

Abstract
Genetic factors have long been recognized as important determinants of interindividual variability in drug efficacy and toxicity. However, despite the increasing number of established gene–drug associations, candidate polymorphisms can only explain a fraction of the genetically encoded functional variability in drug disposition. Advancements in genetic profiling methods now allow to analyse the landscape of human pharmacogenetic variations comprehensively, which opens new opportunities to identify novel factors that could explain the “missing heritability.” Here, we provide an updated overview of the landscape of pharmacogenomic variability based on recent analyses of population‐scale sequencing projects. We then summarize the current state‐of‐the‐art how the functional consequences of variants with unknown effects can be quantitatively estimated while discussing challenges and peculiarities that are specific to pharmacogenes. In the last sections, we discuss the importance of considering ethnogeographic diversity to provide equitable benefits of pharmacogenomics and summarize current roadblocks for the implementation of sequencing‐based guidance of clinical decision‐making. Based on the current state of the field, we conclude that testing is likely to gradually shift from the interrogation of selected candidate polymorphisms to comprehensive sequencing, which allows to consider the full spectrum of pharmacogenomic variations for a true personalization of genomic prescribing.
Keywords: computational prediction, next‐generation sequencing, population diversity, precision medicine, rare variants
1. INTRODUCTION
Pharmacogenetics is a scientific discipline with a long history. The first description of interindividual differences in adverse event risk after ingestion of fava beans dates back to around 510 bc. However, it would take more than two millennia until those differences were linked to heritable factors. Since the beginning of the 20th century, progress in the field drastically accelerated with important milestones including the concept “inborn errors of metabolism,” 1 coining the term “pharmacogenetics” 2 and the identification of an ever increasing number of functionally relevant polymorphism in drug‐metabolizing enzymes, such as TPMT, CYP2D6, CYP2C19 and NAT1/2. These findings were enabled using forward genetics approaches, i.e., the identification of patients with abnormal drug reactions followed by their genetic interrogation. 3 , 4 Later, the emergence of genome‐wide association study (GWAS) designs facilitated the further identification of significant pharmacogenetic biomarkers, including CYP2C9 rs1057910 for phenytoin‐related severe cutaneous adverse reactions, 5 SLCO1B1 rs4363657 for statin‐induced myopathy 6 and NUDT15 p.R139C for thiopurine‐induced early leukopenia. 7
The increasing number of pharmacogenetic associations reported identified by diverse methodologies conducted by a multiple of different labs entailed considerable heterogeneity in variant nomenclature and reporting, which hampered further progress. To increase comparability between studies and increase accessibility of report for non‐experts, a systematic star allele (*) nomenclature system was established with the aim to simplify the names of these well‐characterized pharmacogenetic alleles. The first consolidated online database for cytochrome P450 (CYP) star alleles was established in 1999 hosted by Karolinska Institutet, which provided a summary of alleles and their associated effects and facilitated rapid online dissemination of new alleles. 8 More recently, the resource was transitioned into the Pharmacogene Variation Consortium website. 9
While it is estimated that 20% to 30% of interindividual variability in drug response results from genetic factors, 10 commonly interrogated polymorphisms could explain around 70% to 80% of such variations. 11 , 12 The origin of the remaining so‐called “missing heritability” remains unclear. The increasing capability of sequencing methods revealed the tremendous complexity of pharmacogenomic variation and identified a plethora of rare variants with unknown functional effects. These rare variations are plausible candidates to contribute at least in part to this missing heritability. 12 In this review, we discuss experimental and computational advances for pharmacogenomic variant identification and interpretation. We furthermore highlight current roadblocks and future opportunities for how these might improve clinical decision to refine personalized medicine.
2. ADVANCES IN GENETIC AND GENOMIC PROFILING METHODS THAT ENABLE PGx
Genetic profiling technologies underwent impressive developments over the last decades. Conceptually, pharmacogenomic profiling methods can be divided into (i) panel‐based approaches that interrogate individual candidate variations and (ii) sequencing‐based approaches that comprehensively interrogate predefined genomic areas and can also identify novel variations. Panel‐based approaches are most commonly used in clinical PGx. These methods either rely on PCR or mass spectrometry to identify candidate variants and can vary in scope between the interrogation of one or few variants up to multiple million variations. Mass spectrometric methods are typically cost‐effective for mid‐throughput applications, typically testing up to 36 markers in 384 individuals. 13 In contrast, arrays are highly variable in their gene coverage, inclusion of copy number variations (CNVs) and mitochondrial mutations between different models with current genome‐wide arrays differing between 240 000 variants and 4.1 million. 14 , 15 Furthermore, a growing number of pharmacogene‐specific arrays is available that comprise “only” few hundred to thousand variants; however, as these are focused exclusively on genes involved in pharmacokinetics, pharmacodynamics and drug safety, their coverage of clinically relevant pharmacogenetic variation is nevertheless more dense than in genome‐wide arrays. As such, the selection of genotyping array should thus be done in coordination with the scope of the research question at hand.
Irrespective of the choice of genotyping array, all panel approaches have in common that they only cover limited predefined sets of variants. Consequently, such methods cannot identify variations in genomic positions not covered by the array. This limits the utility of array‐based approaches to clinical genotyping of variants with unknown functional consequences and to pharmacogenetic GWASs that aim to find genetic markers for drug‐related phenotypes. To comprehensively profile the pharmacogenomic landscape, including rare and novel variations, sequencing of the relevant loci is required. In the past three decades, sequencing methods have developed from a low‐throughput technology that could profile around 1000 bases per day to massively parallelized next‐generation sequencing (NGS) or short‐read platforms that allow for the generation of around 1 Tb of sequence per day on a single state‐of‐the‐art instrument, which constitutes a 109‐fold increase. 16 , 17
While NGS has been a major catalyst for pharmacogenomic research in recent years, short‐read sequencing methods cannot accurately profile complex or repetitive genetic loci, which include multiple genes of high pharmacogenomic relevance, such as CYP2B6, CYP2D6 and HLAs. 18 Long‐read sequencing methods, also referred to as “third generation sequencing,” aspire to overcome these technological limitations. While short‐read sequencing is based on the release of pyrophosphate upon extension of a nascent DNA strand, which typically results in read lengths of 100–600 bp, long‐read sequencing relies on the monitoring of polymerase activity on single template molecules in real‐time, resulting in reads that commonly exceed 10 kb. For a detailed overview of the technological basis of long‐read sequencing, we refer the interested reader to excellent reviews on this matter. 19 , 20 Long‐read sequencing facilitates the exact identification of CNVs and structural rearrangements and has already demonstrated considerable advantages compared to short‐read methods for the profiling and phasing of complex pharmacogenomic loci. 21 Both short‐read and long‐read sequencing have contributed to the identification of pharmacogenomic variant and allele distributions at the population scale. 22 , 23 These projects have resulted in the identification of tens of thousands of different single‐nucleotide variations (SNVs), indels and CNVs. This pharmacogenomic landscape and current approaches for its functional interpretation are discussed in the following sections.
3. ETHNOGEOGRAPHIC PHARMACOGENOMIC DIVERSITY
Evaluation of pharmacogenomic variability between human populations is receiving increasing interest. Over the last two decades, studies have pinpointed numerous clinically relevant single‐nucleotide polymorphisms (SNPs) and CNVs with distinct ethnogeographic frequency profiles. 24 Some well‐studied population‐specific variations in CYP2D6, CYP2C19 and HLA‐B are illustrated below.
