

Abstract
Objective
To assess the causality of the associations of rheumatoid arthritis (RA) with coronary artery disease (CAD) and stroke using the Mendelian randomization approach.
Methods
Independent single‐nucleotide polymorphisms strongly associated with RA (n = 70) were selected as instrumental variables from a genome‐wide association meta‐analysis including 14,361 RA patients and 43,923 controls of European ancestry. Summary‐level data for CAD, all stroke, any ischemic stroke and its subtypes, intracerebral hemorrhage (ICH), and subarachnoid hemorrhage were obtained from meta‐analyses of genetic studies, international genetic consortia, the UK Biobank, and the FinnGen consortium. We obtained summary‐level data for common cardiovascular risk factors and related inflammatory biomarkers to assess possible mechanisms.
Results
Genetic liability to RA was associated with an increased risk of CAD and ICH. For a 1‐unit increase in log odds of RA, the combined odds ratios were 1.02 (95% confidence interval [1.01, 1.03]; P = 0.003) for CAD and 1.05 (95% confidence interval [1.02, 1.08]; P = 0.001) for ICH. Genetic liability to RA was associated with increased levels of tumor necrosis factor and C‐reactive protein (CRP). The association with CAD was attenuated after adjustment for genetically predicted CRP levels. There were no associations of genetic liability to RA with the other studied outcomes.
Conclusion
This study found that genetic liability to RA was associated with an increased risk of CAD and ICH and that the association with CAD might be mediated by CRP. The heightened cardiovascular risk should be actively monitored and managed in RA patients, and this may include dampening systemic inflammation.
INTRODUCTION
Rheumatoid arthritis (RA) is the most common autoimmune arthritis, with a prevalence of 1%, and cardiovascular disease (CVD) is the leading cause of mortality worldwide (1, 2). Interestingly, CVD risk is substantially increased in RA and to a similar extent as other established risk factors such as diabetes mellitus (3). In meta‐analyses, both cardiovascular morbidity and mortality have been found to be 1.5‐fold elevated in RA compared to the general population (4, 5). The reasons for this remain poorly understood but may relate to shared risk factors (e.g., obesity and smoking) or an influence of RA on traditional cardiovascular risk factors (e.g., side effects of antirheumatic therapies or reduced physical activity due to pain). Importantly though, traditional risk factors do not fully explain the augmented CVD risk in RA, and observational studies suggest that RA may be a novel and independent risk factor for coronary disease (6, 7, 8, 9). CVD and RA have overlapping pathophysiologic mechanisms which may contribute, such as systemic inflammation, with cytokines raised in RA known to be important in driving atherosclerotic diseases (10). Consistent with this, systemic markers of inflammation are associated with cardiovascular risk in RA (11, 12). However, previous observational studies may have been limited by residual confounding or reverse causality. As such, whether RA is an independent and causal risk factor for CVDs and cardiometabolic risk factors remains equivocal.
Mendelian randomization (MR) analysis is an epidemiologic approach that can strengthen causal inference by using genetic variants as instrumental variables for the exposure (13). The method can minimize the influence of residual confounding, since genetic variants are randomly distributed at conception and are therefore unrelated to self‐adopted lifestyle and environmental confounders (13). In addition, the method can diminish reverse causality because the germline genotype cannot be modified by the onset and progression of the disease (13). Here, we conducted a 2‐sample MR study to examine the associations of genetic predisposition to RA with coronary artery disease (CAD), stroke, and its subtypes and cardiometabolic risk factors. We aimed to provide important evidence regarding the causal role of RA in causing a range of CVD and whether this could be through influencing traditional risk factors or systemic inflammation.
MATERIALS AND METHODS
Study design
We first examined the genetic correlations and MR associations of genetic predisposition to RA with CAD and stroke and its subtypes. To assess potential mechanisms, we investigated the associations of genetic predisposition to RA with common cardiovascular risk factors and related inflammatory biomarkers. We then conducted multivariable MR analysis to examine the mediation effects of RA‐associated factors in the associations between genetic predisposition to RA and the cardiovascular end points. This study was based on summary‐level data from international consortia, the UK Biobank, and the FinnGen consortium. All included studies had obtained ethical permits from corresponding ethics committees. The UK Biobank received ethical permits from the North West Multi‐centre Research Ethics Committee, the National Information Governance Board for Health and Social Care in England and Wales, and the Community Health Index Advisory Group in Scotland. All participants provided written informed consent. The present MR analyses were approved by the Swedish Ethical Review Authority (no. 2019‐02793). This study was conducted in accordance with the MR guideline (14).
Instrumental variable selection
Single‐nucleotide polymorphisms (SNPs) strongly associated with RA (P < 5 × 10−8) were obtained from a genome‐wide association meta‐analysis that included 14,361 RA patients and 43,923 controls of European ancestry (15). All RA cases were defined by the 1987 criteria of the American College of Rheumatology for RA diagnosis (16) or by a rheumatologist (15). Linkage disequilibrium in selected SNPs was estimated using the 1000 Genomes European reference panel. SNPs in high linkage disequilibrium (r 2 > 0.01 or clump windows <10,000 kb) were excluded, and the SNP with the lowest P value for the genome‐wide association with RA was retained. A total of 70 independent SNPs with beta and SE coefficients scaled to log‐transformed odds of RA were used as instrumental variables (Supplementary Table 1, on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42239). To provide estimates with a more intuitive interpretation, we estimated absolute genetic associations with RA using linear regression and used these summary‐level data for SNP–RA associations in a supplementary analysis (Supplementary Table 1). This enables the calculation of MR estimates that represent odds ratios (ORs) for the studied CVDs per 1% increase in the absolute probability of RA. Genetic associations were estimated in participants of genetic European descent in the UK Biobank. The outcome was defined using electronic health records and International Classification of Diseases codes (ICD‐9 714.0, ICD‐10: M05 or M06). Linear regression was performed with adjustment for age, sex, and 10 genomic principal components.
