

Abstract
Background
T‐cell activation is associated with an adverse outcome in COVID‐19, but whether T‐cell activation and exhaustion relate to persistent respiratory dysfunction and death is unknown.
Objectives
To investigate whether T‐cell activation and exhaustion persist and are associated with prolonged respiratory dysfunction and death after hospitalization for COVID‐19.
Methods
Plasma and serum from two Norwegian cohorts of hospitalized patients with COVID‐19 (n = 414) were analyzed for soluble (s) markers of T‐cell activation (sCD25) and exhaustion (sTim‐3) during hospitalization and follow‐up.
Results
Both markers were strongly associated with acute respiratory failure, but only sTim‐3 was independently associated with 60‐day mortality. Levels of sTim‐3 remained elevated 3 and 12 months after hospitalization and were associated with pulmonary radiological pathology after 3 months.
Conclusion
Our findings suggest prolonged T‐cell exhaustion is an important immunological sequela, potentially related to long‐term outcomes after severe COVID‐19.
Keywords: NOR‐Solidarity, pulmonary function, SARS‐CoV‐2, T‐cell activation, T‐cell exhaustion
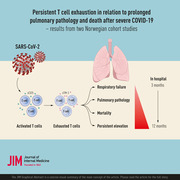
Introduction
Although COVID‐19 is primarily a viral respiratory disease, aberrant immune responses to the causative virus SARS‐CoV‐2—which lead to systemic inflammation—are important features for developing severe and critical disease [1]. Several reports have demonstrated elevated levels of central inflammatory cytokines, such as interleukin (IL) 6, tumor necrosis factor, and various inflammatory chemokines such as CXCL10 in patients with severe COVID‐19, when compared to patients with mild to moderate disease [2]. Lymphopenia—in particular, T‐cell lymphopenia—is also a frequent finding in those with severe disease [3].
T cells contribute to viral clearance, but persistent activation may trigger harmful effects to the host, including the development of T‐cell exhaustion [4, 5]. Several inhibitory molecules and check‐point receptors such as T‐cell immunoglobulin and mucin domain‐3 (Tim‐3) and programmed cell death protein‐1 (PD‐1) [6] are typically upregulated during T‐cell activation to prevent potentially harmful, persistent, and disproportionate T‐cell activation. However, if these receptors are persistently activated, they may lead T cells into a state of exhaustion [6]. Human Tim‐3 is a transmembrane protein, expressed on several cell types, including T cells and monocytes [7, 8]. Tim‐3 can be cleaved from the cell surface by certain matrix metalloproteinases to a soluble (s) form [9]. This circulating form correlates with the level of membrane‐bound Tim‐3 [10], and could represent a soluble marker of T‐cell exhaustion. We have previously reported that elevated plasma Tim‐3 levels were associated with disease severity in 39 hospitalized COVID‐19 patients, implicating persistent T‐cell activation and T‐cell exhaustion in the pathogenesis of severe COVID‐19 [11]. However, whether T‐cell activation and exhaustion persist and are associated with prolonged respiratory dysfunction and death after initial infection is largely unknown.
We herein report findings from two prospective cohort studies in Norway, analyzing the circulating levels of sTim‐3 as a putative marker of T‐cell exhaustion, in 414 hospitalized COVID‐19 patients in relation to disease severity and 60‐day mortality. In addition, we also include measurement of sCD25 as an established marker of T‐cell activation [12]. The study includes data collected during three waves of the COVID‐19 pandemic between March 2020 and September 2021, thus encompassing data before and after the introduction of systemic corticosteroids in the WHO recommendations for treatment of severe COVID‐19 in August 2020 [13]. We also report the relationship between these T‐cell markers and pulmonary function, as well as chest pathology assessed by computed tomography (CT) at outpatient follow‐up 3 months after hospitalization.
Materials and methods
Study design and participants
Data from two prospective cohort studies in Norway were pooled and assessed (Fig. S1). Cohort 1 was the NOR‐Solidarity trial, a multicenter, open‐label, adaptive randomized controlled trial (RCT) evaluating the effect of antiviral drugs on hospitalized COVID‐19 patients admitted to 23 Norwegian hospitals (NCT04321616) [14] as an add‐on to the WHO Solidarity trial [15]. Cohort 2 was the Norwegian SARS‐CoV‐2 study, an observational study of hospitalized COVID‐19 patients admitted to five Norwegian hospitals (NCT04381819), conducted as part of the International Severe Acute Respiratory and Emerging Infection Consortium World Health Organization Clinical Characterization Protocol study (doi: https://doi.org/10.1101/2020.07.17.20155218).
Participants in cohort 1 were included from 28 March until 5 October 2020, encompassing the first two COVID‐19 waves, while participants in cohort 2 were included from 10 March 2020 until 1 September 2021, encompassing three COVID‐19 waves. In Norway, the waves were estimated as the following periods: wave one, 8 March 2020 to 31 July 2020; wave two, 1 August 2020 to 17 February 2021; and wave three, 18 February 2021 to 31 July 2021. From February 2021, Alpha was the dominating variant, replaced by the Delta variant from July 2021. For both cohorts, all participants ≥18 years admitted to the hospital with polymerase chain reaction (PCR)‐confirmed SARS‐2‐CoV‐2 infection were eligible for inclusion, and between one and three blood samples were obtained from each patient from within the first 48 h of admission and up to 10 days during hospitalization, and in subgroups also at 3‐ and 12‐month follow‐ups. Viral load in oropharyngeal specimens was determined by real time (RT)‐PCR analysis, as previously described [14]. All participants gave informed consent before inclusion, either by themselves or by a legally authorized representative. Both studies were approved by the Regional Committee for Medical and Health Research Ethics in South‐Eastern Norway (cohort 1 reference no. 118684, cohort 2 reference nos. 106624 and 2019/306) and cohort 1 also by the Norwegian Medicines Agency (20/04950‐23).
