

Abstract
Bimetallic motifs mediate the selective activation and functionalization of CO2 in metalloenzymes and some recent synthetic systems. In this work, we build on the nascent concept of bimetallic frustrated Lewis pairs (FLPs) to investigate the activation and reduction of CO2. Using the Fe0 fragment [(depe)2Fe] (depe=1,2‐bis(diethylphosphino)ethane) as base, we modify the nature of the partner Lewis acid to accomplish a divergent and highly chemoselective reactivity towards CO2. [Au(PMe2Ar)]+ irreversibly dissociates CO2, Zn(C6F5)2 and B(C6F5)3 yield different CO2 adducts stabilized by push‐pull interactions, while Al(C6F5)3 leads to a rare heterobimetallic C−O bond cleavage, and thus to contrasting reduced products after exposure to dihydrogen. Computational investigations provide a rationale for the divergent reactivity, while Energy Decomposition Analysis‐Natural Orbital for Chemical Valence (EDA‐NOCV) method substantiates the heterobimetallic bonding situation.
Keywords: Bimetallic, CO2 Activation, Cooperativity, Lewis Acids, Push-Pull Interactions
Contrasting CO2 activation reactivity using bimetallic Fe0/Lewis acid combinations is reported. The use of gold promotes irreversible CO2/N2 exchange, whereas push‐pull CO2 fixation structures emerge from the use of acidic zinc and borane compounds. The use of an alane, Al(C6F5)3, promotes bimetallic C−O bond cleavage. Computational studies support the observed divergent reactivity and substantiate the bonding scenario.
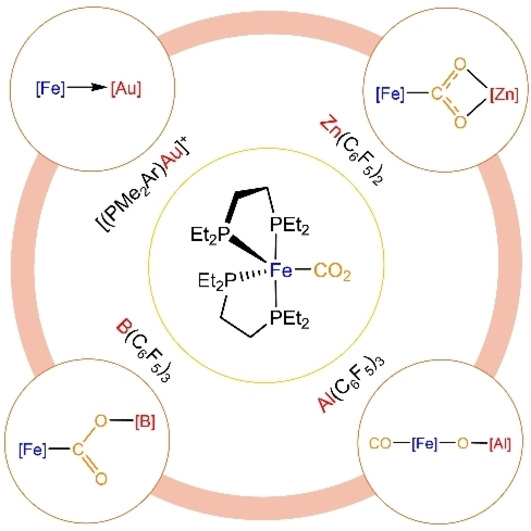
Introduction
The selective transformation of CO2 into reduced C1 products is one of the most challenging and environmentally appealing reactions pursued by chemists in the last decades. [1] Inspiring designing principles come out from nature, where CO‐dehydrogenase (CODH) enzymes mediate the redox interconversion between CO2 and CO. Crucial for their efficiency are their Ni/Fe and Mo/Cu bimetallic active sites, [2] capable of cooperatively activating carbon dioxide by means of complementary Lewis acidic/Lewis basic behaviour of the two metals. Not surprisingly, artificial bioinspired approaches that rely on this type of bimetallic synergism have attracted great attention in recent times.[ 3 , 4 ] In a similar vein, Frustrated Lewis pairs (FLPs) operate upon analogous premises for the activation of CO2, [5] including metal‐containing FLP‐type structures [6] that resemble as well the active site of CODH enzymes. [7] It has also been well recognized that Lewis acidic metals drastically influence the capacity of redox active sites to reduce CO2, [8] either by serving as Z‐type ligands [9] or by directly contributing to stabilize key intermediates during CO2 reduction. [10]
Regardless of the precise mode of action, the use of bimetallic combinations based on non‐precious metals to reduce CO2 remains an important challenge. Mankad and co‐workers have very recently described a heterobimetallic complex based on the two most abundant metals in the Earth crust, iron and aluminium, which activate CO2 via radical intermediates. [11] The use of aluminium has indeed shown success in other bimetallic combinations, both with transition [12] and main group [13] metals. In fact, a recent study by Camp et al. evidenced the cleavage of a C−O bond of CO2 by induced umpolung reactivity at an Ir/Al system. [14] Reverse polarity has been exploited by Aldridge and Goicoechea for group 11‐aluminyl bimetallic complexes in CO2 activation [15] (Figure 1a). Inspired by these results and building on our prior studies on bimetallic FLPs, [16] we decided to investigate the cooperative activation of CO2 using the synthon [(depe)2Fe] (depe=1,2‐bis(diethylphosphino)ethane) as a Fe0 metallic base. We report divergent CO2 reactivity resulting from varying the nature of the partner Lewis acid. This spans from CO2 dissociation in the case of gold to different modes of bimetallic activation for fluorinated zinc and borane Lewis acids and even C−O bond cleavage in the case of the highly electrophilic Al(C6F5)3 (Figure 1b).
Figure 1.
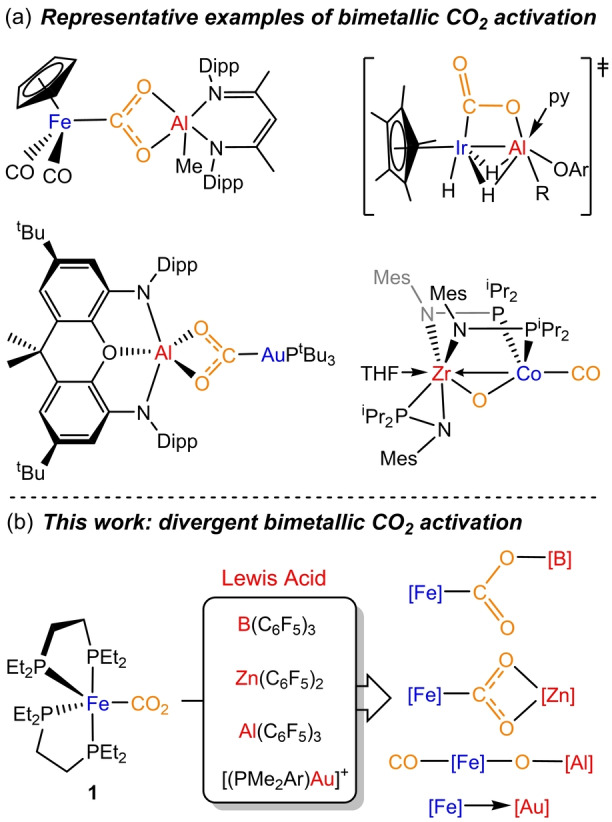
a) Representative examples of bimetallic CO2 activation pathways relevant to the present work; b) divergent bimetallic CO2 activation routes as a function of the Lewis acid (this work).