Data sources for outcomes
Summary‐level data for the associations of RA‐associated SNPs with CAD, all stroke, any ischemic stroke and its subtypes, ICH, and subarachnoid hemorrhage were obtained from meta‐analyses of genetic studies, international genetic consortia (17, 18, 19, 20), the UK Biobank, and the FinnGen consortium (21). There was minimal sample overlap between the exposure and outcome data sets. Detailed information, including case and control number and covariates adjusted for in the genome‐wide association analysis, is shown in Table 1. The associations of RA‐associated SNPs with the above outcomes are presented in Supplementary Table 2 (https://onlinelibrary.wiley.com/doi/10.1002/art.42239).
Table 1.
Included studies and consortia*
| Data source/outcome | Outcome | Ancestry | No. of patients | No. of controls | Adjustments in the GWAS |
|---|---|---|---|---|---|
| CARDIoGRAMplusC4D and UKBB | |||||
| CAD | Mixed | 122,733 | 424,528 | Not reported | |
| MEGASTROKE consortium | |||||
| All stroke | European | 40,585 | 406,111 | Age and sex | |
| Any ischemic stroke | European | 34,217 | 406,111 | Age and sex | |
| Large artery stroke | European | 3,373 | 406,111 | Age and sex | |
| Small vessel stroke | European | 5,386 | 406,111 | Age and sex | |
| Cardioembolic stroke | European | 7,193 | 406,111 | Age and sex | |
| ISGC Intracerebral hemorrhage | European | 3,223 | 3,725 | Age, sex, and principal components | |
| GWAS by Bakker et al Subarachnoid hemorrhage | European | 5,140 | 71,952 | Not reported | |
| UKBB | |||||
| All stroke | European | 12,036 | 355,525 | Age, sex, and 10 genetic principal components | |
| Any ischemic stroke | European | 6,566 | 360,995 | Age, sex, and 10 genetic principal components | |
| Intracerebral hemorrhage | European | 1,504 | 366,057 | Age, sex, and 10 genetic principal components | |
| Subarachnoid hemorrhage | European | 1,292 | 366,269 | Age, sex, and 10 genetic principal components | |
| FinnGen consortium | |||||
| CAD | European | 30,952 | 187,840 | Age, sex, first 10 genetic principal components, and genotyping batch | |
| All stroke | European | 18,661 | 166,201 | Age, sex, first 10 genetic principal components, and genotyping batch | |
| Any ischemic stroke | European | 10,551 | 202,223 | Age, sex, first 10 genetic principal components, and genotyping batch | |
| Intracerebral hemorrhage | European | 1,687 | 201,146 | Age, sex, first 10 genetic principal components, and genotyping batch | |
| Subarachnoid hemorrhage | European | 1,338 | 201,230 | Age, sex, first 10 genetic principal components, and genotyping batch |
GWAS = genome‐wide association study; CARDIoGRAMplusC4D = Coronary Artery Disease Genome‐wide Replication and Meta‐analysis plus The Coronary Artery Disease Genetics consortium; UKBB = UK Biobank; CAD = coronary artery disease; ISGC = International Stroke Genetic Consortium.
Data sources for cardiovascular risk factors, inflammatory biomarkers, and inflammatory bowel disease (IBD)
We obtained summary‐level data for cardiovascular risk factors (including body mass index [22], blood pressure [23], fasting glucose and insulin [24], high‐density and low‐density lipoprotein cholesterol and triglyceride [25], smoking initiation [26], and moderate‐to‐vigorous physical activity [the Neale Lab data, http://www.nealelab.is/uk-biobank] and inflammatory biomarkers such as interleukin‐6 [IL‐6] [27], tumor necrosis factor [TNF] [27], and C‐reactive protein [CRP] [28]) from international consortia and the UK Biobank (the Neale Lab data). Summary‐level data on IBD were obtained from a genome‐wide association meta‐analysis study including 59,957 individuals of European descent (29). Detailed information on the studies used are shown in Supplementary Table 3 (https://onlinelibrary.wiley.com/doi/10.1002/art.42239).
Genetic correlation analysis
Genome‐wide pairwise correlations between RA and studied CVD outcomes based on consortia data were estimated using linkage disequilibrium score regression (LDSC) that leverages genome‐wide association analysis summary‐level data and linkage disequilibrium to estimate genetic correlation (30). This method estimates universal genetic correlation by measuring correlation of effect size between SNP exposure and SNP outcome associations across all genetic variants in the genome. A genetic correlation (rg) of >0.7 was deemed a strong correlation. P values less than 0.006 (0.05 for 8 outcomes) was treated as significant in LDSC analysis.
Statistical analysis
We aligned the SNPs based on allele letter and allele frequency. SNPs that were missing in the outcome data sets were replaced by proxy SNPs, which were searched in https://ldlink.nci.nih.gov/ by implementing a setting of r 2 > 0.8 and using European populations as reference groups. Missing SNPs without proxies were excluded from the analysis. We searched phenotypes associated with RA‐associated SNPs at the genome‐wide significance level in PhenoScanner V2, a database of human genotype–phenotype associations (31).
The inverse variance–weighted method under the multiplicative random effects model was used as the main method to calculate the associations of genetic liability to RA with cardiovascular outcomes, cardiovascular risk factors, and inflammatory biomarkers. This method can provide the most precise estimate; however, it is sensitive to horizontal pleiotropy and outliers. Several sensitivity analyses, including the weighted median (32), MR‐Egger (33), MR‐PRESSO (34), and contamination mixture (35) methods, were used to examine the consistency of results and detect and correct for horizontal pleiotropy. The weighted median analysis can provide consistent causal estimates, given that more than half of weight derives from valid SNPs (32). The MR‐Egger regression can detect the horizontal pleiotropy by its intercept test and provide estimates after correcting for pleiotropic effects; however, the analysis is less powerful for most scenarios (33). In a comparative study, power to detect causal effect is usually greater for the inverse variance–weighted method compared to the MR‐Egger method in scenarios of different status of pleiotropy and satisfaction of the Instrument Strength Independent of Direct Effect assumption (33). The MR‐PRESSO method can also correct for horizontal pleiotropy by identifying and removing outlying SNPs (34). The contamination mixture method is good at analysis based on multiple genetic instruments and can generate causal estimates even when instruments contain invalid SNPs (35).