Intervention and outcomes
In cohort 1 (the NOR‐Solidarity trial, n = 162), participants were randomized and allocated to one of three treatment arms: (1) local standard of care (SoC); (2) SoC plus 800 mg of oral hydroxychloroquine (HCQ) twice daily on day 1, then 400 mg twice daily up to 9 days; or (3) SoC plus 200 mg of intravenous remdesivir on day 1, then 100 mg daily up to 9 days. All study treatments were stopped at hospital discharge or if contraindicated during the study. Since the interventions did not affect clinical outcome, viral clearance, or systemic inflammation [14], data from the different intervention arms were in this substudy pooled together with samples from cohort 2 (the Norwegian SARS‐CoV‐2 study, n = 252) to examine whether levels of sCD25 and sTim‐3 were associated with: (i) acute respiratory failure (RF), defined as PaO2/FiO2 (P/F ratio) <26.6 kPa (<200 mmHg) during hospitalization; (ii) the need for treatment at the intensive care unit (ICU) during hospitalization; and (iii) 60‐day post‐admission total mortality.
Follow‐up
Biobanking. As illustrated in the flow chart (Fig. 1), 257 participants (cohort 1, n = 100; cohort 2, n = 157) attended a follow‐up visit 3 months after hospital discharge, which included blood sampling for routine clinical biochemistry and biobanking. In cohort 2, 62 participants also attended a 12‐month follow‐up visit that included blood sampling for routine clinical biochemistry and biobanking.
Fig. 1.
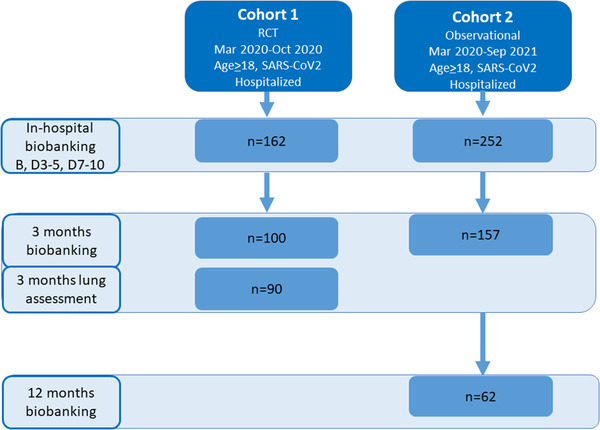
Flow chart showing the assessment of participants from the two cohorts during hospitalization, at 3‐month follow‐up, and at 12‐month follow‐up. Additionally, at 3‐month follow‐up, T cells from six participants in cohort 1 were included for transcriptomic analyses. B, baseline; D, day.
Pulmonary assessment. In cohort 1, lung function tests (n = 90, Fig. S1)—consisting of spirometry and diffusion capacity of the lungs for carbon monoxide (DLCO)—were performed as previously described [16]. DLCO was selected as a measure of pulmonary function as this has been shown to be the most frequently affected after hospitalization for COVID‐19 [17]. DLCO in percent of predicted value and the lower limit of normal (LLN) were calculated according to the Global Lung Function Initiative Network [16]. Persistent respiratory dysfunction was defined as DLCO below the LLN.
In addition, low‐dose, thin‐section chest CT images were obtained in supine and prone positions during breath holding in deep inspiration as described [16]. For the purpose of this study, we assessed the prevalence of any ground‐glass opacities (GGO), GGO ≥10% in at least one of the four lung zones, or any mosaic pattern as potentially reversible changes and grouped together. Any consolidations, reticular pattern, parenchymal bands, interlobular septal thickening, or any bronchiectasis were interpreted as potentially irreversible changes and grouped together. CT changes that were classified as possible reversible changes were interpreted to reflect inflammation, and changes that were classified as potentially irreversible were interpreted to reflect fibrosis [16].
Blood sampling protocol and biochemical analyses
Peripheral venous blood was drawn into pyrogen‐free blood collection tubes with (cohort 1) or without ethylenediamine tetraacetic acid (EDTA) (cohort 2) as an anticoagulant (i.e., plasma and serum, respectively), immediately immersed in melting ice, and centrifuged at 2500 × g for 30 min within 20 min to obtain platelet‐poor plasma or allowed to clot before centrifugation (serum, 1500 × g for 10 min). All samples were stored at −80°C and thawed less than three times.
Soluble levels of sCD25 and sTim‐3 were measured in duplicate by enzyme immunoassays (EIA) using commercially available antibodies (R&D Systems, Minneapolis, MN) in a 384‐format using a combination of a SELMA (Jena, Germany) pipetting robot and a BioTek (Winooski, VT, USA) dispenser/washer. Absorption was read at 450 nm with wavelength correction set to 540 nm using an EIA plate reader (Bio‐Rad, Hercules, CA, USA). The intra‐assay coefficient of variation (%), based on data from our laboratory, was <10%. When comparing serum and plasma levels of sCD25 and sTim‐3 in 16 healthy controls, we found no significant differences (sCD25 p = 0.54, sTim‐3 p = 0.16). Samples from 21 age‐ and sex‐matched healthy controls (mean age ± standard deviation [SD] 55 ± 12; 55% men) were used for reference values.
Routine laboratory variables (i.e., C‐reactive protein [CRP], ferritin, and total leukocyte, neutrophil, lymphocyte, and monocyte counts) were measured at the biochemical laboratories at the participating hospitals.
Statistics
Patient characteristics were compared using Student's t‐test or Mann–Whitney U‐test depending on the distribution or chi‐square for continuous and categorical variables, respectively.
The association between admission levels of sCD25 and sTim‐3 and RF/ICU admission or 60‐day all‐cause mortality, respectively, was first assessed by receiver operating characteristic (ROC) analysis. The association with 60‐day all‐cause mortality was assessed by Kaplan–Meier analysis according to a cut off identified by Youden's index and Cox regression. For 60‐day mortality, the number of events did not justify more than four adjustment variables, and the adjustment variables were comprised in three models using propensity scores (M1: age, COVID wave, and dexamethasone treatment; M2: variables in M1 + chronic cardiac and pulmonary disease, neutrophil and lymphocyte counts, and estimated glomerular filtration rate [eGFR]; M3: variables in M2 + CRP).
Soluble levels of sCD25 and sTim‐3 were non‐normally distributed and thus transformed using log10 for temporal comparisons between groups with the linear mixed model analysis. Linear mixed models were performed with the subject as the random effect, and time and RF, ICU admission, or 60‐day mortality as fixed effects (also as interaction) in addition to adjustment variables (RF/ICU admission: COVID wave, dexamethasone treatment, obesity, and neutrophil and lymphocyte counts). For 60‐day mortality, M2 as described above was used for initial adjustment, as well as M3 (i.e., +CRP). Data are presented as back‐transformed estimated marginal means with 95% confidence intervals from the mixed models, and levels reflect the adjustment from covariates. Post‐hoc analysis (sequential Sidak) between groups is reported if the group or group interaction term were significant. Similar models were used for evaluating the effects of randomized treatment and DLCO and chest CT in cohort 1 (adjusting for randomized treatment, age, sex, and neutrophil counts), and “COVID wave” or dexamethasone were used in cohort 2 (adjusting for age and sex).
