

Abstract
In marine species experiencing intense fishing pressures, knowledge of genetic structure and local adaptation represent a critical information to assist sustainable management. In this study, we performed a landscape genomics analysis in the American lobster to investigate the issues pertaining to the consequences of making use of putative adaptive loci to reliably infer population structure and thus more rigorously delineating biological management units in marine exploited species. Toward this end, we genotyped 14,893 single nucleotide polymorphism (SNPs) in 4190 lobsters sampled across 96 sampling sites distributed along 1000 km in the northwest Atlantic in both Canada and the USA. As typical for most marine species, we observed a weak, albeit highly significant genetic structure. We also found that adaptive genetic variation allows detecting fine‐scale population structure not resolved by neutral genetic variation alone. Using the recent genome assembly of the American lobster, we were able to map and annotate several SNPs located in functional genes potentially implicated in adaptive processes such as thermal stress response, salinity tolerance and growth metabolism pathways. Taken together, our study indicates that weak population structure in high gene flow systems can be resolved at various spatial scales, and that putatively adaptive genetic variation can substantially enhance the delineation of biological management units of marine exploited species.
Keywords: adaptive markers, fisheries, genetic units, genomics, lobster
1. INTRODUCTION
Marine species and their ecosystems play fundamental roles in supporting human populations and societies (Béné et al., 2016). Seafood represents one of the most traded sources of animal proteins, and it plays a critical role in food security and poverty reduction for many vulnerable coastal communities (Guillen et al., 2019; McIntyre et al., 2016; Pauly et al., 2002). This critical role hinges on authorities' capacity to protect and manage the global health of seafood in the face of ever‐increasing anthropic pressures such as overfishing, climate change, invasive species and habitat degradation (McCauley et al., 2015; Ormerod, 2003). Efficient management requires an understanding of the biological principles underlying the dynamics of exploited species, which is defined by the “stock” concept in fishery science (Pita et al., 2016). The toolbox for assessment of marine stocks has expanded considerably in recent years and now comprises an extended spectrum of methods, including acoustic telemetry, otolith sciences, and demographic modelling (reviewed by Hawkins et al., 2016). Modern genomic methods represent a powerful addition to the fishery management toolbox, allowing the genotyping of thousands of markers on thousands of individuals in single studies. Indeed, new genomic methods can provide fundamental knowledge of marine populations and inform decision‐making by delineating biologically distinct populations, assessing connectivity between populations, documenting the extent of population adaptive divergence, performing commercial traceability, and assessing the impact of fishery‐induced evolution (reviewed in Bernatchez et al., 2017).
Documenting the rate and scale of population connectivity is important to fish stock assessment. However, the estimation of population connectivity is often challenging in many marine species, which typically exhibit a combination of life history traits (e.g., large effective population size – N e, high fecundity, and high dispersal rate) resulting in weak population structure (Gagnaire et al., 2015; Waples, 1998). The limit between demographic dependence and demographic independence among populations has rarely been investigated. In one of the few studies on this topic, and based on mathematical modelling, Hastings (1993) suggested that it occurs when dispersal rate (or m, the proportion of effective migrants per generation between populations) is approximately 10%. This level of connectivity is challenging to the identification of population genetic structure of marine species, since a 10% dispersal rate (m), combined with large effective population size (N e), results in a high rate of gene flow “N e m” (Palumbi, 2003; Waples, 1998), which in turn leads to very low levels of population genetic differentiation that are not readily detected by traditional genetic measures (e.g., F ST) due to insufficient statistical power (Waples et al., 2008; Waples & Gaggiotti, 2006). This statistical limitation may thus lead to erroneous conclusions regarding the degree of demographic independence among populations, which may hamper the usefulness of population genomics for fishery management.
So far, while neutral genetic markers have been considered essential to properly document population structure (Landguth & Balkenhol, 2012; Luikart et al., 2003), another important consideration pertains to the interpretation of “outlier loci” for delineating populations (reviewed in Bernatchez, 2016). Although it has been common practice to discard such markers when inferring population genetic structure and connectivity, some have argued in favour of retaining such markers, especially when studying weakly differentiated systems such as those involving marine (Attard et al., 2018; Gagnaire et al., 2015; Milano et al., 2014; Tran Lu Y et al., 2022) or terrestrial organisms, such as many plant species (Keller et al., 2018; Tyrmi et al., 2020) but also a number of terrestrial animal species (Batista et al., 2016). Indeed, genomic regions under divergent selection (including adjacent genomic regions in linkage disequilibrium [LD] with the targets of selection) are often characterized by stronger restriction of gene flow among populations (locally affecting N e and m) compared to the genome‐wide background, which can promote evolution of reproductive isolation (Barton & Bengtsson, 1986). Therefore, adaptive loci and genomic regions in LD with them may be more likely to reveal biologically meaningful population differentiation than neutral markers (Gagnaire et al., 2015). Moreover, adaptive markers can be particularly important for conservation purposes, for instance in assisting in the design of Marine Protected Areas to ensure a better protection of adaptive variability and potential response to environmental change (Xuereb et al., 2020). Depending on views and contexts, the issue of how adaptive loci should be considered for delineating population structure is still highly debated (Funk et al., 2012; Hohenlohe et al., 2021).
The American lobster (Homarus americanus) is the most valuable exploited species in the Northwest Atlantic Ocean. To date, studies based either on a few genetic (i.e., microsatellites) or many genomic (i.e., SNPs) markers have shown that the species exhibits a weak genetic population structure (global F ST < 0.005), mainly involving the delineation of two large‐scale genetic clusters separated by a latitudinal break between northern versus southern sampling sites (Benestan et al., 2015; Kenchington et al., 2009). Other studies revealed that the environment, particularly temperature, may play a role in shaping this “north–south” broad‐scale structure in the North‐Western Atlantic area (Lehnert et al., 2019; Stanley et al., 2018). Yet, the delineation of fine‐scale population structure within each of these genetic clusters remains unclear, either due to limited resolution (e.g., microsatellite markers) or to the sparse distribution of the sampling design (D'Aloia et al., 2020; Takeshima et al., 2016).
In this study, we conducted one of the most extended sampling efforts applied to a marine species for genomics analysis. First, using this high‐resolution sampling design we precisely located the north–south genetic break in American lobster, and improved inferences concerning the species' fine‐scale genetic structure within both Northern and Southern genetic groups. Next, we performed genotype‐environment‐association (GEA) analyses to infer the extent of adaptive differentiation and examine whether adaptive genetic variation may provide useful insights for delineating lobster population units at different spatial scales. Lastly, thanks to a recently published reference genome for the American lobster, we investigated the role of SNPs putatively under selection among a set of annotated functional genes potentially related to environmental adaptation.
