

Abstract
HIDEA syndrome is caused by biallelic pathogenic variants in P4HTM. The phenotype is characterized by muscular and central hypotonia, hypoventilation including obstructive and central sleep apneas, intellectual disability, dysautonomia, epilepsy, eye abnormalities, and an increased tendency to develop respiratory distress during pneumonia. Here, we report six new patients with HIDEA syndrome caused by five different biallelic P4HTM variants, including three novel variants. We describe two Finnish enriched pathogenic P4HTM variants and demonstrate that these variants are embedded within founder haplotypes. We review the clinical data from all previously published patients with HIDEA and characterize all reported P4HTM pathogenic variants associated with HIDEA in silico. All known pathogenic variants in P4HTM result in either premature stop codons, an intragenic deletion, or amino acid changes that impact the active site or the overall stability of P4H‐TM protein. In all cases, normal P4H‐TM enzyme function is expected to be lost or severely decreased. This report expands knowledge of the genotypic and phenotypic spectrum of the disease.
Keywords: genes, HIDEA, intellectual disability, P4HTM, recessive

1. INTRODUCTION
Hypotonia, hypoventilation, impaired intellectual development, dysautonomia, epilepsy, and eye abnormalities (HIDEA) (OMIM #618493) is an autosomal recessive neurodevelopmental disorder caused by biallelic pathogenic variants in prolyl 4‐hydroxylase, transmembrane (P4HTM). Functional characterization of the pathogenic P4HTM variants revealed an improper folding of the corresponding protein, suggesting a loss‐of‐function disease mechanism. 1 , 2
Eukaryotic prolyl 4‐hydroxylases (P4Hs) are key enzymes in the synthesis of collagens and the regulation of oxygen homeostasis. 3 P4H‐TM is localized to the endoplasmic reticulum (ER) membrane. It contains a short N‐terminal cytoplasmic region, a transmembrane helix, and within the ER lumen an EF‐hand domain and a P4H domain. The active site is composed of two His and one Asp residues that together with the co‐substrate 2‐oxoglutarate coordinate the Fe2+ which is central to the P4H‐TM activity. Recently, P4H‐TM was found to participate in gliotransmission in astrocytes, raising the question of whether this might be linked to the intellectual disability (ID) phenotype observed in HIDEA. 4
To the authors' knowledge, 24 HIDEA patients with 12 different disease‐associated P4HTM variants have been described in the medical literature. 1 , 2 , 5 , 6 , 7 , 8 Here, we review the clinical and molecular data from all the published patients and describe six previously unpublished patients with HIDEA caused by five different biallelic pathogenic P4HTM variants.
2. MATERIALS AND METHODS
2.1. Patient recruitment
Patients were enrolled from three centers: Oulu University Hospital, Oulu, Finland (Families 1, 3–4), Next Generation Polyclinic, Mashhad, Iran (Families 2 and 6), and University of Sorbonne, Paris, France (Family 5). Families 1, 3, and 4 were identified after genetic testing or databank query in Centogene (Rostock, Germany). Families 2 and 6 were identified through GeneMatcher, 9 and Family 5 was identified through ERN‐ITHACA call for collaboration. Detailed clinical data of Patients 1–6 and pedigrees are provided in the supplementary case histories and Figure S1.
2.2. Molecular genetics
Genomic DNA was extracted from peripheral blood samples using standard methods. WES was performed for all index cases and the parents of Families 1 and 3. Targeted Sanger sequencing was used for segregation analysis of the identified P4HTM variants in siblings of Family 1 and 2, and the parents of Families 2, 4, 5, and 6. Details of WES and haplotype analysis are included in the supplementary methods.
3. RESULTS
3.1. Clinical data
We review the clinical details of both the new (N = 6) and previously reported HIDEA patients (N = 24; Figure 1, Table 1, Table S1).
FIGURE 1.