We further used linear mixed models to model the association between P/F ratio (outcome) and sCD25 and sTim‐3, included individually in separate models, with time treated as a factor variable. A random intercept by subject was used to control for repeated measures, with each subject having between one and three measured follow‐up periods.
Results
Baseline characteristics
Demographic data and clinical characteristics from the combined cohort according to clinical outcomes are given in Table 1 and in the two separate cohorts in Table S1. Patients with outcomes had more comorbidities (ICU or RF: obesity; nonsurvivors: chronic cardiac and pulmonary disease); were more often treated with dexamethasone; received oxygen therapy; and had lower P/F ratio, hemoglobin, and lymphocyte counts and higher CRP, ferritin, WBC, and neutrophil counts. In addition, patients who died were older and had lower eGFR. Comparing the two cohorts, patients were an average of 60 (cohort 1) and 57 years old (cohort 2), mostly men (64% and 63%, respectively), and hypertension was the major comorbidity. Apart from the different treatment modalities within cohort 1 (RCT) and frequent use of dexamethasone and higher rate of ICU admission among patients in cohort 2, the two cohorts had comparable baseline characteristics.
Table 1.
Demographic, clinical, and biochemical characteristics in 414 patients hospitalized for COVID‐19, stratified according to acute‐phase (ICU admission or RF) outcomes or 60‐day death
| ICU or RF | Death | |||
|---|---|---|---|---|
| No, n = 238 | Yes, n = 176 | No, n = 377 | Yes, n = 37 | |
| Age, years | 57.2 ± 15.2 | 59.1 ± 15.5 | 56.7 ± 15.0 | 71.5 ± 12.2 * |
| Male gender (%) | 151 (63) | 111 (63) | 235 (62) | 27 (73) |
| Body mass index, kg/m2 | 28.1 ± 5.0 | 29.1 ± 4.7 | 28.6 ± 4.9 | 27.4 ± 4.6 |
| Wave | ||||
| 1 | 143 (60) * | 73 (42) | 201 (53) | 15 (41) |
| 2 | 56 (24) | 62 (35) * | 102 (27) | 16 (43) * |
| 3 | 39 (16) | 41 (23) | 74 (20) | 6 (16) |
| Treatment group | ||||
| SoC (%) | 185 (78) | 148 (84) | 300 (80) | 33 (89) |
| SoC + hydroxychloroquine (%) | 29 (12) | 14 (8) | 41 (11) | 2 (5) |
| SoC + remdesivir (%) | 24 (10) | 14 (8) | 36 (10) | 2 (5) |
| Vaccinated ≥1 dose | 4 (2) | 2 (1) | 5 (1) | 1 (3) |
| Dexamethasone (%) | 47 (20) | 89 (51) * | 114 (30) | 22 (60) * |
| Oxygen therapy (%) | 131 (55) | 154 (88) | 253 (67) | 32 (87) * |
| Comorbidities | ||||
| Chronic cardiac disease (%) | 40 (17) | 31 (18) | 52 (14) | 19 (51) * |
| Hypertension (%) | 77 (34) | 58 (34) | 121 (33) | 14 (40) |
| Chronic pulmonary disease (%) | 54 (23) | 44 (25) | 84 (23) | 14 (38) * |
| Obesity (%) | 54 (24) | 59 (34) * | 104 (29) | 9 (24) |
| Diabetes (%) | 48 (21) | 37 (22) | 75 (21) | 10 (29) |
| Current smoker (%) | 12 (5) | 8 (5) | 19 (5) | 1 (3) |
| HIV/AIDS (%) | 7 (4) | 3 (2) | 8 (3) | 2 (7) |
| Outcomes | ||||
| ICU admission (%) | 0 (0) | 110 (63) | 83 (22) | 27 (73) * |
| RF (%) | 0 (0) | 125 (72) | 101 (27) | 24 (67) * |
| 60‐day death (%) | 4 (2) | 33 (19) | 0 (0) | 37 (100) |
| P/F ratio at admission, kPa |
45.2 (37.9, 52.7) |
30.4 (21.9, 43.2) * |
41.3 (31.6, 49.8) |
27.1 (13.8, 48.3) |
| Hemoglobin, g/dl | 13.3 ± 1.7 | 12.7 ± 1.7 * | 13.1 ± 1.6 | 12.0 ± 1.8 * |
| C‐reactive protein, mg/L |
48 (24, 102) |
91 (42, 154) * |
56 (26, 120) |
108 (75, 162) * |
| Ferritin, μg/L |
514 (256, 853) |
875 (455, 1436) * |
600 (301, 1107) |
878 (532, 1435) * |
| White blood cell count, ×109/L | 6.1 ± 2.6 | 7.6 ± 3.5 * | 6.6 ± 3.0 | 7.5 ± 3.3 * |
| Neutrophils, ×109/L | 4.3 ± 2.4 | 6.2 ± 3.4 * | 5.0 ± 3.0 | 6.2 ± 2.8 * |
| Lymphocytes, ×109/L | 1.2 ± 0.6 | 1.0 ± 0.5 * | 1.2 ± 0.5 | 0.8 ± 0.4 * |
| eGFR |
94 (77, 107) |
90 (72, 105) |
94 (78, 108) |
72 (37, 90) * |
Note: Continuous variables are shown as mean ± standard deviation (SD) or median (25th, 75th) percentile depending on distribution.
Abbreviations: eGFR, estimated glomerular filtration rate; ICU, intensive care unit; P/F ratio, PaO2/FiO2 ratio; RF, respiratory failure; SoC, standard of care.
* p < 0.05.