In addition, we used scatter plots to visualize the heterogeneity in estimates of the SNPs used and to determine whether the association was driven by certain SNPs. Estimates from different data sets, but for the same CVD, were combined using the fixed‐effects meta‐analysis method in which study‐specific estimates were weighted based on the amount of information captured by that study (i.e., more weight was given to a large study with many patients than a small study with few patients). Given that the HLA gene regions are shared by RA and other autoimmune disorders (36), we performed a sensitivity analysis after removal of SNPs in these gene regions (including HLA–A, HLA–B, HLA–C, HLA–DPA1, HLA–DPB1, HLA–DQA1, HLA–DQB1, HLA–DRA, HLA–DRB1, and HLA–DRB3). We used the multivariable MR analysis to estimate the mediation effects of RA‐associated factors in the associations between RA and cardiovascular outcomes. The multivariable MR analysis was based on the same set of genetic instruments (SNPs for RA), and the model was based on summary‐level beta coefficients and the corresponding standard error for RA, the outcome, and the mediator. In addition, we conducted a multivariable MR analysis to adjust for genetic liability to IBD (a common autoimmune disease) to minimize its influence. Likewise, this analysis used the same genetic variants as the main analysis, and MR estimates were obtained from a multivariable inverse variance–weighted analysis on the association between genetic liability to RA with a CVD outcome with adjustment for genetic liability to IBD.
Cochran's Q statistic and P value for MR‐Egger intercept were used to assess the heterogeneity and horizontal pleiotropy, respectively. The Bonferroni correction was used to account for multiple testing in examining the association between RA and CVDs. Associations with a P value less than 0.006 (0.05 for 8 outcomes) were deemed significant in order to correct for multiple testing. All tests were 2‐sided and were conducted using the TwoSampleMR and MendelianRandomization package (37, 38).
RESULTS
Results of the search in PhenoScanner V2 are presented in Supplementary Table 4 (https://onlinelibrary.wiley.com/doi/10.1002/art.42239). Several RA‐associated SNPs were found to be associated with other autoimmune diseases, including IBD, systemic lupus erythematosus, and type 1 diabetes, at the genome‐wide significance levels. A few other traits, such as immune cells, were identified to be associated with the SNPs that were used. There were few strong genetic correlations between RA and the studied cardiovascular outcomes (Supplementary Table 5, https://onlinelibrary.wiley.com/doi/10.1002/art.42239). RA showed a weak significant association with overall stroke (rg = 0.20; P = 0.003).
Genetic liability to RA was associated with an increased risk of CAD and ICH (Figure 1) consistently across sources. For a 1‐unit increase in log odds of RA, the combined odds ratios (ORs) were 1.02 (95% confidence interval [95% CI] 1.01, 1.03; P = 0.003) for CAD and 1.05 (95% CI 1.02, 1.08; P = 0.001) for ICH. The results were stable in all sensitivity analyses (Supplementary Table 6, https://onlinelibrary.wiley.com/doi/10.1002/art.42239). In a supplementary analysis in which estimates for the CVD outcomes were scaled per 1% increase in genetic liability to RA on the risk difference scale, the OR was 1.03 (95% CI 1.01, 1.05) for CAD and 1.06 (95% CI, 1.01, 1.11) for ICH (Supplementary Table 7, https://onlinelibrary.wiley.com/doi/10.1002/art.42239).
Figure 1.
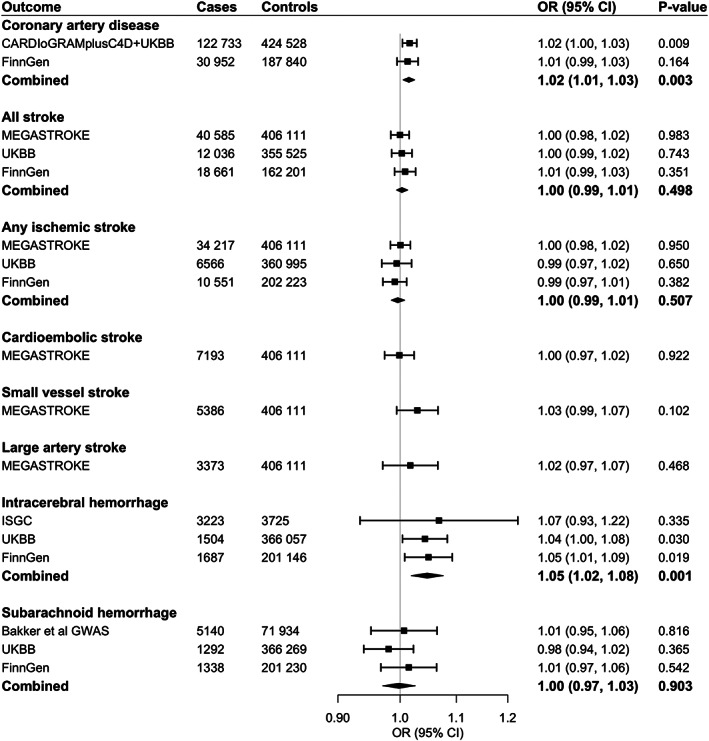
Associations of genetic liability to rheumatoid arthritis with coronary artery disease and stroke. OR = odds ratio; 95% CI = 95% confidence interval; CARDIoGRAMplusC4D = Coronary Artery Disease Genome‐wide Replication and Meta‐analysis plus The Coronary Artery Disease Genetics consortium; UKBB = UK Biobank; ISGC = International Stroke Genetic Consortium; GWAS = genome‐wide association study.
We detected moderate heterogeneity in the analyses for CAD and no horizontal pleiotropy (P for MR‐Egger intercept test > 0.4) (Supplementary Table 6). Even though a few outliers were detected in the MR‐PRESSO analyses for CAD, the associations remained consistent after removal of these outliers (Supplementary Table 6). As for associations with ICH in the 3 data sets, we observed no or modest heterogeneity, no indication of horizontal pleiotropy in the MR‐Egger intercept tests, and no outliers were detected by the MR‐PRESSO analyses (Supplementary Table 6). In scatter plots of associations with CAD and ICH, we did not observe any SNPs that drove the overall positive associations (Figure 2). Otherwise, there were no associations of genetic liability to RA with all stroke, any ischemic stroke and its subtypes, or subarachnoid hemorrhage (Figure 1 and Supplementary Table 6).