2. MATERIALS AND METHODS
2.1. Sample collection, rapture sequencing and variant calling
Genomic DNA was extracted from muscle tissue of 4664 ovigerous female lobsters collected during the fishing season between 2012 and 2017 at 96 localities along the northwestern Atlantic coast from Cape cod (USA) to Newfoundland (Figure 1 and Table S1). DNA extraction and Rapture library preparation were conducted following the procedure previously adapted for the American lobster and described in Dorant et al. (2019). The Rapture approach was initially developed by Ali et al. (2016) and successfully adapted to the American lobster by Dorant et al. (2019). This approach represents a highly flexible genotyping method protocol allowing hundreds to thousands of individuals to be sequenced simultaneously with a high sequencing depth (Ali et al., 2016). The sequence capture step allows targeting a panel of informative and high‐quality RAD loci, which have been discovered and selected from an initial RADseq experiment. In this study, we used the same 9818 targeted loci previously identified for the American lobster by Dorant et al. (2019). Rapture libraries were multiplexed with 384 individuals and sequenced on the Ion Torrent p1v3 chip at the Plateforme d'analyses génomiques of the Institute of Integrative and Systems Biology (IBIS, Université Laval, Québec, Canada). Two rounds of sequencing (i.e., two separate chips) were conducted for all Rapture libraries.
FIGURE 1.
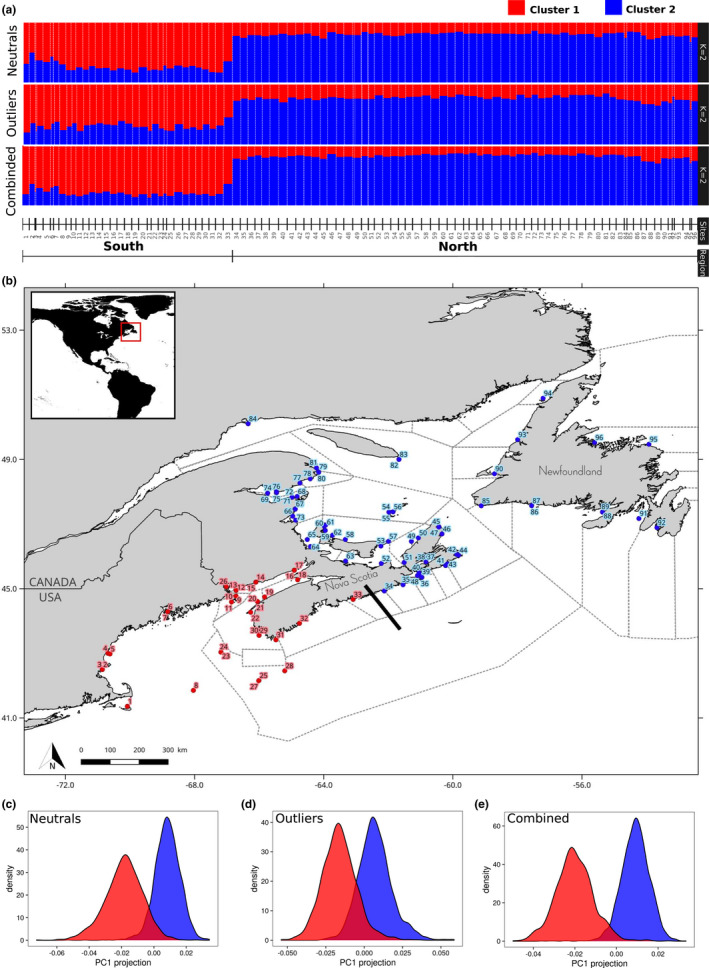
Genome‐wide population structure of the American lobster. (a) The inferred ancestral population admixture across the 96 lobster sampling sites (K = 2) for data set of neutral SNPs (top), data set of outlier SNPs based on genome scan analyses (i.e., F ST based approach and GEA) (middle) and the combined data set of all SNPs. Each colour bar indicates individual membership probability at each cluster (K), averaged for each sampling site (x‐axis). (b) Map of lobster sampling sites in the northwest Atlantic area, with sites from the north (blue) versus south (red) regions being identified by different colours according to the most prominent cluster membership displayed in (a) (i.e., proportion of cluster membership >50%). Site labels correspond to numbers attributed for each sampling site in A and Table S1 Dotted squares represent the delineation of the 41 Lobster Fishing Areas (LFAs) in Canada. The black line represents the geographical position of the split between lobster sampling sites in the south (red) and north (blue) regions. (c) Density plot of individuals along the first principal component from PCA based on neutral SNP data set (4190 individuals, 13,912 SNPs). (d) Density plot of individuals along the first principal component from PCA based on outliers SNP data set (4190 individuals, 981 SNPs). (e) Density plot of individuals along the first principal component from PCA based on the combined SNP data set (4190 individuals, 14,893 SNPs). For (c–e), groups are displayed by different colours according to their membership to the genetic clusters (blue, North; red, South) depicted by the model‐based Admixture analysis in (a).
Sequenced data were processed following the RADseq pipeline stacks_workflow available at https://github.com/enormandeau/stacks_workflow. Raw reads were trimmed to 80 bp and shorter reads were discarded with cutadapt (Martin, 2011) allowing for an error rate of 0.2. Individuals were demultiplexed using the module process_radtag from stacks version 1.48 (Catchen et al., 2013). Only individuals with at least 150,000 sequencing reads were retained for downstream bioinformatic processes for maximizing the expected locus read depth and remove poorly sequenced individuals. Reads were mapped to the Rapture base catalogue (9818 targeted loci, see Dorant et al., 2019) using bwa‐mem with default parameters (Li, 2013). Then aligned reads were processed with stacks version 1.48 for SNP calling and genotyping. A minimum stack depth of four (pstacks module) and a maximum of three nucleotide mismatches (cstacks module) were allowed. We used the populations module of stacks to first retain SNPs with a minimum stack depth of four and present in at least 20 sampling locations with at least 60% of data in each location. Next, individuals and SNPs were filtered using an iterative procedure and custom scripts available in stacks_workflow and previously described in Dorant et al. (2020). Complete procedure of the SNPs filtering process is provided in the Appendix S1: Materials and Methods, Tables S2 and S3 and Figures S1–S3. Briefly, we used prior filters to remove poor quality SNPs (i.e., putatively due to sequencing errors or bioinformatic errors) as well as individuals showing high rate of missing data or DNA contamination. Then, we discriminated nonduplicated SNPs from duplicated SNPs (i.e., SNPs related to paralog sequences) using the same procedure proposed by Dorant et al. (2020). The resulting data set of nonduplicated SNPs was postfiltered by keeping all unlinked SNPs.