Levels of sCD25 and sTim‐3 in relation to comorbidities
We first assessed the temporal profile of sCD25 and sTim‐3 during hospitalization according to comorbid disease. As shown in Fig. S1A, there were no effects of comorbidities including chronic cardiac or pulmonary disease, hypertension, obesity, or diabetes, or any effect of accumulated comorbidities (i.e., adding together within each patient, Fig. S1B). Dividing chronic pulmonary disease in chronic obstructive pulmonary disease (COPD) and asthma yielded no differences in the temporal profile of sCD25. For sTim‐3, patients with chronic cardiac disease and diabetes had higher levels at 7–10 days, while obese patients had higher levels at admission and 3–5 days. No other differences were detected for sTim‐3.
Plasma sCD25 and sTim‐3 levels in relation to RF during hospitalization
Figure 2a,b shows ROC analysis of admission levels of sCD25 and sTim‐3 in relation to RF and ICU admission in the total cohort. Although highly significant, the discriminatory abilities were modest in identifying patients with RF or ICU admission.
Fig. 2.
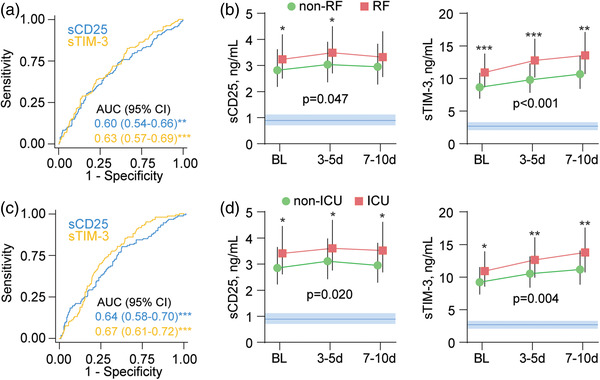
sCD25 and sTim‐3 in relation to respiratory failure (RF) and intensive care unit (ICU) admission. Receiver operating characteristic (ROC) analysis of admission levels of sCD25 and sTim‐3 in relation to (a) RF and (c) ICU admission. Temporal profile of sCD25 and sTim‐3 according to (b) RF or (d) ICU admission during the first 10 days after admission shown as estimated marginal means and 95% confidence intervals (CI), shown as red or green area. The p‐values reflect the group (outcome) effect from the linear mixed models adjusted for COVID wave, dexamethasone treatment, obesity, neutrophil count, and lymphocyte count. Blue areas reflect reference ranges from 21 healthy controls. *p < 0.05, **p < 0.01, ***p < 0.001 between groups.
In longitudinal samples during hospitalization, patients with RF maintained higher levels of sTim‐3 and sCD25 than patients without RF in a linear mixed model adjusting for treatment modalities, obesity, COVID wave, neutrophil and lymphocyte counts, and vaccination status (Fig. 1b). While sCD25 levels were relatively stable during follow‐up, sTim‐3 increased during the hospital stay. The main statistical effect of RF on the temporal profile of sCD25 was attenuated upon additional adjustment for CRP (p = 0.26), whereas sTim‐3 remained significant (p = 0.001). Analysis of the correlation between the P/F ratio and T‐cell markers during the first 10 days revealed a negative correlation with sCD25 levels (estimate −0.18, t = −5.5, p < 0.001) and sTim‐3 (estimate −0.23, t = −6.4, p < 0.001).
As shown in Fig. 1c, a similar temporal profile of sCD25 and sTIM‐3 was seen in relation to ICU admission as for RF with the same adjustment strategy. Further adjustment with CRP revealed overall attenuated results with nonsignificant p‐values for sCD25 (p = 0.18) but sTIM‐3 remained significant (p = 0.042).
The viral load in admission oropharynx samples (only measured in cohort 1), determined as SARS‐CoV‐2 RNA copies per 1000 human cells, did not correlate with baseline levels of sTim‐3 (r = −0.10, p = 0.33) or sCD25 (r = 0.03, p = 0.78).
Levels of sCD25 and sTim‐3 in relation to 60‐day total mortality
A total of 37 patients died within 60 days after hospital admission (Table 1). As shown in Fig. 3a, ROC analysis revealed that admission levels of sTIM‐3 had a decent discriminatory power in detecting patients who died during 60‐day follow‐up. A Kaplan–Meier (Fig. 3b) analysis of cut off determined by Youden's index (above or below 3.8 ng/ml and 8.1 ng/ml for sCD25 and sTim‐3, respectively) confirmed a higher risk of death (p < 0.001 for both T‐cell markers). When evaluated in Cox regression using these cut offs, having an above‐threshold level of sCD25 was associated with a 2.62 (95% confidence interval [CI] 1.32–5.19) times higher risk of death (p = 0.006) in age, COVID wave, and treatment modality adjusted analysis (model 1). The association was not significant following adjustment with chronic cardiac and pulmonary comorbidity, neutrophil and lymphocyte counts, and eGFR (model 2; hazard ratio [HR] 1.65, p = 0.21), and was further attenuated when adding CRP as a covariate (model 3; HR 1.27, p = 0.55). Using above‐threshold levels of sTim‐3 was associated with a 5.18 (1.76–15.25)–times higher risk of 60‐day death in model 1 (p = 0.003) and remained significant in model 2 (HR 4.32, p = 0.009) and model 3 (HR 4.26, p = 0.010). This threshold gave a good sensitivity (86%) but modest specificity (59%), with high negative predictive value (98%) and poor positive predictive value (17%) in identifying patients at risk for 60‐day death.
Fig. 3.
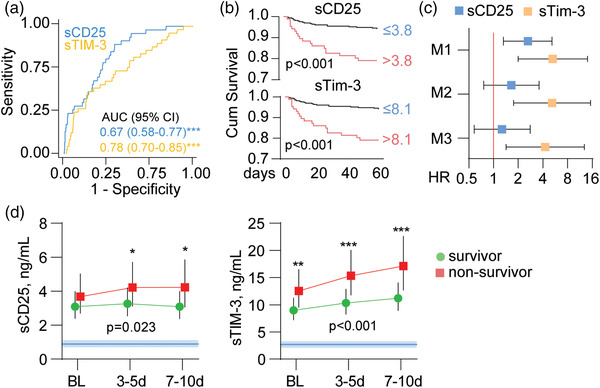
sCD25 and sTim‐3 and 60‐day mortality in severe COVID‐19. (a) Receiver operating characteristic (ROC) analysis of admission levels of sCD25 and sTim‐3 in relation to 60‐day mortality, (b) Kaplan–Meier analysis of 60‐day mortality (n = 31) according to dichotomized admission levels of sCD25 (Youden's index cut off: 3.8 ng/ml) and sTim‐3 (Youden's index cut off: 8.1 ng/ml). (c) Cox regression of admission levels of sCD25 and sTim‐3 (dichotomized according to Youden's index as in b) in relation to 60‐day mortality with different levels of adjustment (M1: age, COVID wave, and dexamethasone treatment; M2: M1 + chronic cardiac and pulmonary disease, neutrophil count and lymphocyte count, and estimated glomerular filtration rate; M3: M2 + C‐reactive protein). (d) Temporal profile of sCD25 and sTim‐3 during the first 10 days after admission according to 60‐day mortality shown as estimated marginal means and 95% confidence intervals (CI) with adjustment for M2 from c. The p‐values reflect the group (outcome) effect from the linear mixed models. Blue areas reflect reference ranges from 21 healthy controls. **p < 0.01, ***p < 0.001 between groups.