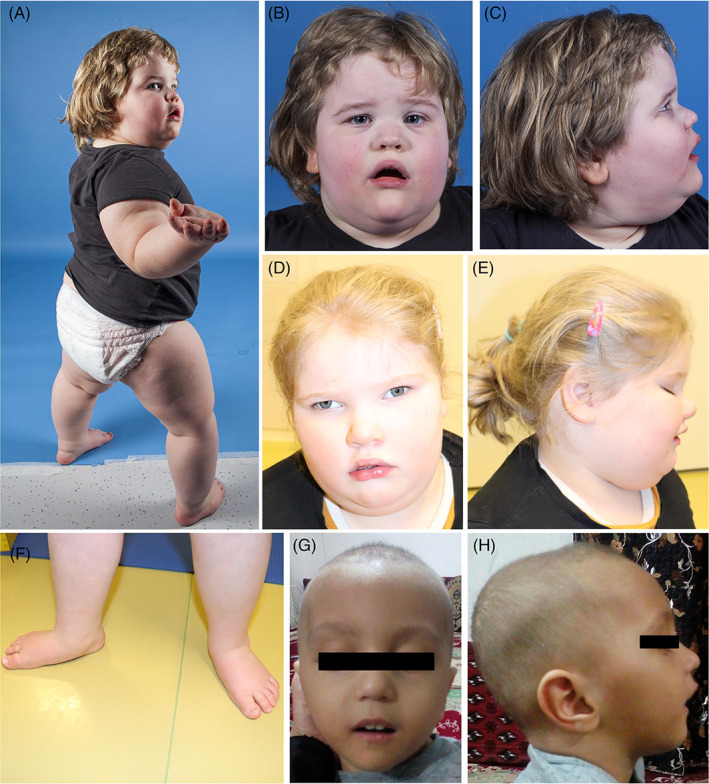
Clinical characteristics of the patients. All the patients show facial hypotonia with an open‐mouth appearance, tented upper lip vermilion, and a low nasal bridge. Strabismus (D), retrognathia (G, H), and pes planus (A, F) are shown [Colour figure can be viewed at wileyonlinelibrary.com]
TABLE 1.
Main clinical features of Patients 1–6 described in this report, and the clinical features of previously published HIDEA patients
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Freq in this report | Freq in publ cases | Freq in all cases | |
|---|---|---|---|---|---|---|---|---|---|
| Obesity | Yes | No | Yes | Yes | Yes | No | 4/6 | 13/23 | 59% (17/29) |
| Hypotonia | Yes | Yes | Yes | No | Yes | Yes | 5/6 | 24/24 | 97% (29/30) |
| ID/GDD | Yes | Yes | Yes | Yes | Yes | Yes | 6/6 | 24/24 | 100% (30/30) |
| Learned to walk | Yes | No | Yes | Yes | Yes | No | 4/6 | 11/19 | 60% (15/25) |
| Verbal | Yes | No | Yes | Yes | Yes | Yes | 5/6 | 4/14 | 45% (9/20) |
| Epilepsy/seizures | No | Yes | Yes | Yes | Yes | Yes | 5/6 | 12/24 | 57% (17/30) |
| Nystagmus | No | No | Yes | No | No | NA | 1/6 | 7/20 | 31% (8/26) |
| Strabismus | No | No | Yes | No | Yes | Yes | 3/6 | 13/18 | 67% (16/24) |
| Other ophthalmological findings | No | No | Yes | Yes | No | NA | 2/5 | 20/22 | 81% (22/27) |
| MRI brain abnormalities | No | Yes | No | No | No | No | 1/6 | 5/13 | 32% (6/19) |
| Obstructive sleep apnea | Yes | Yes | NA | No | NA | No | 2/4 | 7/17 | 43% (9/21) |
| Central sleep apnea | Yes | Yes | NA | No | NA | No | 2/4 | 8/18 | 45% (10/22) |
| BiPAP or other assistive therapy | Yes | NA | No | No | No | No | 1/5 | 12/21 | 50% (13/26) |
| Parasomnia | No | No | NA | Yes | Yes | Yes | 3/5 | 5/17 | 36% (8/22) |
| Impaired thermoregulation | Yes | Yes | No | No | No | No | 2/6 | 4/16 | 27% (6/22) |
| Constipation | No | No | Yes | No | No | Yes | 2/6 | 7/20 | 35% (9/26) |
| Valgus knees | No | No | Yes | No | No | No | 1/6 | 6/6 | 58% (7/12) |
| Varus knees | Yes | No | No | No | No | Yes | 2/6 | 0/6 | 17% (2/12) |
| Flexion/extension of the knees a | Yes | No | NA | No | No | NA | 1/4 | 5/5 | 67% (6/9) |
| Pes planus | Yes | No | Yes | No | No | No | 2/6 | 8/8 | 71% (10/14) |
| Gait abnormality | Yes | NA | Yes | Yes | No | NA | 3/4 | 5/5 | 89% (8/9) |
Abbreviations: freq, frequency; GDD, global developmental delay; ID, intellectual disability; NA, not available; publ, published.