Finally, we investigated for the presence of a pattern of missingness associated with library batches (that is, referring to sequencing lanes of 384 individuals each). Indeed, missing data can influence clustering of individuals based on biases in the absence of data rather than true biological relationship. While low level of missing data (i.e., average missing proportion <5%) is generally results in only a slight background noise in well‐structured populations, the influence of biases might be critical when the level of genetic differentiation is very weak (Yi & Latch, 2022). To do this, pairwise identity‐by‐missingness distances among individuals were calculated using plink version 1.9. Results were then visualized across a multidimensional scaling approach to determine clustering based on missing data. SNPs strongly associated with any pattern of missingness among sequencing batches were excluded until removing the observed pattern of missingness. Complete description of this filtering step is available in Figures S2–S4.
2.2. Screening for SNPs under selection
To investigate the possible occurrence of adaptive divergence between lobster populations, we applied two different approaches for outlier (candidate adaptive SNP) detection, the F ST‐based statistic implemented in outflank and a genotype‐environment association (GEA) performed using a redundancy discriminant analysis (RDA). Marine environmental variables were extracted from bio‐oracle version 2.1 public database (Assis et al., 2017) for the period 2000–2014 (spatial resolution of 5 arcmin) using the R library sdmpredictors. Three environmental layers were considered: the mean annual sea surface temperature (SST), the mean annual sea surface salinity (SSS) and the mean annual sea surface chlorophyll concentration (SSC). For each environmental layer, we defined a circular buffer of 15 km radius around each sampling site and values within this buffer were averaged to minimize pixel anomalies, which are known to provide abnormal values in remote sensing, especially in coastal areas and even after correction algorithms (Smit et al., 2013). RDA was performed to investigate the association between the genetic variation and each environmental variable considered using the vegan R library (Oksanen et al., 2018) and following Forester et al. (2018) (see https://popgen.nescent.org/2018‐03‐27_RDA_GEA.html for details). Statistical significance of each RDA model was assessed using a permutation‐based analysis of variance (ANOVA) procedure with 1000 permutations. Once markers were loaded against the RDA axes, candidates for an association with environmental variables were determined as those exhibiting a loading >2.25 standard deviations from the mean loading (p < .024) (Forester et al., 2018).
2.3. Population structure
Patterns of broad‐scale population genetic structure were examined using three data sets: (1) The combined data set which include all SNPs; (2) data set of putatively neutral SNPs; and (3) data set of SNPs identified as putatively under selection. First, we conducted admixture analysis using the model‐based mixture method implemented in admixture version 1.3.0 (Alexander et al., 2009). Ancestry and admixture proportions inferred for each individual were carried out with 1000 bootstrap replicates for each population cluster (K) values ranging from two to five. Population structure was also investigated through a principal component analysis (PCA) based on the individual genotype matrix. As PCA require complete genotype matrix (i.e., no missing data), missing genotypes for a given SNP were imputed using an ancestry‐based missing data imputation implemented in stacks_workflow. Lastly, pairwise genetic differentiation among sampling sites was estimated with the unbiased F ST estimator θ (Weir & Cockerham, 1984) using the R package stampp (Pembleton et al., 2013). Significance or F ST values was estimated for 1000 bootstraps.
2.4. Within‐region population structure
Fine‐scale genetic structure within both North and South regions was examined by means of PCA performed using neutral SNPs, as well as candidate adaptive SNPs that were associated with variation of the three environmental variables considered (SST, SSS and SSC), which were identified by the RDA as described above. PCAs were based on SNP allele frequencies measured for each sampling site. To minimize potential bias in allele frequency estimation due to small sample size, we only considered sampling sites represented by at least 30 individuals (i.e., 59/63 sites for the North and 29/33 sites for the South). Rare SNPs that were only found in one sample were discarded (Linck & Battey, 2019). Results of each PCA were then compared with an RGB composite habitat layer, which represents a synthetic characterization of the three environmental variables considered within each region.
2.5. Demographic modelling
To test for putative demographic dependence/independence among large scale population structure groups, we estimated the rate of gene flow and the ancestral and present effective population size among populations from demographic modelling using the diffusion approximation framework implemented in dadi (Gutenkunst et al., 2009). Demographic inference was performed using the joint site frequency spectrum (jSFS) from 50 randomly selected samples associated with each genetic cluster identified. Note that SNPs were also filtered for nonmissing data. One 10‐parameter demographic model was built to test the general historical demography of the populations: Isolation‐with‐migration with continuous gene flow since divergence (IMAG). This model was chosen because it seems the most likely given that any previous studies on this species reported any kind of evidence for scenarios that would involve long‐term isolation followed by say no genetic exchange or secondary contact. For this model, we allowed dynamic effective population size change across generations (i.e., contraction or expansion of the N e). We also implemented asymmetric migration rate (m1 ← 2, m2 ← 1) between the northern and southern groups. A total of 30 replicated runs was performed and the best replicate was chosen based on the Akaike information criterion (AIC).
Demographic parameters values were then extrapolated from model parameters assuming an average genome‐wide mutation rate (μ) of 1e‐08 μ/bp/generation (Tine et al., 2014). We estimated the effective genome length by multiplying the total base pairs of our data set by the fraction of all SNPs included in the JSFS. Estimated migration rates were divided by 2N ref to infer the proportion of migrants in every generation for each subpopulation.
2.6. Functional annotation
Rapture tag sequences were mapped to the H. americanus reference genome recently published (NCBI Accession PRJNA655509; Polinski et al., 2021), using gmap version 2021.08.25 (Wu & Watanabe, 2005). Sequences showing multiple hits (that is, putative repetitive DNA sequences) or a mapped quality score below 30 were then excluded. Functional annotations were assessed for Rapture loci which contained SNPs showing significant association with the three environmental factors considered (i.e., SST, SSS, and SSC), and were extracted from the gene‐finding format (GFF) file associated with the H. americanus reference genome.
3. RESULTS
3.1. Data generation
stacks variant calling (Catchen et al., 2013) across 4400 individuals (excluding 264 with too low coverage) initially yielded 76,863 SNPs distributed across 8879 loci before any filtering process (Table S2). Presence of duplicated SNPs (i.e., putative paralogues) was assessed following the approach proposed by Dorant et al. (2020) to retain 19,868 nonduplicated SNPs and putatively unlinked (Figure S1, Tables S2 and S3). Ultimately, after bias corrections due to missing data (Figures S2–S5), 4190 individuals and 14,893 high quality nonduplicated SNPs were retained for downstream analyses. The average read depth over all nonmissing genotype calls was 34× (SD = 17.36) and the median of missing genotypes was 2% (SD = 0.027) across all individuals (Figure S5).