The association with 60‐day mortality was more pronounced in patients not receiving dexamethasone for both sCD25 (HR 14.3, p < 0.001) and sTim‐3 (HR 9.12, p = 0.004) compared with attenuated (sTim‐3, HR 5.99, p = 0.004) or absent (sCD25, HR 1.28, p = 0.59) association in patients treated with dexamethasone.
Evaluation of the temporal profile during the first 10 days after inclusion revealed that patients who died displayed increasing levels of both sCD25 and sTim‐3, with the largest differences seen at the end of the observation period in a linear mixed model adjusting for model 2. For sTim‐3, this association was also significant after further adjustment for CRP (main effect p = 0.001), but not for sCD25 (main effect p = 0.15).
Temporal profile of circulating sCD25 and sTim‐3 in relation to treatment modalities
In cohort 1, we found no significant effect of investigational drugs (HCQ or remdesivir) on plasma levels of sCD25 or sTim‐3, or interaction between time and treatment (sCD25 treatment p = 0.39, interaction treatment time p = 0.17; sTim‐3 treatment p = 0.51, interaction treatment time p = 0.49).
In cohort 2, dexamethasone‐treated patients had higher levels of sCD25 and sTim‐3 compared to patients not receiving dexamethasone at admission and at days 3–5, but the groups had similar levels at days 7–10 (Fig. 4a). The higher levels at admission in dexamethasone users most likely reflect more severe disease in these patients.
Fig. 4.
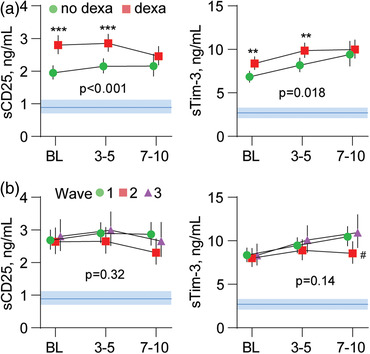
Temporal profile of sCD25 and sTim‐3 according to (a) dexamethasone treatment and (b) COVID‐19 wave, shown. The p‐values reflect the group (outcome) effect from the linear mixed models. Data are presented as estimated marginal means and 95% confidence intervals (CI) in age‐ and sex‐adjusted analysis. Blue areas reflect reference ranges from 21 healthy controls. **p < 0.01, ***p < 0.001 between groups; #p < 0.05 versus waves 1 and 3.
When evaluating the temporal course of the markers in relation to the COVID‐19 wave in the combined cohort, no overall group effects were observed for sCD25 (p = 0.32) and sTim‐3 (p = 0.14) (Fig. 4b).
Levels of sCD25 and sTim‐3 in relation to pulmonary function and chest CT at 3‐month follow‐up
In cohort 1, pulmonary testing (n = 108) and chest CT (n = 107) were performed in a subgroup of patients (Table S2) 3 months after admission to the hospital. We examined the association between sCD25 and sTim‐3 levels both during hospitalization and at 3 months and the occurrence of (i) pulmonary function impairment, (ii) reversible CT changes, and (iii) irreversible CT changes at 3‐month follow‐up. Patients with impaired pulmonary function (i.e., DLCO < LLN) at 3 months had significantly higher levels of sCD25 during hospitalization (Fig. 5a) and at 3‐month follow‐up (Fig. 5b), but not for sTim‐3 compared to those without impairment. Patients with reversible CT changes at 3 months had increased sTim‐3 levels during hospitalization (Fig. 4c), but not at 3 months (Fig. 5d), compared to those without reversible CT changes. No such differences were observed between patients with and without irreversible CT changes at 3 months, and levels with sCD25 during hospitalization (data not shown) and 3 months (Fig. 5b) did not correlate with any of the CT changes.
Fig. 5.
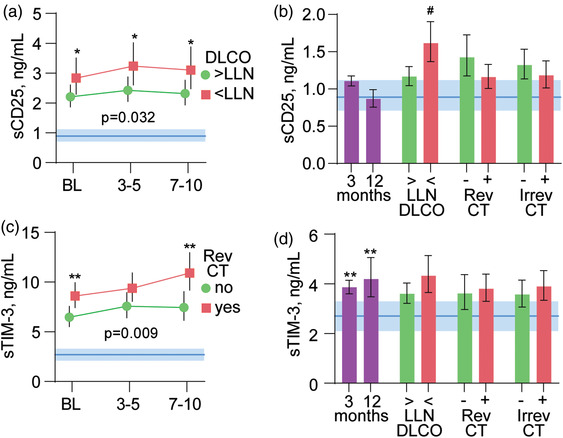
sCD25 and sTim‐3 at follow‐up and in relation to pulmonary pathology. Temporal profile of (a) sCD25 and (c) sTim‐3 during the first 10 days after admission according to impaired diffusing capacity of the lungs for carbon monoxide (DLCO, above or below the lower limit of normal [LLN]) or reversible (Rev) or irreversible (Irrev) computed tomography (CT) changes at 3‐month follow‐up. The p‐values reflect the group (outcome) effect from the linear mixed models adjusting for randomized treatment, age, sex, and neutrophil counts. Blue areas in panels a and c reflect the reference value range. *p < 0.05, **p < 0.01 between groups. Panels b and d (left part) show levels of sCD25 (b) and sTim‐3 (d) at 3 and 12 months (n = 257) compared with healthy controls (n = 21). The right parts of panels b and d show levels of sCD25 (b) and sTim‐3 (d) at 3 months in relation to DLCO below or above LLN and reversible and irreversible CT changes at 3 months. #p < 0.01 versus DLCO < LLN.
