

Abstract
New N‐heterocyclic compounds for organic functional materials and their efficient syntheses are highly demanded. A surprising entropy‐induced selectivity switch in the gold‐catalyzed intramolecular hydroarylation of 2‐ethynyl N‐aryl indoles was found and its exploitation led to straightforward syntheses of indolo[1,2‐a]quinolines. Experimental and computational mechanistic investigations gave insight into this uncommon selectivity phenomenon and into the special reactivity of the indolo[1,2‐a]quinolines. The high functional group tolerance of this methodology enabled access to a diverse scope with high yields. In addition, bidirectional approaches, post‐functionalization reactions, and π‐extension of the core structure were feasible. An in‐depth study of the photophysical properties explored the structure‐effect relationship for different derivatives and revealed a high potential of these compounds for future applications as functional materials.
Keywords: gold catalysis, material science, nitrogen heterocycles, photophysics, selectivity switch
An entropy‐induced selectivity switch in the gold‐catalyzed hydroarylation of 2‐ethynyl N‐aryl indoles was discovered, and the investigation by experimental and computational mechanistic studies led to a full explanation of the observed temperature‐dependent selectivity. The in this context introduced methodology allows versatile access to indolo[1,2‐a]quinolines, which are highly fluorescent dyes with appealing photophysical properties making them promising candidates for OLED applications.

Introduction
A temperature‐controlled selectivity, which determines the product distribution, is often found among chemical reactions, where a kinetic product is competing with a thermodynamic product. Here, selectivity for the thermodynamic product is possible if the formation of the kinetic product is reversible or if interconversion between products can occur. If this is not the case, the selectivity can still be temperature‐controlled, when the formation of an intermediate has a lower activation barrier than its conversion to the final product. Thus, the intermediate can become the predominating species at lower temperatures whereas at higher temperatures conversion to the final product is achieved. [1] Much rarer are reactions where the temperature significantly changes the entropy of the transition state, caused by for example different conformations or molarities. [2] The temperature‐dependence of the entropic contribution to free enthalpies can then be used for controlling the reaction outcome. For gold‐catalyzed reactions, entropy‐induced selectivity switches are unknown, the selectivity usually depends on other factors (e. g., catalyst, substrate, solvent, additive). [3]
Indole and the isomeric indolizine along with their reduced and oxidized counterparts are important scaffolds found in many natural products, [4] biologically active compounds, [5] and pharmaceuticals [6] (Figure 1a). In addition, they can serve as ligands [7] and applications in organic functional materials are reported. [8] Indolo[1,2‐a]quinolines contain a bridgehead nitrogen and combine structural motifs of indoles, indolizines, and quinolines in a unique tetracyclic framework. Their appealing optical and electronical properties make them candidates for organic functional materials, which is demonstrated by examples for solar cell photosensitizers, [9] applications in cell imaging, [10] and in organic semiconductors [11] (Figure 1b).
Figure 1.

a) Selected examples of molecules bearing the indolizine substructure with significance in biology, pharmacology or as fluorophore. b) Parent structure of indolo[1,2‐a]quinoline and selected examples of molecules bearing the indolo[1,2‐a]quinoline substructure with application in the field of organic functional materials.
The reported synthetic methods towards the indolo[1,2‐a]quinoline framework remain limited. After some early reports in the 1980s applying photochemistry [12] and flash vacuum pyrolysis [13] with lacking selectivity, Lautens et al. developed a Pd‐catalyzed annulation of strained alkenes followed by a retro‐Diels‐Alder reaction to get synthetic access (Scheme 1a). [14] Later, transition metal‐, [15] iodine‐, [16] and radical‐ [17] mediated hydroarylations of alkynes or haloalkenes containing N‐arylated indoles (Scheme 1b) and strategies utilizing C−H‐activation (Scheme 1c/d) [18] along with a few other methods [19] were established. All these examples ‐ with the exception of some non‐versatile methods ‐ have in common that they result in products with a C6 (or C5 and C6) substitution, while manipulations of the C1‐C4 positions are difficult, based on the accessibility of the starting materials (Scheme 1e). Due to the importance of the addressed target, alternative synthetic protocols are still highly desirable, especially to address substitution patterns or to assure functional group tolerances that are difficult to achieve by known methodologies.
Scheme 1.

Examples for known syntheses of indolo[1,2‐a]quinolines and our approach to the structural motif.
To get more versatility for the construction of indolo[1,2‐a]quinolines, we envisioned a synthesis starting from 2‐ethynyl‐N‐aryl‐1H‐indoles (Scheme 1f). These starting materials are easily accessible and the addressed hydroarylation of the indole‐tethered alkyne would allow a flexible approach to new substitution patterns.
Intramolecular hydroarylations on alkynes are an established method for the building of higher molecular complexities via atom economic cycloisomerization reactions and are usually catalyzed by Brønsted or Lewis acids or transition metal complexes. [20] In recent years, gold catalysts were established as a superior tool for the activation of alkynes and alkyne‐involving transformations. [21] Gold‐catalyzed hydroarylations can often be achieved at room temperature [22] and higher temperatures are only required for structures with a more rigid tether and a larger distance between the involved molecule parts. [23] For the combination of a non‐activated aryl moiety and a bridge consisting of a five‐membered aromatic ring (pyrrole, indole, thiophene etc.) with accompanying large angle and an increased distance between the two reacting carbons, only few examples can be found in the literature and due to the challenging substrates, these hydroarylation steps require at least 100 °C [24] (70 °C for a thiophene bridge [25] ).
We herein present the gold‐catalyzed synthesis of indolo[1,2‐a]quinolines via intramolecular hydroarylation of 2‐ethynyl N‐aryl indoles. In the course of this investigation we discovered an unusual entropic shift of the product distribution, which partly turned our focus on the investigation of this rarely observed phenomenon. Results on this discovery as well as a broad scope of the addressed targets and their photophysical properties will be discussed within this work.
Results and Discussion
As our intended starting materials were considered as systems with unfavorable geometry for a cyclization event, we decided to utilize gold catalysis as well to achieve the desired hydroarylation. For our initial attempts we chose 1‐phenyl‐2‐(phenylethynyl)‐1H‐indole (1 a) as starting material and IPrAuNTf2 (IPr=1,3‐bis(2,6‐diisopropylphenyl)imidazol‐2‐ylidene) as catalyst under varying reaction conditions using dichloromethane (DCM), chloroform, 1,2‐dichloroethane (DCE), 1,1,2,2‐tetrachloroethane (TCE), chlorobenzene or toluene as solvent at 40–130 °C. While these combinations showed virtually no reactivity, using DCE or chlorobenzene under microwave irradiation in a pressure tube at 150 °C resulted in full conversion of the starting material after 30 min (Scheme 2a). To our delight, we could identify the desired 5‐phenylindolo[1,2‐a]quinoline (2 a) in the product mixture in up to 45 % yield, but also discovered large amounts of two major (3 a/3 b) and two minor (3 c/3 d) side‐products, which seemed to be unsymmetrical dimers of the starting material (as derived by 1H NMR and HR‐MS spectra). In addition, the reproducibility of the microwave reactions was inconsistent and a slow decomposition of the product was observed. By vapor diffusion of MeOH into a concentrated solution of 2 a in chloroform we were able to grow specimen suitable for single crystal X‐ray analysis and the crystallographic structure undoubtedly confirms the connectivity of the product (Scheme 2b). [26]
Scheme 2.

