body glucose disposal. Very few studies have assessed the effects of sulphonylureas with the gold standard euglycaemic‐hyperinsulinaemic clamp technique. In a previous study of 11 obese subjects with type 2 diabetes, it was shown that treatment with glimepiride for 4 months was associated with minimal changes in insulin sensitivity, as assessed by euglycaemic‐hyperinsulinaemic clamp study (4.6 ± 0.7 vs. 4.3 ± 0.7 μmol × kg−1 × min−1 × pmol−1, respectively). 38 The small sample studied and the shorter duration of treatment with glimepiride may explain the different results observed between the present and the prior study. However, another study assessing the effect of glimepiride treatment for 14 weeks on a patient with type A insulin resistance syndrome resulting from a heterozygous missense mutation in the insulin receptor gene, showed a significant reduction in insulin sensitivity, as assessed by euglycaemic‐hyperinsulinaemic clamp (5.25 vs. 2.90 mg/kg/min, respectively). 39 In both studies, treatment with glimepiride was associated with a significant increase in plasma insulin levels. It is conceivable that glimepiride‐induced hyperinsulinaemia may affect insulin sensitivity by downregulating insulin receptor expression and its downstream signalling molecules, mediating its metabolic effects. 40
4.3. Cardiac geometry and systolic function
As secondary endpoints, we found that empagliflozin significantly reduced left atrial diameter (−0.19 cm), LVESV (−4.98 ml) and LVEDV (−5.74 ml) compared with glimepiride, thus suggesting a reduction in cardiac preload and afterload. Strikingly, these favourable echocardiographic modifications were associated with a significant improvement in LVEF (2.36%), with a consequent reduction in circulating NT‐proBNP levels and s‐PAP. These differences between the two treatments remained significant, even after adjusting for changes in SBP and DBP, thus arguing against the possibility that a reduction in blood pressure has a major role in changes in cardiac geometry and function. Our results are in agreement with those of a recent meta‐analysis that evaluated the effects of treatment with SGLT2is on LV volumes and function assessed by cardiac magnetic resonance imaging or transthoracic echocardiography. 41 This meta‐analysis showed that SGLT2i treatment was significantly associated with a reduction in LVESV (−8.44 ml) and LVEDV (−9.13 ml), and an increase in LVEF (2.45%), compared with placebo or other antidiabetic agents. 41 Comparing the results of the present study with those of prior randomized clinical trials that have assessed LV volumes and function by transthoracic echocardiography, we found that our results on LV volumes are in agreement with those of some, 42 , 43 but not with other studies. 44 , 45 , 46 Additionally, we found that empagliflozin treatment was associated with a numerical but not significant reduction in LVMI. Two prior studies examining the effects of SGLT2is on LV mass assessed by transthoracic echocardiography in patients with type 2 diabetes have shown a significant reduction in LVMI, 42 , 45 whereas another study failed to do so. 44 It is probable that these divergent results are a result of differences in the clinical features of the recruited patients (subjects without a history of coronary heart disease or HF vs. subjects with established CV disease), duration of type 2 diabetes and duration of treatment with SGLT2is between the studies. Further randomized clinical trials of adequate duration carried out in larger cohorts of patients with type 2 diabetes, and including patients with HF, are required to confirm the observed changes in cardiac geometry and function.