Results and Discussion
We decided to use the Fe0 fragment [(depe)2Fe] as a metallic Lewis base because its corresponding CO2 adduct (1) is readily accessible [17a] and exhibits a rich carbon dioxide functionalization chemistry.[ 17b , 17c ] Besides, Szymczak and co‐workers used the latter to investigate the activation of dinitrogen by push‐pull interactions. [18] In line with that work, we started our investigations by treating complex [(depe)2Fe(CO2)] (1) with the acidic borane B(C6F5)3 (Scheme 1a). The addition of the borane to toluene or benzene solutions of 1 promotes an immediate change in colour from orange to pink solutions due to the formation of complex 2, which could be isolated in 82 % yield after the workup.
Scheme 1.
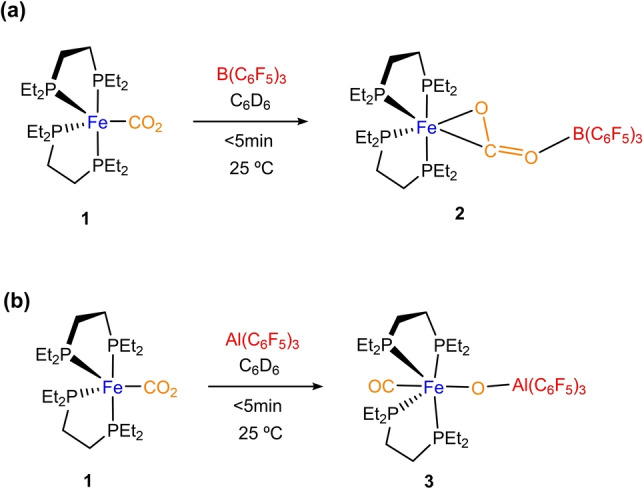
Activation of CO2 by treating Fe−CO2 adduct 1 with Lewis acids B(C6F5)3 (a) and Al(C6F5)3 (b).
Monitoring the reaction by 31P{1H} NMR we observed the complete consumption of the starting iron species (87.0, 79.0, 70.0 and 62.6 ppm) and the appearance of a new set of three broad signals (85.1, 80.4 and 70.4 ppm, with relative intensities 1 : 1 : 2 in benzene‐d 6). The corresponding 11B{1H} NMR spectrum exhibited a broad signal at −1.6 ppm similar to the chemical shift observed for the somewhat related [(depe)2Fe(N2)B(C6F5)3] product (−6.3 ppm) from N2‐activation. [18] IR spectroscopy provided relevant information, as two stretch bands were found at 1641 cm−1 and 1251 cm−1 indicative of a CO2 unit.[ 7 , 8 ] Moreover, a 13C{1H} NMR signal at 214.8 ppm in thf‐d 8 evidences that a carbonyl group is still retained in complex 2. [19]
We were able to confirm its structure as the binuclear product [(depe)2Fe(μ‐CO2)B(C6F5)3] (2) (Figure 2a), in which the activated CO2 bridges the Fe/B pair in a μ‐CO2−1κO 1:C 1:2κO 2 fashion. The molecular structure is comparable to the other four crystallized bridging CO2‐adducts stabilized by push‐pull interactions between a basic metal (Ni, Pt, Mo and Re) and a borane.[ 6a , 6b , 8 ] η 2 ‐ Coordination to iron is evidenced by the corresponding Fe1−O2 (2.212(2) Å) and Fe1−C21 (1.861(3) Å) bond lengths. The other oxygen atom is linked to the borane with an O1−B1 bond length of 1.531(5) Å. The O2−C21−O1 angle of 126.61(3)° is practically identical to that one found for precursor 1 (124(2)°) (Figure 2a). [17] Nonetheless, the presence of the borane results in the elongation of the connected C21−O1 bond (1.305(4) Å) compared to the one bound to iron (C21−O2, 1.249(3) Å), being also the former C21−O1 bond longer than in adduct 1 (1.25(3) and 1.28(2) Å).
Figure 2.
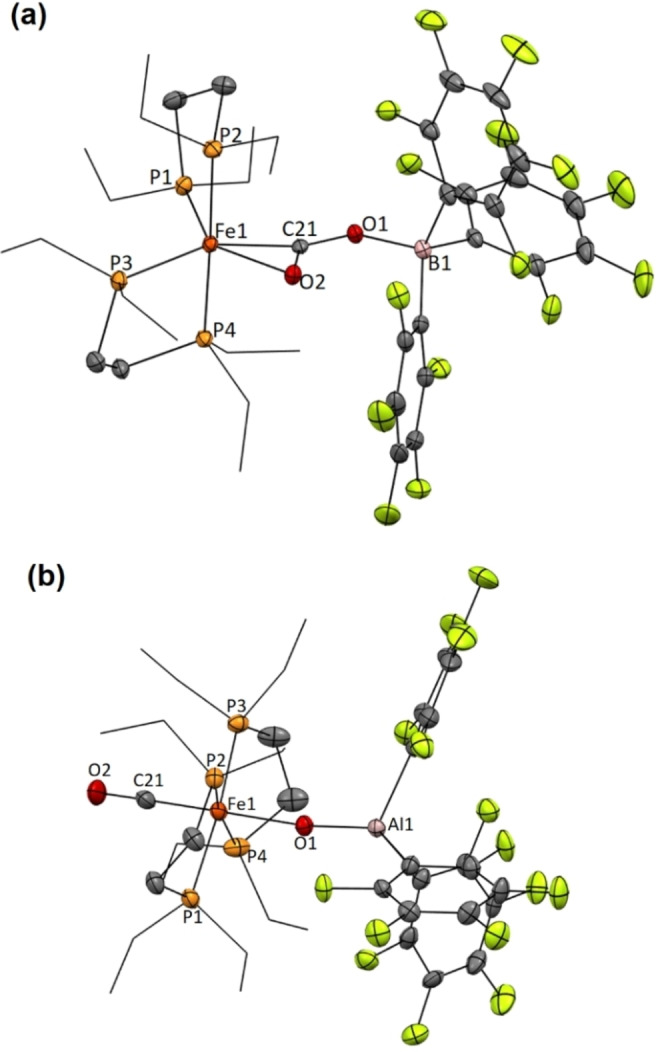
ORTEP diagrams for complexes 2 (a) and 3 (b). Hydrogen atoms have been excluded, and ethyl groups of depe ligand are represented in wire‐frame format for clarity. Thermal ellipsoids are set at 50 % probability.