a) Initial attempts for the cycloisomerization of 1 a. b) Solid‐state molecular structure of 2 a. The thermal ellipsoids are set at a 50 % probability level and hydrogen atoms are omitted for clarity. [a] The yields refer to several batches of the same reaction under microwave conditions with low reproducibility.
As a next step, an extensive catalyst and conditions screening was conducted to enhance the selectivity and improve the yield of the monomeric product (see Supporting Information for a detailed description of the screening). In summary, o‐dichlorobenzene (ODCB) was deemed the most appropriate solvent and for the optimization of the selectivity JohnPhosAu(MeCN)SbF6 at 160 °C gave the best result. After further optimization of the substrate concentration and catalyst loading as well as using oxygen‐free conditions to reduce decomposition of the product, we established following optimized standard conditions (SC): 5–15 μmol/mL substrate concentration in degassed ODCB, JohnPhosAu(MeCN)SbF6 with a 2.5 mol % catalyst loading and 4 h reaction time at 170 °C under oxygen‐free conditions. Although some catalyst decomposition was observed during the reaction (e. g. by formation of a gold mirror), the catalyst was still stable enough at this elevated temperature to achieve full conversion with this relatively low catalyst loading.
In the course of the screening, we discovered an interesting temperature dependency in the product distribution between 2 a and 3 a–d. Depending on the applied catalyst, a temperature change of 40–60 °C completely shifted the selectivity from >95 % 3 a–d to >95 % 2 a. For example, using 10 mol % IPrAuNTf2 at 140 °C resulted in a mixture of almost exclusively 3 a–d with only trace amounts of 2 a; at 160 °C an almost equal product ratio of 2 a to 3 a–d was observed, at 180 °C 2 a became the major product with less than 3 % of 3 a–d. Exchanging the gold catalyst had a much less significant influence on the product distribution than the temperature change. By using different catalysts, the point of equal product distribution changed to lower temperatures (80–160 °C), but to achieve sufficiently high ratios of 2 a : 3 a–d (>9 : 1), teperatures of at least 140 °C were necessary. Note, for the ratio of the major side‐products 3 a to 3 b the catalyst was the main deciding factor with phosphine catalysts favoring 3 a and NHC catalysts favoring 3 b (3 c/3 d occur only as minor side‐products; <10 %).
This phenomenon warranted further investigation. We hypothesized that this temperature‐dependent selectivity switch may be caused by the different entropies along the reaction pathways for the monomer and dimers, which should be significantly affected by a temperature change due to the involvement of a different number of molecules.
As a first step in shedding light into this subject, we tried to identify compounds 3 a–d. The separation of this mixture was very problematic due to virtually identical R f ‐values and solubilities. Only by a repeated use of preparative thin layer chromatography were we able to enrich 3 a and generate specimen suitable for single crystal X‐ray analysis by vapor diffusion of MeOH into a concentrated chloroform solution (Scheme 3a). [26] The solid‐state molecular structure shows that 3 a consists of the intramolecular hydroarylation product 2 a coupled to the starting material 1 a via a subsequent intermolecular hydroarylation.
Scheme 3.

a) Identified dimer 3 a and the corresponding solid‐state molecular structure. The thermal ellipsoids are set at a 50 % probability level and hydrogen atoms are omitted for clarity. b) Suggested structures of 3 b–d. c) Control experiments to explain the product formation.
Next, a series of control experiments were conducted to get a first insight into the mechanisms that lead to the dimers (Scheme 3c). First, a reaction of the mixture 3 a–d with JohnPhosAu(MeCN)SbF6 under standard conditions (at 170 °C) and a reaction of 2 a with the same catalyst at 100 °C resulted in no conversion, which excludes an equilibrium between these structures. To find the lowest temperature necessary for the reasonably fast formation of 2 a, 1 a and IPrAuNTf2 were stirred in ODCB for 3 d at 65 °C and in a second batch at 40 °C. At 65 °C 75 % conversion to a mixture of mostly dimers was observed, while at 40 °C less than 2 % conversion occurred. Using the same conditions (40 °C) but adding a surplus of 2 a to the reaction resulted in full conversion of 1 a in less than 16 h to a dimer mixture with a similar distribution as observed before. However, a 1 : 1 ratio of 1 a and 2 a at 180 °C resulted in almost exclusively 2 a with only small amounts of dimers. These control experiments show that the observed side‐products are formed by the intermolecular hydroarylation of 2 a onto 1 a and that this step is much faster at lower temperatures than the intramolecular hydroarylation. The unidentified side‐products 3 b–d are therefore highly likely cis/trans and/or regioisomers of the identified dimer 3 a (Scheme 3b). This is also supported by NMR spectra, which show only small variations of the chemical shifts between the different compounds 3 a–d.
To get a deeper understanding of the mechanism, a density functional theory (DFT) study was conducted. Calculations were performed in the Jaguar 9.1 suite [27] of ab initio quantum chemistry programs at the B3LYP‐D3/6‐31G**/LACVP(Au)//cc‐pVTZ(−f)/LACV3P(Au) [28] level of theory with the Poisson‐Boltzmann solvation model for ODCB (ϵ=9.93). [29] Calculations of the respective addition barriers of the addition reaction of π‐complexes 1 a [Au] to 2 a with the simplified ligand system 1,3‐dimethylimidazolylidene (IMe) suggest that 3 b is the major component and 3 a, which we were able to isolate, is the second most produced isomer (see Scheme 3). Both 3 a and 3 b appear kinetically favored over the monomer 2 a assuming a 1 M concentration of all reaction partners at 40 °C in ODCB (Figure 2). The initial formation of 2 a [Au] has a barrier of 21.1 kcal/mol (2 a [Au]‐TS) at 40 °C. The subsequent deprotonation and protodeauration recover [Au]+ and yield 2 a in a strongly exergonic process (−32.5 kcal/mol). The addition of another molecule of 1 a [Au] to the product 2 a leads to the dimers 3 a–d. The barriers of this step at 40 °C are computed to be 20.1 (3 a [Au]‐TS), 17.0 (3 b [Au]‐TS), 22.8 (3 c [Au]‐TS), and 26.0 (3 d [Au]‐TS) kcal/mol, respectively. Due to the larger activation entropy involved in the formation of the dimers 3 a–d, they become increasingly disfavored at elevated temperatures. At 140 °C, we find the free energy of 3 b [Au]‐TS at 22.9 kcal/mol. The lower involved transition entropy of the initial cyclization of 1 a [Au] leads to a minor impact of heating on the barrier 2 a [Au]‐TS (21.5 kcal/mol). This renders the monomer (2 a) formation favored energetically on top of the intrinsic statistic preference of the intramolecular pathway over intermolecular reactions. Deprotonation and protodeauration to yield [Au]+ and 3 a–d herein are estimated in the range of −0.03 to −5.62 kcal/mol at 40 °C. This indicates that these reactions should be reversible at the reaction temperature, supporting that the switch to the dimerization is primarily kinetically driven. These terminating steps (deprotonation/protodeauration) become more favorable at 140 °C with a range of free energies of −5.62 to −10.9 kcal/mol. The backreaction barriers are found to be 30.8 to 38.2 kcal/mol after deauration speaking against reversibility from the dimeric products in agreement with our experimental evidence.