Individuals of European descent are more likely to carry loss‐of‐function variant CYP2D6*3 and *4, whereas the decreased function allele CYP2D6*10 is the main cause of decreased CYP2D6 activity in East Asia. 25 In contrast, the gain‐of‐function variations CYP2D6*1xN and *2xN are most abundant in Oceania, East Africa and the Middle East. 26 Increased CYP2C19 activity due to the CYP2C19*17 allele is frequent in Europe (MAF = 23.1%), the Middle East (MAF = 22.8%) and Africa (MAF = 20.9%) but very rare in East Asia (MAF = 0.7%). 27 Interestingly, differences in CYP2D6 and CYP2C19 allele frequencies not only differ between major populations but can also be remarkably different between relatively close ethnogeographic groups. For instance, within Europe, frequencies of the inactive CYP2C19*2 allele differ between 8% in the Czech Republic and 21% in Cyprus, while CYP2D6*4 varies between 10% in Finland and 33.4% on the Faroe Islands. 28 The resulting functional differences at the population scale emphasize the potential utility of leveraging ancestry information for pharmacological treatment decisions.
Besides PK gene variability, specific variants in HLA genes that constitute established risk factors of severe or life‐threatening drug hypersensitivity reactions, including the Stevens–Johnson syndrome (SJS), the toxic epidermal necrolysis (TEN), drug reaction with eosinophilia and systemic symptoms (DRESS) and maculopapular eruption (MPE) have pronounced ethnogeographic differences. The most clinically established case concerns the associations of HLA‐B*1502 with carbamazepine and oxcarbazepine hypersensitivity. HLA‐B*1502 is highly prevalent in Asian populations with allele frequencies up to 22%, whereas it is almost absent outside of Asia, 29 resulting in population‐stratified recommendations for pre‐emptive genotyping in the labels of these drugs. Similarly, the frequency of the HLA‐B*58:01 allele that is associated with allopurinol‐induced SJS/TEN/DRESS is substantially higher across Asia and Africa, suggesting that genotyping of HLA‐B*58:01 in these populations might be considered before initiating therapy for the treatment of gout. 29
Importantly, however, the ancestry information is not sufficient to accurately guide pharmacological treatment. As such, ethnicity can only serve as a weak‐at‐best proxy of an individual's genotype in the absence of additional data and cannot depict the uniqueness of an individual's pharmacogenetic makeup. 30 In this context, we find it important to highlight the recent policy statement by the American Academy of Pediatrics (AAP) for the “Eliminations of Race‐based medicine”. 31 Specifically, the authors of the white paper state that “race is a social, not a biologic, construct, and the use of race as a proxy for factors such as genetic ancestry is scientifically flawed”. It is therein underlined that the inclusion of race as a guide for therapeutic decision‐making in many of the current clinical algorithms or practice guidelines is rather inferred, and not adequately supported by solid epidemiologic evidence, which calls the notion of “equitable care assertion” into question. In an effort to fix these inaccuracies, the medical guidance that incorporates race assignment is under re‐examination and reconsideration not only by the AAP but also by pharmacogenetic expert groups, such as the Clinical Pharmacogenetics Implementation Consortium (CPIC).
In summary, it has become increasingly clear that population studies cannot inform about an individual's genetic fingerprint with sufficient accuracy to guide the selection of appropriate pharmacotherapy. From a scientific perspective and abstracting from practical constraints, we argue that it is therefore time for population pharmacogenomic advice to be complemented, if not superseded, by genomic evaluations at the level of the individual for an equitable and true personalization of medicine.
4. PHARMACOGENOMIC VARIABILITY BEYOND WELL‐CHARACTERIZED POLYMORPHISMS
Pharmacokinetic (PK) genes are involved in drug absorption, distribution, metabolism and excretion (ADME). Notably, these genes are commonly under low evolutionary pressure at least in part due to the lack of endogenous substrates and thus harbour a large repertoire of genetic variants. Using large‐scale pharmacogenomic sequencing data of clinically relevant PK genes, we and others have identified more than 69 000 variants, of which common variants, including the well‐characterized star alleles, with minor allele frequencies (MAF) ≥ 1% accounted for less than 2%. 12 , 32 , 33 , 34 Across the 57 members of the human CYP gene family, sequencing data from 6503 individuals revealed 4025 SNVs that resulted in amino acid alterations, of which 93% were rare with frequencies <1%. 35 , 36 Furthermore, using more recent consolidated large‐scale sequencing data from 141 614 unrelated individuals identified 6016 exonic variants in the eight clinically most important CYP genes alone, 98.8% and 96.8% of which were rare with frequency below 1% and 0.1%, respectively. 27 Surprisingly, other important PK gene families, such as ABC, SLC and SLCO transporters carried similar numbers of rare genetic variations 37 , 38 , 39 (Table 1). However, while CYP genes harbour >30 common decreased and loss‐of‐function alleles, deleterious variations in drug transporters were generally rare.
TABLE 1.
Pharmacogenomic variation characterized by large‐scale sequencing data
| Gene category | Gene number | Sequencing method | Sequenced individuals | Variants identified | Rare variants identified (in %) | References |
|---|---|---|---|---|---|---|
| CYPs | 8 | WGS and WES | 141 614 | 6016 variants in exons | 98.8 | Zhou and Lauschke 27 |
| SLC transporters | 401 | WGS and WES | 141 456 | 204 287 variants in exons | 99.8 | Schaller and Lauschke 37 |
| SLCO transporters | 7 | WGS and WES | 138 632 | 9966 variants in exons | 99.3 | Zhang and Lauschke 38 |
| ABC transporters | 48 | WGS and WES | 138 632 | 62 793 variants in exons | 98.5 | Xiao et al. 39 |
| GPCRs | 108 | WES | 60 706 | 14 192 missense variants | 97.2 | Hauser et al. 40 |
| Drug target genes | 202 | Target gene sequencing | 14 002 | 1 variant per 17 bp | >95 | Nelson et al. 41 |
| Drug target genes | 606 | WGS and WES | 138 632 | 479 860 variants in exons | 98.1 | Zhou et al. 42 |
| Pharmacogenes | 208 | WES | 60 706 | 69 923 variants | 98.5 | Ingelman‐Sundberg et al. 12 |
| Pharmacogenes | 146 | WGS and WES | >6500 | 12 152 variants in exons | 92.9 | Kozyra et al. 32 |
Abbreviations: WES, whole‐exome sequencing; WGS, whole‐genome sequencing.
