

Abstract
Dear Editor, X‐linked ichthyosis (XLI) is the second most common ichthyosis after ichthyosis vulgaris (IV). 1 Despite their differences, it can be challenging to clinically distinguish the different patient groups. Whereas IV is mainly caused by loss‐of‐function variations in the filaggrin gene (FLG), 2 , 3 XLI is predominantly (85–90%) caused by deletion of the entire steroid sulfatase gene (STS). 1 , 4 Nevertheless, other pathogenic variants are found, currently encompassing 81 unique pathogenic variants in the Leiden Open Variation Database (LOVD v.3.0).
In this large Dutch cohort, 109 male patients clinically suspected of having XLI were included for genetic diagnosis at the Maastricht UMC+ by the patient’s dermatologist or clinical geneticist affiliated with various Dutch hospitals. Only limited clinical information on the patients’ phenotypes was available. The institutional review board of the Maastricht UMC+ approved the study. All DNA samples were first analysed by multiplex ligation‐dependent probe amplification (MLPA) (kit P160‐C1; MRC Holland, Amsterdam, the Netherlands) for copy number variant detection to identify entire gene deletions of STS, often found in XLI. If MLPA was normal, subsequent Sanger sequencing of the STS coding exons and splice sites was performed. Enzymatic STS activity was tested in patients’ leucocytes (Amsterdam UMC, Laboratory Genetic Metabolic Diseases) to further classify variants, when predictive computational analyses was not conclusive (Alamut Visual v.2.15; SOPHiA GENETICS, Saint Sulpice, Switzerland). Sequential targeted single‐molecule molecular inversion probe analysis for FLG 3 and/or whole‐exome sequencing 5 was requested in some patients only when considered necessary by the patient’s physician.
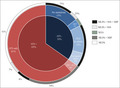
In 71 patients (65%) an STS pathogenic variant was detected (Figure 1a). The majority of patients with XLI (57, 80%), showed the frequently found genomic deletion of the entire STS gene and flanking PUDP sequence containing at least chrX.hg19:g.(?_6968202)_(7269849_?)del. This finding more accurately portrays the frequency in comparison with previous, smaller cohort studies. 1 , 4 Detection of copy number variants further uncovered a deletion of the complete STS gene without the PUDP sequence, an exon 6 deletion and a duplication of exons 8–9 in three individual patients. Subsequent Sanger sequencing identified one patient with a splice‐site variant, six patients with missense variants, one patient with a nonsense variant, and three patients with a frameshift variant (Figure 1b). The variants were classified according to the American College of Medical Genetics guidelines. 6 The variant c.1253A>G p.(Asp418Gly) was classified as likely to be benign due to normal STS enzymatic activity in the patient’s blood. As a result, 11 novel STS (probable) pathogenic variants were identified, of which four resulted in no enzymatic activity (Figure 1b). The remaining novel missense variants were primarily classified based on variants published as pathogenic at equivalent positions in paralogue sulfatase genes (GALNS, ARSL). In total, the number of known mutations in LOVD was increased by 14%.
Figure 1.

(a) Distribution of pathogenic variants in the Dutch cohort. The inner circle represents the proportion of patients with variants in STS. The middle circle shows patients with only STS variants, concomitant variants, FLG variants, autosomal recessive congenital ichthyosis (ARCI)‐associated variants, or no pathogenic variants. The outermost circle represents the genetic analysis performed within subgroups of the cohort. MIP: FLG single‐molecule molecular inversion probe analysis, MLPA: multiplex ligation‐dependent probe amplification, SSS: Sanger sequencing STS, WES: whole‐exome sequencing. (b) Distribution of novel pathogenic variants in STS. Gene structure (scaled) of STS: 10 exons (with alternate exons 1a and 1b representing different isoforms) and two deletions (red), one duplication (blue), and nucleotide variants (arrows) for each patient. Data are based on NCBI RefSeq:NM_000351.5. #Steroid sulfatase (STS) enzymatic activity absent. All variants were submitted to the public Leiden Open Variation Database with their corresponding classification.
Additional analysis of FLG was performed in 66 patients (61%). In 23 patients (21% of the total cohort) loss‐of‐function variants in FLG were detected, which had a similar variant distribution to the Dutch population (72% frequent, 28% rare), 3 as well as one novel FLG variant, NM_002016.2:c.4297G>T p.(Glu1433*). Of these, 19 patients (17%, six of 19 heterozygotes) were incorrectly clinically diagnosed with XLI while actually having IV. This emphasizes the challenges that physicians encounter in clinically distinguishing the different forms of ichthyosis. In at least four cases (4%) the FLG variant was concomitant with an STS pathogenic variant (Figure 1a), potentially exacerbating XLI in these patients. 2 The modifying effect could not be evaluated in these patients, as an intrafamilial reference patient only having the STS variant was not available.