Figure 2.

DFT calculations of the gold‐catalyzed intramolecular cyclization of 1 a compared to the intermolecular addition of 1 a to 2 a modelled at 40 °C (left) and 140 °C (right).
To probe the translation of these results into more sterically demanding ligand frameworks, we computed the discussed minima with the full IPr ligand as well. The formation of 2 a [IPrAu] is found to be slightly more disfavored than with IMe (12.3 kcal/mol at 40 °C; 12.7 kcal/mol at 140 °C). The formation of the dimer 3 b [IPrAu] is more favored, likely due to the vinylic position leading to less bulk around the large ligand (−15.8 kcal/mol at 40 °C; −10.2 kcal/mol at 140 °C). The increased gap in the intermediate stability is expected to be followed by a similar trend in the reaction barriers. These results are indicative for an even stronger temperature sensitivity of the reaction outcome with larger ligands, aligning well with our experimental observations.
Using the previously determined optimized conditions, we studied the scope and limitations of our reaction protocol (Scheme 4). A broad range of 2‐ethynyl‐N‐aryl‐1H‐indoles (1 a–r) mainly with variations on the N‐aryl (Ar) and alkyne (R2) moiety was synthesized. Depending on the starting material and accessibility of the intermediates, different strategies involving established Cu‐ and/or Pd‐catalyzed cross coupling methods were utilized to build the corresponding ethynylindoles (see Supporting Information for more details). The substrate scope was initially explored with starting materials lacking substitution on the indole‐C3 position (R1). After the gold catalysis, all these substrates showed the occurrence of dimers (assigned by characteristic signals in the 1H NMR and HR‐MS spectra) as minor impurities, which could be separated chromatographically as a mixture from the monomers. First, variations on the N‐aryl moiety were explored by using different 2‐(phenylethynyl)‐1H‐indoles. The reference substrate 1 a with N‐phenyl substitution resulted in 2 a in 91 % yield, while electron‐rich (methoxy, trimethoxy), electron‐deficient (CF3), and halogen (Br) substituents on the phenyl ring gave the corresponding products in 61–87 % yield (2 b–e). Substrate 1 f with a nitro substituent was readily converted to 2 f in 61 % yield, although more catalyst and a prolonged reaction time were needed. Aryl moieties with strong electron‐withdrawing groups are often difficult substrates for these kinds of transformations and to the best of our knowledge, it was never reported for nitrophenyl derivatives. It is noteworthy, that for this substrate no dimeric side‐products and no oxidation of the product were observed. As expected, the similar hydroarylation with an electron‐rich five membered ring (thiophene) was also possible and the thienopyridoindole 2 g was synthesized in 85 % yield. Concerning variations on the alkyne moiety, N‐phenyl‐1H‐indoles with distinct 2‐alkynyl substitutions were used next. The n‐butyl substituent resulted in 91 % yield and phenyl rings with electron‐rich (methoxy) and electron‐deficient (CF3) groups as well as halogens (F and Br) all gave good results (2 h–l, 74–92 % yield). Thiophene as heteroaromatic substituent was tolerated and 2 m could be achieved in 80 % yield. We were even able to convert pyridine substrate 1 n, although more catalyst and reaction time was needed in this case (2 n, 58 % yield). This is of note, as pyridine moieties are challenging substrates in gold catalysis due to the formation of a stable pyridine‐gold complex, which often inhibits the catalysis. [30] Substitution of the reference substrate at the C3 position (R1) with either an aryl or a methyl group completely suppressed the formation of dimers and hardly any decomposition of the product was observed, which resulted in excellent isolated yields for these compounds (2 o, 94 % yield; 2 p, 91 % yield). The corresponding substrate bearing a methyl group on the C3 position and a terminal alkyne on C2 (1 q) delivered 2 q in only 38 % yield, accompanied by the formation of unidentified oligomers (as derived by mass spectrometry). The reaction of 2‐ethynyl‐1‐phenyl‐1H‐indole and its silyl‐protected alkyne analogues (1 r–H, 1 r‐TMS, and 1 r‐TIPS) always gave the same non‐substituted product 2 r in low yields (≤18 %), alongside with a major side‐product and again a mixture of oligomers (Scheme 4b). It is noteworthy that the TMS or TIPS groups were never detected in the corresponding product mixtures and must have been cleaved during the catalysis or after product formation, possibly due to the high temperature. The major side‐product for this reaction could be identified as dimer 4 (up to 63 % yield) and no other of the possible dimers where observed. By lowering the substrate concentration of 1 r–H from 15.3 to 2.51 μmol/mL the yield of 2 r could be raised to 45 % while lowering the yield of 4 to 30 %. The building of dimer 4 seems to be more favored than the dimer formation of the other derivatives, which implies that either 1 r–H or 2 r (or both) account for a higher reactivity in the intermolecular hydroarylation. To test which is the case, a trapping experiment with 1 r–H and a surplus of 2 d at 150 °C was conducted, which selectively resulted in the formation of trapping product 5, without any detectable amounts of 4 (Scheme 4c). From this it can be deduced that for terminal alkynes the competing intermolecular reaction is much faster than for internal alkynes.
Scheme 4.

a) Gold‐catalyzed cycloisomerization of 1 forming indolo[1,2‐a]quinoline derivatives 2. b) Occurrence of dimeric side‐product 4 for derivatives of 1 r. c) Trapping experiment to explain the abundance of 4. [a] 150 μmol scale: 91 % yield; 3.48 mmol scale: 87 % yield. [b] SC for 4 h, then,+2.5 mol % catalyst, 180 °C, 12 h. [c] SC for 4 h, then,+7.5 mol % catalyst, 180 °C, 16 h, then,+10 mol % catalyst, 180 °C, 24 h. [d] Using SC with c(1 r‐H)=15.3 μmol/mL: yield (2 r)=13 %, yield (4)=63 %; with c(1 r‐H)=2.51 μmol/mL: yield (2 r)=45 %, yield (4)=30 %. [e] 20 mol % of catalyst was used and the mixture was stirred at 180 °C for 16 h; 50 % of the starting material 1 r‐TIPS was recovered. [f] 150 °C.
