

Abstract
The PET tracer [18F]F‐AraG, an arabinosyl guanine analog, has shown promise for visualizing activated T cells in multiple diseases. Herein, a practitioner's protocol is described, in which the PET tracer is prepared using minimal equipment and manual actions, making it widely accessible for preclinical applications.
Keywords: [18F]F‐AraG; AraG; nucleophilic fluorination; nucleophilic substitution; practitioner's protocol; SPE purification, T cell imaging
1. INTRODUCTION
F‐AraG is a fluorinated arabinosyl guanine analog of the drug nelarabine, which exhibits selective T‐lymphocyte and T‐lymphoblastoid cytotoxicity. The compound was first radiolabeled with fluorine‐18 in 2011 to enable in vivo imaging of activated T cells using positron emission tomography (PET). 1 Since then, [18F]F‐AraG has been used in both preclinical and clinical imaging research in multiple diseases, including rheumatoid arthritis, graft‐versus‐host disease, multiple sclerosis, and cancer. 2 , 3 , 4 , 5
Despite the many described applications of [18F]F‐AraG in literature, to the best of our knowledge, its synthesis has not been revisited since it was first reported. In the original procedure reported by Namavari et al., 1 [18F]fluoride was eluted from a quarternary methylammonium (QMA) cartridge with potassium carbonate/kryptofix in an aqueous solution and dried under vacuum at 65°C. The dry [18F]fluoride was then reacted with a protected triflate precursor to produce an 18F‐labeled AraG intermediate, which was isolated using high‐performance liquid chromatography (HPLC). Following two consecutive deprotection reactions, the crude product was purified by a second HPLC and formulated, yielding [18F]F‐AraG in 7–10% radiochemical yield (decay corrected), 30‐48 GBq/μmol molar activity, >95% radiochemical purity in a total synthesis time of 140–160 min. 1
Though robust, the above‐described method includes two HPLC purification steps and vacuum evaporation in multiple steps of the synthesis, making the synthesis procedure impractical or even impossible to implement in some synthesis modules. We therefore set out to develop a new, simplified method for the preparation [18F]F‐AraG that would be accessible to the wider radiochemistry community.
2. PROCEDURE
2.1. [18F]fluoride production, trapping and elution
[18F]Fluoride was produced in a PETtrace 800 (GE Healthcare, Uppsala, Sweden) cyclotron by irradiating 1.5 ml of [18O]water (Rotem Industries Ltd, Beer Sheva, Israel) at 35 μA for 15 min. The [18F]fluoride processing and synthesis were completed in a custom‐built synthesis module (DM Automation, Nykvarn, Sweden). The [18F]fluoride was separated from the [18O]water using an anion exchange Oasis® WAX 1cc 30 mg cartridge (Waters Corporation, Milford, Massachusetts USA) conditioned with sodium bicarbonate solution (aq. 0.5 M, 3 ml) and subsequently Milli‐Q water (3 ml). The [18F]fluoride was released from the SPE cartridge by elution with tetraethylammonium bicarbonate solution (0.01 M in 97:3 acetonitrile/Milli‐Q water, 1 ml). The eluted [18F]tetraethylammonium fluoride was azeotropically dried by heating the vial to 110°C under an increasing flow of nitrogen (0–100 ml/min) during 10 min before the addition of 400 μl of acetonitrile and repetition of the drying procedure (Figure 1, described in blue).
FIGURE 1.