Increasing the acidity of the Lewis acid partner by treating 1 with the analogous Al(C6F5)3, [20] likewise resulted in an immediate reaction leading to a yellow solution in benzene‐d 6. However, the first hint pointing to a contrasting reactivity arose from 31P{1H} NMR analysis, where a single signal was recorded at 67.9 ppm. At variance with compound 2, this suggests the formation of a new compound (3) with high symmetry (Scheme 1b). Moreover, its corresponding IR spectrum presents an intense band at 1914 cm−1, characteristic of a CO moiety. Crystals suitable for X‐ray diffraction studies were grown by placing a concentrated toluene solution of 3 in the glovebox freezer at −30 °C for one day (27 % yield), revealing the bimetallic cleavage of a C−O bond to yield [(depe)2(CO)Fe(μ‐O)Al(C6F5)3] (Figure 2b). The 13C{1H} NMR experiment of complex 3 (in thf‐d 8) features a signal at 213.6 ppm which further confirms the presence of a CO ligand. [19] For comparison, we performed the analogous reaction using AlEt3 as the electrophilic fragment. Although the resulting product could not be isolated, we were able to record a 31P{1H} NMR spectrum (Figure S13) which exhibited the same pattern and multiplicity as complex 2, suggesting the formation of a similar CO2‐adduct and pointing out to the need of the perfluorinated substituents to accomplish the cleavage the C−O bond.
The formation of 3 constitutes a highly unusual event from different standpoints. First, while examples of bimetallic CO2 activation have grown in number in the last decade, [3] those in which the cleavage of a C−O bond is achieved are more limited. [21] In like manner, heterobimetallic versions are even scarcer.[ 14 , 22 ] Structurally, compound 3 represents the first example of an oxo‐bridged Fe/Al complex, which is of interest considering the limited accessibility but high reactivity of oxo‐bridged heterobimetallic motifs, for instance in the context of C−H bond functionalization. [23] The Fe1−O1−Al1 angle is almost linear (171.9(4)°), while the O1−Al1 and Fe1−O1 bonds feature distances of 1.685(7) and 2.007(7) Å, respectively. Note that the latter is larger than those reported for oxo‐bridged diiron complexes, [24] probably as a result of the interaction with the highly acidic and crowded alane fragment. The Fe(CO) group presents the characteristic structural data for an iron carbonyl fragment [25] and is in trans position to the Fe−(μ‐O)Al bond.
We next wondered whether the contrasting reactivity of Fe/B vs Fe/Al pairs would result in dissimilar reduction pathways for CO2. Firstly, it is important to remark that the reaction of 1 with dihydrogen leads to the quantitative formation of the previously described FeII dihydride species [(depe)2Fe(H)2], [26] a result that is in stark contrast to the reaction outcomes in the presence of both borane and alane. Thus, exposure of 2 to dihydrogen under mild conditions (1 bar, 25 °C) resulted in the formal hydrogenation of the Fe−C bond towards compound 4 in a quantitative manner (Scheme 2a). This ion pair exhibits distinctive resonances at 92.3 ppm and −3.0 ppm in the 31P{1H} and 11B{1H} NMR spectra, respectively. Besides, a characteristic 1H NMR signal at 8.6 ppm due to the formate and two low‐frequency signals at −11.0 and −15.1 ppm due to the dihydrogen and hydride ligands respectively, which match prior data for the individual fragments of 4, [27] further confirm its formulation (Figures S14–S16). In accordance, its IR spectrum reveals a band at 1643 cm‐1 assigned to the ν(C=O) stretching vibration (Figure S17).[ 27c , 28 ]
Scheme 2.
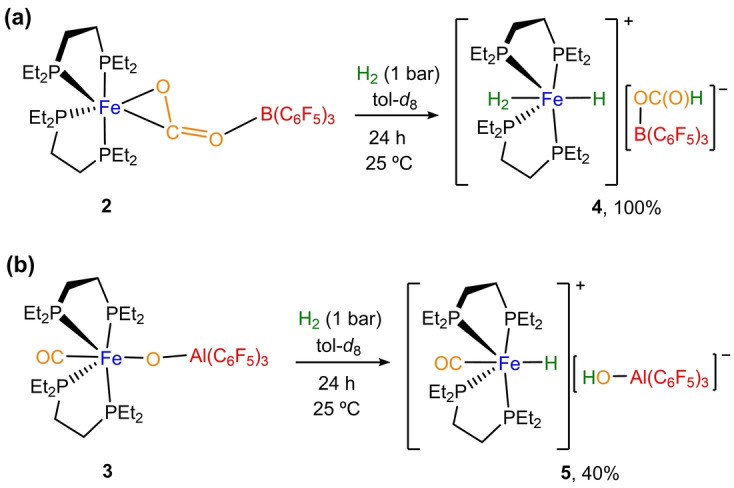
Reactivity of compounds 3 and 4 with H2. Spectroscopic yields were measured by 31P{1H} NMR using triphenyl phosphine oxide as internal standard.
Interestingly, when complex 3 is exposed to dihydrogen atmosphere under the same reaction conditions, the oxo Fe(μ‐O)Al bridge is broken resulting in the appearance of a new Fe−H fragment inferred from a distinctive low‐frequency signal in the 1H NMR spectrum at −14.7 ppm (br). This hydride ligand belongs to a new major species (5) that is formed in around 40 % spectroscopic yield and resonates in the 31P{1H} NMR spectrum at 65.4 ppm (Figures S18–S19). IR spectroscopy of the reaction mixture revealed two bands at 1902 and 3396 cm−1 (Figure S20). While we attribute the former to the corresponding CO ligand of 5, the latter broad band accounts for the formation of a new OH bond. We attribute the moderate yield measured for compound 5, at least in part, to its poor solubility in toluene. In fact, performing the reaction in a more polar solvent such as CD2Cl2, we could achieve full conversion of 3 and formation of 5 in better yields (ca. 75 %), although accompanied by the appearance of other minor unidentified species (Figure S21). These results are in agreement with the hydrogenolysis of the Fe−O bond in 3 to yield compound 5 (Scheme 2b), once more matching the spectroscopic data available for related ion pairs based on the [(depe)2Fe(H)(CO)]+ cation. [29] Overall, this sequential transformation is reminiscent of that mediated by CODH enzymes, where bimetallic CO2 activation is followed by reduction with an electron transfer reagent, dihydrogen in the present case.
The complete selectivity towards either CO2 activation or C−O bond cleavage resulting from an apparently innocent modification of the Lewis acidic site prompted us to investigate the bimetallic mechanism in more detail. To this end, Density Functional Theory (DFT) calculations were carried out at the dispersion corrected PCM(toluene)‐BP86‐D3/def2‐TZVPP//BP86‐D3/def2‐SVP level. Figure 3 shows the computed reaction profiles for the reactions of [(depe)2Fe(CO2)] (1) with B(C6F5)3 and Al(C6F5)3.
Figure 3.