An upscaling of the reaction was also possible, which was demonstrated by converting a 1.02 g batch of substrate 1 a, which gave access to 883 mg (87 %) of 2 a. Efforts in introducing a halogen to the C7 position of the final product were made, but neither the hydroarylation under standard conditions starting with the corresponding 3‐bromo or 3‐iodo ethynyl indole (1 s or 1 t), nor the attempted post‐halogenation of 2 a were effective (Scheme 5a). Instead, with N‐iodosuccinimide (NIS) the completely air‐stable C7 dimer 6 was formed quite selectively (70 % yield), while N‐bromosuccinimide (NBS) produced the same product along with major unidentified impurities (<30 % yield). This oxidative dimerization is a process that is rarely reported in literature for simple indoles and has never been mentioned for larger indole containing systems. [31] To extend the scope, bidirectional approaches were conducted next (Scheme 5b). Starting with bidirectional, butadiyne‐bridged substrate 7 the C5‐linked dimer 8 was obtained in 42 % yield. The reduced yield can be explained by the occurrence of more dimeric and oligomeric side‐products as well as a higher rate of decomposition of the final product. Finally, bidirectional, benzene‐bridged substrate 9 most probably resulted in the formation of dimer 10, but due to high instability in solution, the characterization was limited to a 1H NMR and a mass spectrum. By comparison of the calculated highest occupied molecular orbital (HOMO) energies (see Supporting Information) of 2 a, 6, 8 and 10 the higher instability towards oxidation of 8 and especially 10 can be rationalized. The HOMO of 2 a lies at −5.19 eV, 8 at −5.06 eV and 6 as well as 10 much higher at −4.75 eV. The trend of higher calculated HOMO energies correlates with the observed instabilities towards oxidation with the exception of 6, where the reactive C7 position is blocked and therefore non‐reactive. With the bromine‐substituted product 2 l, a further post‐functionalization by Sonogashira cross coupling to compound 11 was demonstrated (Scheme 5c). A subsequent gold‐catalyzed hydroarylation under standard conditions gave access to the yet unknown core structure of a π‐extended indolo[1,2‐f]phenanthridine 12. [32]
Scheme 5.

a) Post‐functionalization / dimerization of 2 a. b) Synthesis of dimers 8 and 10. c) Postfunctionalization of 2 l / synthesis of 12. Conditions: i) N‐iodosuccinimide (NIS; 1.1 equiv), DCM, rt, 16 h; ii) N‐bromosuccinimide (NBS; 1.1 equiv), DCM, rt, 16 h; iii) phenylacetylene (3 equiv), Pd(Ph3P)4 (5 mol %), CuI (10 mol %), Et3N, 90 °C, 2 d. [a] 10 showed high instability, therefore characterization was limited to a 1H NMR and a HR‐MS spectrum.
Regarding the stability of indolo[1,2‐a]quinolines, we discovered some interesting aspects. If no C7‐substituent is present (especially for electron‐rich substrates) the products tend to slowly turn green in solution under ambient air. This is often already observed when dissolving the product in non‐purified CDCl3 (containing traces of acid). This process is not occurring under inert conditions and is slow under an atmosphere of oxygen in the absence of moisture, but the addition of acids accelerates the decomposition significantly. For 2 a, the decomposition compounds were separated from the product by chromatography and HR‐MS experiments suggest that a mixture of oxidized decomposition products (2 a–ox) is present. This mixture consists mainly of an oxidized derivative of 2 a and oligomerization of this compound with varying amounts of oxygen/oxidizations (m/z (2 a)=293.12; m/z (2 a–ox)=308.11/597.20/599.21/888.30 etc.). Addition of trifluoroacetic acid to solutions of 2 a under inert conditions results in a protonation of the compound without a similar oligomerization and is reversible by addition of a surplus of base (see Supporting Information, Figure S126/S127 for UV spectra of 2 a–ox and protonation experiments). Although these decompositions can be reduced by working under inert conditions when feasible, using 3‐substituted indoles as starting materials decreases unwanted side‐reactions effectively.
All of the observed unusual aspects in the chemistry of indolo[1,2‐a]quinolines (side‐product formation, dimerization to 6, instability towards oxygen) seem to be caused by the high reactivity/nucleophilicity of its C7 position on the five‐membered ring. To theoretically investigate and to rationalize this behavior, we computed the atomic Fukui indices for 2 a from the HOMO and from the lowest unoccupied molecular orbital (LUMO) (Figure 3). Atomic Fukui indices were derived from the MO coefficients of the respective orbitals, predicting the positions’ reactivity. [33] The atomic Fukui indices show that the observed reactions at C7 are strongly favored by the electron distribution in the HOMO with a Fukui f of 0.24. The position that is expected to be the second strongest nucleophile is in a distal full aromatic 6‐ring with an f of only 0.10.
Figure 3.
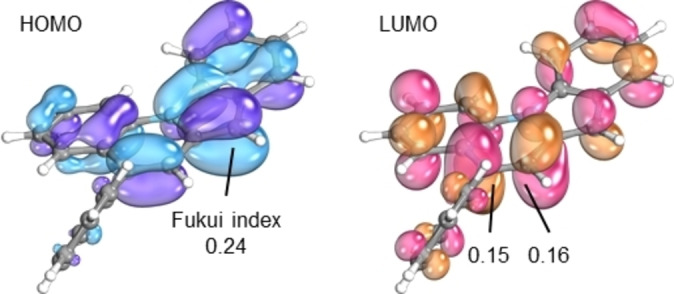
Atomic Fukui indices from the frontier orbitals of 2 a.
Indolo[1,2‐a]quinolines 2 a–r and its derived compounds 4, 6, 8, and 12 were studied by UV‐Vis and fluorescence spectroscopy in DCM (Table 1, Figure 4). Almost all synthesized compounds are yellow solids (2 h: oil) and exhibited a strong fluorescence in solution (blue to blue‐green in DCM) as well as in the solid state. 2 a–e and 2 g–r show large absorption peaks in the mid UV range with maxima at 280–303 nm and several smaller absorption peaks at the near UV and visible range (corresponding to π‐π* transitions) with maxima of the longest absorption wavelength (λmax,abs) at 417–443 nm equating in measured optical gaps between 2.65 eV and 2.86 eV. The different substituents on the aromatic rings have only small influences on the absorption spectra, whereas extending (2 o) and shortening (2 h/2 r) the π‐system results as expected in a bathochromic shift and in a hypsochromic shift, respectively. Interestingly, the two dimers 6 and 8 show significant differences in the UV spectra. While C5‐linked 8 exhibits a similar absorption to the monomeric counterparts, the spectra of C7‐linked 6 is considerably red‐shifted (by about 40 nm) resulting in a lower optical gap of 2.45 eV. This different behavior can be intuitively rationalized through the dihedral angles between the two linked hemispheres (calculated from DFT‐optimized geometries). For 8 the angle is close to vertical (108°) and for 6 the dihedral angle is 135°, therefore more in‐plane resulting in a better overlap of the π‐system and a red‐shift of the spectrum.
Table 1.