While PK variations are predominantly studied in the germline genome, variants in pharmacodynamic (PD) genes are commonly interrogated in oncology where treatments are available that specifically target certain somatic mutations. As of the writing of this review, 74 somatic pharmacogenetic biomarkers in oncological drug targets are recognized by the U.S. Food and Drug Administration (FDA), and we refer the interested reader to recent reviews on the topic. 43 , 44 Curiously, with notable exceptions, PD germline variability has received considerably less attention. Among the well‐characterized PD associations are links between variants in VKORC1 and warfarin response, CFTR variants with drug selection for the treatment of cystic fibrosis and associations between variations in β‐adrenergic receptors and response to anti‐asthmatics. The landscape of PD gene variability has only been recently analysed comprehensively. In G‐protein coupled receptors (GPCRs), which constitute the targets of 34% of approved drugs, sequencing has identified 14 192 missense variants, covering approximately 25% of all nucleotide positions across the entire GPCRome. 40 Further drug target sequencing projects showed that rare variants are predominant also in other PD genes with around 800 000 genetic variants being identified across all FDA‐approved drug targets (98.1% of which were rare with MAF < 1%). 41 , 42 , 45
5. FUNCTIONAL INTERPRETATION OF RARE PHARMACOGENOMIC VARIANTS
Given these vast numbers of identified variations in both PK and PD genes, their correct functional interpretation is of high importance if those variants are supposed to be used to improve clinical decision‐making. Heterologous variant expression in cell lines, such as HEK293 cells, followed by functional assays using appropriate endpoints is considered as the gold‐standard method to evaluate pharmacogenetic variant function. In addition, epidemiological association studies can provide another layer of evidence in determining variant impact on patients. However, these approaches are not suitable for the comprehensive interrogation of the pharmacogenomic variability landscape for multiple reasons. First, in vitro assays are generally low throughput and testing hundreds of thousands of rare variants using conventional assays would require excessive financial resources. Second, experimental assays are time‐consuming and require trained laboratory staff, which makes them unsuitable to rapidly deliver variant function results at the point of care. And third, epidemiological analyses require sufficiently large sample sizes to yield statistically significant results. However, obtaining sufficiently large numbers of rare variant carriers is not feasible or even impossible for rare variants as impractical numbers of individuals would need to be screened.
Given this preamble, it is thus not surprising that computational predictions have emerged as the most commonly used go‐to method to assess the function of otherwise uncharacterized variants. Computational methods are often specialized for different variant classes (missense, synonymous, frameshift, etc.) or types of functional impacts (structural alterations, splice effects, effects on gene regulation, etc.) and consider a variety of features and parameters, including sequencing conservation, structure stabilities and functional genomic data to derive their classifications (Figure 1). The arguably most commonly used prediction methods are SIFT, 47 PolyPhen‐2 48 and CADD. 49 Readers are referred to recent reviews for a detailed discussion of variant effect prediction principles and a comprehensive overview of currently available computational tools. 46 , 50
FIGURE 1.
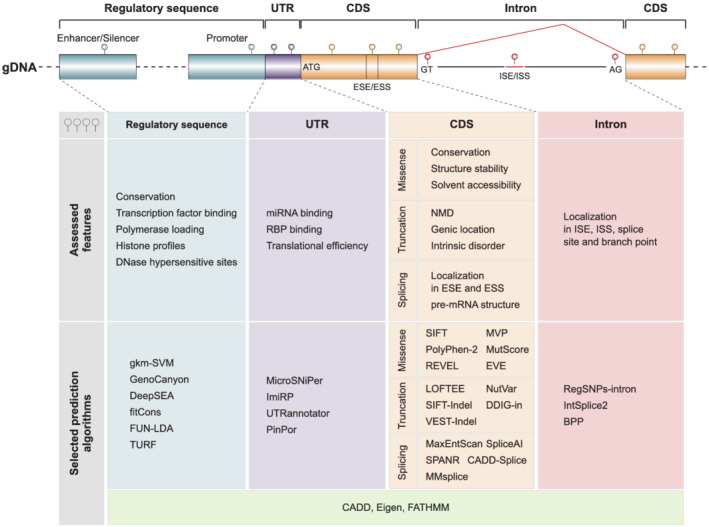
Computational algorithms assess different genetic and genomic features. Predictions are commonly performed on variants located in coding sequences (CDS), introns, untranslated regions (UTR) and putatively regulatory sequences and evaluate a multitude of different evolutionary, structural, biochemical and functional genomic parameters. ESE/ESS, exonic splicing enhancer/silencer; ISE/ISS, intronic splicing enhancer/silencer; NMD, nonsense‐mediated decay; RBP, RNA‐binding protein. Figure modified with permission from Zhou et al. 46
Importantly, however, these computational methods generally underperform on pharmacogenetic variant sets (Figure 2). The main reason is related to the critical dependency of machine learning‐based methods on the quality of training datasets. With very few notable exceptions discussed below, computational algorithms use pathogenic, that is, disease‐associated, variants as positive training sets and common variants with frequencies >5% to 10%, which are not likely to be pathogenic, as negative sets. However, pathogenicity and variant deleteriousness are different concepts. While they overlap in genes associated with genetic disease, pharmacogenes are rarely associated with diseases, and thus, deleterious pharmacogenetic variants are rarely pathogenic. As discussed above, multiple deleterious pharmacogenomic variants, including CYP2C19*2, CYP2D6*4 and CYP3A5*3, are very common in the general population 25 resulting in misclassifications already during model training. Related to the focus of computational methods on pathogenicity rather than deleteriousness predictions and based on the assumption that conserved genomic regions are more important for organismal fitness, sequence conservation constitutes the most commonly used key parameter for variant effect predictions. 54 However, many pharmacogenetic loci are only poorly conserved and even deletions of the entire gene body of a pharmacogene is relatively common (allele frequency of deletions of CYP2D6 are 1% to 6%).
FIGURE 2.