3.2. Genomic signature of natural selection
For subsequent analyses of population structure, we separated loci into two data sets: putatively neutral and putatively influenced by divergent selection. Genome scans were conducted using two different approaches: an F ST‐based method implemented in outflank version 2 (Whitlock & Lotterhos, 2015) and a genetic‐environment association (GEA) based on redundancy discriminant analysis (RDA). Overall, genome scans identified 981 outlier SNPs, of which 22 (2.2%) were detected by both approaches considered. outflank identified 33 SNPs that showed higher F ST than expected under neutrality. RDA conducted over three environmental variables (sea surface temperature [SST], sea surface salinity [SSS] and concentration of sea surface chlorophyll [SSC]) revealed highly significant GEAs (p < .001; ANOVA 1000 permutation), which explained 0.22%, 0.18% and 0.013% of the total genetic variation for association with SST, SSS, and SSC, respectively. In total, the RDA analysis identified 970 SNPs significantly associated with at least one of the three environmental variables considered. Among the set of candidates for environmental association, SNPs associated with SSS and SST shared the largest number of SNPs (i.e., 146 SNPs, representing 15% of overall GEA candidates, Figure S6A). Candidate SNPs associated with SSC were mostly represented by rare variants, with only ~13% showing a MAF above 0.1. In contrast, 38.4% and 39.8% of the candidate SNPs associated with SST and SSS showed a MAF higher than 0.1, respectively (Figure S6B,C). Finally, 13,912 SNPs were considered as neutral for further analyses.
3.3. Population structure and admixture
Broad‐scale population structure was examined using unsupervised model‐based clustering implemented in the program admixture (Alexander et al., 2009) for a total data set of 14,893 SNPs including 13,912 neutral and 981 SNPs putatively under selection. We ran admixture for K = 2–5 and obtained CV errors, determined that the best K (the one with lowest cross‐validation error) was K = 2 (Figure S7). In the three cases, the proportion of individual ancestry averaged by sampling sites showed a major split between sites 33 and 34, which were located on the Scotian Shelf, near the middle of the east cost of Nova Scotia (Figure 1a). Two genetic clusters (K = 2) clearly stood out, delineating a Southern region (sites 1–33) and a Northern region (site 34–96) (Figure 1b). The same signal was also recovered using individual‐based PCA with the same data sets (Figure 1c–e and Figure S11). Admixture plots for K = 2–5 are available in Figures S8–S10 for neutral, outlier and combined SNP data sets respectively.
The AMOVA revealed that, as typically observed in any study, most of the variance (>98.6%) was explained within‐sample variation (Table 1). A higher proportion of the remaining genetic variance was explained by the North versus South regional grouping than among sampling sites within each of these groups, and this applied to both neutral and outliers SNP data sets. However, the proportion of explained variance between the North and South regions was one order of magnitude higher for outlier SNPs (proportion of variance = 1.20%, p < .01) compared to that of neutral markers (0.124%, p < .01). Also, the AMOVAs provided evidence of significant genetic variation among sites within regions for outlier markers (p < .01), but not for neutral ones (p = .28). The proportion of explained variance among sites within regions was also more than one order of magnitude higher for outlier SNPs (0.13%) compared to neutral ones (0.0034%). Furthermore, an AMOVA based exclusively on the 12 sites that were sampled twice showed that there was no significant variation between temporal samples of a same site (p = .16 and p = .73 for outlier and neutral SNPs, respectively), indicating that genetic variation among sampling sites within a region was more important than temporal variation (Table S4). In summary, these results support the contention that putatively adaptive markers offer more resolution than neutral markers in delineating population differentiation in the weakly structured American lobster.
TABLE 1.
Hierarchical analysis of molecular variance (AMOVA)
| Source of variance | df | MSD | Variance component | % of variation | ϕ | p‐Value |
|---|---|---|---|---|---|---|
| Neutral | ||||||
| Between regions (north vs. south) | 1 | 1416.08 | 0.544 | 0.124 | 0.0012 | <.01 |
| Between sites within regions | 94 | 438.44 | 0.015 | 0.0034 | 3 × 10−5 | .29 |
| Between individuals within sites | 4094 | 437.79 | 437.794 | 99.8 | – | <.01 |
| Total variations | 4189 | 438.04 | 438.353 | 100.00 | 0.0276 | – |
| Outliers | ||||||
| Between regions (north vs. south) | 1 | 1485.19 | 0.788 | 1.20 | 0.0120 | <.01 |
| Between sites within regions | 94 | 68.64 | 0.087 | 0.13 | 0.0013 | <.01 |
| Between individuals within sites | 4094 | 64.84 | 64.841 | 98.66 | – | <.01 |
| Total variations | 4189 | 65.26 | 65.717 | 100.00 | 0.0133 | – |
Note: ϕ provides the “Phi” population differentiation statistics. These are used to test hypotheses about population differentiation.
Abbreviations: df, degree of freedom; MSD, mean squared deviation.
Analysis of pairwise population differentiation showed that the strongest genetic differences were also observed when comparing sites from the northern genetic cluster to sites from the southern one (average F ST = 0.00064; Figure 2a). Conversely, very weak or no level of genetic differentiation was observed among sites within both North and South regions (average F ST of 0.00014 and 0.00010 for northern and southern genetic units, respectively). Indeed, the majority of pairwise F ST measures within each region was not significant (Figure 2b), which corroborated the results also observed from the AMOVA analysis.
FIGURE 2.
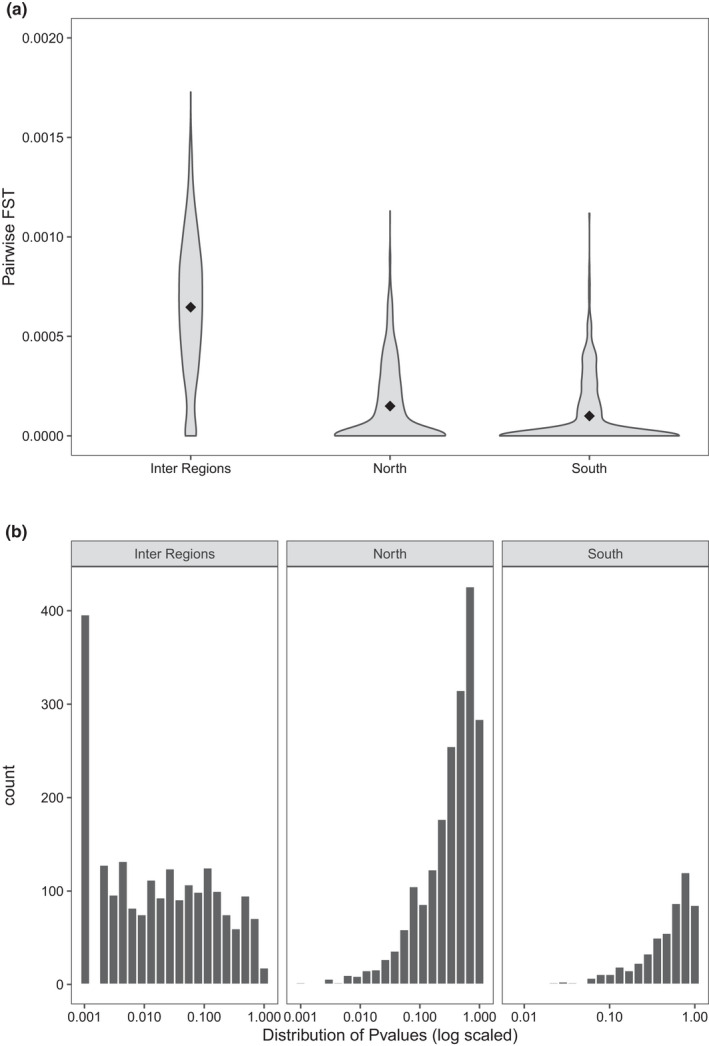
Estimation of pairwise genetic differentiation. (a) Distribution of pairwise F ST measure for comparisons of sites belonging from the north against south genetic unit regions, sites belonging from the northern region only and sites belonging from the southern region only. Black diamonds represent the average F ST value for each group of comparisons. (b) F ST significance distribution for each population pairwise comparison displayed in (a).