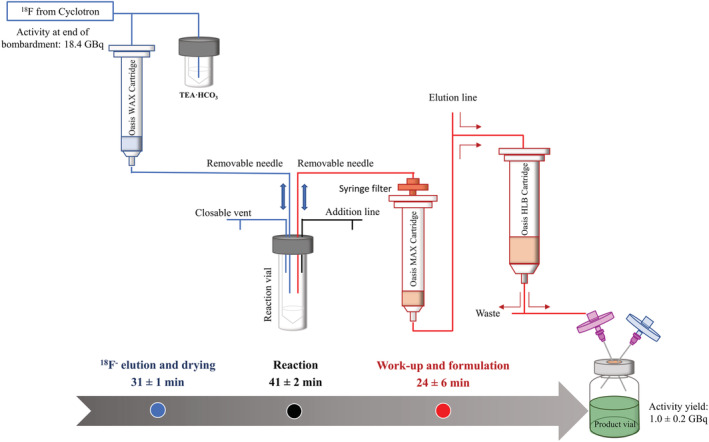
[18F]F‐AraG production protocol, where blue describes the [18F]Fluoride production, trapping, and elution; black the one‐pot reaction; and red the work‐up and formulation of the product
2.2. The one‐pot reaction
2‐N‐Acetyl‐6‐O‐([4‐nitrophenyl]ethyl)‐9‐(3,5‐di‐O‐trityl‐2‐trifyl‐β‐d‐ribofuranosyl)guanine (6.0 ± 0.1 mg, prepared as previously described 1 ) was dissolved in 500 μl of anhydrous dimethyl sulfoxide (Sigma‐Aldrich, Merck, Burlington, MA, USA) and added by syringe to the reaction vial containing the [18F]tetraethylammonium fluoride. The reaction vial was heated at 85°C for 10 min, after which it was cooled to 40°C. Sodium hydroxide (aq. 0.5 M, 0.5 ml) was added through the installed addition line, and the reaction was heated at 120°C for 10 min. The reaction mixture was subsequently cooled to 40°C before adding hydrochloric acid (aq. 1 N, 0.5 ml) through the addition line (Figure 1, described in black). The reaction mixture was heated at 120°C for 10 min before cooling to 40°C prior to purification.
2.3. Work‐up and formulation
It was observed that the pH is critical for product retention on the SPE. Therefore, the reaction mixture was neutralized with sodium bicarbonate solution (aq. 0.05 M, 10 ml). The reaction mixture was passed through a series of filters and cartridges, consisting of an Acrodisc® 13 mm syringe filter with 0.2 μm nylon membrane (Pall, New York, USA), an Oasis®MAX 1 cc 10 mg cartridge (Waters Corporation, Milford, Massachusetts USA, conditioned with 3 ml sodium bicarbonate solution [aq. 0.5 M] and 3 ml Milli‐Q water) and lastly an OASIS®HLB 6cc 500 mg cartridge (Figure 1, described in red). By this arrangement, any undissolved particles in the neutralized reaction mixture were caught on the syringe filter; radioactive apolar impurities were trapped on the MAX cartridge (Figure S1), the product was trapped on the HLB cartridge, and the free [18F]fluoride was collected in a waste bottle behind lead shielding. The valve between the MAX and HLB cartridges was thereafter redirected, enabling the washing of the HLB cartridge with Milli‐Q water (20 ml), phosphoric acid buffer (0.05 M, pH 5.75, 20 ml), and Milli‐Q water (20 ml, see Figure S2 for an example of the washing eluate). The product was eluted from the HLB cartridge using 50% ethanol/saline solution (2 ml), followed by saline (10 ml) before being sterile‐filtered through a 0.22 μm Millex‐GV Syringe Filter Unit (Sigma‐Aldrich, Merck, Burlington, MA, USA).
2.4. Quality control procedures
The chemical and radiochemical purity of [18F]F‐AraG was determined by analytical HPLC (Figure S3). The HPLC was run on an Agilent 1220 Infinity LC (California, US) equipped with a Flow‐Count 2Ch radiodetector (Eckert & Ziegler Radiopharma Inc, Massachusetts, USA), using an XBridge BEH C18 column (150 × 4.6 mm, 5 μm, Waters Corporation, Massachusetts, USA) and acetonitrile in ammonium formate (aq. 0.1 M) using the following gradient: 1% for 1 min; 1 to 5% in 4 min; 5% held for 2 min at 2.5 ml/min; and UV detection wavelength of 254 nm. The product identity was verified by co‐injection with the cold reference compound (Figure S4). The activity was measured on a CRC‐120 radioisotope calibrator (Capintec, now Mirion Technologies, New Jersey, USA).
3. RESULTS AND DISCUSSION
The synthesis of [18F]F‐AraG can be divided into three distinct parts: (i) elution and drying of [18F]fluoride, (ii) 18F‐fluorination and deprotections, and (iii) work‐up and formulation (Figure 1). In the first part, vacuum evaporation was eliminated by trapping and releasing [18F]fluoride from a suitable solid‐phase extraction (SPE) cartridge using a solution containing a low water concentration, which enabled the evaporation to be carried out under a stream of nitrogen. The 18F‐fluorination and deprotection reactions were thereafter carried out as a one‐pot reaction, thus obviating the first HPLC purification. The second HPLC purification of the crude product was also replaced by a series of solid‐phase extractions, enabling direct formulation from the SPE instead of evaporation and dissolution as described in the original publication. By this simplified procedure, 1019 ± 218 MBq [18F]F‐AraG was obtained in a radiochemical yield of 10 ± 2% (n = 3, decay corrected to end of 18F‐production), 47 ± 19 GBq/μmol molar activity, >99% radiochemical purity, in a total synthesis time of 97 ± 5 min. The product was radiochemically stable for up to 4 h post‐synthesis.