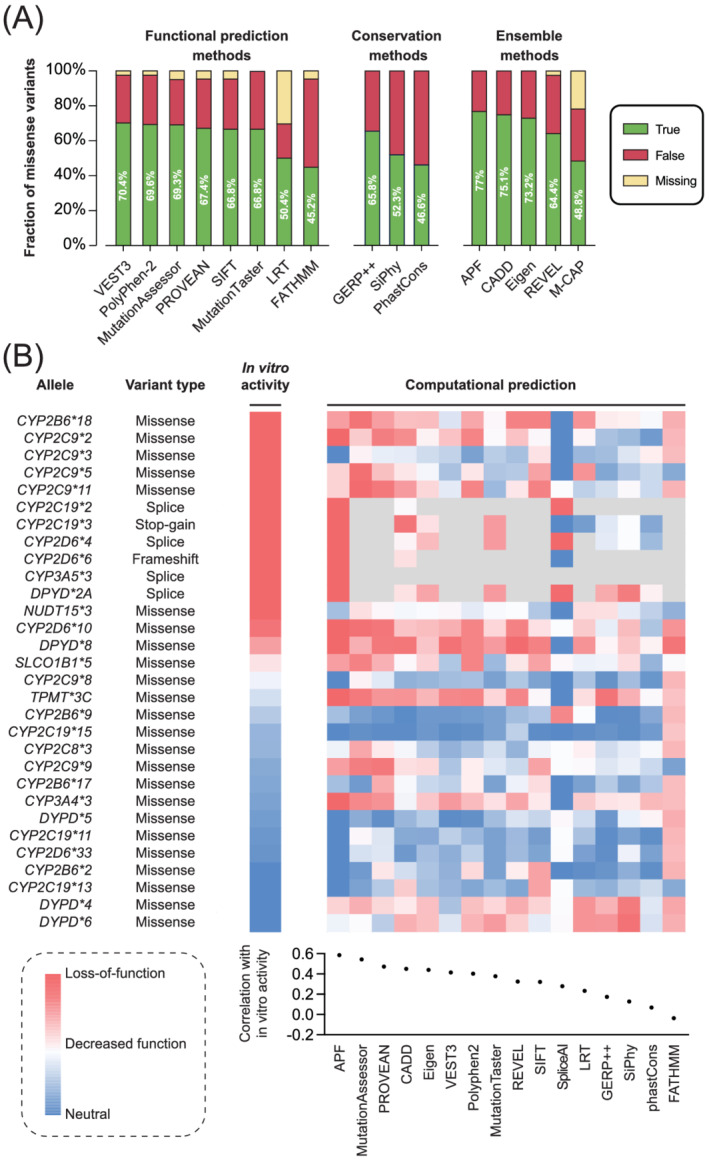
The predictive performance of computational methods on pharmacogenomic variants remains overall limited. (A) Predictive accuracy of commonly used functional prediction, conservation and ensemble methods when applied to pharmacogenomic variants with experimentally characterized functions. Dataset adapted from Zhou et al. 51 and Russell et al. 52 (B) Quantitative functional prediction of well‐characterized pharmacogenomic star alleles and their correlation (Pearson's r) to in vitro activity. Variants whose function could not be predicted with the algorithm in question are shown in grey. Figure modified with permission from Zhou and Lauschke 53
To overcome these limitations, computational methods have been developed that were specifically trained on pharmacogenes. Using 337 experimentally characterized variants across 44 pharmacogenes as training dataset, we optimized the performance of 18 partly orthogonal machine learning algorithms and integrated the best performing tools into an ensemble score termed ADME Prediction Framework (APF). 55 Notably, APF achieved 93% accuracy when predicting loss‐of‐function and neutral pharmacogenomic variants and outperformed conventional variant predictors based on five‐fold cross‐validations. Furthermore, unlike most other methods that provide only binary classifications or risk propensities, APF provided scores that are significantly correlated with enzyme activity (R 2 = 0.9, p = 2.9 × 10−5), opening possibilities for quantitative assessments of variant impact. Notably, APF also performed well on predictions for DPYD despite the fact that no DPYD variations were utilized for model training. 51 In contrast, APF performance on the disease‐associated drug transporter SLC10A1 (NTCP) was not higher than other algorithms. 52 Similar to APF, another machine learning‐based model was recently developed with good performance in prioritizing NGS‐derived pharmacogenomic variants. 56
Besides those prediction methods applicable to the entire pharmacogenome, several gene‐specific predictors have been developed. The DPYD‐specific variant classifier DPYD‐Varifier was trained using in vitro functional data of 156 missense DPYD variants and achieved 85% of predictive accuracy. 57 Recently, a convolutional neural network approach has been used to build prediction tools for CYP2D6. 58 By leveraging CYP2D6 long‐read sequencing data, the model predicted CYP2D6 function in a continuous scale and demonstrated its performance superior to conventional predictions that are based on diplotype/phenotype categories or gene activity scores. 59
Overall, computational tools constitute versatile and effective means to rapidly evaluate the function of uncharacterized or novel pharmacogenomic variants. However, while their performance improved considerably in recent years, it remains questionable whether their accuracy is currently sufficient to warrant their use for clinical applications. With increasing available experimental data for model training and advances in machine learning, computational approaches hold promise to further improve, thereby paving the way for the clinical implementation of sequencing‐based PGx.
6. IMPLEMENTATION AND PRECISION MEDICINE
It is evident that NGS technologies can offer much broader information on pharmacogenetic variability compared to a panel of selected variants with established functional impact. In the clinics, the usage of such genetic information aspires to introduce a paradigm shift from traditional prescribing to genome‐considerate precision drug prescription (Figure 3). Utilizing variations for which actionable information is available can provide a first step, while further inclusion of uncharacterized or private variations based on NGS aspires to provide further possibilities for treatment individualization. It is critical to consider though that the clinical implementation of NGS can only add value if there are rules and frameworks in place regarding how to handle novel or even unique variants for which the function is only predicted based on computational models rather than experimentally established using PK in vivo data. As such, extensive clinical validations are required that carefully scrutinize whether NGS can add value to the patient and the healthcare system. This is particularly true if NGS data is intended to alter the therapeutic regimen for a given patient. We thus do not envision that novel uncharacterized variations can directly guide prescribing in the near future. However, we believe that lower intensity interventions, such as increasing monitoring frequency and surveillance for carriers of rare, putatively deleterious but otherwise uncharacterized variations might be a viable way forward that could allow to add value to patients already in the short‐term without leaving the boundaries of established prescribing practices. Importantly though, already today, PGx‐based dosing is subject to cautious interpretations in clinical practice, as the overall relationship between diplotypes and concrete dose advice is dependent on parallel clearance pathways, concomitant drug treatment with possible drug–drug interactions and intolerability issues arising from PD variability.
FIGURE 3.
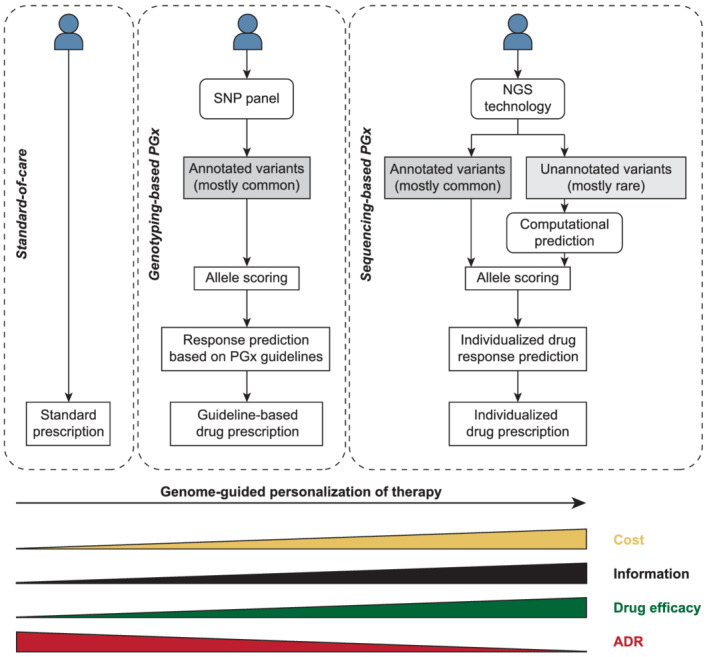
Schematic depiction of different strategies for genome‐considerate prescribing. The standard of care does not consider the genetic information of a given patient in the vast majority of cases. Leveraging information of annotated, mostly common variations for which actionable pharmacogenomic guidelines are available can help to refine treatment and improve patient outcomes. Eventually, next‐generation sequencing (NGS) coupled to computational predictions of unannotated rare or novel variations might allow for full personalization based on the entire pharmacogenomic signature of the individual patient. ADR: adverse drug reaction
From an immediate clinical perspective, we discuss in the following multiple important considerations. Issues that need to be addressed include considerations of (1) which patients will qualify for a broader pharmacogenomic investigation, (2) how will these patients best be informed about the underlying purpose of the PGx investigation and its corresponding implications, (3) how will secondary findings be managed, (4) who will be responsible for data management and interpretation of the results within healthcare, (5) how should PGx data be presented in a clinically useful format, (6) can we achieve the necessary turnaround times to achieve effective decision support, (7) how can we ensure that important findings are utilized at the point of care and (8) how to deal with novel or even patient‐unique genetic variants without any functional correlate. For the discussion of further issues related to reimbursement, privacy and PGx education, we refer the interested reader to previous reviews. 60 , 61 , 62 While many of these items are general, we do acknowledge that some aspects are country‐ and healthcare system‐specific and discussion of those is provided from a Swedish perspective.