Persistently elevated sTim‐3 levels but not sCD25 at 3‐ and 12‐month follow‐ups
At follow‐up visits 3 months (combined cohort, n = 257) and 12 months (cohort 2, n = 62) after hospitalization, sCD25 levels were normalized compared to healthy controls (Fig. 4b), while sTim‐3 levels were still elevated even after 12 months (Fig. 4d).
Discussion
The present study investigated whether circulating markers of T‐cell activation and exhaustion were related to acute RF and the need for ICU admission during hospitalization for COVID‐19, as well as 60‐day mortality and pulmonary pathology 3 months after hospital admission. Our main findings were as follows: (i) elevated levels of sCD25 and, in particular, sTim‐3 during hospitalization were strongly associated with both RF and need of ICU treatment; (ii) both markers measured during hospitalization were associated with 60‐day mortality in univariate analyses, but only sTim‐3 remained significant in multivariate analyses; (iii) elevated levels of sCD25 and sTim‐3 were associated with impaired lung function and pathological changes on chest CT, respectively, at 3‐month follow‐up; and (iv) levels of sTim‐3 increased during hospitalization and remained elevated after 3 and even 12 months, whereas levels of sCD25 plateaued during hospitalization with levels comparable to healthy controls thereafter.
In a previous pilot study (n = 39), we found that elevated sTim‐3 levels were associated with severe COVID‐19 in hospitalized patients [11]. Subsequent small‐to‐moderate size studies have found lymphopenia and T‐cell exhaustion, as assessed by flow cytometry, to be associated with disease severity and to persist for several months after severe COVID‐19 [18, 19, 20]. One of these studies suggested that sTim‐3 was the most important marker of CD4+ T‐cell exhaustion in COVID‐19 [19]. The present study confirms and extends these previous findings in a much larger cohort that spans three consecutive waves of the COVID‐19 pandemic, and relates sTim‐3 levels to persistent pulmonary changes and mortality. We further show that levels of sTim‐3 remain elevated even 12 months after hospitalization. Whereas sCD25 was associated with mortality in univariate but not in multivariate analyses, the association with sTim‐3 was not affected by adjustment for age, COVID wave, dexamethasone treatment, chronic cardiac and lung disease, CRP, neutrophil and lymphocyte counts, or renal function. Of note, the temporal profiles of sTim‐3 during hospitalization were also associated with 60‐day mortality, even in multivariate analyses.
An even more striking finding was the pattern of increasing levels of sTim‐3 during hospitalization with no signs of normalization, even for as long as 12 months after discharge. In contrast, levels of sCD25 plateaued during hospitalization and reached normal levels at 3‐ and 12‐month follow‐ups. One interpretation could be that T‐cell activation in the acute phase is accompanied and followed by persisting T‐cell exhaustion and dysfunction as immunological sequelae after severe COVID‐19 [20]. There are some data suggesting that sTim‐3 may reflect the degree of membrane‐expressed Tim‐3 and as such, be a marker of T‐cell exhaustion [21]. However, whether membrane‐expressed Tim‐3 is downregulated after release of sTim‐3 and whether this influences the degree of T‐cell exhaustion, as well as whether sTim‐3 has any function on its own, is still being discussed [21, 22].
Interestingly, a previous study found that SARS‐CoV2‐specific CD4+ T cells expressed exhaustion markers PD‐1 and sTIM‐3 at 12 months post‐infection, with a loss of the multifunctional phenotype reported to dominate at earlier time points [23]. Thus, whereas a transient and short‐lived upregulation of checkpoint inhibitors such as sTim‐3 could be beneficial to dampen overwhelming immune responses, a persistent upregulation—as shown in the present study—could be harmful for the host. Importantly, given the association between sTim‐3 and pathological pulmonary CT scans after 3 months shown in our study, this persistent T‐cell exhaustion and dysfunction should be investigated in relation to long‐term pulmonary sequelae and other long‐COVID manifestations in future studies.
We have previously reported persistently elevated sTim‐3 levels in chronic HIV‐infected individuals [24], and chronic viral replication is a known contributor to persistent T‐cell exhaustion [5]. However, in the present study, we found no association between viral load in upper airways and sTim‐3 levels, and persistent SARS‐CoV‐2 replication over several months has not been demonstrated. Thus, whereas elevated sTim‐3 levels in acute COVID‐19 may be triggered by SARS‐CoV‐2, the sustained elevation of this marker most likely reflects other mechanisms such as an overshooting and prolonged checkpoint stimulation following persistent, but transient, T‐cell activation. Interestingly, IL‐7 and other immunomodulators—such as PD‐1 blockade—have been suggested to reverse immune abnormalities in COVID‐19 disease [25]. Hence, our data could potentially provide support for the rationale of testing such an approach even in patients with severe long‐COVID manifestations [26].
Comorbidities have been shown to influence patient outcome in COVID‐19 disease [27]. In the present study, however, we found no effects of comorbidities including chronic cardiac or pulmonary disease, hypertension, obesity, or diabetes, or any effect of accumulated comorbidities on levels of sTim‐3 and sCD25, further supporting that these markers reflect distinct COVID‐19 pathology. To this end, several markers of disease severity have been established in hospitalized COVID‐19 patients, such as CRP and high neutrophil and low lymphocyte counts [28]. These factors were, therefore, adjusted for in the present study without having a major influence on the results. However, some of the findings were markedly attenuated when adjusting for CRP, a reliable marker of upstream inflammatory pathway, indicating that circulating CRP and sCD25 at least partly reflect overlapping pathways in relation to COVID‐19 pathology.
Our study has some limitations, including the lack of baseline samples before the start of dexamethasone treatment, and that assessment of pulmonary function at 3 months was performed in only cohort 1, and we have no data on pulmonary function before COVID‐19. Although previous reports have shown that circulating sTim‐3 levels correlate with expression of the membrane‐bound form [10], the present dataset did not include specimens for analyses of cellular expression of Tim‐3 on T cells and monocytes. Moreover, associations do not necessarily mean any causal relationship. The study also has several strengths, including two well‐characterized prospective cohorts with common standardized protocols for biobanking, as well as long‐term follow‐up with clinical assessment and continued biobanking. The study also included samples obtained during three waves of the COVID‐19 pandemic.