Figure 2.
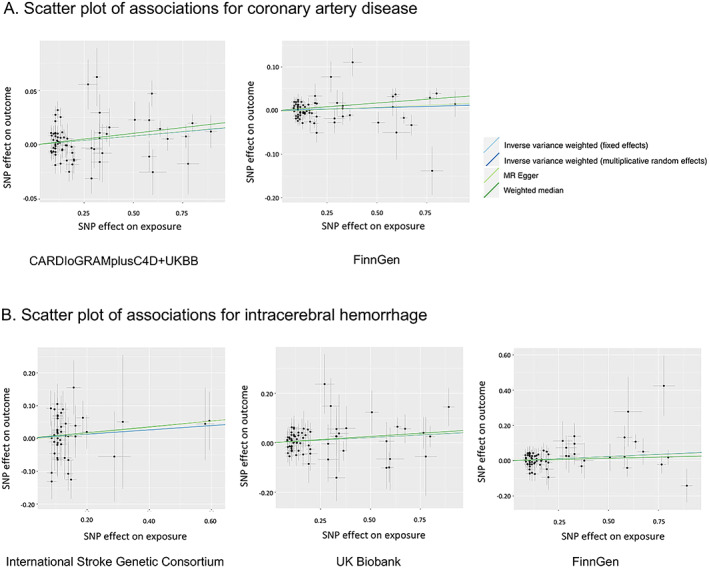
Scatter plots of associations with coronary artery disease and intracerebral hemorrhage. SNP = single‐nucleotide polymorphism; CARDIoGRAMplusC4D = Coronary Artery Disease Genome‐wide Replication and Meta‐analysis plus The Coronary Artery Disease Genetics consortium; MR = Mendelian randomization.
The observed associations with CAD and ICH remained stable in the sensitivity analysis after removal of SNPs in HLA gene regions (Supplementary Table 8, https://onlinelibrary.wiley.com/doi/10.1002/art.42239). The associations were also stable in the multivariable MR analysis with adjustment for genetic liability to IBD (Supplementary Table 9, https://onlinelibrary.wiley.com/doi/10.1002/art.42239).
With respect to cardiometabolic risk factors, genetic liability to RA was associated with reduced log odds ratio of smoking initiation and increased levels of high‐density lipoprotein cholesterol, TNF, and CRP (Figure 3). The associations remained directionally consistent in sensitivity analyses (Supplementary Table 10, https://onlinelibrary.wiley.com/doi/10.1002/art.42239). There were no associations of genetic liability to RA with the other cardiovascular risk factors and inflammatory biomarkers studied (Figure 3).
Figure 3.
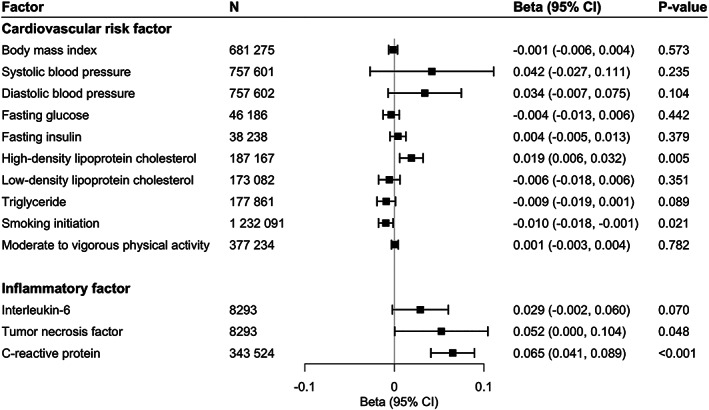
Associations of genetic liability to rheumatoid arthritis with cardiometabolic risk factors and inflammatory cytokines. 95% CI = 95% confidence interval.
Multivariate MR analyses were conducted to adjust for genetically predicted levels of TNF and CRP levels. The association between RA and CAD attenuated in the analysis with adjustment for genetically predicted CRP levels but not in the analysis with adjustment for genetically predicted TNF. The association between RA and ICH changed only slightly in the multivariable MR analyses (Figure 4).
Figure 4.
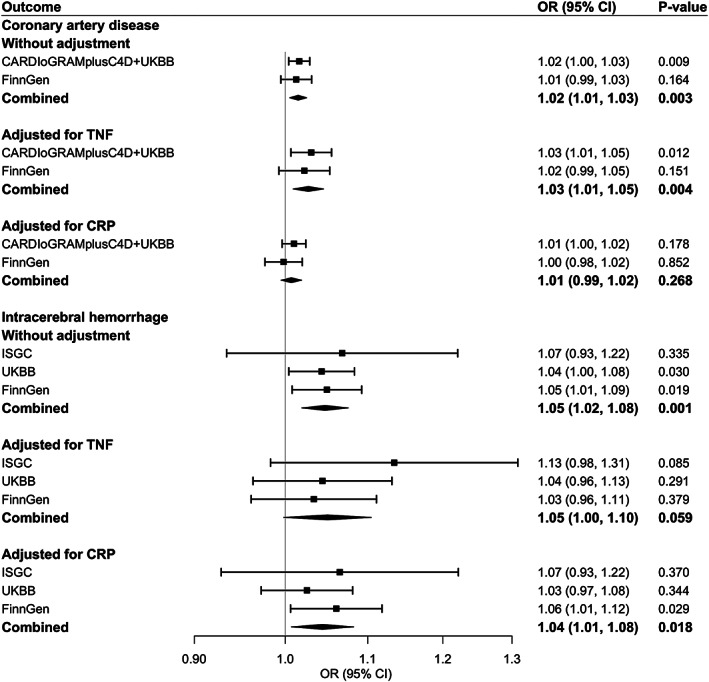
Associations of genetic liability to rheumatoid arthritis with coronary artery disease and intracerebral hemorrhage after adjustment for tumor necrosis factor (TNF) and C‐reactive protein (CRP). See Figure 1 for other definitions.