4.4. Myocardial efficiency
Additionally, we found that empagliflozin significantly reduced blood pressure and resting HR compared with glimepiride. These changes might be contributing to improvements in LV myocardial MEE. LV myocardial MEE can be calculated as the ratio between cardiac workload and oxygen consumption, and a decrease in LV myocardial MEE reflects a state in which oxygen consumption increases more than cardiac work, 47 as observed in patients with HF and early diabetic cardiomyopathy. 48 , 49 We observed that empagliflozin significantly reduced both cardiac workload, estimated by SBP × echocardiographic SV, and oxygen consumption, estimated using the ‘double product’ of SBP × HR, compared with glimepiride, resulting in a significant improvement in LV myocardial MEE. Several previous studies have validated the double product as an index of MVO2 using direct assessment of MVO2 by employing an indicator and catheterization of the coronary sinus, showing a high correlation between MVO2 and the double product (r = 0.86‐0.88). 50 , 51 , 52 Two previous studies assessing cardiac oxygen consumption using PET methods led to results that are different from our findings, the first showing no change in MVO2 in 13 individuals with type 2 diabetes treated for 4 weeks with empagliflozin, 11 and the other a reduction (−0.30 [−0.49, −0.12] J/g/min) in 25 subjects with type 2 diabetes treated for 4 weeks with dapagliflozin, although these changes were not statistically significant compared with placebo. 12 The use of indirect measures of oxygen consumption, the larger sample studied and the longer duration of treatment with SGLT2is may explain the different results observed between the present study and those reported by Lauritsen et al. 11 and Oldgren et al. 12
It is important to note that the evaluation of myocardial energy efficiency was not performed during the euglycaemic‐hyperinsulinaemic clamp study. It is important to note that evaluation of myocardial energy efficiency were not performed during the euglycaemiac‐hyperinsulinaemic clamp study, but under fasting conditions, unlike the MVO2 that we measured both during fasting and during the clamp study. However, the effect of physiological hyperinsulinaemia on myocardial energetics in subjects treated with SGLT2is remains to be determined.
Finally, as expected from prior CVOT studies, 1 , 2 , 3 we found that empagliflozin significantly increased red blood cells, haematocrit and haemoglobin levels compared with glimepiride. A mediation analysis from the EMPA‐REG OUTCOME trial has suggested that increases in haematocrit and haemoglobin accounted for approximately 50% of the observed clinical benefit of the empagliflozin treatment. 53 Thus, a higher haematocrit induced by empagliflozin treatment might contribute to deliver more oxygen to tissues, improving the myocardial oxygen demand.
4.5. Strengths and limitations
The FIORE trial has some strengths and limitations that merit consideration. One main strength of the present study is the use of gold standard methods to assess myocardial and whole‐body metabolism by cardiac 18F‐FDG‐PET combined with the euglycaemic‐hyperinsulinaemic clamp technique, which allows evaluation of myocardial glucose uptake under uniform experimental conditions of euglycaemia and physiological hyperinsulinaemia, thus removing the confounding factor of different circulating glucose and insulin levels. As a matter of fact, there is significant interindividual variability in plasma glucose and insulin levels under fasting conditions, causing heterogeneity in myocardial MrGlu, and assessment of myocardial glucose uptake during an oral glucose load does not produce a metabolic steady‐state, resulting in variable and unstable metabolic conditions. 54 , 55 , 56 , 57 Other strengths include the recruitment of patients by a single study centre, which minimized variability in the imaging results, the crossover design that allowed each subject to act as their own control, and the active‐controlled study design that allowed examining the effects of empagliflozin on primary and secondary endpoints without the confounding effect of differences in glucose control between treatment groups.
The present study also has some limitations. First, because of the design of the study, the overall sample size was small and the follow‐up was brief. However, the effects of empagliflozin on myocardial MrGlu and cardiac geometry and function in the current study were observed early, over a 6‐month treatment period, consistent with the early separation of the Kaplan–Meier curves for CV death and HHF observed in the EMPA‐REG OUTCOME trial. 1 Furthermore, our planned sample size of 23 subjects allows for adequate statistical power, and going beyond this number would have resulted in additional subjects being exposed to unnecessary amounts of radiation. Second, because we recruited participants with comparatively early type 2 diabetes in monotherapy treatment with metformin to investigate the cardiac effects of SGLT2i treatment as second‐line therapy, the results may not necessarily be applicable to patients with more advanced disease. Third, we restricted our inclusion criteria to patients with type 2 diabetes without a history of coronary artery disease or HF, and, therefore, the current results could have been different if patients with established CV disease, especially HF, had been included. Fourth, we only recruited Caucasian subjects living in a homogenous geographical region in the south of Italy, thus it remains to be addressed whether the present results can be extended to other ethnic groups. Fifth, ketone bodies were measured using a blood ketone testing method that, although it is not the most accurate method, is recommended for its ability to quantify β‐hydroxybutyric acid. 58 Sixth, we did not measure FFA and β‐hydroxybutyrate levels during the clamp study, and therefore the effects of ketone bodies on insulin‐stimulated myocardial MrGlu in subjects treated with SGLT2is remain to be determined. Furthermore, we did not measure endogenous glucose production during the clamp study. Finally, LV oxygen consumption and MEEi were estimated by indirect validate measures rather than by cardiac PET or magnetic resonance spectroscopy.