Patient selection for NGS. This complex question involves both organization of patient care in different therapeutic areas, medical needs and the priority of PGx with regard to different treatment regimens in the frame of the allocation of limited healthcare resources. At present, precision medicine and the broader use of genomics has focused primarily on cancer treatment and the ambition to include all or at least the majority of cancer patients has been expressed in several countries, including Sweden. 63 Utilizing these data for pharmacogenetic interpretations would be justified for the simple reason that the data are already available and that these patients are likely to benefit from the respective results during the years to come within highly specialized care. However, it is currently difficult to integrate sequencing into established routines outside of oncology due to the lack of downstream analytics. Notably, in geriatrics or psychiatry, the potential value of PGx characterization may be even higher due to increased frequency of polypharmacy. 64 , 65 , 66
Understanding the purpose of the PGx investigation. Patient education and empowerment constitute important issues of pharmacogenetic testing. It will not be possible to carry out laboratory analyses that the patient never approved or understood the purpose of. As such, selection of specific genetic variants for pharmacogenetic panel testing will be inappropriate as neither the physician nor the patient can be expected to understand the details and limitations of the conducted tests as well as the relevance of generated results. Thus, a more paedagogic way might be to perform pharmacogenetic testing using predefined strategies for specific umbrella terms, such as “metabolic drug elimination capacity” with regard to drug‐metabolizing enzymes or “drug hypersensitivity profile” with regard to immune‐mediated events and corresponding HLA markers.
Incidental or secondary findings. The issue of how to handle incidental genetic findings of potential relevance to disease or disease prognosis is always important when it comes to broader genetic investigations, and we refer to more detailed discussions elsewhere. 67 In principle, two classes of incidental findings can be distinguished. The first class is related to the pleiotropic effects of some pharmacogenes. Examples are testing of UGT1A1 to predict irinotecan response, which can reveal carrier status of variants causing Crigler–Najjar syndrome, or tests of VKORC1 variability to guide warfarin dosing, which can return secondary findings regarding the risks of familiar coagulopathies. 68 While the second is a consequence of testing strategies that evaluate not only a given locus of interest, but might evaluate the entire pharmacogenome, exome or genome. While targeted pharmacogenomic sequencing rarely overlaps with analyses of strong markers of disease risk or prognosis, the likelihood of incidental findings increases if WES or WGS are employed. As such, there need to be clear guidelines in place as to how to manage secondary findings with consideration of patient preference.
Data management. Based on regulatory and ethical arguments, patients are expected to have full access to their personal healthcare data. However, with an increasing use of advanced diagnostics and data rich analyses, this might turn out to be practically difficult. The analytical results of such tests will only be relevant for the individual patient after extensive processing and translation into functional consequence, the latter being a suitable task for the discipline of clinical pharmacology. Nevertheless, it needs to be clearly defined who owns the data in the individual case.
PGx result reporting. For the clinical implementation of pharmacogenetic tests, it is imperative that results are integrated into electronic medical records in a format that is transparent and easily understandable for clinical staff who might not be PGx experts. It is important that test reports follow established guidelines for nomenclature and result reporting. 69 This should include a list of the investigated genes and allelic variants as well as the translation of the genetic findings into predicted phenotypes and corresponding clinical interpretations for the individual patient. A table summarizing the investigated genes, the detected variants and the predicted individual activity of different metabolic pathways, as compared to the general population or average patient, should help the responsible physician to understand whether the patient might be at an increased risk of non‐response or toxicity using standard doses. In this respect, international work on consensus guidelines on how to interpret and quantify the impact of different variants is important. 70 , 71 Given the life‐long relevance of PGx results, we recommend that the PGx profile of a given patient is kept separate in a specific folder of the patient records rather than in a single post as part of a consecutive list of laboratory results, which is unfortunately common practice today, at least in the leading medical centres of Sweden.
Turnaround times. Sample preparation, sequencing and data analysis typically entail turnaround times of a few weeks. 72 As a consequence, the use of NGS for the pre‐emptive guidance of personalized prescribing is not realistic for acute cases. Notably however, substantially faster turnaround times down to three business days have recently been reported for NGS‐based testing of molecular panels in a community hospital setting, 73 raising hopes that application for sub‐acute cases might become realistic in the near future. It is important to emphasize that once a pharmacogenomic profile is generated for a given patient, this information will be rapidly available for future occasions, meaning potential access even on an acute basis. For example, for a patient initially subjected to pharmacogenomic profiling for oncological treatment, genotype data should be at hand later in life that on an acute basis helps to guide treatment with antiplatelets after the placement of coronary artery stents.
Clinical decision support systems. No single professional can learn to manage and practice the differential impact of many PGx variants on numerous prescription drugs. Analogous to the situation with drug–drug interactions, 74 drug‐gene interactions would be ideal for database‐driven, clinical decision support tools to be used at the point of care. The system should provide warnings if drugs or dosages are prescribed to a given patient that are contrary to current pharmacogenomic guidelines. In this context it is critical to note that guidelines between regulatory agencies feature notable discrepancies and achieving evidence‐based consensus is important to enable their efficient use in the clinics. 75 , 76 Importantly, decision support should not only utilize genetic information, but should integrate such information with other patient‐specific data of relevance for drug treatment, such as PK drug–drug interactions, body weight and kidney function.
Novel genetic variants. As described in detail in previous sections, NGS can be expected to uncover pharmacogenetic variations for which no functional data based on epidemiological or experimental evaluations exist. Computational models that predict the functional correlate may be the principal way forward by allowing to flag carriers of variations with putative deleterious impacts for intensified follow‐up and, if applicable, a recommendation for therapeutic drug monitoring.
In addition to these direct clinical considerations, it is of paramount importance to determine whether sequencing for pharmacogenetic applications constitutes an efficient allocation of healthcare resources. In such health economic evaluations, the costs and patient effects of sequencing‐guided therapy is compared to the standard of care. These analyses can be conducted assuming two different perspectives. First, it can be evaluated whether sequencing is cost‐effective for guiding the treatment of a condition the respective patient was diagnosed with. Alternatively, the frame of evaluation of cost‐effectiveness of sequencing can be extended to include the entire lifetime of the patient. However, while more accurate, the latter drastically increases the complexity of the evaluation due to the added uncertainty. While most studies that evaluated the economics of pharmacogenomic interventions concluded that testing was cost‐effective, 77 it is important to note that these studies focused exclusively on the genotyping of candidate variations. Furthermore, economic calculations are highly sensitive to healthcare system‐specific parameters and, thus, require resource‐intensive modelling efforts for each country separately. However, recently developed generic models hold promise to facilitate such analyses. 78 To date, no trials have been published that evaluate the cost‐effectiveness of pharmacogenomic sequencing outside of oncology. Prospective clinical trials that evaluate the cost‐effectiveness of NGS coupled to computational variant predictions are thus of critical importance to provide patient benefits without overburdening the healthcare system.