Although the SPE purification reported herein completely removed all apolar radioactive impurities and significantly reduced the amount of UV‐absorbing impurities (see the supporting information), not all of the UV impurities could be completely eliminated. The current procedure is, therefore, likely limited to preclinical studies as the amount of UV‐absorbing impurities may exceed acceptable levels when the injection volume is larger (i.e., >2 ml). Since the UV impurities were not isolated and characterized, it is impossible to assess their influence on the biodistribution of [18F]F‐AraG. However, it should be noted that this synthesis procedure could be modified to incorporate a final HPLC purification step that would eliminate this risk for clinical applications. As no data on the chemical purity were reported in the publication of Namavari et al., 1 comparing the HPLC and SPE purification methods is impossible. Nonetheless, the reported procedure is well suited for preclinical studies where smaller volumes are typically injected. A preclinical injection of 15 MBq, 20 min after the end of synthesis, would result in an injected carrier mass of 0.12 ± 0.04 μg and an estimated 0.90 ± 0.10 μg of chemical impurities. For a non‐human primate, an injection of 150 MBq would have resulted in an injected carrier mass of 1.2 ± 0.4 μg and an estimated 9.0 ± 1.0 μg of chemical impurities.
CONFLICT OF INTEREST
Magnus Schou is an employee and shareholder at AstraZeneca.
Supporting information
Figure S1. a) Example of a radioactivity trace showing the impurities trapped on the Oasis®MAX 1 cc 10 mg cartridge, where Rt = 0.667 is unreacted [18F]fluoride (55%), Rt = 2.517 product (28%), Rt = 4.067 (7%) and 5.400 (10%) are radioactive impurities. b) Example of a radioactivity trace after SPE purification where Rt = 2.517 product (100%). HPLC parameters: XBridge BEH C18 (150 x 4.6 mm, 5 μm); acetonitrile in ammonium formate (aq. 0.1 M); gradient: 1% for 1 minute; 1 to 90% in 4 minutes; 90% held for 2 minutes at 2.5 ml/min.
Figure S2. Example of an HPLC chromatogram showing the UV impurities washed off the OASIS®HLB 6 cc 500 mg cartridge with the first 20 ml water. HPLC parameters: XBridge BEH C18 (150 x 4.6 mm, 5 μm); acetonitrile in ammonium formate (aq. 0.1 M); gradient: 1% for 1 minute; 1 to 5% in 4 minutes; 5% held for 2 minutes at 2.5 ml/min and UV detection wavelength of 254 nm.
Figure S3. [18F]F‐AraG analytical HPLC chromatogram showing the UV‐absorbing impurities present in the formulated product. HPLC parameters: XBridge BEH C18 (150 x 4.6 mm, 5 μm); acetonitrile in ammonium formate (aq. 0.1 M); gradient:); gradient: 1% for 1 minute; 1 to 15% in 4 minutes; 15% held for 2 minutes at 2.5 ml/min and UV detection wavelength of 254 nm.
Figure S4. [18F]F‐AraG analytical HPLC chromatogram with co‐injection of reference F‐AraG. HPLC parameters: XBridge BEH C18 (150 x 4.6 mm, 5 μm); acetonitrile in ammonium formate (aq. 0.1 M); gradient: 1% for 1 minute; 1 to 5% in 4 minutes; 5% held for 2 minutes at 2.5 ml/min and UV detection wavelength of 254 nm.
ACKNOWLEDGEMENTS
Antonia Högnäsbacka and Miguel A. Cortés González contributed equally to this work and are co‐first authors of this publication. We are grateful to all members of the PET group at the Karolinska Institutet. The study was funded by AstraZeneca. MS is grateful for financial support from the Knut and Alice Wallenbergs Foundation (Dnr: 2018.0066).