In conclusion, the present study provides evidence that empagliflozin treatment in subjects with type 2 diabetes without coronary artery disease leads to a significant reduction in the insulin‐stimulated myocardial MrGlu. Furthermore, empagliflozin treatment improves cardiac geometry, myocardial mechano‐energetic efficiency and cardiac function.
AUTHOR CONTRIBUTIONS
ES designed the study, researched and analysed data, wrote and edited the manuscript; PV analysed the data from the cardiac PET scans; AP and FC performed cardiac PET scans; SM performed the echocardiographic examinations; TVF, AS and FA researched data and reviewed the manuscript; PV and GLC contributed to study design and to the discussion and reviewed the manuscript; GS designed the study, analysed data, wrote and reviewed and edited the manuscript. GS is the guarantor of the study, conceived the study and takes full responsibility for the work. All the authors read and approved the final manuscript.
CONFLICT OF INTEREST
No potential conflicts of interest relevant to this article were reported.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/dom.14816.
Supporting information
Appendix S1. Changes in myocardial glucose metabolic rate, insulin sensitivity and clinical and metabolic parameters after treatment for 26 weeks after first period.
Figure S1: Study design.
Figure S2: Flowchart of the study participants.
ACKNOWLEDGMENT
Open Access Funding provided by Universita degli Studi Magna Graecia di Catanzaro within the CRUI‐CARE Agreement.
Succurro E, Vizza P, Papa A, et al. Effects of 26 weeks of treatment with empagliflozin versus glimepiride on the myocardial glucose metabolic rate in patients with type 2 diabetes: The randomized, open‐label, crossover, active‐comparator FIORE trial. Diabetes Obes Metab. 2022;24(12):2319‐2330. doi: 10.1111/dom.14816
Funding informationThis research received no external funding.
DATA AVAILABILITY STATEMENT
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117‐2128. [DOI] [PubMed] [Google Scholar]
- 2. Wiviott SD, Raz I, Bonaca MP, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347‐357. [DOI] [PubMed] [Google Scholar]
- 3. Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644‐657. [DOI] [PubMed] [Google Scholar]
- 4. Zelniker TA, Wiviott SD, Raz I, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta‐analysis of cardiovascular outcome trials. Lancet. 2019;393:31‐39. [DOI] [PubMed] [Google Scholar]
- 5. Cannon CP, Pratley R, Dagogo‐Jack S, et al. Cardiovascular outcomes with ertugliflozin in type 2 diabetes. N Engl J Med. 2020;383:1425‐1435. [DOI] [PubMed] [Google Scholar]
- 6. McMurray JJV, Solomon SD, Inzucchi SE, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381:1995‐2008. [DOI] [PubMed] [Google Scholar]
- 7. Packer M, Anker SD, Butler J, et al. Cardiac and renal outcomes with empagliflozin in heart failure. N Engl J Med. 2020;383:1413‐1424. [DOI] [PubMed] [Google Scholar]
- 8. Anker SD, Butler J, Filippatos G, et al. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021;385:1451‐1461. [DOI] [PubMed] [Google Scholar]
- 9. Lopaschuk GD, Verma S. Mechanisms of cardiovascular benefits of sodium glucose co‐transporter 2 (SGLT2) inhibitors: a state‐of‐the‐art review. JACC Basic Transl Sci. 2020;5:632‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA‐REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care. 2016;39:1108‐1114. [DOI] [PubMed] [Google Scholar]
- 11. Lauritsen KM, Nielsen BRR, Tolbod LP, et al. SGLT2 inhibition does not affect myocardial fatty acid oxidation or uptake, but reduces myocardial glucose uptake and blood flow in individuals with type 2 diabetes: a randomized double‐blind, placebo‐controlled crossover trial. Diabetes. 2021;70:800‐808. [DOI] [PubMed] [Google Scholar]