Computed reaction profiles for the reactions of [(depe)2Fe(CO2)] (1) with B(C6F5)3 and Al(C6F5)3. Relative free energies (ΔG, at 298 K) are given in kcal mol−1. All data have been computed at the PCM(toluene)‐BP86‐D3/def2‐TZVPP//BP86‐D3/def2‐SVP level.
In both cases, the reaction begins with the highly exergonic formation of the dative bond between the lone‐pair of the O(=C) moiety of 1 and the vacant p atomic orbital of the group 13 atom of the Lewis acid leading to the observed species 2 and the analogous complex 2‐Al. Although both processes are highly exergonic, the higher Lewis acidity of Al(C6F5)3 becomes evident from the much higher exergonicity computed for the formation of 2‐Al (ΔG R=−46.6 kcal mol−1 vs −23.0 kcal mol−1). Species 2‐Al is then transformed into INT1 through TS1, a saddle point associated with the slippage of the bridging CO2 ligand with a feasible activation barrier of 17.7 kcal mol−1 in a slightly endergonic transformation (ΔG R=7.4 kcal mol−1). Intermediate INT1 evolves to INT2 via TS2, which is associated with the rupture of the OC−O bond. This crucial step requires a low barrier of only 9.1 kcal mol−1 and is again slightly endergonic (ΔG R=2.9 kcal mol−1). The transformation ends up with the isomerization of INT2, where the carbonyl ligand is placed cis to the O−Al moiety, into the corresponding trans‐3 complex. This final step is highly exergonic (ΔG R=−16.6 kcal mol−1) and compensates the previous endergonic steps therefore driving the complete transformation forward towards the formation of the observed complex 3. A similar reaction profile was computed for the boron counterpart 2, but at variance, the formation of the related species 3‐B is thermodynamically not favoured. Despite the computed barriers involving TS1‐B and TS2‐B are again feasible at room temperature (ΔG ≠=22.4 and 15.6 kcal mol−1, respectively), a similar thermodynamic driving force to that commented above for the profile involving Al(C6F5)3 is lacking for the process involving B(C6F5)3 and for this reason, the experimentally observed complex 2 remains the global minimum on the potential energy surface. The transient formation of 2‐Al could be substantiated by monitoring the reaction between 1 and Al(C6F5)3 at −80 °C, which starts with the formation of a new species characterized by 31P{1H} NMR resonances at 87.2, 80.3, 74.1 and 70.0 ppm that cleanly evolves to compound 3 upon mild warm‐up.
The above divergent reactivity encouraged us to examine the role of other metallic Lewis acids to be combined with CO2‐adduct 1. We first investigated the addition of the highly electrophilic AuI compound [(PMe2ArXyl2)Au(NTf2)], [30] where ArXyl2 stands for C6H3‐2,6‐(C6H3‐2′,6′‐(CH3)2)2 and NTf2 for [N(SO2CF3)2]−. This AuI species has been exploited in our group for the design of bimetallic FLPs, [16] since it contains a sterically shielding terphenyl phosphine and a weakly‐coordinating triflimidate ligand that facilitates access to the aimed electrophilic [(PMe2ArXyl2)Au]+ moiety. Treating 1 with an equimolar amount of [(PMe2ArXyl2)Au(NTf2)] under nitrogen atmosphere (Scheme 3) resulted in dissociation of the CO2 ligand and formation of an Fe0→AuI metal‐only Lewis pair (6). [31] In fact, the same compound can be easily prepared by adding [(PMe2ArXyl2)Au(NTf2)] to the nitrogen adduct [(depe)2Fe(N2)].
Scheme 3.
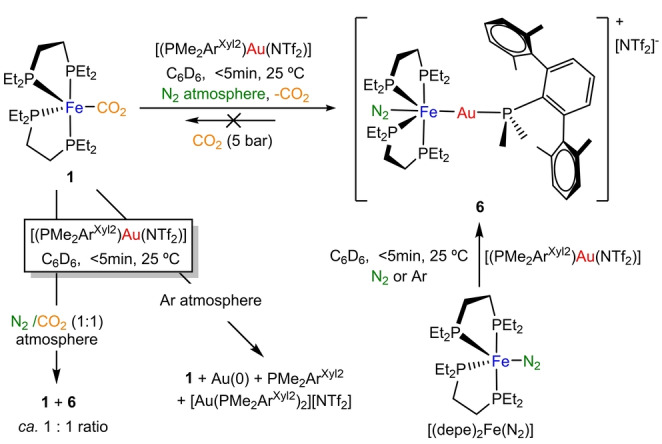
Reactivity of 1 with gold complex [(PMe2ArXyl2)Au(NTf2) as a function of the atmosphere.
Interestingly, the former reaction entails an irreversible ligand substitution defined by the coordination of a N2 molecule from the reaction atmosphere, in a trans position towards the Fe−Au bond. It is important to remark that substitution of CO2 by the very weakly coordinating N2 ligand is rare [7] and, for this system, it has only been observed in the presence of gold, further certifying the crucial influence exerted by coordination of Lewis acids to transition metals. Formation of 6 from either [(depe)2Fe(CO2)] or [(depe)2Fe(N2)] contrasts with prior studies from Simonneau where AuI compounds are prone to participate in push‐pull interactions to activate dinitrogen, [32] while herein the formation of the Fe0→AuI dative bond prevails. Changing the atmosphere to an approximate equimolar N2/CO2 mixture led to the presence of both precursor 1 and bimetallic adduct 6, along with other decomposition products. In contrast, under argon atmosphere we could only observe decomposition of the gold complex, forming a black solid precipitate, free terphenyl phosphine and [Au(PMe2ArXyl2)2][NTf2], as detected by 31P{1H} NMR monitoring experiments, which evinces the surprisingly important stabilizing role played by the dinitrogen ligand. Nonetheless, complex 6 is thermally unstable and decomposes to metallic gold and free phosphine after minutes in benzene or toluene solutions. Attempts to prepare analogous species with bulkier phosphine ligands (PMe2ArDipp2 or PCyp2ArXyl2) directly led to decomposition products.
Despite its reduced stability, we managed to characterize compound 6 by spectroscopic means (1H, 31P NMR and IR spectroscopy) and authenticate its molecular structure by X‐ray diffraction studies (Figure 4). It exhibits two characteristic signals in the 31P{1H} NMR spectrum, a doublet at 72.4 ppm (3 J PP=42 Hz) and a quintet at 31.0 pm (3 J PP=42 Hz). The N≡N bond of the coordinated dinitrogen is slightly shorter (1.12(2) Å) than in its monometallic parent species [(depe)2Fe(N2)] (1.139(13) Å), [33] which we attribute to reduced donation from Fe0 to a π*(NN) orbital as a result of the competing donation from iron to gold (see below). In fact, the Fe−N bond in 6 is elongated to 1.83(1) Å compared to 1.748(8) Å in [(depe)2Fe(N2)], while the formation of a dative Fe→Au bond is characterized by a distance of 2.530(2) Å (Figure 4). All these metrics are in accordance with an IR stretching band for N2 in complex 6 sharply shifted to 2057 cm−1 with respect to 1955 cm−1 found for [(depe)2Fe(N2)]. [33]
Figure 4.