When walking.
Twenty‐three patients are alive (age at last examination from 9 months to 59 years) and seven are deceased (age of death 7 months–61 years). The most common cause of death was respiratory tract infection (N = 4/7, 57%). Nineteen patients are male, and 11 are female. All patients have global developmental delay (DDD)/ID (N = 30/30, 100%). Common features include hypotonia (N = 29/30, 97%), epilepsy (N = 17/30, 57%), strabismus (N = 16/24, 67%), nystagmus (N = 8/26, 31%) or other ophthalmological abnormalities such as abnormal eye movements, cortical blindness, refractive errors, or achromic fundi (N = 22/27, 81%). Central (N = 10/22, 45%) and/or obstructive sleep apneas (N = 9/21, 43%) are common associated features and many patients (N = 13/26, 50%) require bilevel positive airway pressure ventilation (BiPAP) or other forms of respiratory support.
Most patients (N = 17/29, 59%) are or have been obese (>95th percentile). Six patients (N = 6/22, 27%) show dysautonomia of thermoregulation, including recurrent hypothermia or hyperthermia and reduced sweating. Facial dysmorphisms such as tented upper lip vermilion and low nasal bridge, are common (N = 22/23, 97%), but individual features vary. Brain MRIs are normal in most patients (N = 13/19, 68%), but three have brain atrophy, two have abnormalities of the white matter, and one patient has both. A majority of patients learned to walk (N = 15/25, 60%), walking age ranging from 18 months to 4 years. Patients who have achieved independent walking frequently present with gait abnormalities (N = 8/9, 89%). The age of first words ranges from 1 to 4 years, while 11 patients out of 20 (55%) were nonverbal at the time of study.
3.2. Molecular genetics
Five P4HTM (NM_177939.3) variants were observed in a homozygous or compound heterozygous state in the six patients in the current study: c.1238C>T, p.(Pro413Leu); c.1371G>A, p.(Trp457*); c.1073G>A, p.(Arg296Ser;Val297_Arg358del); c.1082C>T, p.(Thr361Ile); and c.934G>A, p.(Glu312Lys).
Haplotype analysis of the two recurrent variants revealed shared haplotypes extending approximately 7 Mb around the P4HTM p.(Pro413Leu) variant and 6.9 Mb around the P4HTM p.(Arg296Ser;Val297_Arg358del) variant.
We characterized the HIDEA causing variants using the recently published multiple sequence alignments and crystal structure showing the residues from 107 to 481 of the prevalent 502‐residue form of P4H‐TM (Figure 2A). 10 Nonsense, frameshift, and in‐frame deletion variants in P4HTM (Figure 2B–G) are likely be degraded by nonsense‐mediated decay, or degraded due to protein misfolding and will not retain any P4H‐TM enzyme activity. The P4HTM missense variants p.(Thr361Ile) and p.(Pro413Leu) are residues conserved in P4Hs, while p.(Glu312Lys) is conserved in collagen prolyl 4‐hydroxylases but not in the hypoxia‐inducible factor prolyl 4‐hydroxylases. Glu312 and Thr361 are in the vicinity of the active site and interact with central active site residues (Figure 2H). The missense substitutions would lose these interactions and disrupt the positions of the central residues. The Pro413 side chain is positioned in a hydrophobic pocket near Lys451 and Tyr365 (Figure 2I). Leucine substitution here would disrupt the neighboring residues that help to coordinate the co‐substrate 2‐oxoglutarate (Figure 2I). Pathogenic missense variants resulting in the loss of the conserved residues are likely to decrease or completely abolish P4H‐TM enzyme activity.