Högnäsbacka AA, Cortés González MA, Halldin C, Schou M. Simplified and accessible [18F]F‐AraG synthesis procedure for preclinical PET. J Label Compd Radiopharm. 2022;65(10‐11):288‐291. doi: 10.1002/jlcr.3997
Antonia Högnäsbacka and Miguel Cortés González contributed equally to this work.
Funding information AstraZeneca Knut and Alice Wallenbergs Foundation, Grant/Award Number: 2018.0066
Contributor Information
Antonia A. Högnäsbacka, Email: antonia.hognasbacka@ki.se.
Miguel A. Cortés González, Email: miguel.cortes.gonzalez@ki.se.
DATA AVAILABILITY STATEMENT
Data available in article supplementary material.
REFERENCES
- 1. Namavari M, Chang YF, Kusler B, Yaghoubi S, Mitchell BS, Gambhir SS. Synthesis of 2′‐Deoxy‐2′‐[18F]fluoro‐9‐β‐D‐arabinofuranosylguanine: a novel agent for imaging T‐cell activation with PET. Mol Imaging Biol. 2011;13(5):812‐818. doi: 10.1007/s11307-010-0414-x [DOI] [PubMed] [Google Scholar]
- 2. Ronald JA, Kim BS, Gowrishankar G, et al. A PET imaging strategy to visualize activated T cells in acute graft‐versus‐host disease elicited by allogenic hematopoietic cell transplant. Cancer Res. 2017;77(11):2893‐2902. doi: 10.1158/0008-5472.CAN-16-2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Franc BL, Goth S, MacKenzie J, et al. In vivo PET imaging of the activated immune environment in a small animal model of inflammatory arthritis. Mol Imaging. 2017;16:1‐9. doi: 10.1177/1536012117712638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levi J, Lam T, Goth SR, et al. Imaging of activated T cells as an early predictor of immune response to anti‐PD‐1 therapy. Cancer Res. 2019;79(13):3455‐3465. doi: 10.1158/0008-5472.CAN-19-0267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guglielmetti C, Levi J, Huynh TL, et al. Longitudinal imaging of T cells and inflammatory demyelination in a preclinical model of multiple sclerosis using 18F‐FAraG PET and MRI. J Nucl Med. 2022;63(1):140‐146. doi: 10.2967/jnumed.120.259325 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. a) Example of a radioactivity trace showing the impurities trapped on the Oasis®MAX 1 cc 10 mg cartridge, where Rt = 0.667 is unreacted [18F]fluoride (55%), Rt = 2.517 product (28%), Rt = 4.067 (7%) and 5.400 (10%) are radioactive impurities. b) Example of a radioactivity trace after SPE purification where Rt = 2.517 product (100%). HPLC parameters: XBridge BEH C18 (150 x 4.6 mm, 5 μm); acetonitrile in ammonium formate (aq. 0.1 M); gradient: 1% for 1 minute; 1 to 90% in 4 minutes; 90% held for 2 minutes at 2.5 ml/min.
Figure S2. Example of an HPLC chromatogram showing the UV impurities washed off the OASIS®HLB 6 cc 500 mg cartridge with the first 20 ml water. HPLC parameters: XBridge BEH C18 (150 x 4.6 mm, 5 μm); acetonitrile in ammonium formate (aq. 0.1 M); gradient: 1% for 1 minute; 1 to 5% in 4 minutes; 5% held for 2 minutes at 2.5 ml/min and UV detection wavelength of 254 nm.
Figure S3. [18F]F‐AraG analytical HPLC chromatogram showing the UV‐absorbing impurities present in the formulated product. HPLC parameters: XBridge BEH C18 (150 x 4.6 mm, 5 μm); acetonitrile in ammonium formate (aq. 0.1 M); gradient:); gradient: 1% for 1 minute; 1 to 15% in 4 minutes; 15% held for 2 minutes at 2.5 ml/min and UV detection wavelength of 254 nm.
Figure S4. [18F]F‐AraG analytical HPLC chromatogram with co‐injection of reference F‐AraG. HPLC parameters: XBridge BEH C18 (150 x 4.6 mm, 5 μm); acetonitrile in ammonium formate (aq. 0.1 M); gradient: 1% for 1 minute; 1 to 5% in 4 minutes; 5% held for 2 minutes at 2.5 ml/min and UV detection wavelength of 254 nm.
Data Availability Statement
Data available in article supplementary material.