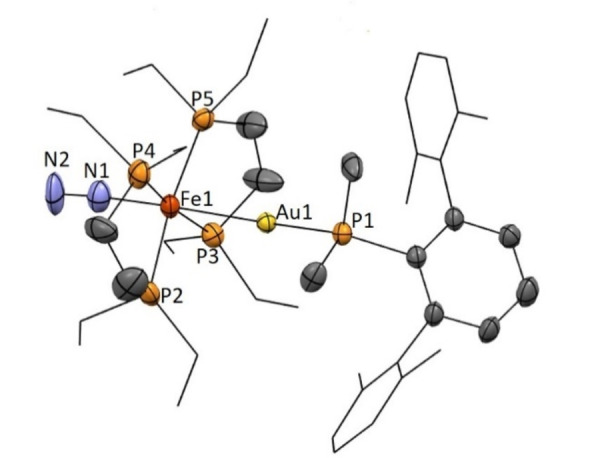
ORTEP diagram for complex 6+ . Hydrogen atoms have been excluded, and ethyl groups of depe ligand and flanking aryl rings of the terphenyl phosphine are represented in wire‐frame format for clarity. Thermal ellipsoids are set at 50 % probability.
The particular bonding situation of the newly prepared complex 6 in comparison with the parent species [(depe)2Fe(N2)] deserves further analysis. With the help of the Energy Decomposition Analysis‐Natural Orbital for Chemical Valence (EDA‐NOCV) method, we first compared the nature of the Fe‐N2 bond in [(depe)2Fe(N2)] and the bimetallic cation [(N2)(depe)2Fe−Au(PMe2ArXyl2)]+ (6+ ) at the relativistic and dispersion corrected ZORA‐BP86‐D3/TZ2P//BP86‐D3/def2‐SVP level. Our calculations indicate that the presence of the gold(I) fragment makes the Fe‐N2 bond weaker as confirmed by the computed lower interaction energy in 6+ (see Table S1 in the Supporting Information). The partitioning of the ΔE int term indicates that the strength of the orbital and electrostatic interactions is comparable being the former slightly stronger in both species. Interestingly, three different orbital interactions dominate the total ΔE orb term, namely the σ‐donation from the N2 to the transition metal fragment (denoted as ΔE(ρ 1)) and two π‐backdonations from the transition metal into both π* molecular orbitals of the N≡N ligand (denoted as ΔE(ρ 2) and ΔE(ρ 3), respectively, Figure 5). From the data in Table S1 and Figure 5, it becomes evident that the out‐of‐plane π‐backdonation (ρ 2) is significantly weaker (ΔΔE(ρ 2)=20 kcal mol−1) in the cationic complex 6+ as a result of the reduced electronic density at the Fe0 centre by coordination to the AuI fragment. As a consequence, the N≡N bond is stronger in 6 which is reflected in the observed higher IR stretching and shorter distance. Finally, we also analyzed the Fe−Au bond in 6+ with the EDA‐NOCV method. Although the Fe−Au interaction is mainly electrostatic (the ΔE elstat term contributes ca. 56 % to the total ΔE int), our calculations confirm the dative nature of the Fe0→AuI bond (associated ΔE(ρ)=−68.4 kcal mol−1, see Table S1) involving the donation from a doubly‐occupied d atomic orbital of Fe0 to the vacant s atomic orbital of AuI. [34]
Figure 5.

Contour plots of NOCV deformation densities Δρ and associated energies ΔE(ρ) (computed at the ZORA‐BP86‐D3/TZ2P//BP86‐D3/def2‐SVP level) in [(depe)2Fe(N2)] (a) and 6+ (b). Electron‐density charge flows in the direction red→blue.
As aforementioned, we were intrigued by the ability of gold to promote irreversible ligand substitution between CO2 and N2. Although exposure of 6 to CO2 under more forcing pressure conditions (5 bar) did not offer any hint of CO2 coordination (Scheme 3), the addition of one equivalent of B(C6F5)3 under CO2 atmosphere (0.5 bar) immediately generates complex 2. Our calculations are in line with this observation as the highly endergonic reaction 6+ (NTf2)−+CO2→1+AuNTf2+N2 (ΔG R=+14.6 kcal mol−1) becomes exergonic in the presence of B(C6F5)3 to produce 2 (ΔG R=−8.4 kcal mol−1). This behaviour contrasts with the ability of the gold complex to displace the borane in [(depe)2Fe(N2)B(C6F5)3], which readily occurs to form complex 6 upon mixing the two species. Analogously, we have proved the inertness of the Fe/Au bimetallic compound 6 towards B(C6F5)3 in the absence of carbon dioxide, which according to our calculations is indeed a highly endergonic (thus, unfeasible) process (ΔG R=+43.8 kcal mol−1). These results suggest that the most thermodynamically stable compound of the series is compound 2.
To complete these studies, we finally examined the use of Zn(C6F5)2 as a Lewis acid, owing to its higher oxophilicity compared to gold [35] and thus a presumed enhanced ability to activate CO2. In fact, treating a thf solution of precursor 1 with Zn(C6F5)2 in equimolar amounts resulted in a rapid colour change from orange to pink. The formation of a new compound 7 (Scheme 4a) is evidenced by 31P{1H} NMR, where a broad signal at 77.8 ppm was recorded in thf‐d 8 at 25 °C. This signal becomes a set of four broad multiplets (89.1, 80.9, 74.7 and 67.9 ppm) when the temperature was decreased to −20 °C. Single‐crystals suitable for X‐ray diffraction analysis were grown from a concentrated toluene solution at −30 °C (35 % yield), unveiling the anticipated activation of CO2 in a bimetallic manner (Figure 6). At variance with B(C6F5)3, the CO2 molecule now bridges the two metals in a μ‐CO2−1κC 1:2κO 1:O 2 fashion. The corresponding structural parameters are comparable to previous examples that exhibit this type of binding.[ 4 , 12 , 13c ] η 2‐coordination to zinc forces the O−C−O angle to diminish to 114(2)° (c.f. 124(2)° in 1). Activation of CO2 upon zinc coordination is also discernible by a slight elongation of the C−O bonds to an average value of 1.29 Å (1.27(2) for O1−C21 and 1.31(2) Å for O2−C21 distances) compared to the average value of 1.26 Å in complex 1 (1.28(2) and 1.25(3) Å values for the two C−O distances). [17] Intriguingly, a molecule of N2 coordinates to the Fe0 centre upon CO2 activation to yield a highly unusual structure, namely the first organometallic species structurally characterized in which both N2 and CO2, typically very poorly coordinating ligands, are bound to the same metal. Their presence is in accordance with its corresponding IR spectrum, where bands at 1633 and 2105 cm−1 are assigned to CO2 and N2, respectively. The function of the iron centre as a Lewis base for the activation of CO2 results in a reduced N≡N bond length of 1.07(2) Å, even shorter than in compound 6 (1.12(2) Å).