FIGURE 2.

The residues targeted by the P4H‐TM HIDEA variants are presented in a cartoon and stick model of the P4H‐TM crystal structure. (A) An overview of the P4H‐TM crystal structure (residues 107–481). The side chains of the residues impacted by different variants are shown as sticks and colored magenta. The active site residues and Fe2+ are shown in orange and the 2‐oxoglutarate analog N‐oxalylglycine (NOG) in pink. Ca2+ ions are shown as green spheres. The N‐ and C‐termini and the variant residues are labeled. The deleted regions are shown in blue for the (B) Gln190Leufs*9, (C) Trp220*, (D) Asn274Glufs*11, (E) Arg296Ser;Val297_Arg358del, (F) Val317Phefs*30, and (G) Trp457* variants. The environment and interacting residues of (H) Glu312, Thr361, and Trp457 and (I) Arg410 and Pro413 are presented as a cartoon model where the interacting residues are shown as sticks and labeled. Hydrogen bonds and electrostatic interactions generated by the targeted residues are shown as green dashed lines. (G) Pathogenic P4HTM variants reported in the current study (above the gene) and in the literature (underneath the gene) are distributed throughout the gene (NM_177939.3). Similar HIDEA phenotype can be caused by pathogenic missense, nonsense, splice‐site, and frameshift variants, as well as in‐frame deletions [Colour figure can be viewed at wileyonlinelibrary.com]
Details of pathogenic P4HTM variants (Figure 2J), their in silico characterization, and haplotype analysis are provided in the supplementary results and Tables S2 and S3.
4. DISCUSSION
Here, we report six new unrelated patients and review the clinical details of all 24 previously published HIDEA patients. The phenotype of the patients identified in the current study is comparable to the phenotype described in the literature. 1 , 2 , 5 , 6 , 7 , 8 One patient in this study has dystonia, which has not previously been described in HIDEA. Dystonia is a movement disorder thought to result from an abnormality or damage to the basal ganglia or other brain regions controlling movement. The patient had generalized brain atrophy and cerebellar atrophy has previously been associated with dystonia. 11 In addition, P4HTM is expressed in the basal ganglia, 12 and P4THM deficiency may predispose to basal ganglia dysfunction leading to dystonia. Further research is needed to confirm the possible association between HIDEA and dystonia.
One third of all reported patients had history of pneumonias, and respiratory tract infection were the most common cause of death. Almost half of the HIDEA patients had central and/or obstructive sleep apneas and half required BiPAP treatment at nights or during respiratory infections. Thus, performing polysomnography and assessing the need for noninvasive ventilatory support is advisable.
The clinical presentation of HIDEA is variable, even for the same variant, and no clear genotype–phenotype correlation has been observed. 2 , 5 In the current study, patients with pathogenic homozygous P4HTM p.Glu312Lys missense and homozygous P4HTM p.Trp457* nonsense variants had similarly severe ID, confirming that both missense and truncating variants in P4HTM can result in a severe HIDEA phenotype. It is likely that background genetic factors, environmental factors, and stochastic factors modify the severity of the phenotype.
Haplotype analysis of the two recurring P4HTM variants, p.(Arg296Ser;Val297_Arg358del) and p.(Pro413Leu), revealed shared haplotypes, suggesting that both P4HTM variants are embedded within founder haplotypes. This is often seen in the Finnish population due to historical population bottlenecks, genetic drift events, and recent population expansion. In contrast, we identified two pathogenic novel and unique homozygous P4HTM variants, p.(Trp457*) and p.(Glu312Lys), in two consanguineous Iranian families, where consanguineous marriages are common increasing the risk for children with autosomal recessive disorders.