DISCUSSION
We conducted a 2‐sample MR study to investigate the causal associations of RA with CAD and stroke using data from large consortia and genetic studies. Few strong genetic correlations were observed between RA and studied cardiovascular outcomes. We found that genetic liability to RA was associated with elevated risk of CAD and ICH but not ischemic stroke or subarachnoid hemorrhage. These associations were consistent across different data sources, after removal of SNPs in HLA gene regions, and in the multivariable MR analysis with adjustment for genetic liability to IBD. Genetic liability to RA was associated with elevated levels of TNF and CRP. The increased levels of CRP appeared to mediate the association with CAD. We thus provide important genetic evidence supporting the link between RA and some CVDs and underscore the role of inflammation in driving CAD specifically.
RA has widely been reported as an important risk factor for CAD and impaired vascular function (33). A higher prevalence, extent, and severity of coronary plaque measured by coronary calcification (6) is found in RA patients, and this is related to disease duration, being increased in established compared to early RA (31). Similarly, invasive angiographic studies have also demonstrated RA to be associated with an increased extent of coronary atherosclerosis, with a higher prevalence of multivessel CAD, even after adjustment for some traditional risk factors (9). Importantly, this accelerated coronary atherosclerosis also appears to confer a substantially elevated risk of cardiovascular events, with incident myocardial infarction and CAD‐related mortality increased by 68% and 59% in RA, respectively, according to large meta‐analyses (3, 4).
Our findings support a causal role for RA in driving CAD, although we report a more modest effect size of 2% in the main analysis. This likely relates to differences in outcome definition (myocardial infarction versus the softer end point of CAD), more healthy populations included, and the calculation of risk according to log odds of RA. Overall, though, the totality of evidence suggests CAD to be increased in RA, and our study strongly suggests a causal role in this. There are ~14 million people globally with RA (39). As lifetime risk of CAD for people in general is already high (40), even a small increase in odds of CVD raises the expected number of CAD events in this population by tens of thousands compared to that expected for a similar sized group without RA. This potentially impacts public health policy around targeting of education, screening, and treatment at RA patients. Second, it provides insight into potential mechanisms of CAD. Even though we identified that chronic inflammation might mediate the association between RA and CAD, future research may be able to pinpoint exactly which metabolic or inflammatory changes occur in people with RA that lead to this increase in CAD, potentially identifying treatment or screening options for the whole population.
Stroke risk, of both ischemic and hemorrhagic types, has also widely been reported in observational studies to be increased in RA patients (41). Our study did not detect an association between genetic liability to RA and risk of stroke overall or ischemic stroke. This discrepancy may be related to confounding in observational studies, or increased stroke risk may not be caused by RA per se but by certain features of RA patients. Consistent with this, incident adverse events, including serious infections and insufficient treatment of CVDs, have been found to be drivers of the increased risk of stroke in RA patients (42). These null MR findings might be caused by inadequate power. Our study did, however, suggest a causal role for RA in causing ICH and supports the 68% increased risk reported in meta‐analysis of observational studies (41). Our consistent findings across 3 data sets highly suggest the validity of this association, and the underlying mechanisms warrant further investigation.
Several mechanisms have been proposed to explain the increased risk of CVD in RA patients. First, it has been suggested that RA may influence the development of traditional cardiovascular risk factors. We did not find genetic liability to RA to be associated with the majority of cardiometabolic risk factors, and only high‐density lipoprotein cholesterol, a protective factor, was significantly increased. We therefore provide some mechanistic evidence against the role of traditional risk factors, although it is possible that RA may indirectly influence traditional risk factors, such as due to side effects of antirheumatic or antiinflammatory medications. In addition, our null MR findings for the associations of genetic liability to RA with cardiometabolic risk factors could not completely rule out the effects of shared nongenetic factors on these associations. Furthermore, although far from significant, relatively large effect sizes were found for blood pressure in our study. In a recent published MR analysis including 461,880 hypertension patients and 337,653 controls, RA was associated with a high risk of hypertension (43), which is an important risk factor for CAD and the main cause of hemorrhagic stroke. However, a key hypothesis is that elevated systemic inflammation and remarkably overlapping inflammatory processes between the 2 conditions lead to progression of CVD (11, 44). In agreement with this, circulating levels of inflammatory markers such as CRP, erythrocyte sedimentation rate, and IL‐6 in RA patients are associated with a risk of cardiovascular events (11, 45) and with radiologic measures of coronary atherosclerosis (46).
Our study supports the notion that chronic inflammation drives CAD risk in RA, as CRP mediates the association between RA and CAD risk. However, our multivariable MR analysis did not suggest an important role of TNF for CAD or for RA‐associated inflammation on ICH. Other RA‐related abnormalities that may predispose one to CAD or ICH may include endothelial dysfunction, oxidative stress, lipid alterations, and posttranslational modifications of peptides (45). Further investigation is required into the mechanisms underlying the association between RA and ICH.
Elevated CVD in RA has long been recognized, and in recent years this has been incorporated into European clinical guidelines written for use by both rheumatology (European Alliance of Associations for Rheumatology) (47) and cardiology (European Society of Cardiology) (48) clinicians. In particular, the importance of regular cardiovascular risk assessment every 5 years is emphasized, as is the use of a 1.5‐fold multiplication factor to account for RA in risk scores based solely on traditional risk factors. However, evidence exists that cardiovascular risk factors remain undiagnosed in RA patients, and, even when detected, they may be undermanaged compared to patients with other risk factors like diabetes (49). Our study provides the first MR evidence supporting a causal role of RA in driving heightened cardiovascular risk. This not only emphasizes the importance of monitoring this high‐risk population, but also supports the notion in clinical guidelines that combating rheumatic disease activity is also integral to reducing cardiovascular risk. Current strategies to do this remain controversial, as some therapies have been associated with adverse cardiovascular effects (47, 50).