Scheme 4.
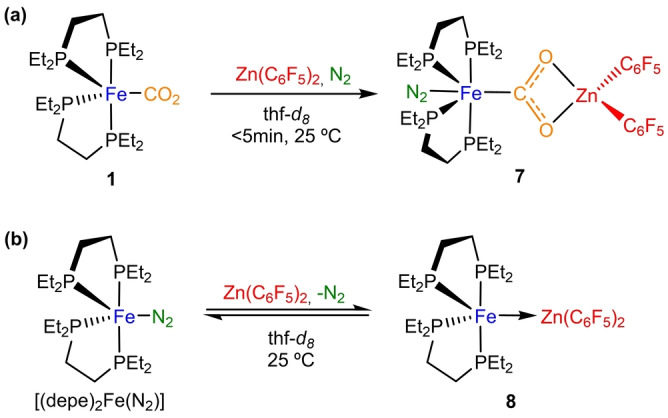
Reactivity of Fe0 compounds with Zn(C6F5)2.
Figure 6.
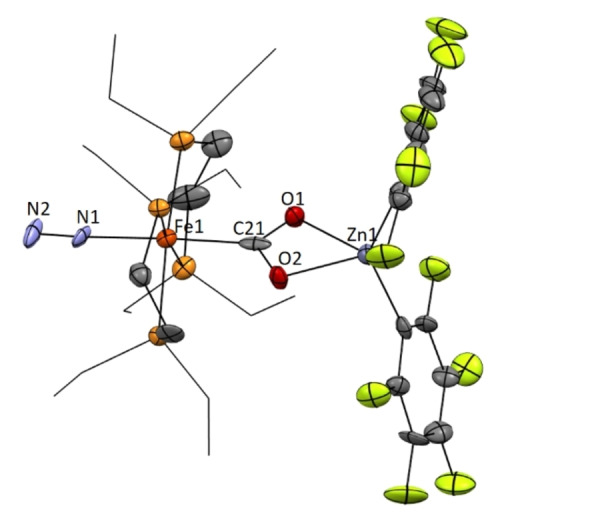
ORTEP of complex 7. Hydrogen atoms have been excluded, and ethyl groups of depe ligand are represented in wire‐frame format for clarity. Thermal ellipsoids are set at 50 % probability. [37]
The unexpected coordination of dinitrogen in 7 led us to examine the reactivity of [(depe)2Fe(N2)] with Zn(C6F5)2 (Scheme 4b). Conversely, in the absence of CO2, the metal only Lewis adduct 8 characterized by a dative Fe0→ZnII bond (dFeZn=2.561 Å; see Figure S36) is produced as the major species, with concomitant release of dinitrogen. Nonetheless, complex 8 exhibits a dynamic equilibrium in solution with its separated fragments, being the Fe−Zn bond cleavage favoured at low temperatures (see Supporting Information). In fact, compound 8 could be isolated as green crystals at −30 °C in 28 % yield. The lability of the bimetallic bond is also confirmed by the addition of one equivalent of B(C6F5)3 under CO2 atmosphere, which results in quantitative formation of 2. However, exposure of 8 to CO2 (1 atm) results in the formation of unidentified products and phosphine ligand dissociation.
Comparing the C−Zn−C angle of both zinc‐based structures, it diminishes from 135.6(7)° in 7 to 100.2(1)° in 8, significantly reduced compared to other transition metal base‐zinc Lewis adducts. [36] Once more, this result contrasts with the push‐pull activation of N2 observed before for the same iron fragment in the presence of B(C6F5)3 [18] and attests the importance of tuning the nature of Lewis acids in bimetallic architectures as a crucial designing principle for bond activation studies.
Conclusion
We have demonstrated that the choice of Lewis acid for the binuclear activation of carbon dioxide through push‐pull forces is of paramount importance to modulate reaction selectivity. In fact, maintaining a common Lewis basic Fe0 synthon ([(depe)2Fe(CO2)]), we disclose a completely divergent reactivity that span from irreversible CO2 dissociation, to its activation by two dissimilar modes and up to the rare bimetallic cleavage of one of the C−O bonds. We have performed computational studies that provide solid understanding on the observed divergent reactivity and substantiate the bonding scenario by means of state‐of‐the‐art methods. Overall, the results of this work offer fundamental knowledge on the way to developing bimetallic efficient strategies for the valorisation of carbon dioxide.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
This work was supported by the European Research Council (ERC Starting Grant, CoopCat, Project 756575), Spanish MCIN/AEI/10.13039/501100011033 (Projects PID2019‐110856GA−I00, PID2019‐106184GB−I00 and RED2018‐102387‐T) and Junta de Andalucia (P18‐FR‐4688). F. de la C.‐M. acknowledges the Spanish Ministry of Universities for a Margarita Salas postdoctoral fellowship.
H. Corona, M. Pérez-Jiménez, F. de la Cruz-Martínez, I. Fernández, J. Campos, Angew. Chem. Int. Ed. 2022, 61, e202207581; Angew. Chem. 2022, 134, e202207581.
Contributor Information
Dr. Israel Fernández, Email: israel@quim.ucm.es.
Dr. Jesús Campos, Email: jesus.campos@iiq.csic.es.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.