7. CONCLUSION
The development of sequencing methods in the past 20 years has facilitated the discovery of tens of thousands of rare pharmacogenomic variants. Consideration of this complexity beyond well‐characterized polymorphisms promises to eventually improve the personalization of pharmacogenetic recommendations. However, to leverage its added value, routines and workflows are required that establish if, when and how such data can be utilized to guide clinical decisions. In this context, computational methods provide versatile and rapid means to interpret the functional impact of previously uncharacterized pharmacogenomic variations. However, before NGS can be meaningfully used for clinical applications, rigorous trials are required that evaluate whether current tools are sufficiently accurate to cost‐effectively improve patient care.
Even after extensive clinical trials, the pre‐emptive generation of NGS data for clinical applications appears at present unrealistic outside of life‐threatening diseases that are associated with high healthcare costs. This includes various cancers but could also include certain genetic diseases for which genetic information can guide therapy. However, the more widespread availability of NGS data, for example, via business‐to‐consumer sequencing outside of direct medical indications or generated in the context of oncological therapy entails that such data can be increasingly repurposed or applied to less costly diseases or applications where pharmacogenomic information can add value, such as the guidance of prescribing of psychiatric medications. While multiple hurdles need to be overcome, it thus seems realistic to envision a future clinical context where broad PGx data will be easily accessible and incorporated into clinical decision‐making, especially regarding the determination of starting doses for drugs with clear pharmacogenetic associations, as well as for the identification of patients that require more intense monitoring.
CONFLICT OF INTEREST
YZ and VML are co‐founders and shareholders of PersoMedix AB. In addition, VML is CEO and shareholder of HepaPredict AB. EE is vice‐chair of the Genomic Medicine Sweden Pharmacogenomics work package, supported by grants from The Swedish Innovation Agency. The other authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
EE acknowledges funding from the Stockholm Region, CIMED and the Medical Diagnostics function at Karolinska University Hospital. SK acknowledges funding from the Onassis Foundation.
Zhou Y, Koutsilieri S, Eliasson E, Lauschke VM. A paradigm shift in pharmacogenomics: From candidate polymorphisms to comprehensive sequencing. Basic Clin Pharmacol Toxicol. 2022;131(6):452‐464. doi: 10.1111/bcpt.13779
Funding information Onassis Foundation; Stockholm Region, CIMED and the Medical Diagnostics function at Karolinska University Hospital
REFERENCES
- 1. Bearn AG. Inborn errors of metabolism: Garrod's legacy. Mol Med. 1996;2(3):271‐273. doi: 10.1007/BF03401624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vogel F. Moderne Probleme der Humangenetik. In: Heilmeyer L, Schoen R, de Rudder B, eds. Ergebnisse der Inneren Medizin und Kinderheilkunde. Vol.12. Springer; 1959. doi: 10.1007/978-3-642-94744-5_2. [DOI] [Google Scholar]
- 3. Johansson I, Lundqvist E, Bertilsson L, Dahl ML, Sjöqvist F, Ingelman‐Sundberg M. Inherited amplification of an active gene in the cytochrome P450 CYP2D locus as a cause of ultrarapid metabolism of debrisoquine. Proc Natl Acad Sci U S A. 1993;90(24):11825‐11829. doi: 10.1073/pnas.90.24.11825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Morais SM, Wilkinson GR, Blaisdell J, Meyer UA, Nakamura K, Goldstein JA. Identification of a new genetic defect responsible for the polymorphism of (S)‐mephenytoin metabolism in Japanese. Mol Pharmacol. 1994;46(4):594‐598. [PubMed] [Google Scholar]
- 5. Chung WH, Chang WC, Lee YS, et al. Genetic variants associated with phenytoin‐related severe cutaneous adverse reactions. JAMA. 2014;312(5):525‐534. doi: 10.1001/jama.2014.7859 [DOI] [PubMed] [Google Scholar]
- 6. Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin‐induced myopathy‐‐a genomewide study. N Engl J Med. 2008;359(8):789‐799. doi: 10.1056/NEJMoa0801936 [DOI] [PubMed] [Google Scholar]
- 7. Yang SK, Hong M, Baek J, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine‐induced leukopenia. Nat Genet. 2014;46(9):1017‐1020. doi: 10.1038/ng.3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sim SC, Ingelman‐Sundberg M. The Human Cytochrome P450 (CYP) Allele Nomenclature website: a peer‐reviewed database of CYP variants and their associated effects. Hum Genomics. 2010;4(4):278‐281. doi: 10.1186/1479-7364-4-4-278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gaedigk A, Ingelman‐Sundberg M, Miller NA, Leeder JS, Whirl‐Carrillo M, Klein TE. The Pharmacogene Variation (PharmVar) Consortium: incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin Pharmacol Ther. 2018;103(3):399‐401. doi: 10.1002/cpt.910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sim SC, Kacevska M, Ingelman‐Sundberg M. Pharmacogenomics of drug‐metabolizing enzymes: a recent update on clinical implications and endogenous effects. Pharmacogenomics J. 2013;13(1):1‐11. doi: 10.1038/tpj.2012.45 [DOI] [PubMed] [Google Scholar]
- 11. Matthaei J, Brockmöller J, Tzvetkov MV, et al. Heritability of metoprolol and torsemide pharmacokinetics. Clin Pharmacol Ther. 2015;98(6):611‐621. doi: 10.1002/cpt.258 [DOI] [PubMed] [Google Scholar]
- 12. Ingelman‐Sundberg M, Mkrtchian S, Zhou Y, Lauschke VM. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum Genomics. 2018;12(1):26. doi: 10.1186/s40246-018-0157-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Millis MP. Medium‐throughput SNP genotyping using mass spectrometry: multiplex SNP genotyping using the iPLEX® Gold assay. Methods Mol Biol. 2011;700:61‐76. doi: 10.1007/978-1-61737-954-3_5 [DOI] [PubMed] [Google Scholar]
- 14. Ha NT, Freytag S, Bickeboeller H. Coverage and efficiency in current SNP chips. Eur J Hum Genet. 2014;22(9):1124‐1130. doi: 10.1038/ejhg.2013.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Verlouw JAM, Clemens E, de Vries JH, et al. A comparison of genotyping arrays. Eur J Hum Genet. 2021;29(11):1611‐1624. doi: 10.1038/s41431-021-00917-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Heather JM, Chain B. The sequence of sequencers: the history of sequencing DNA. Genomics. 2016;107(1):1‐8. doi: 10.1016/j.ygeno.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shendure J, Balasubramanian S, Church GM, et al. DNA sequencing at 40: past, present and future. Nature. 2017;550(7676):345‐353. doi: 10.1038/nature24286 [DOI] [PubMed] [Google Scholar]
- 18. Lauschke VM, Milani L, Ingelman‐Sundberg M. Pharmacogenomic biomarkers for improved drug therapy‐recent progress and future developments. AAPS J. 2017;20(1):4. doi: 10.1208/s12248-017-0161-x [DOI] [PubMed] [Google Scholar]