We provide the causal genetic evidence that inflammation drives CAD risk in RA and implicate this as an effective therapeutic target. TNF inhibitors are commonly used in clinical practice, and although some evidence exists for reduced cardiovascular risk in patients on such treatments (51), our results do not support this. CRP is a broad inflammatory marker raised by many pathways, including the IL‐1/IL‐6 axis. IL‐1 inhibition reduced cardiovascular events in the Canakinumab Antiinflammatory Thrombosis Outcome Study trial, and IL‐6 inhibition has been found to have beneficial effects on markers of atherosclerosis such as carotid intima‐media thickness (47) and to reduce cardiovascular events in RA (52). Although antiinflammatory treatments may prove useful in cardiovascular prevention in RA, studies to date have had inadequate follow‐up times and have been confounded by therapies being allocated to those with the most severe disease. Well‐designed clinical trials studying the impact of antiinflammatory therapies on cardiovascular risk in RA are required.
Given that autoimmune diseases have some overlapping genetic architecture, whether the observed associations between RA and CAD and between RA and ICH in our MR analysis were exclusive to RA remained undetermined, even though we employed several approaches to examine this. First, the results from the search of phenotypes associated with RA‐associated SNPs in PhenoScanner V2 showed no clear pattern that these used RA‐associated SNPs could systematically mimic the effects of other immune‐mediated disorders, although several RA‐associated SNPs were associated with several other immune‐mediated diseases at the genome‐wide significance level. Second, the observed associations with CAD and ICH remained stable in the sensitivity analysis after removal of SNPs in the HLA gene regions shared by autoimmune disorders (36), which indicated that the effects of most shared genes among autoimmune diseases did not drive the associations. Third, the associations remained in the multivariable MR analysis with adjustment for IBD. However, we could not perform this analysis to adjust for genetic liability to other common autoimmune diseases due to lack of data or too many missing SNPs in corresponding analysis. Even though our exploration implies the observed associations with CAD and ICH are likely to be specific to RA, further studies are needed to confirm our hypothesis.
The present study has several strengths, including MR design, the use of multiple genetic instruments, the use of different outcome data sources, the use of the multivariable MR analysis to explore possible mechanisms, and the population confinement to individuals of European descent (reducing population structure bias). In addition, lack of strong genetic correlations between RA and studied outcomes suggest that the observed associations with CAD and ICH may not be driven by shared genetic risk.
Several limitations should be considered when interpreting our findings. We observed moderate heterogeneity in the analyses for CAD in the CARDIoGRAMplusC4D (Coronary Artery Disease Genome‐wide Replication and Meta‐analysis plus The Coronary Artery Disease Genetics) consortium, UK Biobank, and FinnGen data sets. However, the corresponding MR‐Egger regression analysis did not detect any indication of horizontal pleiotropy, which suggests possible balanced horizontal pleiotropy that is unlikely to bias the MR estimate (53). In addition, the associations with CAD in 2 data sets were consistent across different sensitivity analyses with different assumptions. Even though there were a few outliers detected by MR‐PRESSO analyses, the associations remained after removal of these outliers. We did not take anti‐RA treatments into consideration in the current analysis. Nonetheless, whether corresponding treatments, such as anti‐TNF drugs and nonsteroidal antiinflammatory drugs, are associated with cardiovascular risk is unclear (44, 54). In addition, these treatments should not bias our causal estimation, as their use follows the diagnosis of RA and would therefore be classified as vertical pleiotropy (53).
The population confinement to European populations might limit the generalizability of our findings to other populations. In addition, whether the null findings for stroke and its subtypes (except for ICH) could be robustly held are uncertain, as the lack of significant associations might be caused by inadequate power despite the large sample size, at least for ischemic stroke. A power calculation for the current analysis was not possible due to the lack of information on phenotypic variance in RA explained by the SNPs used in the analysis, as this information cannot be calculated for a binary phenotype. Thus, future studies are needed to confirm these null findings. Whether the observed associations could be applied to subgroups defined by sex and status of anti–citrullinated protein autoantibodies could not be assessed due to lack of data.
In conclusion, this MR study found positive associations of genetic liability to RA with CAD and ICH, and the association with CAD appeared to be mediated by high levels of CRP. These findings highlight the importance of active monitoring and prevention of cardiovascular risk to combat CAD and ICH in RA patients. We further suggest that dampening inflammation might be a preventive strategy for CAD in RA patients, and well‐designed clinical trials are required to assess this.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Yuan had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Yuan, Carter, Larsson.
Acquisition of data
Yuan, Carter, Mason, Burgess, Larsson.
Analysis and interpretation of data
Yuan, Carter, Mason, Yang, Burgess, Larsson.
Supporting information
Disclosureform
Table S1 Genetic instruments for rheumatoid arthritis
Table S2. Associations of RA‐associated SNPs with studied outcomes
Table S3. Data sources for cardiovascular risk factors and inflammatory biomarkers
Table S4. Phenotypes assiciated with genetic instruments at the genome‐wide significance level
Table S5. Genetic correlations between RA and studied outcomes
Table S6. Associations of genetic liability to rheumatoid arthritis with coronary artery disease and stroke in sensitivity analyses
Table S7. Odds ratios of coronary artery disease and stroke per 1% increase in genetic liability to RA in the inverse variance weighted method
Table S8. Associations of genetic liability to rheumatoid arthritis with coronary artery disease and intracerebral hemorrhage after removal of SNPs in HLA gene regions
Table S9. Associations of genetic liability to rheumatoid arthritis with CAD and intracerebral hemorrhage in the multivariable MR analysis with adjustment for genetic liability to inflammatory bowel disease
Table S10. Associations of genetic liability to rheumatoid arthritis with cardiovascular risk factors and inflammatory biomarkers in sensitivity analyses
ACKNOWLEDGMENTS
We thank Mr. Bowen Tang (Department of Medical Epidemiology and Biostatistics, Karolinska Institutet) for assisting with data analysis. Genetic association estimates for coronary artery disease were obtained from a genome‐wide association meta‐analysis of CARDIoGRAMplusC4D (Coronary Artery Disease Genome‐wide Replication and Meta‐analysis plus The Coronary Artery Disease Genetics) and the UK Biobank study. Genetic association estimates for stroke were obtained from data sets from the MEGASTROKE study, the International Stroke Genetics Consortium, genome‐wide associations study by Bakker et al, the UK Biobank, and the FinnGen consortium. The authors thank all investigators for sharing these data. The MEGASTROKE project received funding from sources specified at http://www.megastroke.org/acknowledgments.html. The author list of MEGASTROKE is listed in https://www.megastroke.org/authors.html. Analyses of UK Biobank data were performed under application no. 29202.