All known pathogenic variants in P4HTM are predicted to lose or decrease P4H‐TM enzyme activity. 5′ prime nonsense variants and the deletion of exon 6 lose critical parts of the P4H domain. Nonsense variants at positions 457 and 471 lose the ER retention signal and disrupt protein folding. Missense variants of conserved amino acids of the P4H domain disrupt the active site coordination or the binding of substrate or co‐substrate. Other missense variants are predicted to disrupt protein folding.
To the authors' knowledge, P4HTM p.(Glu312Lys), p.(Thr361Ile), and p.(Trp457*) variants have not previously been reported as disease‐causing, hence expanding the genotypic spectrum of the disease. To date, including the cases reported in the present study, there are 15 different pathogenic variants of P4HTM reported to cause HIDEA syndrome. 1 , 2 , 5 , 6 , 7 , 8
5. CONCLUSIONS
HIDEA syndrome is a recognizable neurodevelopmental disorder caused by pathogenic rare or founder P4HTM variants that are likely to disrupt the P4H‐TM activity. Greater knowledge of the genotypic and phenotypic spectrum of HIDEA will support the development of tailored therapies benefiting the patients.
AUTHOR CONTRIBUTIONS
Conceptualization: Leila Soikkonen, Minna Kraatari‐Tiri, Elisa Rahikkala. Writing—original draft: Leila Soikkonen, Minna Kraatari‐Tiri, Matti Myllykoski, Elisa Rahikkala. Writing—review and editing: Minna Kraatari‐Tiri, Leila Soikkonen, Matti Myllykoski, Yalda Jamshidi, Ehsan G. Karimiani, Jonna Komulainen‐Ebrahim, Hanna Kallankari, Cyril Mignot, Boris Keren, Hanna Kallankari, Marie‐Christine Nougues, Zahra Alsahlawi, Antonio Romito, Javier Martini, Mehran B. Toosi, Christopher J. Carroll, Kornelia Tripolszki, Peter Bauer, Johanna Uusimaa, Aida M. Bertoli‐Avella, Peppi Koivunen, Elisa Rahikkala.
CONFLICT OF INTEREST
Antonio Romito, Javier Martini, Kornelia Tripolszki, Peter Bauer, Aida M. Bertoli‐Avella are employees of CENTOGENE GmbH. Other authors declare no conflicts of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/cge.14203.
ETHICS STATEMENT
The study is approved by the Ethics Committee of the Northern Ostrobothnia Hospital District (EETTMK: 186/2020). Written informed consent was obtained from all parents or guardians of the patients. Written informed consent was obtained to publish patient photos.
Supporting information
Appendix S1 Supporting Information
Table S1 Clinical characteristics of patients described in this report (Patients 1–6) and all the previously published HIDEA patients to date.
Table S3 Results of the haplotype analysis showing the shared haplotype around the P4HTM (NM_177939.3) p.(Pro413Leu) variant and P4HTM p.(Arg296Ser;Val297_Arg358del) variant. The shared haplotype extended approximately 7 Mb (GRCh38 g.3:45758441–52 818 394) around the P4HTM (NM_177939.3) p.(Pro413Leu) variant and approximately 6.9 Mb (GRCh38 g.3:46372893–53 235 071) around the P4HTM (NM_177939.3) p.(Arg296Ser;Val297_Arg358del) variant.
Video S1 The gait of Patient 1. The video demonstrates the typical waddling gait associated with HIDEA syndrome.
Video S2 Dystonic movements of Patient 2.
ACKNOWLEDGEMENTS
The authors thank all the families who participated in this study. This study was supported by the Academy of Finland (decision number 338446) to Elisa Rahikkala. Some authors of this publication are members of the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability (ERN‐ITHACA). [EU Framework Partnership Agreement ID: 3HP‐HP‐FPA ERN‐01‐2016/739516].
Kraatari‐Tiri M, Soikkonen L, Myllykoski M, et al. HIDEA syndrome is caused by biallelic, pathogenic, rare or founder P4HTM variants impacting the active site or the overall stability of the P4H‐TM protein. Clinical Genetics. 2022;102(5):444‐450. doi: 10.1111/cge.14203
Minna Kraatari‐Tiri, Leila Soikkonen, Matti Myllykoski, and Yalda Jamshidi contributed equally to this work.