- 19. Van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. The third revolution in sequencing technology. Trends Genet. 2018;34(9):666‐681. doi: 10.1016/j.tig.2018.05.008 [DOI] [PubMed] [Google Scholar]
- 20. Logsdon GA, Vollger MR, Eichler EE. Long‐read human genome sequencing and its applications. Nat Rev Genet. 2020;21(10):597‐614. doi: 10.1038/s41576-020-0236-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van der Lee M, Rowell WJ, Menafra R, Guchelaar HJ, Swen JJ, Anvar SY. Application of long‐read sequencing to elucidate complex pharmacogenomic regions: a proof of principle. Pharmacogenomics J. 2022;22(1):75‐81. doi: 10.1038/s41397-021-00259-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldfeder RL, Wall DP, Khoury MJ, Ioannidis JPA, Ashley EA. Human genome sequencing at the population scale: a primer on high‐throughput DNA sequencing and analysis. Am J Epidemiol. 2017;186(8):1000‐1009. doi: 10.1093/aje/kww224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Coster W, Weissensteiner MH, Sedlazeck FJ. Towards population‐scale long‐read sequencing. Nat Rev Genet. 2021;22(9):572‐587. doi: 10.1038/s41576-021-00367-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Y, Lauschke VM. Population pharmacogenomics: an update on ethnogeographic differences and opportunities for precision public health. Hum Genet. 2021;141(6):1113‐1136. doi: 10.1007/s00439-021-02385-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhou Y, Ingelman‐Sundberg M, Lauschke VM. Worldwide distribution of cytochrome P450 alleles: a meta‐analysis of population‐scale sequencing projects. Clin Pharmacol Ther. 2017;102(4):688‐700. doi: 10.1002/cpt.690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gaedigk A, Sangkuhl K, Whirl‐Carrillo M, Klein T, Leeder JS. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19(1):69‐76. doi: 10.1038/gim.2016.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou Y, Lauschke VM. The genetic landscape of major drug metabolizing cytochrome P450 genes—an updated analysis of population‐scale sequencing data. Pharmacogenomics J. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Petrović J, Pešić V, Lauschke VM. Frequencies of clinically important CYP2C19 and CYP2D6 alleles are graded across Europe. Eur J Hum Genet. 2020;28(1):88‐94. doi: 10.1038/s41431-019-0480-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Y, Krebs K, Milani L, Lauschke VM. Global frequencies of clinically important HLA alleles and their implications for the cost‐effectiveness of preemptive pharmacogenetic testing. Clin Pharmacol Ther. 2021;109(1):160‐174. doi: 10.1002/cpt.1944 [DOI] [PubMed] [Google Scholar]
- 30. Shah RR, Gaedigk A. Precision medicine: does ethnicity information complement genotype‐based prescribing decisions? Ther Adv Drug Saf. 2018;9(1):45‐62. doi: 10.1177/2042098617743393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wright JL, Davis WS, Joseph MM, Ellison AM, Heard‐Garris NJ, Johnson TL. Eliminating race‐based medicine. Pediatrics. 2022;150(1). doi: 10.1542/peds.2022-057998 [DOI] [PubMed] [Google Scholar]
- 32. Kozyra M, Ingelman‐Sundberg M, Lauschke VM. Rare genetic variants in cellular transporters, metabolic enzymes, and nuclear receptors can be important determinants of interindividual differences in drug response. Genet Med. 2017;19(1):20‐29. doi: 10.1038/gim.2016.33 [DOI] [PubMed] [Google Scholar]
- 33. Ahn E, Park T. Analysis of population‐specific pharmacogenomic variants using next‐generation sequencing data. Sci Rep. 2017;7(1):8416. doi: 10.1038/s41598-017-08468-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wright GEB, Carleton B, Hayden MR, Ross CJD. The global spectrum of protein‐coding pharmacogenomic diversity. Pharmacogenomics J. 2018;18(1):187‐195. doi: 10.1038/tpj.2016.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fujikura K, Ingelman‐Sundberg M, Lauschke VM. Genetic variation in the human cytochrome P450 supergene family. Pharmacogenet Genomics. 2015;25(12):584‐594. doi: 10.1097/FPC.0000000000000172 [DOI] [PubMed] [Google Scholar]
- 36. Gordon AS, Tabor HK, Johnson AD, et al. Quantifying rare, deleterious variation in 12 human cytochrome P450 drug‐metabolism genes in a large‐scale exome dataset. Hum Mol Genet. 2014;23(8):1957‐1963. doi: 10.1093/hmg/ddt588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schaller L, Lauschke VM. The genetic landscape of the human solute carrier (SLC) transporter superfamily. Hum Genet. 2019;138(11–12):1359‐1377. doi: 10.1007/s00439-019-02081-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang B, Lauschke VM. Genetic variability and population diversity of the human SLCO (OATP) transporter family. Pharmacol Res. 2019;139:550‐559. doi: 10.1016/j.phrs.2018.10.017 [DOI] [PubMed] [Google Scholar]
- 39. Xiao Q, Zhou Y, Lauschke VM. Ethnogeographic and inter‐individual variability of human ABC transporters. Hum Genet. 2020;139(5):623‐646. doi: 10.1007/s00439-020-02150-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hauser AS, Chavali S, Masuho I, et al. Pharmacogenomics of GPCR drug targets. Cell. 2018;172(1‐2):41‐54.e19. doi: 10.1016/j.cell.2017.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nelson MR, Wegmann D, Ehm MG, et al. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337(6090):100‐104. doi: 10.1126/science.1217876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou Y, Arribas GH, Turku A, et al. Rare genetic variability in human drug target genes modulates drug response and can guide precision medicine. Sci Adv. 2021;7(36):eabi6856. doi: 10.1126/sciadv.abi6856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chan HT, Chin YM, Low SK. The roles of common variation and somatic mutation in cancer pharmacogenomics. Oncol Ther. 2019;7(1):1‐32. doi: 10.1007/s40487-018-0090-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Russell LE, Zhou Y, Almousa AA, Sodhi JK, Nwabufo CK, Lauschke VM. Pharmacogenomics in the era of next generation sequencing—from byte to bedside. Drug Metab Rev. 2021;53(2):253‐278. doi: 10.1080/03602532.2021.1909613 [DOI] [PubMed] [Google Scholar]
- 45. Schärfe CPI, Tremmel R, Schwab M, Kohlbacher O, Marks DS. Genetic variation in human drug‐related genes. Genome Med. 2017;9(1):117. doi: 10.1186/s13073-017-0502-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou Y, Fujikura K, Mkrtchian S, Lauschke VM. Computational methods for the pharmacogenetic interpretation of next generation sequencing data. Front Pharmacol. 2018;9:1437. doi: 10.3389/fphar.2018.01437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863‐874. doi: 10.1101/gr.176601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248‐249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310‐315. doi: 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hassan MS, Shaalan AA, Dessouky MI, Abdelnaiem AE, ElHefnawi M. A review study: computational techniques for expecting the impact of non‐synonymous single nucleotide variants in human diseases. Gene. 2019;680:20‐33. doi: 10.1016/j.gene.2018.09.028 [DOI] [PubMed] [Google Scholar]