The views expressed are those of the author(s) and not necessarily those of the NIHR or the Department of Health and Social Care.
Supported by the Swedish Heart‐Lung Foundation (Hjärt‐Lungfonden, grant 20210351), the Swedish Research Council for Health, Working Life and Welfare (Forte, grant 2018‐00123), the Swedish Research Council (Vetenskapsrådet, grant 2019‐00977), the United Kingdom Research and Innovation Medical Research Council (grant MC_UU_00002/7), the British Heart Foundation (grants RG/13/13/30194 and RG/18/13/33946), and NIHR Cambridge Biomedical Research Centre. Dr. Mason's work was supported by the EU/EFPIA Innovative Medicines Initiative Joint Undertaking BigData@Heart (grant 116074). Dr. Burgess's work was supported by a Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (grant 204623/Z/16/Z).
Drs. Yuan and Carter contributed equally to this work.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42239&file=art42239‐sup‐0001‐Disclosureform.pdf.
REFERENCES
- 1. Safiri S, Kolahi AA, Hoy D, Smith E, Bettampadi D, Mansournia MA, et al. Global, regional and national burden of rheumatoid arthritis 1990‐2017: a systematic analysis of the Global Burden of Disease study 2017. Ann Rheum Dis 2019;78:1463–71. [DOI] [PubMed] [Google Scholar]
- 2. Joseph P, Leong D, McKee M, Anand SS, Schwalm JD, Teo K, et al. Reducing the global burden of cardiovascular disease, part 1: the epidemiology and risk factors. Circ Res 2017;121:677–94. [DOI] [PubMed] [Google Scholar]
- 3. Van Halm VP, Peters MJ, Voskuyl AE, Boers M, Lems WF, Visser M, et al. Rheumatoid arthritis versus diabetes as a risk factor for cardiovascular disease: a cross‐sectional study, the CARRE Investigation. Ann Rheum Dis 2009;68:1395–400. [DOI] [PubMed] [Google Scholar]
- 4. Avina‐Zubieta JA, Thomas J, Sadatsafavi M, Lehman AJ, Lacaille D. Risk of incident cardiovascular events in patients with rheumatoid arthritis: a meta‐analysis of observational studies. Ann Rheum Dis 2012;71:1524–9. [DOI] [PubMed] [Google Scholar]
- 5. Aviña‐Zubieta JA, Choi HK, Sadatsafavi M, Etminan M, Esdaile JM, Lacaille D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta‐analysis of observational studies. Arthritis Rheum 2008;59:1690–7. [DOI] [PubMed] [Google Scholar]
- 6. Karpouzas GA, Malpeso J, Choi TY, Li D, Munoz S, Budoff MJ. Prevalence, extent and composition of coronary plaque in patients with rheumatoid arthritis without symptoms or prior diagnosis of coronary artery disease. Ann Rheum Dis 2014;73:1797–804. [DOI] [PubMed] [Google Scholar]
- 7. Løgstrup BB, Olesen KK, Masic D, Gyldenkerne C, Thrane PG, Ellingsen T, et al. Impact of rheumatoid arthritis on major cardiovascular events in patients with and without coronary artery disease. Ann Rheum Dis 2020;79:1182–8. [DOI] [PubMed] [Google Scholar]
- 8. Del Rincón ID, Williams K, Stern MP, Freeman GL, Escalante A. High incidence of cardiovascular events in a rheumatoid arthritis cohort not explained by traditional cardiac risk factors. Arthritis Rheum 2001;44:2737–45. [DOI] [PubMed] [Google Scholar]
- 9. Warrington KJ, Kent PD, Frye RL, Lymp JF, Kopecky SL, Goronzy JJ, et al. Rheumatoid arthritis is an independent risk factor for multi‐vessel coronary artery disease: a case control study. Arthritis Res Ther 2005;7:R984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuan S, Lin A, He QQ, Burgess S, Larsson SC. Circulating interleukins in relation to coronary artery disease, atrial fibrillation and ischemic stroke and its subtypes: a two‐sample Mendelian randomization study. Int J Cardiol 2020;313:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang J, Chen L, Delzell E, Muntner P, Hillegass WB, Safford MM, et al. The association between inflammatory markers, serum lipids and the risk of cardiovascular events in patients with rheumatoid arthritis. Ann Rheum Dis 2014;73:1301–8. [DOI] [PubMed] [Google Scholar]
- 12. Navarro‐Millán I, Yang S, DuVall SL, Chen L, Baddley J, Cannon GW, et al. Association of hyperlipidaemia, inflammation and serological status and coronary heart disease among patients with rheumatoid arthritis: data from the National Veterans Health Administration. Ann Rheum Dis 2016;75:341–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burgess S, Thompson SG. Mendelian randomization: methods for using genetic variants in causal estimation. London: Chapman and Hall/CRC; 2015. [Google Scholar]
- 14. Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res 2019;4:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014;506:376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 17. Van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res 2018;122:433–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, et al. Multiancestry genome‐wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 2018;50:524–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Woo D, Falcone GJ, Devan WJ, Brown WM, Biffi A, Howard TD, et al. Meta‐analysis of genome‐wide association studies identifies 1q22 as a susceptibility locus for intracerebral hemorrhage. Am J Hum Genet 2014;94:511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bakker MK, van der Spek RA, van Rheenen W, Morel S, Bourcier R, Hostettler IC, et al. Genome‐wide association study of intracranial aneurysms identifies 17 risk loci and genetic overlap with clinical risk factors. Nat Genet 2020;52:1303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. consortium TF . R5 release of FinnGen consortium genome‐wide association analysis data. 2021. URL: https://finngen.gitbook.io/documentation/.