3.4. Demographic modelling
The demographic model for isolation‐with‐migration (IMAG) fitted well the lobster data with small residual between modelled jSFS and empirical data (log‐likelihood = −1829.04; AIC = 3678.08; Table S5 and Figure S12). From biological parameters inference, the “contemporary” effective population sizes were estimated to 120,336 and 281,937 for northern and southern genetic units respectively with evidence that populations current sizes were largely due to strong growth (Table S5). Note that the estimated “contemporary” population size takes into account population dynamics (growth rate) across generations at the end of the model run and should not be interpreted as real N e value per se but rather as an approximation of the order of magnitude of the real N e value. Demographic inference from the IMAG model reported a weak level of historical migration rate between northern and southern genetic units (m < 1e−3; Table S5). Interestingly, we observed that historical migration rate was higher from south to north with m south → north of 0.00134 and m north → south of 0.00071.
3.5. Within‐region population structure: putative neutral versus adaptive markers
To characterize fine‐scale population structure within the northern and southern regions, we conducted two principal component analyses (PCA) of population allele frequencies, one based on (1) putative neutral SNPs and the second based on (2) putative candidate adaptive SNPs associated with one or several of our environmental variables.
For the northern region, putatively adaptive genetic variation revealed a west‐to‐east distribution of sampling sites along the first principal component (PC1), which explained 4.47% of the adaptive genetic variance isolated from our data (Figure 3c). This geographic gradient includes three distinct groups according to their habitat characteristics (Figure 3a,c,e). First, a western group that is composed of sampling sites distributed along the Gaspé Peninsula and the Baie‐des‐Chaleurs, corresponding to a more estuarine environment mostly represented by higher values of SSC and lower values of SSS and SST (i.e., sites of varying shades of green). Second, an eastern group that is composed of sites distributed along the coastline of Newfoundland in a more marine environment, mostly represented by lower SSC, low to medium SST and higher SSS (i.e., dark blue to dark purple site coloration). Third, a median group, composed of sites in a marine environment represented by relatively higher values of SST and lower values of SSS and SSC compared to the other two groups (i.e., dark red site coloration), mostly distributed in the east‐southern part of the Gulf of St. Lawrence and the east coast of Nova Scotia.
FIGURE 3.
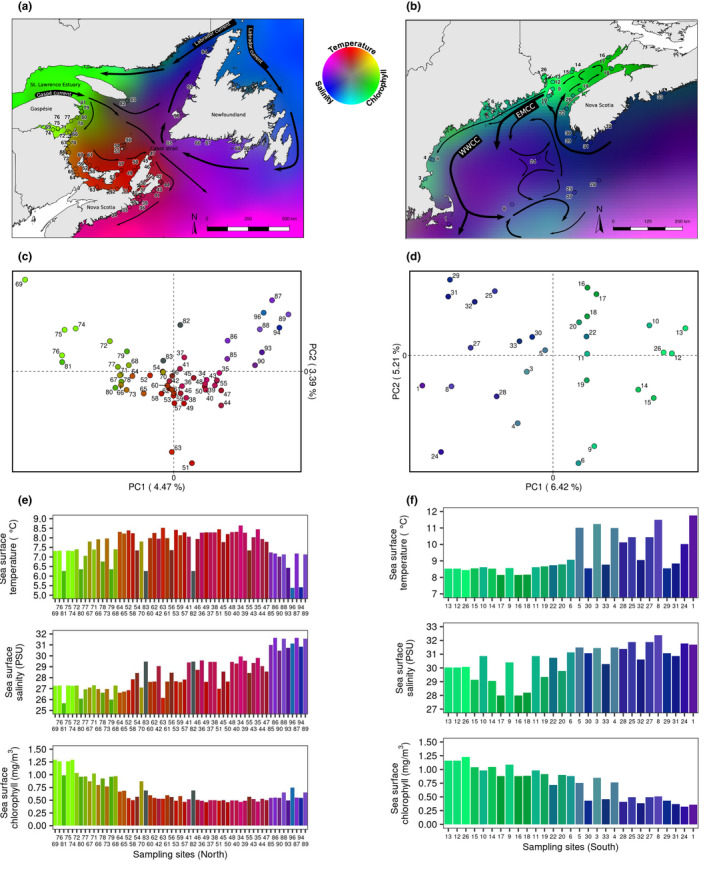
Fine‐scale adaptive population genetic structure. (Upper panels) RGB composite habitat layer for the (a) northern and (b) southern genetic clusters. The red, green and blue colour channels represent the intensities of the mean annual sea surface temperature (SST), sea surface chlorophyll concentration (SSC) and sea surface salinity (SSS) at each pixel, respectively. Environmental layers were normalized between 0 and 1 before RGB projection, and normalized layers were “contrast stretched” to enhance visual clarity (min quantile = 0.05 and max quantile = 0.95). For instance, a pixel coloured by magenta means that the intensity of SST and SSS are elevated and equivalent between the two variables. On the opposite, the intensity of SSC is very low or even zero (R = 0.99; G ~ 0, B = 0.99). Sampling sites are represented by circles coloured according to an RGB habitat value averaged over a buffer of two map units (2*5 arcmin; ~18.4 km radius). Black arrows represent major current circulation within the two regions. EMCC and WMCC indicate the “Eastern Maine Coastal Current” and the “Western Maine Coastal Current”, respectively. (Middle panels) PCA biplot based on allele frequencies of candidate loci for sampling sites in the (c) northern (940 SNPs) and (d) southern (952) study regions, where each circle represents a sampling site coloured according to its RGB projection (a, b). (Lower panels) Regional distribution of environmental conditions among sampling sites in the (e) northern and (f) southern study regions. Vertical bars represent the absolute value of each environmental variable for a given site (y‐axis) averaged over years 2000–2014 (biooracle marine database version 2.1) and coloured according to its RGB habitat projection above. Sampling sites are sorted according to their position along the PCA axis.