- 51. Zhou Y, Dagli Hernandez C, Lauschke VM. Population‐scale predictions of DPD and TPMT phenotypes using a quantitative pharmacogene‐specific ensemble classifier. Br J Cancer. 2020;123(12):1782‐1789. doi: 10.1038/s41416-020-01084-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Russell LE, Zhou Y, Lauschke VM, Kim RB. In vitro functional characterization and in silico prediction of rare genetic variation in the bile acid and drug transporter, Na+‐taurocholate cotransporting polypeptide (NTCP, SLC10A1). Mol Pharm. 2020;17(4):1170‐1181. doi: 10.1021/acs.molpharmaceut.9b01200 [DOI] [PubMed] [Google Scholar]
- 53. Zhou Y, Lauschke VM. Computational tools to assess the functional consequences of rare and noncoding pharmacogenetic variability. Clin Pharmacol Ther. 2021;110(3):626‐636. doi: 10.1002/cpt.2289 [DOI] [PubMed] [Google Scholar]
- 54. Peterson TA, Doughty E, Kann MG. Towards precision medicine: advances in computational approaches for the analysis of human variants. J Mol Biol. 2013;425(21):4047‐4063. doi: 10.1016/j.jmb.2013.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou Y, Mkrtchian S, Kumondai M, Hiratsuka M, Lauschke VM. An optimized prediction framework to assess the functional impact of pharmacogenetic variants. Pharmacogenomics J. 2019;19(2):115‐126. doi: 10.1038/s41397-018-0044-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pandi MT, Koromina M, Tsafaridis I, et al. A novel machine learning‐based approach for the computational functional assessment of pharmacogenomic variants. Hum Genomics. 2021;15(1):51. doi: 10.1186/s40246-021-00352-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shrestha S, Zhang C, Jerde CR, et al. Gene‐specific variant classifier (DPYD‐Varifier) to identify deleterious alleles of dihydropyrimidine dehydrogenase. Clin Pharmacol Ther. 2018;104(4):709‐718. doi: 10.1002/cpt.1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McInnes G, Dalton R, Sangkuhl K, et al. Transfer learning enables prediction of CYP2D6 haplotype function. PLoS Comput Biol. 2020;16(11):e1008399. doi: 10.1371/journal.pcbi.1008399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Van der Lee M, Allard WG, Vossen R, et al. Toward predicting CYP2D6‐mediated variable drug response from CYP2D6 gene sequencing data. Sci Transl Med. 2021;13(603). doi: 10.1126/scitranslmed.abf3637 [DOI] [PubMed] [Google Scholar]
- 60. Garrison LP Jr, Carlson RJ, Carlson JJ, Kuszler PC, Meckley LM, Veenstra DL. A review of public policy issues in promoting the development and commercialization of pharmacogenomic applications: challenges and implications. Drug Metab Rev. 2008;40(2):377‐401. doi: 10.1080/03602530801952500 [DOI] [PubMed] [Google Scholar]
- 61. Lauschke VM, Ingelman‐Sundberg M. Requirements for comprehensive pharmacogenetic genotyping platforms. Pharmacogenomics. 2016;17(8):917‐924. doi: 10.2217/pgs-2016-0023 [DOI] [PubMed] [Google Scholar]
- 62. Chang WC, Tanoshima R, Ross CJD, Carleton BC. Challenges and opportunities in implementing pharmacogenetic testing in clinical settings. Annu Rev Pharmacol Toxicol. 2021;61(1):65‐84. doi: 10.1146/annurev-pharmtox-030920-025745 [DOI] [PubMed] [Google Scholar]
- 63. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372(9):793‐795. doi: 10.1056/NEJMp1500523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rollinson V, Turner R, Pirmohamed M. Pharmacogenomics for primary care: an overview. Genes. 2020;11(11):1337. doi: 10.3390/genes11111337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Van Schaik RHN, Müller DJ, Serretti A, Ingelman‐Sundberg M. Pharmacogenetics in psychiatry: an update on clinical usability. Front Pharmacol. 2020;11:575540. doi: 10.3389/fphar.2020.575540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pardiñas AF, Owen MJ, Walters JTR. Pharmacogenomics: a road ahead for precision medicine in psychiatry. Neuron. 2021;109(24):3914‐3929. doi: 10.1016/j.neuron.2021.09.011 [DOI] [PubMed] [Google Scholar]
- 67. Westbrook MJ, Wright MF, van Driest SL, et al. Mapping the incidentalome: estimating incidental findings generated through clinical pharmacogenomics testing. Genet Med. 2013;15(5):325‐331. doi: 10.1038/gim.2012.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haga SB. Revisiting secondary information related to pharmacogenetic testing. Front Genet. 2021;12:741395. doi: 10.3389/fgene.2021.741395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kalman LV, Agúndez J, Appell ML, et al. Pharmacogenetic allele nomenclature: international workgroup recommendations for test result reporting. Clin Pharmacol Ther. 2016;99(2):172‐185. doi: 10.1002/cpt.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Relling MV, Klein TE, Gammal RS, Whirl‐Carrillo M, Hoffman JM, Caudle KE. The clinical pharmacogenetics implementation consortium: 10 years later. Clin Pharmacol Ther. 2020;107(1):171‐175. doi: 10.1002/cpt.1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Abdullah‐Koolmees H, van Keulen AM, Nijenhuis M, Deneer VHM. Pharmacogenetics guidelines: overview and comparison of the DPWG, CPIC, CPNDS, and RNPGx guidelines. Front Pharmacol. 2020;11:595219. doi: 10.3389/fphar.2020.595219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee Y, Clark EW, Milan MS, et al. Turnaround time of plasma next‐generation sequencing in thoracic oncology patients: a quality improvement analysis. JCO Precis Oncol. 2020;4(4):1098‐1108. doi: 10.1200/PO.20.00121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sheffield BS, Beharry A, Diep J, et al. Point of care molecular testing: community‐based rapid next‐generation sequencing to support cancer care. Curr Oncol. 2022;29(3):1326‐1334. doi: 10.3390/curroncol29030113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Böttiger Y, Laine K, Andersson ML, et al. SFINX—a drug‐drug interaction database designed for clinical decision support systems. Eur J Clin Pharmacol. 2009;65(6):627‐633. doi: 10.1007/s00228-008-0612-5 [DOI] [PubMed] [Google Scholar]
- 75. Shekhani R, Steinacher L, Swen JJ, Ingelman‐Sundberg M. Evaluation of current regulation and guidelines of pharmacogenomic drug labels: opportunities for improvements. Clin Pharmacol Ther. 2020;107(5):1240‐1255. doi: 10.1002/cpt.1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Koutsilieri S, Tzioufa F, Sismanoglou DC, Patrinos GP. Unveiling the guidance heterogeneity for genome‐informed drug treatment interventions among regulatory bodies and research consortia. Pharmacol Res. 2020;153:104590. doi: 10.1016/j.phrs.2019.104590 [DOI] [PubMed] [Google Scholar]
- 77. Verbelen M, Weale ME, Lewis CM. Cost‐effectiveness of pharmacogenetic‐guided treatment: are we there yet? Pharmacogenomics J. 2017;17(5):395‐402. doi: 10.1038/tpj.2017.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pandi MT, Koromina M, Vonitsanos G, van der Spek PJ, Patrinos GP, Mitropoulou C. Development of an optimized and generic cost‐utility model for analyzing genome‐guided treatment data. Pharmacol Res. 2022;178:106187. doi: 10.1016/j.phrs.2022.106187 [DOI] [PubMed] [Google Scholar]