- 22. Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, et al. Meta‐analysis of genome‐wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet 2018;27:3641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Evangelou E, Warren HR, Mosen‐Ansorena D, Mifsud B, Pazoki R, Gao H, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet 2018;50:1412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010;42:105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet 2013;45:1274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 2019;51:237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ahola‐Olli AV, Wurtz P, Havulinna AS, Aalto K, Pitkanen N, Lehtimaki T, et al. Genome‐wide association study identifies 27 loci influencing concentrations of circulating cytokines and growth factors. Am J Hum Genet 2017;100:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Folkersen L, Gustafsson S, Wang Q, Hansen DH, Hedman ÅK, Schork A, et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat Metab 2020;2:1135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, et al. Genome‐wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 2017;49:256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bulik‐Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015;47:1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, et al. PhenoScanner V2: an expanded tool for searching human genotype‐phenotype associations. Bioinformatics 2019;35:4851–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 2016;40:304–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bowden J, Smith GD, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50:693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Burgess S, Foley CN, Allara E, Staley JR, Howson JM. A robust and efficient method for Mendelian randomization with hundreds of genetic variants. Nat Commun 2020;11:376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet 2009;54:15–39. [DOI] [PubMed] [Google Scholar]
- 37. Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR‐Base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol 2017;46:1734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Almutairi K, Nossent J, Preen D, Keen H, Inderjeeth C. The global prevalence of rheumatoid arthritis: a meta‐analysis based on a systematic review. Rheumatol Int 2021;41:863–77. [DOI] [PubMed] [Google Scholar]
- 40. Lloyd‐Jones DM, Larson MG, Beiser A, Levy D. Lifetime risk of developing coronary heart disease. Lancet. 1999;353:89–92. [DOI] [PubMed] [Google Scholar]
- 41. Wiseman SJ, Ralston SH, Wardlaw JM. Cerebrovascular disease in rheumatic diseases: a systematic review and meta‐analysis. Stroke 2016;47:943–50. [DOI] [PubMed] [Google Scholar]
- 42. Meissner Y, Richter A, Manger B, Tony HP, Wilden E, Listing J, et al. Serious adverse events and the risk of stroke in patients with rheumatoid arthritis: results from the German RABBIT cohort. Ann Rheum Dis 2017;76:1583–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Qiu S, Li M, Jin S, Lu H, Hu Y. Rheumatoid arthritis and cardio‐cerebrovascular disease: a mendelian randomization study. Front Genet 2021;12:745224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yuan S, Carter P, Bruzelius M, Vithayathil M, Kar S, Mason AM, et al. Effects of tumour necrosis factor on cardiovascular disease and cancer: a two‐sample Mendelian randomization study. EBioMedicine 2020;59:102956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. England BR, Thiele GM, Anderson DR, Mikuls TR. Increased cardiovascular risk in rheumatoid arthritis: mechanisms and implications. BMJ 2018;361:k1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chung CP, Oeser A, Raggi P, Gebretsadik T, Shintani AK, Sokka T, et al. Increased coronary‐artery atherosclerosis in rheumatoid arthritis: relationship to disease duration and cardiovascular risk factors. Arthritis Rheum 2005;52:3045–53. [DOI] [PubMed] [Google Scholar]
- 47. Agca R, Heslinga SC, Rollefstad S, Heslinga M, McInnes IB, Peters MJ, et al. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis 2017;76:17–28. [DOI] [PubMed] [Google Scholar]
- 48. Piepoli MF, Hoes AW, Agewall S, Albus C, Brotons C, Catapano AL, et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: the Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J 2016;37:2315–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Desai SS, Myles JD, Kaplan MJ. Suboptimal cardiovascular risk factor identification and management in patients with rheumatoid arthritis: a cohort analysis. Arthritis Res Ther 2012;14:R270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Semb AG, Ikdahl E, Wibetoe G, Crowson C, Rollefstad S. Atherosclerotic cardiovascular disease prevention in rheumatoid arthritis. Nat Rev Rheumatol 2020;16:361–79. [DOI] [PubMed] [Google Scholar]
- 51. Westlake SL, Colebatch AN, Baird J, Curzen N, Kiely P, Quinn M, et al. Tumour necrosis factor antagonists and the risk of cardiovascular disease in patients with rheumatoid arthritis: a systematic literature review. Rheumatology (Oxford) 2011;50:518–31. [DOI] [PubMed] [Google Scholar]
- 52. Singh S, Fumery M, Singh AG, Singh N, Prokop LJ, Dulai PS, et al. Comparative risk of cardiovascular events with biologic and synthetic disease‐modifying antirheumatic drugs in patients with rheumatoid arthritis: a systematic review and meta‐analysis. Arthritis Care Res (Hoboken) 2020;72:561–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Holmes MV, Ala‐Korpela M, Smith GD. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol 2017;14:577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Antman EM. Evaluating the cardiovascular safety of nonsteroidal anti‐inflammatory drugs. Circulation 2017;135:2062–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosureform
Table S1 Genetic instruments for rheumatoid arthritis
Table S2. Associations of RA‐associated SNPs with studied outcomes
Table S3. Data sources for cardiovascular risk factors and inflammatory biomarkers
Table S4. Phenotypes assiciated with genetic instruments at the genome‐wide significance level
Table S5. Genetic correlations between RA and studied outcomes
Table S6. Associations of genetic liability to rheumatoid arthritis with coronary artery disease and stroke in sensitivity analyses
Table S7. Odds ratios of coronary artery disease and stroke per 1% increase in genetic liability to RA in the inverse variance weighted method
Table S8. Associations of genetic liability to rheumatoid arthritis with coronary artery disease and intracerebral hemorrhage after removal of SNPs in HLA gene regions
Table S9. Associations of genetic liability to rheumatoid arthritis with CAD and intracerebral hemorrhage in the multivariable MR analysis with adjustment for genetic liability to inflammatory bowel disease
Table S10. Associations of genetic liability to rheumatoid arthritis with cardiovascular risk factors and inflammatory biomarkers in sensitivity analyses