- 12. Oldgren J, Laurila S, Åkerblom A, et al. Effects of 6 weeks of treatment with dapagliflozin, a sodium‐ glucose co‐transporter‐2 inhibitor, on myocardial function and metabolism in patients with type 2 diabetes: a randomized, placebo‐controlled, exploratory study. Diabetes Obes Metab. 2021;23:1505‐1517. [DOI] [PubMed] [Google Scholar]
- 13. Salerno A, Fragasso G, Esposito A, et al. Effects of short‐term manipulation of serum FFA concentrations on left ventricular energy metabolism and function in patients with heart failure: no association with circulating bio‐markers of inflammation. Acta Diabetol. 2015;52:753‐761. [DOI] [PubMed] [Google Scholar]
- 14. Rosenstock J, Kahn SE, Johansen OE, et al. Effect of linagliptin vs glimepiride on major adverse cardiovascular outcomes in patients with type 2 diabetes the CAROLINA randomized clinical trial. JAMA. 2019;322:1155‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Succurro E, Pedace E, Andreozzi F, et al. Reduction in global myocardial glucose metabolism in subjects with 1‐h postload hyperglycemia and impaired glucose tolerance. Diabetes Care. 2020;43:669‐676. [DOI] [PubMed] [Google Scholar]
- 16. DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214‐E223. [DOI] [PubMed] [Google Scholar]
- 17. Carson RE. Tracer kinetic modeling in PET. In: Bailey DL, Townsend DW, Valk PE, and Maisey MN, (eds.), Positron Emission Tomography. London, UK: Springer; 2005:127‐159. [Google Scholar]
- 18. Vizza P, Guzzi PH, Veltri P, et al. Experiences on quantitative cardiac pet analysis. 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). Shenzen, China; 2016:1148‐1153.
- 19. Lang RM, Badano LP, Mor‐Avi V, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2015;28:1‐39.e14. [DOI] [PubMed] [Google Scholar]
- 20. Devereux RB, Alonso DR, Lutas EM. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings. Am J Cardiol. 1986;57:450‐458. [DOI] [PubMed] [Google Scholar]
- 21. de Simone G, Kizer JR, Chinali M, et al. Normalization for body size and population‐attributable risk of left ventricular hypertrophy: the Strong Heart Study. Am J Hypertens. 2005;18:191‐196. [DOI] [PubMed] [Google Scholar]
- 22. Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr. 2010;23:685‐713. [DOI] [PubMed] [Google Scholar]
- 23. Losi MA, Izzo R, Mancusi C, et al. Depressed myocardial energetic efficiency increases risk of incident heart failure: the Strong Heart Study. J Clin Med. 2019;8:1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Simone G, Izzo R, Losi MA, et al. Depressed myocardial energetic efficiency is associated with increased cardiovascular risk in hypertensive left ventricular hypertrophy. J Hypertens. 2016;34:1846‐1853. [DOI] [PubMed] [Google Scholar]
- 25. de Simone G, Chinali M, Galderisi M, et al. Myocardial mechano‐energetic efficiency in hypertensive adults. J Hypertens. 2009;27:650‐655. [DOI] [PubMed] [Google Scholar]
- 26. Mancusi C, de Simone G, Best LG, et al. Myocardial mechano‐energetic efficiency and insulin resistance in non‐diabetic members of the strong heart study cohort. Cardiovasc Diabetol. 2019;18:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fiorentino TV, Miceli S, Succurro E, Sciacqua A, Andreozzi F, Sesti G. Nonalcoholic fatty liver disease is associated with a decreased myocardial mechano‐energetic efficiency. J Intern Med. 2020;289:221‐231. [DOI] [PubMed] [Google Scholar]