In conclusion, the T‐cell exhaustion marker sTim‐3 was associated with acute RF, as well as 60‐day mortality and pulmonary pathology after 3 months. As sTim‐3 remained elevated for as long as 12 months of follow‐up, our findings suggest T‐cell exhaustion is a potential immunological sequela after severe COVID‐19. Future studies are warranted to clarify the possibility of long‐term T‐cell exhaustion, including the clinical consequences and the potential therapeutic implications of such immune dysregulation.
Funding
This work received the following funding: the National Clinical Therapy Research in the Specialist Health Services (KLINBEFORSK), Norway; South‐Eastern Norway Regional Health Authority (grant number 2021071); Oslo University Hospital; Research Council of Norway (grant no 312780), and a philanthropic donation from Vivaldi Invest A/S owned by Jon Stephenson von Tetzchner. The funders had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Conflict of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Author contributions
T. U., T. B. D., B. H., P. A., and M. T. were responsible for the study conception and execution of the present substudy and securing the financial support. A. B. D., A. M. D. R., K. N. H., P. A., and M. T. were responsible for the management, coordination, research activity planning, and execution of the NOR‐Solidarity trial. J. C. H., A. M. D. R., L. H., A. B. K., A. A. T., K. T., A. R. H., F. M., and S. B. were responsible for the management, coordination, research activity, planning, and execution of the Norwegian SARS‐CoV‐2 study. T. V. L., O. H. S., and T. M. A. were responsible for the 3‐month follow‐up protocol for pulmonary function and CT scan. J. C. H., A. M. D. R., B. F., B. K. G., L. H., and A. B. K. were responsible for the 12‐month follow‐up collection of bio samples. A. M. D. R., T. B. D., A. B. D., B. H., T. R., P. A., H. H., A. M., B. K. G, and J. C. H. coordinated the collection and storage of the biobank material. T. B. D., T. U., A. E. M., K. Y., S. M., A. Q. J., and B. H. were responsible for the biochemical analyses. All authors revised and approved the final version of the manuscript.
Supporting information
Supplemental Figure 1. sCD25 and sTim‐3 during hospitalization in relation to comorbidities.
Supplemental Table 1. Demographic, clinical, and biochemical characteristics in 414 patients hospitalized for COVID‐19, stratified by two large multi‐centre cohorts in Norway.
Supplemental Table 2. Admission demographic, clinical, and biochemical characteristics in 113 patients who assessed pulmonary function and chest CT at 3‐month follow‐up, stratified according if they had DLCO<LLN or reversible CT‐changes.
Acknowledgments
We thank WHO Solidarity and NOR‐Solidarity study groups for the opportunity to perform this add‐on study. We appreciate the strong collaboration of the Norwegian SARS‐CoV‐2 study group, including clinicians, nurses, bioengineers, mercantile employees, and researchers. We would also like to thank Karoline Hansen Skåra and Azita Rashidi at the Institute of Internal Medicine, Oslo University Hospital, Rikshospitalet, and Sarah Nur at the Department of Infectious Diseases, Oslo University Hospital, Ullevål, for their contributions to the biobank collection, and Mona Skjelland for access to the control plasma biobank. We are grateful to Linda Gail Skeie, Kjerstin Røstad, and Ingeborg Holand at the Department of Infectious Diseases, Oslo University Hospital, Ullevål, who contributed to COVID‐19 clinical patient data. Moreover, we thank Susanne Dudman, Mona Holberg‐Petersen, Anne Katrine Steffensen, and Cathrine Fladeby at the Department of Microbiology, Oslo University Hospital, for access to the viral analysis data.
Trøseid M, Dahl TB, Holter JC, Kildal AB, Murphy SL, Yang K, et al. Persistent T‐cell exhaustion in relation to prolonged pulmonary pathology and death after severe COVID‐19: Results from two Norwegian cohort studies. J Intern Med. 2022;292:816–828.
Tuva B. Dahl and Jan C. Holter contributed equally to this study.
Contributor Information
Marius Trøseid, Email: marius.troseid@medisin.uio.no.
Thor Ueland, Email: thor.ueland@medisin.uio.no.
Data availability statement
Regarding data sharing, Norwegian institutional data privacy regulations prohibit deposition of individual‐level data to public repositories. Participant written consent also does not cover public sharing of data for use for unknown purposes. However, upon contact with Marius Trøseid (marius.troseid@medisin.uio.no) or Thor Ueland (thor.ueland@medisin.uio.no), an institutional data transfer agreement can be established, and data shared if the aims of data use are covered by ethical approval and patient consent. The procedure will involve an update to the ethical approval as well as review by legal departments at both institutions, and the process will typically take 1–2 months from initial contact.