Photophysical properties of the final compounds measured in DCM, if not stated otherwise.
|
Compound |
λmax,abs [nm][a] |
λmax,em [nm][b] |
Stokes Shift [cm−1] |
QY [%][c] |
λonset,abs [nm] |
Eg(opt) [eV][d] |
Eg(calc) [eV][e] |
|---|---|---|---|---|---|---|---|
|
2 a |
424/424[f]/424[g] |
454/451[f]/443[g]/483[h] |
1558/1412[f]/1012[g] |
79/76[f]/57[g] |
446 |
2.78 |
3.48 |
|
2 b |
432 |
464 |
1596 |
70 |
458 |
2.71 |
‐ |
|
2 c |
425 |
454 |
1503 |
90 |
446 |
2.78 |
‐ |
|
2 d |
428 |
460 |
1625 |
72 |
451 |
2.75 |
‐ |
|
2 e |
432 |
463 |
1550 |
42 |
461 |
2.69 |
‐ |
|
2 f |
403/402[f]/405[g] |
–[i]/–[f],[i]/482[g]/–[h],[i] |
3944[g] |
8[g] |
526 |
2.36 |
2.70 |
|
2 g |
436 |
471 |
1704 |
37 |
462 |
2.68 |
‐ |
|
2 h |
418 |
443 |
1350 |
52 |
438 |
2.83 |
3.50 |
|
2 i |
425 |
453 |
1454 |
68 |
449 |
2.76 |
‐ |
|
2 j |
426 |
454 |
1448 |
85 |
450 |
2.76 |
‐ |
|
2 k |
423 |
451 |
1468 |
70 |
446 |
2.78 |
‐ |
|
2 l |
424 |
446 |
1163 |
58 |
447 |
2.77 |
‐ |
|
2 m |
427 |
478 |
2499 |
69 |
458 |
2.71 |
‐ |
|
2 n |
425 |
458 |
1695 |
60 |
449 |
2.76 |
‐ |
|
2 o |
440 |
469 |
1405 |
72 |
463 |
2.68 |
3.38 |
|
2 p |
443 |
474 |
1476 |
77 |
468 |
2.65 |
‐ |
|
2 q |
434 |
459 |
1255 |
75 |
456 |
2.72 |
‐ |
|
2 r |
417 |
433 |
886 |
74 |
433 |
2.86 |
3.60 |
|
4 |
447 |
473 |
1230 |
34 |
469 |
2.64 |
‐ |
|
6 |
436 |
543 |
4520 |
38 |
506 |
2.45 |
2.96 |
|
8 |
423 |
471 |
2409 |
66 |
455 |
2.72 |
3.17 |
|
12 |
407 |
487 |
4036 |
40 |
460 |
2.70 |
3.41 |
[a] Maximum of the longest absorption wavelength. [b] Maximum of the shortest emission wavelength. [c] Fluorescence quantum yield. [d] Optical gap estimated from λonset,abs: Eg(opt)=1239.8/λonset,abs. [e] HOMO‐LUMO gap values computed by DFT, see Supporting Information. [f] In 1,4‐dioxane. [g] In cyclohexane. [h] As powder. [i] No fluorescence observed.
Figure 4.

a) Comparison of normalized absorption and emission spectra of all final compounds in DCM. b) Solvatochromic behavior of 2 a and 2 f. c) Photographs of 2 a as solution in DCM and as solid under irradiation by UV light (365 nm).
Comparable tendencies to the absorption are observed for the fluorescence spectra. Compounds 2 (except 2 f) have the maxima of their shortest emission wavelength (λmax,em) at 433–478 nm, resulting in Stokes shifts of 886–2499 cm−1, with notable hypsochromic shift for 2 h/2 r and bathochromic shift for the thiophene derivatives 2 g/2 m as well as for 2 o/2 p. Again, 6 shows the strongest red‐shift with λmax,em at 543 nm and a larger Stokes shift of 4520 cm−1. Fluorescence quantum yields (QY) for the majority of compounds range around 60–80 % and reach even higher values for 2 c (90 %) and 2 j (85 %). Lower QY are observed for the bromine substituent and for thiophene in the core structure (2 e: 42 %; 2 g: 37 %) as well for most of the larger π‐systems (4: 34 %; 6: 38 %; 12: 40 %).
The solvatochromic properties were examined for 2 a (Table 1, Figure 4b). While the absorption in DCM, 1,4‐dioxane, and cyclohexane show only negligible differences, the emission spectra are slightly blue‐shifted in less‐polar solvents resulting in lower Stokes shifts of 1412 cm−1 in dioxane and 1012 cm−1 in cyclohexane. In the solid state, measured as powder, the emission shows a strong bathochromic shift of 30–40 nm. For 2 a in dioxane a QY of 76 % (DCM: 79 %) was measured and the QY in cyclohexane gave a lower value of 57 %.
For selected compounds (2 a, 2 f, 2 h, 2 o, 2 r, 6, 8, 12) the HOMO‐LUMO energies were calculated using DFT (see Supporting Information). The corresponding HOMO‐LUMO gaps (Eg(calc)) follow the same trend as seen with the optical gap (Eg(opt)). The values for Eg(calc) are higher than for Eg(opt) (about 0.7 eV for most compounds), which is expected due to the different nature of these energy gaps. [34]
An interesting optical behavior was observed for nitro‐derivative 2 f. In the solid state it has a red‐orange color and the UV spectrum shows a bathochromic shift of about 80–100 nm compared to 2 a. The onset manifests in a broad absorption area of low intensity without clear maximum corresponding to a low optical gap of 2.36 eV. 2 f shows no fluorescence in the solid state and in a DCM or dioxane solution, which is a common solvent dependent phenomenon of nitro‐containing fluorophores due to favored radiation‐less deactivation modes of the excited state. [35] In contrast, fluorescence with λmax,em of 482 nm (corresponding to a larger Stokes shift of 3944 cm−1) and a QY of 8 % is displayed in a cyclohexane solution of the compound. DFT calculations of 2 f show that the LUMO is mainly concentrated on the nitrobenzene substituent, which yields an energy level (−2.80 eV) roughly 1 eV lower than in its relatives, where the LUMO is mainly localized on the core structure (see Supporting Information for more details). HOMO and LUMO+1 are localized on the indoloquinoline core structure and correspond to the MOs that are involved in the first excitation in all other computed cases. This agrees well with the much lower observed optical gap.
Conclusion
A highly flexible route for the efficient synthesis of indolo[1,2‐a]quinolines and their derivatives via a gold‐catalyzed cycloisomerization of 2‐ethynyl N‐aryl indoles was developed. A competing formation of dimeric side‐products by intermolecular paths was explained by experimental und computational methods and gave insight into an entropy‐controlled selectivity switch that was unprecedented in gold catalysis. This concept of a temperature‐controlled selectivity switch might find widespread use in the future. The new approach for the synthesis of indolo[1,2‐a]quinolines enables access to formerly unachievable substitution patterns. The functional group tolerance is remarkable, which enables the introduction of further synthetic handles for the possibility of post‐functionalization and π‐extension reactions. This should pave the way for further modifications of the core structure, for example, for introducing push/pull functionalities to fine‐tune the opto‐electronical properties. The impressive photophysical properties, most notably the high fluorescence quantum yield and solid‐state fluorescence, crave for application in OLED or fluorescent dyes. Inspired by the promising potential of this methodology, we are currently working on an expansion of the scope to synthesize larger systems with seven annulated aromatic rings, which could be attractive semiconductors.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Acknowledgements
R.H. is grateful for funding by the Deutsche Forschungs‐gemeinschaft (DFG; SFB 1249 ‐ N‐Heteropolyzyklen als Funktionsmaterialien). F.F.M. is grateful to the Alexander von Humboldt‐Foundation for a Feodor Lynen Research Fellowship and for a Feodor Lynen Return Fellowship and acknowledges the Institute for Basic Science (IBS‐R10‐A1) in Korea for financial support. Open Access funding enabled and organized by Projekt DEAL.