For the southern region, the PCA based on putatively adaptive genetic variation distinguished sampling sites from the Bay of Fundy and the rest of the Gulf of Maine along the first axis, which explained 6.42% of the adaptive genetic variance isolated from our data (Figure 3d). Here, sites in the Bay of Fundy area were characterized by relatively high values of SSC and low values of SSS and SST. Conversely, sites in the Gulf of Maine were characterized by higher values of SSS and SST and lower values of SSC (Figure 3b,f).
In contrast to clear patterns of population structure depicted using candidate SNPs, PCAs based on neutral SNPs revealed no clear pattern of within‐region population structure. Within either the northern or southern region, most of the sampling sites clustered together, with seven and two sites forming “individual clusters” in the North (i.e., sites 51, 63, 65, 69, 87, 89 and 96, Figure S13C) and the South (i.e., sites 9 and 10, Figure S13D), respectively. Within each region, sites that were not part of the main “clusters” had a smaller sample size than those within the main cluster (i.e., median sample size for unique clusters versus main cluster was 37 versus 50 and 32 versus 43 for the North and the South regions, respectively), indicating that their distinct patterns may be the result of less accurate estimate of allele frequency at those sites.
3.6. Functional annotation of adaptive SNPs
Overall, 1304 out of 9818 (13.3%) Rapture tags sequences were properly mapped to the reference genome of H. americanus, passing our quality requirements. Only mapped loci containing candidate SNPs identified by the RDA approach and having annotation for gene sequence were considered. A total of 27 SNPs distributed along to 21 loci confidently mapped to known annotation for gene sequence (Table 2). Their identification and potential adaptive role is interpreted in Section 4.3.
TABLE 2.
Annotation for putatively adaptive SNPs
| Rapture SNP id | Genome scaffold ref | Start bp | Stop bp | GFF gbkey | Gene annotation | Predictor |
|---|---|---|---|---|---|---|
| Locus‐10322_25 | JAHLQT010047199.1 | 6,562,320 | 6,562,399 | XRCC3‐L | DNA repair protein XRCC3 | SSC |
| Locus‐10322_29 | JAHLQT010047199.1 | 6,562,320 | 6,562,399 | XRCC3‐L | DNA repair protein XRCC3 | SSS |
| Locus‐10322_29 | JAHLQT010047199.1 | 6,562,320 | 6,562,399 | XRCC3‐L | DNA repair protein XRCC3 | SST |
| Locus‐10559_37 | JAHLQT010011632.1 | 12,514,636 | 12,514,715 | Hamer_G000906 | Hypothetical protein | SSS |
| Locus‐10559_37 | JAHLQT010011632.1 | 12,514,636 | 12,514,715 | Hamer_G000906 | Hypothetical protein | SST |
| Locus‐12276_14 | JAHLQT010037402.1 | 1,938,579 | 1,938,658 | Psmc6a‐L | 26S Proteasome regulatory subunit 6A‐like | SSS |
| Locus‐19299_61 | JAHLQT010025476.1 | 15,868,499 | 15,868,578 | CP1876‐L8 | Cuticle protein | SST |
| Locus‐20965_53 | JAHLQT010021820.1 | 839,811 | 839,890 | MBTPS1‐L | Membrane‐bound transcription factor site‐1 protease‐like | SSC |
| Locus‐2199_06 | JAHLQT010010005.1 | 46,652 | 46,732 | Hamer_026672 | Hypothetical protein | SST |
| Locus‐23124_25 | JAHLQT010001931.1 | 8,026,397 | 8,026,476 | Hamer_G008307 | Hypothetical protein | SSC |
| Locus‐23444_62 | JAHLQT010011632.1 | 23,500,981 | 23,501,060 | Hamer_001047 | Hypothetical protein | SST |
| Locus‐25514_58 | JAHLQT010014926.1 | 52,578 | 52,657 | NinaC‐L | Neither inactivation nor after potential protein C‐like | SST |
| Locus‐27285_48 | JAHLQT010028013.1 | 5,692,331 | 5,692,410 | Wnt9A‐L | Wnt9A‐L | SSC |
| Locus‐28132_08 | JAHLQT010044460.1 | 1,633,662 | 1,633,741 | FASN‐L | Fatty acid synthase‐like | SSC |
| Locus‐28132_08 | JAHLQT010044460.1 | 1,633,662 | 1,633,741 | FASN‐L | Fatty acid synthase‐like | SST |
| Locus‐31177_36 | JAHLQT010026447.1 | 20,898,423 | 20,898,484 | Hamer_G003026 | Hypothetical protein | SSS |
| Locus‐31177_36 | JAHLQT010026447.1 | 20,898,423 | 20,898,484 | Hamer_G003026 | Hypothetical protein | SST |
| Locus‐32916_66 | JAHLQT010022185.1 | 7,835,317 | 7,835,386 | Hamer_003182 | Putative Chitin binding | SSS |
| Locus‐34964_76 | JAHLQT010007678.1 | 9,260,629 | 9,260,706 | Hamer_003182 | Hypothetical protein | SSS |
| Locus‐34964_76 | JAHLQT010007678.1 | 9,260,629 | 9,260,706 | Hamer_003182 | Hypothetical protein | SST |
| Locus‐35388_46 | JAHLQT010026447.1 | 19,617,013 | 19,617,092 | Hamer_G003011 | Hypothetical protein | SSS |
| Locus‐43522_70 | JAHLQT010037402.1 | 2,133,284 | 2,133,363 | HADHA‐L | Trifunctional enzyme subunit alpha‐like | SSC |
| Locus‐46691_78 | JAHLQT010024345.1 | 10,184,871 | 10,184,950 | Hamer_007219 | Putative Zona pellucida‐like domain‐containing protein 3 | SST |
| Locus‐47156_64 | JAHLQT010020459.1 | 4,767,081 | 4,767,160 | Gal3st1‐L3 | Galactosylceramide sulfotransferase‐like 3 | SSC |
| Locus‐47432_24 | JAHLQT010024908.1 | 601,333 | 601,412 | Slc30a1‐L2 | Zinc transporter 1‐like 2 | SSC |
| Locus‐48351_22 | JAHLQT010011632.1 | 4,764,944 | 4,765,023 | Hamer_G000785 | Hypothetical protein | SSC |
| Locus‐7131_15 | JAHLQT010014436.1 | 3,498,782 | 3,498,861 | Hamer_G01616 | Hypothetical protein | SSC |
Abbreviations: GFF gbkey, gene accession code from GFF reference genome file; SSC, sea surface chlorophyll concentration; SSS, sea surface salinity; SST, sea surface temperature.