- 1a. Burkart M. D., Hazari N., Tway C. L., Zeitler E. L., ACS Catal. 2019, 9, 7937–7956; [Google Scholar]
- 1b. Appel A. M., Bercaw J. E., Bocarsly A. B., Dobbek H., DuBois D. L., Dupuis M., Ferry J. G., Fujita E., Hille R., Kenis P. J. A., Kerfeld C. A., Morris R. H., Peden C. H. F., Portis A. R., Ragsdale S. W., Rauchfuss T. B., Reek J. N. H., Seefeldt L. C., Thauer R. K., Waldrop G. L., Chem. Rev. 2013, 113, 6621–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Jeoung J.-H., Dobbek H., Science 2007, 318, 1461–1464; [DOI] [PubMed] [Google Scholar]
- 2b. Dobbek H., Gremer L., Kiefersauer R., Huber R., Meyer O., Proc. Natl. Acad. Sci. USA 2002, 99, 15971–15976; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Ahmed M. E., Adam S., Saha D., Fize J., Artero V., Dey A., Duboc C., ACS Energy Lett. 2020, 5, 3837–3842; [Google Scholar]
- 2d. Mouchfiq A., Todorova T. K., Dey S., Fontecave M., Mougel V., Chem. Sci. 2020, 11, 5503–5510; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Li Y., Gomez-Mingot M., Fogeron T., Fontecave M., Acc. Chem. Res. 2021, 54, 4250–4261. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Mankad N. P., Trends Chem. 2021, 3, 159–160; [Google Scholar]
- 3b. Sinhababu S., Lakliang Y., Mankad N. P., Dalton Trans. 2022, 51, 6129–6147. [DOI] [PubMed] [Google Scholar]
- 4. Yoo C., Lee Y., Chem. Sci. 2017, 8, 600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Stephan D. W., Erker G., Chem. Sci. 2014, 5, 2625–2641; [Google Scholar]
- 5b. Fontaine F.-G., Courtemanche M.-A., Légaré M.-A., Rochette E., Coord. Chem. Rev. 2017, 334, 124–135. [Google Scholar]
- 6.
- 6a. Jiang Y., Blacque O., Fox T., Berke H., J. Am. Chem. Soc. 2013, 135, 7751–7760; [DOI] [PubMed] [Google Scholar]
- 6b. Forrest S. J. K., Clifton J., Fey N., Pringle P. G., Sparkes H. A., Wass D. F., Angew. Chem. Int. Ed. 2015, 54, 2223–2227; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2251–2255; [Google Scholar]
- 6c. Jian Z., Kehr G., Daniliuc C. G., Wibbeling B., Wiegand T., Siedow M., Eckert H., Bursch M., Grimme S., Erker G., J. Am. Chem. Soc. 2017, 139, 6474–6483; [DOI] [PubMed] [Google Scholar]
- 6d. Devillard M., Declercq R., Nicolas E., Ehlers A. W., Backs J., Saffon-Merceron N., Bouhadir G., Slootweg J. C., Uhl W., Bourissou D. A., J. Am. Chem. Soc. 2016, 138, 4917–4926. [DOI] [PubMed] [Google Scholar]
- 7. Kim Y.-E., Kim J., Lee Y., Chem. Commun. 2014, 50, 11458–11461. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Benson E. E., Kubiak C. P., Sathrum A. J., Smieja J. M., Chem. Soc. Rev. 2009, 38, 89–99; [DOI] [PubMed] [Google Scholar]
- 8b.O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli, M., J. Am. Chem. Soc. 2014, 136, 6716–6723; [DOI] [PubMed]
- 8c. Buss J. A., VanderVelde D. G., Agapie T., J. Am. Chem. Soc. 2018, 140, 10121–10125. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Cammarota R. C., Vollmer M. V., Xie J., Ye J., Linehan J. C., Burgess S. A., Appel A. M., Gagliardi L., Lu C. C. A., J. Am. Chem. Soc. 2017, 139, 14244–14250; [DOI] [PubMed] [Google Scholar]
- 9b. Takaya J., Iwasawa N., J. Am. Chem. Soc. 2017, 139, 6074–6077; [DOI] [PubMed] [Google Scholar]
- 9c. Prat J. R., Gaggioli C. A., Cammarota R. C., Bill E., Gagliardi L., Lu C. C., Inorg. Chem. 2020, 59, 14251–14262. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Bontemps S., Vendier L., Sabo-Etienne S., Angew. Chem. Int. Ed. 2012, 51, 1671–1674; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1703–1706; [Google Scholar]
- 10b. Bontemps S., Sabo-Etienne S., Angew. Chem. Int. Ed. 2013, 52, 10253–10255; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10443–10445; [Google Scholar]
- 10c. Jin G., Werncke C. G., Escudié Y., Sabo-Etienne S., Bontemps S., J. Am. Chem. Soc. 2015, 137, 9563–9566; [DOI] [PubMed] [Google Scholar]
- 10d. Bontemps S., Coord. Chem. Rev. 2016, 308, 117–130; [Google Scholar]
- 10e. Sampson M. D., Kubiak C. P., J. Am. Chem. Soc. 2016, 138, 1386–1393; [DOI] [PubMed] [Google Scholar]
- 10f. Rauch M., Parkin G., J. Am. Chem. Soc. 2017, 139, 18162–18165; [DOI] [PubMed] [Google Scholar]
- 10g. Heimann J. E., Bernskoetter W. H., Hazari N., J. Am. Chem. Soc. 2019, 141, 10520–10529. [DOI] [PubMed] [Google Scholar]
- 11. Sinhababu S., Radzhabov M. R., Tesler J., Mankad N. P., J. Am. Chem. Soc. 2022, 144, 3210–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu H.-Y., Schwamm R. J., Hill M. S., Mahon M. F., McMullin C. L., Rajabi N. A., Angew. Chem. Int. Ed. 2021, 60, 14390–14393; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 14511–14514. [Google Scholar]
- 13.
- 13a. Weetman C., Bag P., Szilvási T., Jandl C., Inoue S., Angew. Chem. Int. Ed. 2019, 58, 10961–10965; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11077–11081; [Google Scholar]
- 13b. Weetman C., Porzelt A., Bag P., Hanusch F., Inoue S., Chem. Sci. 2020, 11, 4817–4827; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Roy M. M. D., Hicks J., Vasko P., Heilmann A., Baston A.-M., Goicoechea J. M., Aldridge S., Angew. Chem. Int. Ed. 2021, 60, 22301–22306; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 22475–22480. [Google Scholar]
- 14. Escomel L., Del Rosal I., Maron L., Jeanneau E., Veyre L., Thieuleux C., Camp C., J. Am. Chem. Soc. 2021, 143, 4844–4856. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Hicks J., Mansikkamäki A., Vasko P., Goicoechea J. M., Aldridge S. A., Nat. Chem. 2019, 11, 237–241; [DOI] [PubMed] [Google Scholar]
- 15b. McManus C., Hicks J., Cui X., Zhao L., Frenking G., Goicoechea J. M., Aldridge S., Chem. Sci. 2021, 12, 13458–13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Campos J., J. Am. Chem. Soc. 2017, 139, 2944–2947; [DOI] [PubMed] [Google Scholar]
- 16b. Hidalgo N., Moreno J. J., Pérez-Jiménez M., Maya C., López-Serrano J., Campos J., Chem. Eur. J. 2020, 26, 5982–5993; [DOI] [PubMed] [Google Scholar]
- 16c. Alférez M. G., Hidalgo N., Moreno J. J., Campos J., Angew. Chem. Int. Ed. 2020, 59, 20863–20867; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 21049–21053; [Google Scholar]
- 16d. Navarro M., Campos J., Chapter 3 in Adv. Organomet. Chem., Vol. 75 (Ed.: J. Pérez Pedro), Elsevier, Amsterdam, 2021. [Google Scholar]
- 17.