R. Heckershoff, G. May, J. Däumer, L. Eberle, P. Krämer, F. Rominger, M. Rudolph, F. F. Mulks, A. S. K. Hashmi, Chem. Eur. J. 2022, 28, e202201816.
Contributor Information
Dr. Florian F. Mulks, Email: ff@mulks.ac.
Prof. Dr. A. Stephen K. Hashmi, Email: hashmi@hashmi.de.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.
- 1a. Liu Y., Zhang Y., Tetrahedron Lett. 2003, 44, 4291–4294; [Google Scholar]
- 1b. Tian S., Wang Z., Gong W., Chen W., Feng Q., Xu Q., Chen C., Chen C., Peng Q., Gu L., Zhao H., Hu P., Wang D., Li Y., J. Am. Chem. Soc. 2018, 140, 11161–11164; [DOI] [PubMed] [Google Scholar]
- 1c. Yu H., Xu Y., Havener K., Zhang M., Zhang L., Wu W., Huang K., Small 2022, 18, 2106893. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Buschmann H., Scharf H.-D., Hoffmann N., Esser P., Angew. Chem. Int. Ed. 1991, 30, 477–515; [Google Scholar]; Angew. Chem. 1991, 103, 480–518; [Google Scholar]
- 2b. Wang S., Xiao J., Li J., Xiang H., Wang C., Chen X., Carter R. G., Yang H., Chem. Commun. 2017, 53, 4441–4444; [DOI] [PubMed] [Google Scholar]
- 2c. Watanabe N., Hiragaki K., Tsurumi K., Ijuin H. K., Matsumoto M., Tetrahedron 2017, 73, 1845–1853. [Google Scholar]
- 3.
- 3a. McLean E. B., Cutolo F. M., Cassidy O. J., Burns D. J., Lee A.-L., Org. Lett. 2020, 22, 6977–6981; [DOI] [PubMed] [Google Scholar]
- 3b. Hansmann M. M., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2013, 52, 2593–2598; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2653–2659; [Google Scholar]
- 3c. Hashmi A. S. K., Wang T., Shi S., Rudolph M., J. Org. Chem. 2012, 77, 7761–7767; [DOI] [PubMed] [Google Scholar]
- 3d. Hashmi A. S. K., Schuster A. M., Gaillard S., Cavallo L., Poater A., Nolan S. P., Organometallics 2011, 30, 6328–6337; [Google Scholar]
- 3e. Alvarez E., Miguel D., García-García P., Fernández-Rodríguez M. A., Rodríguez F., Sanz R., Beilstein J. Org. Chem. 2011, 7, 786–793; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3f. Zeng Z., Jin H., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2018, 57, 16549–16553; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16787–16791; [Google Scholar]
- 3g. Heckershoff R., Maier S., Wurm T., Biegger P., Brödner K., Krämer P., Hoffmann M. T., Eberle L., Stein J., Rominger F., Rudolph M., Freudenberg J., Dreuw A., Hashmi A. S. K., Bunz U. H. F., Chem. Eur. J. 2022, 28, e202104203; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3h. Hashmi A. S. K., Blanco M. C., Eur. J. Org. Chem. 2006, 2006, 4340–4342; [Google Scholar]
- 3i. Sromek A. W., Rubina M., Gevorgyan V., J. Am. Chem. Soc. 2005, 127, 10500–10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Katritzky A. R., Rees C. W., Scriven E. F. V., Comprehensive Heterocyclic Chemistry II, Pergamon, Oxford, 1996; [Google Scholar]
- 4b. Segraves N. L., Robinson S. J., Garcia D., Said S. A., Fu X., Schmitz F. J., Pietraszkiewicz H., Valeriote F. A., Crews P., J. Nat. Prod. 2004, 67, 783–792; [DOI] [PubMed] [Google Scholar]
- 4c. Higuchi K., Kawasaki T., Nat. Prod. Rep. 2007, 24, 843–868; [DOI] [PubMed] [Google Scholar]
- 4d. Ciulla M. G., Zimmermann S., Kumar K., Org. Biomol. Chem. 2019, 17, 413–431; [DOI] [PubMed] [Google Scholar]
- 4e. Michael J. P., Nat. Prod. Rep. 2008, 25, 139–165. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Silva T. S., Rodrigues M. T., Santos H., Zeoly L. A., Almeida W. P., Barcelos R. C., Gomes R. C., Fernandes F. S., Coelho F., Tetrahedron 2019, 75, 2063–2097; [Google Scholar]
- 5b. Schmidt A. W., Reddy K. R., Knölker H.-J., Chem. Rev. 2012, 112, 3193–3328; [DOI] [PubMed] [Google Scholar]
- 5c. Brown D. W., Graupner P. R., Sainsbury M., Shertzer H. G., Tetrahedron 1991, 47, 4383–4408. [Google Scholar]
- 6.
- 6a. Lavrado J., Moreira R., Paulo A., Curr. Med. Chem. 2010, 17, 2348–2370; [DOI] [PubMed] [Google Scholar]
- 6b. Podoll J. D., Liu Y., Chang L., Walls S., Wang W., Wang X., Proc. Natl. Acad. Sci. USA 2013, 110, 15573–15578; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Anderson W. K., Heider A. R., Raju N., Yucht J. A., J. Med. Chem. 1988, 31, 2097–2102; [DOI] [PubMed] [Google Scholar]
- 6d. Kato M., Ito K., Nishino S., Yamakuni H., Takasugi H., Chem. Pharm. Bull. 1994, 42, 2546–2555; [DOI] [PubMed] [Google Scholar]
- 6e. Reddy M. V., Rao M. R., Rhodes D., Hansen M. S., Rubins K., Bushman F. D., Venkateswarlu Y., Faulkner D. J., J. Med. Chem. 1999, 42, 1901–1907; [DOI] [PubMed] [Google Scholar]
- 6f. Kemnitzer W., Kuemmerle J., Jiang S., Sirisoma N., Kasibhatla S., Crogan-Grundy C., Tseng B., Drewe J., Cai S. X., Bioorg. Med. Chem. Lett. 2009, 19, 3481–3484. [DOI] [PubMed] [Google Scholar]
- 7. Grandini C., Camurati I., Guidotti S., Mascellani N., Resconi L., Nifant′ev I. E., Kashulin I. A., Ivchenko P. V., Mercandelli P., Sironi A., Organometallics 2004, 23, 344–360. [Google Scholar]
- 8.