Funding information Academy of Finland, Grant/Award Number: 338446
DATA AVAILABILITY STATEMENT
The data that supports the findings of this study are available in the supplementary material of this article.
REFERENCES
- 1. Kaasinen E, Rahikkala E, Koivunen P, et al. Clinical characterization, genetic mapping and whole‐genome sequence analysis of a novel autosomal recessive intellectual disability syndrome. Eur J Med Genet. 2014;57(10):543‐551. doi: 10.1016/j.ejmg.2014.07.002 [DOI] [PubMed] [Google Scholar]
- 2. Rahikkala E, Myllykoski M, Hinttala R, et al. Biallelic loss‐of‐function P4HTM gene variants cause hypotonia, hypoventilation, intellectual disability, dysautonomia, epilepsy, and eye abnormalities (HIDEA syndrome). Genet Med. 2019;21(10):2355‐2363. doi: 10.1038/s41436-019-0503-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaelin WG, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393‐402. doi: 10.1016/j.molcel.2008.04.009 [DOI] [PubMed] [Google Scholar]
- 4. Byts N, Sharma S, Laurila J, et al. Transmembrane prolyl 4‐hydroxylase is a novel regulator of calcium signaling in astrocytes. eNeuro. 2021;8(1):ENEURO.0253‐20.2020. doi: 10.1523/ENEURO.0253-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maddirevula S, Ben‐Omran T, AlMureikhi M, et al. Further delineation of HIDEA syndrome. Am J Med Genet A. 2020;182(12):2999‐3006. doi: 10.1002/ajmg.a.61885 [DOI] [PubMed] [Google Scholar]
- 6. Järvelä I, Määttä T, Acharya A, et al. Exome sequencing reveals predominantly de novo variants in disorders with intellectual disability (ID) in the founder population of Finland. Hum Genet. 2021;140(7):1011‐1029. doi: 10.1007/s00439-021-02268-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hay E, Wilson LC, Hoskins B, Samuels M, Munot P, Rahman S. Biallelic P4HTM variants associated with HIDEA syndrome and mitochondrial respiratory chain complex I deficiency. Eur J Hum Genet. 2021;29(10):1536‐1541. doi: 10.1038/s41431-021-00932-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lim AM, Tan PL, Visruthan NK, Fong N, Viegelmann GC, Tan YH. HIDEA syndrome: a rare cause of congenital hypoventilation in a premature infant. Pediatr Pulmonol. 2022;11:1826‐1829. doi: 10.1002/ppul.25966 [DOI] [PubMed] [Google Scholar]
- 9. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928‐930. doi: 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Myllykoski M, Sutinen A, Koski MK, et al. Structure of transmembrane prolyl 4‐hydroxylase reveals unique organization of EF and dioxygenase domains. J Biol Chem. 2021;296:100197. doi: 10.1074/jbc.RA120.016542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Le Ber I, Clot F, Vercueil L, et al. Predominant dystonia with marked cerebellar atrophy: a rare phenotype in familial dystonia. Neurology. 2006;67(10):1769‐1773. doi: 10.1212/01.wnl.0000244484.60489.50 [DOI] [PubMed] [Google Scholar]
- 12. Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue‐based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Table S1 Clinical characteristics of patients described in this report (Patients 1–6) and all the previously published HIDEA patients to date.
Table S3 Results of the haplotype analysis showing the shared haplotype around the P4HTM (NM_177939.3) p.(Pro413Leu) variant and P4HTM p.(Arg296Ser;Val297_Arg358del) variant. The shared haplotype extended approximately 7 Mb (GRCh38 g.3:45758441–52 818 394) around the P4HTM (NM_177939.3) p.(Pro413Leu) variant and approximately 6.9 Mb (GRCh38 g.3:46372893–53 235 071) around the P4HTM (NM_177939.3) p.(Arg296Ser;Val297_Arg358del) variant.
Video S1 The gait of Patient 1. The video demonstrates the typical waddling gait associated with HIDEA syndrome.
Video S2 Dystonic movements of Patient 2.
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.