- 28. Succurro E, Miceli S, Fiorentino TV, et al. Sex‐specific differences in left ventricular mass and myocardial energetic efficiency in non‐diabetic, pre‐diabetic and newly diagnosed type 2 diabetic subjects. Cardiovasc Diabetol. 2021;20:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Inoue R, Ohkubo T, Kikuya M, et al. Predictive value for mortality of the double product at rest obtained by home blood pressure measurement: the Ohasama study. Am J Hypertens. 2012;25:568‐575. [DOI] [PubMed] [Google Scholar]
- 30. Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604‐612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Latva‐Rasku A, Honka MJ, Kullberg J, et al. The SGLT2 inhibitor dapagliflozin reduces liver fat but does not affect tissue insulin sensitivity: a randomized, double‐blind, placebo‐controlled study with 8‐week treatment in type 2 diabetes patients. Diabetes Care. 2019;42:931‐937. [DOI] [PubMed] [Google Scholar]
- 32. Ferrannini E, Baldi S, Frascerra S, et al. Shift to fatty substrate utilization in response to sodium‐glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes. 2016;65:1190‐1195. [DOI] [PubMed] [Google Scholar]
- 33. Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA‐REG OUTCOME study? A unifying hypothesis. Diabetes Care. 2016;39:1115‐1122. [DOI] [PubMed] [Google Scholar]
- 34. Daniele G, Xiong J, Solis‐Herrera C, et al. Dapagliflozin enhances fat oxidation and ketone production in patients with type 2 diabetes. Diabetes Care. 2016;39:2036‐2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merovci A, Solis‐Herrera C, Daniele G, et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J Clin Invest. 2014;124:509‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Merovci A, Abdul‐Ghani M, Mari A, et al. Effect of dapagliflozin with and without Acipimox on insulin sensitivity and insulin secretion in T2DM males. J Clin Endocrinol Metab. 2016;101:1249‐1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mudaliar S, Henry RR, Boden G, et al. Changes in insulin sensitivity and insulin secretion with the sodium glucose cotransporter 2 inhibitor dapagliflozin. Diabetes Technol Ther. 2014;16:137‐144. [DOI] [PubMed] [Google Scholar]
- 38. Korytkowski M, Thomas A, Reid L, Tedesco MB, Gooding WE, Gerich J. Glimepiride improves both first and second phases of insulin secretion in type 2 diabetes. Diabetes Care. 2002;25:1607‐1611. [DOI] [PubMed] [Google Scholar]
- 39. Huang Z, Liu J, Ng K, et al. Glimepiride treatment in a patient with type A insulin resistance syndrome due to a novel heterozygous missense mutation in the insulin receptor gene. J Diabetes Invest. 2018;9:1075‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sesti G. Pathophysiology of insulin resistance. Best Pract Res Clin Endocrinol Metab. 2006;20:665‐679. [DOI] [PubMed] [Google Scholar]
- 41. Shi FH, Li H, Shen L, et al. Beneficial effect of sodium‐glucose co‐transporter 2 inhibitors on left ventricular function. J Clin Endocrinol Metab. 2022;107:1191‐1203. [DOI] [PubMed] [Google Scholar]
- 42. Omar M, Jensen J, Ali M, et al. Associations of empagliflozin with left ventricular volumes, mass, and function in patients with heart failure and reduced ejection fraction: a substudy of the empire HF randomized clinical trial. JAMA Cardiol. 2021;6:836‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Thirunavukarasu S, Jex N, Chowdhary A, et al. Empagliflozin treatment is associated with improvements in cardiac energetics and function and reductions in myocardial cellular volume in patients with type 2 diabetes. Diabetes. 