References
- 1. Giamarellos‐Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID‐19 patients with severe respiratory failure. Cell Host Microbe. 2020;27:992–1000.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Del Valle DM, Kim‐Schulze S, Huang H‐H, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID‐19 severity and survival. Nat Med. 2020;26:1636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang I, Pranata R. Lymphopenia in severe coronavirus disease‐2019 (COVID‐19): systematic review and meta‐analysis. J Intensive Care. 2020;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID‐19). Front Immunol. 2020;11:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Klenerman P, Hill A. T cells and viral persistence: lessons from diverse infections. Nat Immunol. 2005;6:873–9. [DOI] [PubMed] [Google Scholar]
- 6. Jin H‐T, Anderson AC, Tan WG, West EE, Ha S‐J, Araki K, et al. Cooperation of Tim‐3 and PD‐1 in CD8 T‐cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010;107:14733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sakhdari A, Mujib S, Vali B, Yue FY, Macparland S, Clayton K, et al. Tim‐3 negatively regulates cytotoxicity in exhausted CD8+ T cells in HIV infection. PLoS One. 2012;7:e40146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim‐3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV‐1 infection. J Exp Med. 2008;205:2763–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Möller‐Hackbarth K, Dewitz C, Schweigert O, Trad A, Garbers C, Rose‐John S, et al. A disintegrin and metalloprotease (ADAM) 10 and ADAM17 are major sheddases of T cell immunoglobulin and mucin domain 3 (Tim‐3). J Biol Chem. 2013;288:34529–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Riva A, Palma E, Devshi D, Corrigall D, Adams H, Heaton N, et al. Soluble TIM3 and its ligands galectin‐9 and CEACAM1 are in disequilibrium during alcohol‐related liver disease and promote impairment of anti‐bacterial immunity. Front Physiol. 2021;12:632502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ueland T, Heggelund L, Lind A, Holten AR, Tonby K, Michelsen AE, et al. Elevated plasma sTIM‐3 levels in patients with severe COVID‐19. J Allergy Clin Immunol. 2021;147:92–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brusko TM, Wasserfall CH, Hulme MA, Cabrera R, Schatz D, Atkinson MA. Influence of membrane CD25 stability on T lymphocyte activity: implications for immunoregulation. PLoS One. 2009;4:e7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Agarwal A, Rochwerg B, Lamontagne FO, Siemieniuk RA, Agoritsas T, Askie L, et al. A living WHO guideline on drugs for covid‐19. BMJ. 2020;370:m3379. [DOI] [PubMed] [Google Scholar]
- 14. Barratt‐Due A, Olsen IC, Nezvalova‐Henriksen K, Kåsine T, Lund‐Johansen F, Hoel H, et al. Evaluation of the effects of remdesivir and hydroxychloroquine on viral clearance in COVID‐19: a randomized trial. Ann Intern Med. 2021;174:1261–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pan H, Peto R, Henao‐Restrepo AM, Preziosi M‐P, Sathiyamoorthy V, Karim QA, et al. Repurposed antiviral drugs for covid‐19—interim WHO solidarity trial results. N Engl J Med. 2021;384:497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lerum TV, Maltzahn NN, Aukrust P, Trã¸Seid M, Henriksen KN, Kåsine T, et al. Persistent pulmonary pathology after COVID‐19 is associated with high viral load, weak antibody response, and high levels of matrix metalloproteinase‐9. Sci Rep. 2021;11:23205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lerum TV, Aalã¸Kken TM, Brã¸Nstad E, Aarli B, Ikdahl E, Lund KMA, et al. Dyspnoea, lung function and CT findings 3 months after hospital admission for COVID‐19. Eur Respir J. 2021;57:2003448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bobcakova A, Petriskova J, Vysehradsky R, Kocan I, Kapustova L, Barnova M, et al. Immune profile in patients with COVID‐19: lymphocytes exhaustion markers in relationship to clinical outcome. Front Cell Infect Microbiol. 2021;11:646688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Modabber Z, Shahbazi M, Akbari R, Bagherzadeh M, Firouzjahi A, Mohammadnia‐Afrouzi M. TIM‐3 as a potential exhaustion marker in CD4(+) T cells of COVID‐19 patients. Immun Inflamm Dis. 2021;9:1707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rha M‐S, Shin E‐C. Activation or exhaustion of CD8+ T cells in patients with COVID‐19. Cell Mol Immunol. 2021;18:2325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clayton KL, Douglas‐Vail MB, Nur‐ur Rahman AKM, Medcalf KE, Xie IY, Chew GM, et al. Soluble T cell immunoglobulin mucin domain 3 is shed from CD8+ T cells by the sheddase ADAM10, is increased in plasma during untreated HIV infection, and correlates with HIV disease progression. J Virol. 2015;89:3723–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grossman TB, Minis E, Loeb‐Zeitlin SE, Bongiovanni AM, Witkin SS. Soluble T cell immunoglobulin mucin domain 3 (sTim‐3) in maternal sera: a potential contributor to immune regulation during pregnancy. J Matern Fetal Neonatal Med. 2021;34:4119–22. [DOI] [PubMed] [Google Scholar]
- 23. Hou H, Zhang Y, Tang G, Luo Y, Liu W, Cheng C, et al. Immunologic memory to SARS‐CoV‐2 in convalescent COVID‐19 patients at 1 year postinfection. J Allergy Clin Immunol. 2021;148:1481–92.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoel H, Ueland T, Hove‐Skovsgaard M, Hartling HJ, Gelpi M, Benfield T, et al. Soluble T‐cell immunoglobulin mucin domain‐3 is associated with hepatitis C virus coinfection and low‐grade inflammation during chronic human immunodeficiency virus infection. Open Forum Infect Dis. 2020;7:ofaa033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loretelli C, Abdelsalam A, D'Addio F, Ben Nasr M, Assi E, Usuelli V, et al. PD‐1 blockade counteracts post‐COVID‐19 immune abnormalities and stimulates the anti‐SARS‐CoV‐2 immune response. JCI Insight. 2021;6:e146701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Phetsouphanh C, Darley DR, Wilson DB, Howe A, Munier CML, Patel SK, et al. Immunological dysfunction persists for 8 months following initial mild‐to‐moderate SARS‐CoV‐2 infection. Nat Immunol. 2022;23:210–6. [DOI] [PubMed] [Google Scholar]
- 27. Ejaz H, Alsrhani A, Zafar A, Javed H, Junaid K, Abdalla AE, et al. COVID‐19 and comorbidities: deleterious impact on infected patients. J Infect Public Health. 2020;13:1833–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kåsine T, Dyrhol‐Riise AM, Barratt‐Due A, Kildal AB, Olsen IC, Nezvalova‐Henriksen K, et al. Neutrophil count predicts clinical outcome in hospitalized COVID‐19 patients: results from the NOR‐Solidarity trial. J Intern Med. 2022;291:241–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. sCD25 and sTim‐3 during hospitalization in relation to comorbidities.
Supplemental Table 1. Demographic, clinical, and biochemical characteristics in 414 patients hospitalized for COVID‐19, stratified by two large multi‐centre cohorts in Norway.
Supplemental Table 2. Admission demographic, clinical, and biochemical characteristics in 113 patients who assessed pulmonary function and chest CT at 3‐month follow‐up, stratified according if they had DLCO<LLN or reversible CT‐changes.
Data Availability Statement
Regarding data sharing, Norwegian institutional data privacy regulations prohibit deposition of individual‐level data to public repositories. Participant written consent also does not cover public sharing of data for use for unknown purposes. However, upon contact with Marius Trøseid (marius.troseid@medisin.uio.no) or Thor Ueland (thor.ueland@medisin.uio.no), an institutional data transfer agreement can be established, and data shared if the aims of data use are covered by ethical approval and patient consent. The procedure will involve an update to the ethical approval as well as review by legal departments at both institutions, and the process will typically take 1–2 months from initial contact.