4. DISCUSSION
4.1. Large‐scale biogeographic structure has a genome wide signature
Both neutral and adaptive genetic variation uncovered a population structure dominated by broad‐scale differentiation between a northern and a southern region. This observation is consistent with previous studies (Benestan et al., 2015; Kenchington et al., 2009), but here, we could precisely locate for the first time the position of this split between 44.67°N and 44.93°N latitude (LFA 32, Nova‐Scotia, Canada), where there is no obvious geographic barrier that could prevent gene flow between the regions. Moreover, this North/South split has also recently been described in four other marine species (Atlantic cod—Gadus morhua, European green crab—Carcinus maenas, Northern shrimp—Pandalus borealis, Sea scallop—Placopecten magellanicus) (Stanley et al., 2018), suggesting this area may represent an important biogeographic transition zone alike the well documented transition zone between the North Sea and Baltic Sea (Wenne et al., 2020). Moreover, we also found that the geographical position of North/South split of lobster populations clearly overlapped the transition range between north and south groups reported by Stanley et al. (2018). Three main hypotheses have been proposed to account for genetic discontinuity in continuously distributed marine species (reviewed by Wares, 2002): (1) ocean circulation causing asymmetric transition zones affecting dispersal patterns, (2) steep cline of selection pressures that strongly affect the distribution of allele frequencies and (3) historical biogeography, vicariance and related variation of ancestral gene flow. Additionally, a fourth hypothesis (4) may to be considered, involving a particular area of lower population density which can be investigated through historical demography from fishery landings data and by modelling historical demography or habitat availability.
Larval connectivity driven by ocean currents is likely to be of critical importance in shaping the population genetic structure of the American lobster. While no biophysical model of larval dispersal has identified an obvious break in ocean circulation between our north and south regions, a recent seascape genomic study revealed that larval connectivity via ocean currents explained 21% of the neutral genetic variation among American lobster from 18 sampling locations across our study domain (Benestan et al., 2016). Importantly, this value almost certainly underestimates, and potentially considerably so, the true contribution of ocean circulation to population structure in this species, given recognized and probably important shortfalls to our current biophysical modelling of larval dispersal (Benestan et al., 2016).
The hypothesis that historical and/or dispersal processes have contributed to shape the discontinuity at this geographic location is supported by the observation that the genetic differentiation between two genetic groups is observed throughout the genome at neutral markers. Yet, neutral processes may not be the only ones at play in such an ecogeographic transition zone. Indeed, our results show that candidate adaptive loci associated with environmental variation better explained the North/South genetic divergence than genetic variation considered putatively neutral. For instance, selective pressures conferring adaptive differences between populations, such as adaptation to freeze conditions or local predation, may potentially lead to immigration resistance through competitive exclusion (Wares, 2002). Hence, natural selection probably contributes to shaping this broad‐scale divergence between these two major lobster genetic groups. In particular, juvenile lobsters inhabiting the northern region live in shallow coastal habitats that are subject to freezing conditions in winter. In this region, post‐larvae settle to the bottom only above the summer thermocline, which separates the “surface water” and the always very cold intermediate layer (Annis, 2005). In the south, juveniles are not exposed to freezing risk and their bathymetric distribution is much more extensive because the thermocline is less steep, or in some regions non‐existent due to tidal mixing (e.g., Bay of Fundy), and deeper water temperature is high enough to allow post‐larvae to settle (Wahle et al., 2013). Interestingly, the North/South division seems to coincide with the historical extension of sea ice along the Scotian Shelf (Hill et al., 2002), which has probably been modified by the closure of the Canso strait in 1952–1955 (McCraken, 1997). Juvenile lobsters may also be exposed to different predators and prey availability in the two regions, including due to the thermal structure of the water column and their bathymetric distribution.
We cannot rule out the possibility that this genetic discontinuity reflect genetic‐environment associations driven by the existence of intrinsic genetic incompatibilities (endogenous barriers) that are revealed when two populations with different genetic backgrounds come into contact and form a tension zone, which can move over space and come to coincide with, and become stabilized by, biogeographic factors (extrinsic barriers) (Barton, 1979; reviewed in Bierne et al., 2011). This can result in the observed spatial genetic discontinuity where the causal factor is an endogenous barrier and the environmental signal results from coupling (Bierne et al., 2011; Shafer & Wolf, 2013). However, in the American lobster, the hypothesis of a putative historical vicariance followed by a secondary contact among two glacial lineages seems unlikely. Instead, this North/South separation may be better explained by a range expansion from south to north following the last Pleistocene glacial event (~10,000 years BP) (Kenchington et al., 2009). Indeed, Kenchington et al. (2009) reported that the marked genetic homogeneity which characterized lobster within the northern area (i.e., Gulf of St. Lawrence and Newfoundland) compared to the southern area (i.e., Gulf of Maine) may suggest a northern‐edge colonization model (Hewitt, 2000). Additionally, several studies in other marine species distributed in the Northwest Atlantic have found evidence of postglacial expansion (Atlantic cod; Gadus morhua, Carr & Marshall, 2008; Wolffish species Anarhichas spp., McCusker & Bentzen, 2010; Atlantic Silverside; Menidia menidia, Lou et al., 2018), although secondary contacts were not always explicitly tested. Our model‐based inferences of past demographic history suggested that North/South populations demographic history mostly support demographic independence in regard to Hasting's criterion of m < 0.1 for demographic independence (Hastings, 1993). Indeed, while the North/South measure of genetic divergence was weak (average F ST = 0.00064), our demographic analysis showed that this threshold was mostly spurred by large effective population size (>120,000) and low migration rate estimations between regions (m ≪ 0.1). Even if the actual Ne was two orders of magnitude less than our approximation, Hasting's criterion to conclude to demographic independence would still be met. Interestingly, the asymmetrical migration rates estimated between North/South populations also support historical population expansion from the southern area to the north as proposed by Kenchington et al. (2009). Overall, these latter results strongly inform that northern and southern lobster populations should be considered as separated units for future management policies of lobster fishery. Nevertheless, note that our demographic analyses were restricted to genetic data from mature females only. Further demographic analyses should be conducted to improve our results by including genetic data from males and other life stages, which may exhibit different features in terms of behaviours and movements (Campbell & Stasko, 1986).
Finally, particular reduction of population density (either due to intensive exploitation or lack of habitat availability) in the middle part of the LFA 32 might contribute to the presence of genetic discontinuity along Nova‐Scotia (N.S.) Atlantic coast. The study conducted by Hudon (1994) reported interesting knowledge about American lobster landings along N.S. coastal areas with relationship to habitat (bottom substrate) and environmental conditions (temperature and wind). While long term (1947–1991) average lobster landings from fisheries showed higher lobster abundance for management units located at both edges of N.S. (i.e., Cape cod, LFA 27 and Lobster bay LFA 34), abundance estimates were homogeneous for all LFAs along the Atlantic coast of N.S. (i.e., LFAs 30–33), which represent 80% of the Atlantic coast of N.S. (~400–500 km). Moreover, Hudon (1994) showed that lobster abundance along Atlantic N.S. was mostly unrelated to the quantity or type of available bottom habitat, and supported the idea that factors other than suitable bottom habitat controlled lobster abundance. Indeed, the author showed that regions with lowest lobster landings are mainly oriented in the axis of dominant southwest winds, which generate coastal upwelling. These environmental features result in thermal instability of water masses (i.e., high year‐to‐year variability), reducing the proportion of degree‐days favourable for lobster movement and egg maturation as well as larval development. It thus seems unlikely that lower density is a main factor explaining the main genetic split between the north and south groups. Moreover, and despite the fact that we cannot rule out this possibility, it seems unlikely that this specific region would correspond to a location of low population density for all five species for which Stanley et al. (2018) documented a transition range between north and south groups.