- 8a. Leontie L., Druta I., Danac R., Rusu G. I., Synth. Met. 2005, 155, 138–145; [Google Scholar]
- 8b. Kim E., Lee Y., Lee S., Park S. B., Acc. Chem. Res. 2015, 48, 538–547; [DOI] [PubMed] [Google Scholar]
- 8c. Wu Y., Li Y., Gardner S., Ong B. S., J. Am. Chem. Soc. 2005, 127, 614–618; [DOI] [PubMed] [Google Scholar]
- 8d. Hendrich C. M., Bongartz L. M., Hoffmann M. T., Zschieschang U., Borchert J. W., Sauter D., Krämer P., Rominger F., Mulks F. F., Rudolph M., Dreuw A., Klauk H., Hashmi A. S. K., Adv. Synth. Catal. 2021, 363, 549–557. [Google Scholar]
- 9. Baik C., Kim D., Kang M.-S., Song K., Kang S. O., Ko J., Tetrahedron 2009, 65, 5302–5307. [Google Scholar]
- 10. Sun K., Zhang Y.-L., Chen X.-L., Su H.-F., Peng Q.-C., Yu B., Qu L.-B., Li K., ACS Appl. Bio Mater. 2020, 3, 505–511. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Ahmed E., Briseno A. L., Xia Y., Jenekhe S. A., J. Am. Chem. Soc. 2008, 130, 1118–1119; [DOI] [PubMed] [Google Scholar]
- 11b. Zhu L., Kim E.-G., Yi Y., Ahmed E., Jenekhe S. A., Coropceanu V., Brédas J.-L., J. Phys. Chem. C 2010, 114, 20401–20409; [Google Scholar]
- 11c. Choi H. H., Najafov H., Kharlamov N., Kuznetsov D. V., Didenko S. I., Cho K., Briseno A. L., Podzorov V., ACS Appl. Mater. Interfaces 2017, 9, 34153–34161. [DOI] [PubMed] [Google Scholar]
- 12. Wald K. M., Nada A. A., Szilágyi G., Wamhoff H., Chem. Ber. 1980, 113, 2884–2890. [Google Scholar]
- 13. Ohsawa A., Kawaguchi T., Igeta H., Synthesis 1983, 1983, 1037–1040. [Google Scholar]
- 14. Hulcoop D. G., Lautens M., Org. Lett. 2007, 9, 1761–1764. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Chai D. I., Lautens M., J. Org. Chem. 2009, 74, 3054–3061; [DOI] [PubMed] [Google Scholar]
- 15b. Panteleev J., Geyer K., Aguilar-Aguilar A., Wang L., Lautens M., Org. Lett. 2010, 12, 5092–5095; [DOI] [PubMed] [Google Scholar]
- 15c. Shukla S. P., Tiwari R. K., Verma A. K., J. Org. Chem. 2012, 77, 10382–10392; [DOI] [PubMed] [Google Scholar]
- 15d. Ye S., Liu J., Wu J., Chem. Commun. 2012, 48, 5028–5030; [DOI] [PubMed] [Google Scholar]
- 15e. Das A., Ghosh I., König B., Chem. Commun. 2016, 52, 8695–8698; [DOI] [PubMed] [Google Scholar]
- 15f. Kiruthika S. E., Nandakumar A., Perumal P. T., Org. Lett. 2014, 16, 4424–4427. [DOI] [PubMed] [Google Scholar]
- 16. Verma A. K., Shukla S. P., Singh J., Rustagi V., J. Org. Chem. 2011, 76, 5670–5684. [DOI] [PubMed] [Google Scholar]
- 17. Sun K., Chen X.-L., Zhang Y.-L., Li K., Huang X.-Q., Peng Y.-Y., Qu L.-B., Yu B., Chem. Commun. 2019, 55, 12615–12618. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Liu X., Li X., Liu H., Guo Q., Lan J., Wang R., You J., Org. Lett. 2015, 17, 2936–2939; [DOI] [PubMed] [Google Scholar]
- 18b. Okada T., Sakai A., Hinoue T., Satoh T., Hayashi Y., Kawauchi S., Chandrababunaidu K., Miura M., J. Org. Chem. 2018, 83, 5639–5649; [DOI] [PubMed] [Google Scholar]
- 18c. Thavaselvan S., Parthasarathy K., Org. Lett. 2020, 22, 3810–3814; [DOI] [PubMed] [Google Scholar]
- 18d. Karthikeyan I., Sekar G., Eur. J. Org. Chem. 2014, 2014, 8055–8063; [Google Scholar]
- 18e. Gao J., Xu B., Zhu J., Shao Y., Chen L., Zhu J., Wang X., Lv X., Arkivoc 2016, 2016, 1–14; [Google Scholar]
- 18f. Kong L., Yao Q., Wang M., Sun R., Li Y., ChemistrySelect 2018, 3, 456–460. [Google Scholar]
- 19.
- 19a. Lee H. S., Kim S. H., Kim Y. M., Kim J. N., Tetrahedron Lett. 2010, 51, 5071–5075; [Google Scholar]
- 19b. Thanetchaiyakup A., Rattanarat H., Chuanopparat N., Ngernmeesri P., Tetrahedron Lett. 2018, 59, 1014–1018; [Google Scholar]
- 19c. Xie C., Zhang Y., Huang Z., Xu P., J. Org. Chem. 2007, 72, 5431–5434; [DOI] [PubMed] [Google Scholar]
- 19d. Guo S., Liu Y., Zhang X., Fan X., Adv. Synth. Catal. 2020, 362, 3011–3020. [Google Scholar]
- 20.
- 20a. Takahashi I., Fujita T., Shoji N., Ichikawa J., Chem. Commun. 2019, 55, 9267–9270; [DOI] [PubMed] [Google Scholar]
- 20b. Wang X., Zhou L., Lu W., Curr. Org. Chem. 2010, 14, 289–307; [Google Scholar]
- 20c. Li H.-J., Guillot R., Gandon V., J. Org. Chem. 2010, 75, 8435–8449; [DOI] [PubMed] [Google Scholar]
- 20d. Martín-Matute B., Nevado C., Cárdenas D. J., Echavarren A. M., J. Am. Chem. Soc. 2003, 125, 5757–5766; [DOI] [PubMed] [Google Scholar]
- 20e. Fürstner A., Mamane V., J. Org. Chem. 2002, 67, 6264–6267; [DOI] [PubMed] [Google Scholar]
- 20f. Molander G. A., Knochel P. Comprehensive Organic Synthesis. II, Elsevier, Amsterdam, 2014. [Google Scholar]
- 21.
- 21a. Heckershoff R., Schnitzer T., Diederich T., Eberle L., Krämer P., Rominger F., Rudolph M., Hashmi A. S. K., J. Am. Chem. Soc. 2022, 144, 8306–8316; [DOI] [PubMed] [Google Scholar]
- 21b. Hendrich C. M., Sekine K., Koshikawa T., Tanaka K., Hashmi A. S. K., Chem. Rev. 2021, 121, 9113–9163; [DOI] [PubMed] [Google Scholar]
- 21c. Campeau D., León Rayo D. F., Mansour A., Muratov K., Gagosz F., Chem. Rev. 2021, 121, 8756–8867; [DOI] [PubMed] [Google Scholar]
- 21d. Zheng Z., Ma X., Cheng X., Zhao K., Gutman K., Li T., Zhang L., Chem. Rev. 2021, 121, 8979–9038; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21e. Dorel R., Echavarren A. M., Chem. Rev. 2015, 115, 9028–9072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21f. Fürstner A., Chem. Soc. Rev. 2009, 38, 3208–3221; [DOI] [PubMed] [Google Scholar]
- 21g. Hashmi A. S. K., Chem. Rev. 2007, 107, 3180–3211; [DOI] [PubMed] [Google Scholar]
- 21h. Hashmi A. S. K., Frost T. M., Bats J. W., J. Am. Chem. Soc. 2000, 122, 11553–11554; [Google Scholar]
- 21i. Hashmi A. S. K., Schwarz L., Choi J.-H., Frost T. M., Angew. Chem. Int. Ed. 2000, 39, 2285–2288; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2382–2385. [Google Scholar]
- 22.