2021;70:2810‐2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rau M, Thiele K, Hartmann NK, et al. Empagliflozin does not change cardiac index nor systemic vascular resistance but rapidly improves left ventricular filling pressure in patients with type 2 diabetes: a randomized controlled study. Cardiovasc Diabetol. 2021;20:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ersbøll M, Jürgens M, Hasbak P, et al. Effect of empagliflozin on myocardial structure and function in patients with type 2 diabetes at high cardiovascular risk: the SIMPLE randomized clinical trial. Int J Cardiovasc Imaging. 2022;38:579‐587. [DOI] [PubMed] [Google Scholar]
- 46. Verma S, Mazer CD, Yan AT, et al. Effect of empagliflozin on left ventricular mass in patients with type 2 diabetes and coronary artery disease: the EMPA‐HEART CardioLink‐6 randomized clinical trial. Circulation. 2019;140:1693‐1702. [DOI] [PubMed] [Google Scholar]
- 47. Hansson NH, Tolbod L, Harms HJ, et al. Evaluation of ECG‐gated [11C] acetate PET for measuring left ventricular volumes, mass, and myocardial external efficiency [erratum published in J Nucl Cardiol 2016;23:1232]. J Nucl Cardiol. 2016;23:670‐679. [DOI] [PubMed] [Google Scholar]
- 48. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356:1140‐1151. [DOI] [PubMed] [Google Scholar]
- 49. Honka H, Solis‐Herrera C, Triplitt C, Norton L, Butler J, DeFronzo RA. Therapeutic manipulation of myocardial metabolism: JACC state‐of‐the‐art review. J Am Coll Cardiol. 2021;77:2022‐2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vanoverschelde JL, Wijns W, Essamri B, et al. Hemodynamic and mechanical determinants of myocardial O2 consumption in normal human heart: effects of dobutamine. Am J Physiol. 1993;265:H1884‐H1892. [DOI] [PubMed] [Google Scholar]
- 51. Gobel FL, Nordstrom LA, Nelson RR, Jorgensen CR, Wang Y. The rate‐pressure product as an index of myocardial oxygen consumption during exercise in patients with angina pectoris. Circulation. 1978;57:549‐556. [DOI] [PubMed] [Google Scholar]
- 52. Nelson RR, Gobel FL, Jorgensen CR, Wang K, Taylor HL. Hemodynamic predictors of myocardial oxygen consumption during static and dynamic exercise. Circulation. 1974;50:1179‐1189. [DOI] [PubMed] [Google Scholar]
- 53. Inzucchi SE, Zinman B, Fitchett D, et al. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA‐REG OUTCOME trial. Diabetes Care. 2018;41:356‐363. [DOI] [PubMed] [Google Scholar]
- 54. Camici PG, Prasad SK, Rimoldi OE. Stunning, hibernation, and assessment of myocardial viability. Circulation. 2008;117:103‐114. [DOI] [PubMed] [Google Scholar]
- 55. Iozzo P, Chareonthaitawee P, Dutka D, Betteridge DJ, Ferrannini E, Camici PG. Independent association of type 2 diabetes and coronary artery disease with myocardial insulin resistance. Diabetes. 2002;51:3020‐3024. [DOI] [PubMed] [Google Scholar]
- 56. Bax JJ, Visser FC, Poldermans D, et al. Feasibility, safety and image quality of cardiac FDG studies during hyperinsulinaemic‐euglycaemic clamping. Eur J Nucl Med Mol. 2002;29:452‐457. [DOI] [PubMed] [Google Scholar]
- 57. Byrne CD, Perseghin G. Non‐alcoholic fatty liver disease: a risk factor for myocardial dysfunction? J Hepatol. 2018;68:640‐642. [DOI] [PubMed] [Google Scholar]
- 58. Goldstein DE, Little RR, Lorenz RA, et al. Tests of glycemia in diabetes. Diabetes Care. 2004;27:S91‐S93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Changes in myocardial glucose metabolic rate, insulin sensitivity and clinical and metabolic parameters after treatment for 26 weeks after first period.
Figure S1: Study design.
Figure S2: Flowchart of the study participants.
Data Availability Statement
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.