- 17a. Hirano M., Akita M., Tani K., Kumagai K., Kasuga N. C., Fukuoka A., Komiya S., Organometallics 1997, 16, 4206–4213; [Google Scholar]
- 17b. Adamson T. T., Kelley S. P., Bernskoetter W. H., Organometallics 2020, 39, 3562–3571; [Google Scholar]
- 17c. Jurd P. M., Li H. L., Bhadbhade M., Field L. D., Organometallics 2020, 39, 2011–2018. [Google Scholar]
- 18. Geri J. B., Shanahan J. P., Szymczak N. K., J. Am. Chem. Soc. 2017, 139, 5952–5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gibson D. H., Ye M., Sleadd B. A., Mehta J. M., Mbadike O. P., Richardson J. F., Mashuta M. S., Organometallics 1995, 14, 1242–1255. [Google Scholar]
- 20.
- 20a. Kaehler T., Melen R. L., Cell Rep. Phys. Sci. 2021, 2, 100595; [Google Scholar]
- 20b. Timoshkin A. Y., Frenking G., Organometallics 2008, 27, 371–380. See also: [Google Scholar]
- 20c. Cabrera-Trujillo J. J., Fernández I., Chem. Eur. J. 2018, 24, 17823–17831. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Kilpatrick A. F. R., Cloke F. G. N., Chem. Commun. 2014, 50, 2769–2771; [DOI] [PubMed] [Google Scholar]
- 21b. Noor A., Qayyum S., Bauer T., Schwarz S., Weber B., Kempe R., Chem. Commun. 2014, 50, 13127–13130; [DOI] [PubMed] [Google Scholar]
- 21c. Saouma C. T., Day M. W., Peters J. C., Chem. Sci. 2013, 4, 4042–4051; [Google Scholar]
- 21d. Shimogawa R., Takao T., Konishi G. I., Suzuki H., Organometallics 2014, 33, 5066–5069; [Google Scholar]
- 21e. Bagherzadeh B., Mankad N. P., Chem. Commun. 2018, 54, 1097–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krogman J. P., Foxman B. M., Thomas C. M., J. Am. Chem. Soc. 2011, 133, 14582–14585. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. DeRosha D. E., Mercado B. Q., Lukat-Rodgers G., Rodgers K. R., Holland P. L., Angew. Chem. Int. Ed. 2017, 56, 3211–3215; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3259–3263; [Google Scholar]
- 23b. Engelmann X., Yao S., Farquhar E. R., Szilvási T., Kuhlmann U., Hildebrandt P., Driess M., Ray K. A., Angew. Chem. Int. Ed. 2017, 56, 297–301; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 303–307. [Google Scholar]
- 24.
- 24a. Davies S. C., Hughes D. L., Richards R. L., Sanders J. R., Dalton Trans. 2002, 1442–1447; [Google Scholar]
- 24b. Weintrob E. C., Tofan D., Bercaw J. E., Inorg. Chem. 2009, 48, 3808–3813; [DOI] [PubMed] [Google Scholar]
- 24c. Becker J. M., Barker J., Clarkson G. J., van Gorkum R., Johal G. K., Walton R. I., Scott P., Dalton Trans. 2010, 39, 2309–2326. [DOI] [PubMed] [Google Scholar]
- 25. Crabtree R. H., The Organometallic Chemistry of the Transition Metals, 7th ed., Wiley, Hoboken, 2019. [Google Scholar]
- 26.
- 26a. Hirano M., Akita M., Morikita T., Kubo H., Fukuoka A., Komiya S., J. Chem. Soc. Dalton Trans. 1997, 3453–3458; [Google Scholar]
- 26b. Baker M. V., Field L. D., J. Am. Chem. Soc. 1986, 108, 7436–7438. [Google Scholar]
- 27.
- 27a. Berkefeld A., Piers W. E., Parvez M., Castro L., Maron L., Eisenstein O., Chem. Sci. 2013, 4, 2152–2162; [Google Scholar]
- 27b. Wang T., Xu M., Jupp A. R., Qu Z.-W., Grimme S., Stephan D. W., Angew. Chem. Int. Ed. 2021, 60, 25771–25775; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 25975–25979; [Google Scholar]
- 27c. Bautista M. T., Cappellani P., Drouin S. D., Morris R. H., Schweitzer C. T., Sella A., Zubkowski J., J. Am. Chem. Soc. 1991, 113, 4876–4887. [Google Scholar]
- 28. Voss T., Mahdi T., Otten E., Fröhlich R., Kehr G., Stephan D. W., Erker G., Organometallics 2012, 31, 2367–2378. [Google Scholar]
- 29.
- 29a. Bancroft G. M., Mays M. J., Prater B. E., Stefanini F. P., J. Chem. Soc. A 1970, 2146–2149; [Google Scholar]
- 29b. Drouin S. D., Maltby P. A., Rennie B. E., Schweitzer C. T., Golombek A., Cappellani P., Morris R. H., Inorg. Chim. Acta 2021, 516, 120124. [Google Scholar]
- 30. Espada M. F., Campos J., López-Serrano J., Poveda M. L., Carmona E., Angew. Chem. Int. Ed. 2015, 54, 15379–15384; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15599–15604. [Google Scholar]
- 31. Bauer J., Braunschweig H., Dewhurst R. D., Chem. Rev. 2012, 112, 4329–4346. [DOI] [PubMed] [Google Scholar]
- 32. Specklin D., Coffinet A., Vendier L., Del Rosal I., Dinoi C., Simonneau A., Inorg. Chem. 2021, 60, 5545–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Komiya S., Akita M., Yoza A., Kasuga N., Fukuoka A., Kai Y., J. Chem. Soc. Chem. Commun. 1993, 787–788. [Google Scholar]
- 34.Additional calculations using [Fe]2+ and [Au]1− as fragments, which is consistent with the relative electronegativities of iron and gold, indicate that the [Fe](0)/[Au]+ fragmentation better describes the bonding situation in complex 6+ (see Supporting Information).
- 35. Kepp K. P. A., Inorg. Chem. 2016, 55, 9461–9470. [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. Hidalgo N., Romero-Pérez C., Maya C., Fernández I., Campos J., Organometallics 2021, 40, 1113–1119; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36b. Liberman-Martin A. L., Levine D. S., Ziegler M. S., Bergman R. G., Tilley T. D., Chem. Commun. 2016, 52, 7039–7042. [DOI] [PubMed] [Google Scholar]
- 37.Deposition Numbers 2172175, 2172176, 2172177, 2172178 and 2172179 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.