- 22a. Qiu Y., Kong W., Fu C., Ma S., Org. Lett. 2012, 14, 6198–6201; [DOI] [PubMed] [Google Scholar]
- 22b. Xiao F., Chen Y., Liu Y., Wang J., Tetrahedron 2008, 64, 2755–2761; [Google Scholar]
- 22c. Pflästerer D., Rettenmeier E., Schneider S., de Las Heras Ruiz E., Rudolph M., Hashmi A. S. K., Chem. Eur. J. 2014, 20, 6752–6755; [DOI] [PubMed] [Google Scholar]
- 22d. Li X., Zhao J., Xie X., Liu Y., Org. Biomol. Chem. 2017, 15, 8119–8133. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Hirano K., Inaba Y., Takahashi N., Shimano M., Oishi S., Fujii N., Ohno H., J. Org. Chem. 2011, 76, 1212–1227; [DOI] [PubMed] [Google Scholar]
- 23b. Hirano K., Inaba Y., Watanabe T., Oishi S., Fujii N., Ohno H., Adv. Synth. Catal. 2010, 352, 368–372; [Google Scholar]
- 23c. Matsuda T., Moriya T., Goya T., Murakami M., Chem. Lett. 2011, 40, 40–41; [Google Scholar]
- 23d. Vacala T., Bejcek L. P., Williams C. G., Williamson A. C., Vadola P. A., J. Org. Chem. 2017, 82, 2558–2569. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Kawada Y., Ohmura S., Kobayashi M., Nojo W., Kondo M., Matsuda Y., Matsuoka J., Inuki S., Oishi S., Wang C., Saito T., Uchiyama M., Suzuki T., Ohno H., Chem. Sci. 2018, 9, 8416–8425; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Baralle A., Yorimitsu H., Osuka A., Chem. Eur. J. 2016, 22, 10768–10772; [DOI] [PubMed] [Google Scholar]
- 24c. Tsai C.-E., Yu R.-H., Lin F.-J., Lai Y.-Y., Hsu J.-Y., Cheng S.-W., Hsu C.-S., Cheng Y.-J., Chem. Mater. 2016, 28, 5121–5130; [Google Scholar]
- 24d. Alam K., Hong S. W., Oh K. H., Park J. K., Angew. Chem. Int. Ed. 2017, 56, 13387–13391; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13572–13576; [Google Scholar]
- 24e. Li Y., Gryn′ova G., Saenz F., Jeanbourquin X., Sivula K., Corminboeuf C., Waser J., Chem. Eur. J. 2017, 23, 8058–8065. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Maeda C., Tanaka Y., Shirakawa T., Ema T., Chem. Commun. 2019, 55, 10162–10165; [DOI] [PubMed] [Google Scholar]
- 25b. Zhao X., Ge C., Yang X., Gao X., Mater. Chem. Front. 2017, 1, 1635–1640; [Google Scholar]
- 25c. Saunthwal R. K., Danodia A. K., Saini K. M., Verma A. K., Org. Biomol. Chem. 2017, 15, 6934–6942; [DOI] [PubMed] [Google Scholar]
- 25d. Yamamoto Y., Matsui K., Shibuya M., Chem. Eur. J. 2015, 21, 7245–7255. [DOI] [PubMed] [Google Scholar]
- 26.Deposition Numbers2169589 (for 2 a, 2169590 for 3 a)contain(s) the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 27. Bochevarov A. D., Harder E., Hughes T. F., Greenwood J. R., Braden D. A., Philipp D. M., Rinaldo D., Halls M. D., Zhang J., Friesner R. A., Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar]
- 28.
- 28a. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104; [DOI] [PubMed] [Google Scholar]
- 28b. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652; [Google Scholar]
- 28c. Lee, Yang, Parr, Phys. Rev. B 1988, 37, 785–789; [DOI] [PubMed] [Google Scholar]
- 28d. Hay P. J., Wadt W. R., J. Chem. Phys. 1985, 82, 299–310; [Google Scholar]
- 28e. Hariharan P. C., Pople J. A., Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar]
- 29. Marten B., Kim K., Cortis C., Friesner R. A., Murphy R. B., Ringnalda M. N., Sitkoff D., Honig B., J. Phys. Chem. 1996, 100, 11775–11788. [Google Scholar]
- 30.
- 30a. Schießl J., Stein P. M., Stirn J., Emler K., Rudolph M., Rominger F., Hashmi A. S. K., Adv. Synth. Catal. 2019, 361, 725–738; [Google Scholar]
- 30b. Henrion G., Chavas T. E. J., Le Goff X., Gagosz F., Angew. Chem. Int. Ed. 2013, 52, 6277–6282; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6397–6402; [Google Scholar]
- 30c. Sekine K., Stuck F., Schulmeister J., Wurm T., Zetschok D., Rominger F., Rudolph M., Hashmi A. S. K., Chem. Eur. J. 2018, 24, 12515–12518; [DOI] [PubMed] [Google Scholar]
- 30d. Khin C., Hashmi A. S. K., Rominger F., Eur. J. Inorg. Chem. 2010, 1063–1069. [Google Scholar]
- 31.
- 31a. Niu T., Zhang Y., Tetrahedron Lett. 2010, 51, 6847–6851; [Google Scholar]
- 31b. Li Y., Wang W.-H., Yang S.-D., Li B.-J., Feng C., Shi Z.-J., Chem. Commun. 2010, 46, 4553–4555; [DOI] [PubMed] [Google Scholar]
- 31c. Wirtanen T., Mäkelä M. K., Sarfraz J., Ihalainen P., Hietala S., Melchionna M., Helaja J., Adv. Synth. Catal. 2015, 357, 3718–3726; [Google Scholar]
- 31d. Umareddy P., Arava V. R., Synth. Commun. 2019, 49, 2156–2167; [Google Scholar]
- 31e. Daniels M., de Jong F., Kennes K., Martín C., Hofkens J., van der Auweraer M., Dehaen W., Eur. J. Org. Chem. 2018, 2018, 4683–4688. [Google Scholar]
- 32.The core structure is mentioned in following patent, but no synthesis or characterization data is provided: Li, Jian (Arizona State University), US20180334459, 2018.
- 33.
- 33a. Chamorro E., Pérez P., J. Chem. Phys. 2005, 123, 114107; [DOI] [PubMed] [Google Scholar]
- 33b. Contreras R. R., Fuentealba P., Galván M., Pérez P., Chem. Phys. Lett. 1999, 304, 405–413. [Google Scholar]
- 34. Bredas J.-L., Mater. Horiz. 2014, 1, 17–19. [Google Scholar]
- 35.
- 35a. Chen M.-C., Chen D.-G., Chou P.-T., ChemPlusChem 2021, 86, 11–27; [DOI] [PubMed] [Google Scholar]
- 35b. Friese D. H., Mikhaylov A., Krzeszewski M., Poronik Y. M., Rebane A., Ruud K., Gryko D. T., Chem. Eur. J. 2015, 21, 18364–18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.