Furthermore, whole‐exome sequencing in three patients identified previously published pathogenic variants in SDR9C7: NM_148897.3:c.[551A>G];[703G>A] p.[(Asp184Gly)];[(Gly235Arg)], ALOXE3: NM_021628.3:c.[1889C>T];[1889C>T] p.[(Pro630Leu)];[(Pro630Leu)], and NIPAL4: NM_001099287.1:c.[527C>A];[527C>A] p.[(Ala176Asp)];[(Ala176Asp)], associated with types of autosomal recessive congenital ichthyosis (ARCI).
For 43 patients (39% of the total) sequential analysis was not requested. In 16 patients (15%) no pathogenic variants were identified with the tests performed. Therefore, it is conceivable that some patients only tested for STS and without variants (four of 16 patients, 25%) could actually have variants associated with IV or ARCI. Moreover, the proportion of patients carrying concomitant pathogenic variants of STS and FLG may be underestimated as only 30 of 71 patients with confirmed XLI (42%) were tested for both genes (Figure 1a).
Confirmation of diagnoses through genetic analyses is important as the clinical consequences are different for patients with XLI and IV. Patients with XLI need genetic counselling as their mothers and future daughters, being obligate carriers, may have a complicated delivery of (another) affected son, 7 while patients with IV are prone to develop atopic dermatitis, allergies and asthma. 8 Considering our results and all these factors influencing the diagnostic process in XLI, we recommend analysis for both STS and FLG in male patients clinically suspected for XLI, and extended analysis should be considered for other ichthyosis subtypes (ARCIs) when genetic diagnosis remains elusive.
Author contributions
Ivo F. Nagtzaam: Conceptualization (equal); data curation (equal); formal analysis (equal); investigation (equal); project administration (equal); resources (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Frank van Leersum: Conceptualization (equal); investigation (equal); resources (equal); writing – review and editing (equal). Laurie C.M. Kouwenberg: Conceptualization (equal); investigation (equal); visualization (equal); writing – original draft (equal). Marinus J. Blok: Conceptualization (equal); data curation (equal); writing – review and editing (equal). Maaike Vreeburg: Conceptualization (equal); resources (equal); writing – review and editing (equal). Peter M. Steijlen: Conceptualization (equal); resources (equal); writing – review and editing (equal). Antoni Gostynski: Conceptualization (equal); formal analysis (equal); investigation (equal); project administration (equal); resources (equal); writing – review and editing (equal). Michel van Geel: Conceptualization (equal); data curation (lead); formal analysis (lead); investigation (lead); methodology (lead); project administration (lead); resources (equal); supervision (lead); visualization (equal); writing – original draft (equal); writing – review and editing (lead).
Acknowledgments
We would like to thank all patients and medical specialists, and the technical support team for genetic testing, who contributed to this Dutch cohort study. This project is part of the European Reference Network – SKIN thematic group Ichthyosis and Palmoplantar Keratoderma (https://ern‐skin.eu).
I.F.N. and F.S.L. contributed equally to this article.
Funding sources: none.
Conflicts of interest: the authors declare they have no conflicts of interest.
Data availability statement: Data available on request from the authors.
References
- 1. Diociaiuti A, Angioni A, Pisaneschi E et al. X‐linked ichthyosis: clinical and molecular findings in 35 Italian patients. Exp Dermatol 2019; 28:1156–63. [DOI] [PubMed] [Google Scholar]
- 2. Sussmuth K, Gruber R, Rodriguez E et al. Increased prevalence of filaggrin deficiency in 51 patients with recessive X‐linked ichthyosis presenting for dermatological examination. J Invest Dermatol 2018; 138:709–11. [DOI] [PubMed] [Google Scholar]
- 3. Van Leersum FS, Nagtzaam IF, van Oosterhoud CN et al. Improving the diagnostic yield for filaggrin: concealed mutations in the Dutch population. J Allergy Clin Immunol 2020; 145:1704–6. [DOI] [PubMed] [Google Scholar]
- 4. Ballabio A, Carrozzo R, Parenti G et al. Molecular heterogeneity of steroid sulfatase deficiency: a multicenter study on 57 unrelated patients, at DNA and protein levels. Genomics 1989; 4:36–40. [DOI] [PubMed] [Google Scholar]
- 5. Corominas J, Smeekens SP, Nelen MR et al. Clinical exome sequencing – mistakes and caveats. Hum Mutat 2022; 43:1041–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elias PM, Williams ML, Choi EH, Feingold KR. Role of cholesterol sulfate in epidermal structure and function: lessons from X‐linked ichthyosis. Biochim Biophys Acta 2014; 1841:353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Traupe H, Fischer J, Oji V. Nonsyndromic types of ichthyoses – an update. J Dtsch Dermatol Ges 2014; 12:109–21. [DOI] [PubMed] [Google Scholar]