4.2. Fine‐scale population structure reflect a balance between high gene flow and putatively adaptive differentiation
The apparent lack of population genetic structure based on neutral SNPs within both the north and south regions most likely reflects the occurrence of pronounced gene flow among sampling sites, as proposed by Benestan et al. (2016). Yet, we identified nearly a thousand candidate SNPs that are associated with three environmental variables, and which reveal a finer regional resolution that neutral markers could not detect. High gene flow has long been thought to limit adaptive divergence, even in situations where selection coefficients are large (Lenormand, 2002). Yet, recent experimental evidence convincingly demonstrated that ecological divergence arises rapidly, even under high levels of gene flow (Tusso et al., 2021).
Our results show that adaptive divergence segregates local populations mostly based on variation in salinity and/or chlorophyll concentration within each region (Figure 3). Salinity has been reported to be a critical driver for local adaptation (DeFaveri & Merilä, 2014), population structure (Wenne et al., 2020), and community composition in marine species (Dupont et al., 2014; Pecuchet et al., 2016). For the north region, fine‐scale adaptive population structure is related to the considerable influence of the St. Lawrence Estuary on the range and spatial pattern of our environmental variables in the Gulf of St. Lawrence (see Blais et al., 2019; Le Fouest et al., 2006, including for covarying variables such as nutrient levels). The St. Lawrence Estuary brings considerable amounts of freshwater and nutrients in the southern Gulf of St. Lawrence through the Gaspé current, and this body of water then follows the dominant cyclonic circulation in the Gulf, staying mostly along the southern part of the Gulf before leaving it through the south side of the Cabot Strait (Koutitonsky & Bugden, 1991). This results in a pronounced salinity gradient, which is reinforced by several large rivers that empty in the Baie‐des‐Chaleurs and before the head of the Northumberland Strait (Koutitonsky & Bugden, 1991). Additionally, local wind conditions alongshore coupled with lateral fluctuations of the Gaspé current can drive intense upwelling/downwelling events in the Baie‐des‐Chaleurs, which have been suggested to enhance phytoplankton production through nutrient enrichment in the upper photic layer (Bonardelli et al., 1993; Legendre, 1973). Overall, salinity tended to increase eastward along the southern side of the Gulf, and it was greatest along the northern side of the Gulf, including around Newfoundland, which is markedly less influenced by the St. Lawrence Estuary. Importantly, this dominant cyclonic circulation results in a mostly west‐to‐east “stepping‐stone” pattern of expected larval connectivity among our sampling sites in the southern Gulf of St. Lawrence, and to no expected larval connectivity between these sites and those around Newfoundland (Quinn et al., 2017). This is consistent with the pattern of adaptive genetic‐environment covariance we found in this region. While temperature appeared somewhat less important than either salinity and primary production as possible driver of local adaptation as detected at the SNP level, Dorant et al. (2020) recently showed that copy number variation (CNV) was associated with annual variance in surface temperature, thus also supporting a role of temperature in driving local adaptation of lobster in the north region.
Putatively adaptive genetic variation in the south region mainly separated the Bay of Fundy from the rest of the Gulf of Maine. The unique funnel shape and depth of the Bay of Fundy drive the highest tidal range in the world (i.e., up to 16 m, Thurston, 1990), and cause considerable mixing within the bay. The immense energy of the tides, which is estimated to be ~2000 times greater than the daily flow measured for the Gulf of St. Lawrence (Thurston, 1990), coupled with discharges of three relatively large rivers, support a highly productive ecosystem (note high primary production on Figure 3b) involving a highly diverse food web (Thomas, 1983). Moreover, ocean circulation and tidal mixing cause some discontinuity between the Bay of Fundy and the Gulf of Maine (see Figure 3b and Katavouta et al., 2016). Then, biophysical models of larval dispersal suggest that virtually all lobster post‐larvae that settle in the Bay of Fundy originate from the Bay of Fundy and southwest Nova Scotia, and very few larvae produced in the Bay of Fundy settle in the Gulf of Maine (Quinn et al., 2017). These models also suggest, on the other hand, that there is considerable larval connectivity between southwest Nova Scotia and different parts of the Gulf of Maine via major coastal currents around the Nova Scotia southern peninsula and on the US coast between Maine and Rhodes Island (i.e., Eastern Maine Coastal Current, EMCC and Western Maine Coastal Current, WWCC) (Incze et al., 2010; Xue et al., 2008). Our results indicating adaptive genetic divergence between lobsters in the Bay of Fundy and the Gulf of Maine are thus consistent with our understanding of ocean circulation and larval connectivity in this part of the species' range. That being said, these two “systems” are certainly not fully isolated from one to another and lobster undoubtedly move between them to some extent, both as larvae in the water column and as adults on the seafloor (Campbell & Stasko, 1986), and this connectivity is apparently sufficient to prevent differentiation of neutral genetic markers between them, as discussed earlier.
4.3. Functional roles of adaptive SNPs
The functional significance of the gene–environment associations we observed in the Gulf of St. Lawrence and the Gulf of Maine is not entirely clear, including whether these represent real causal associations. First, GEAs indicated that salinity is a potential evolutionary driver of population differentiation in American lobster. Previous studies found that salinity affects the behaviour and physiology of lobster larvae and adults (Charmantier et al., 2001; Jury et al., 2019; Torres et al., 2021). In our study area, the mean annual salinity values estimated at all our study sites is relatively high (25.6–32.4 ppt) compared to levels that have been shown to be stressful to lobsters in field and laboratory studies (i.e., 20 ppt and lower). Nonetheless, these mean values probably mask lower levels of salinity that occur throughout the year, and which could impact lobster (particularly larvae) even if exposures are of short duration (Torres et al., 2021). It is noteworthy that three SNPs (Locus‐12276_14, Locus‐32916_66 and Locus‐10322_29) associated with salinity were mapped to annotated genes regulating molecular response to salinity stress such as osmoregulation, cuticle constituents and DNA repair mechanisms. For instance, 26S proteasome regulatory subunit 6A‐like protein (gene annotation for marker Locus‐12276_14), referring to a multiprotein complex involved in the ATP‐dependent degradation of ubiquitinated proteins (Kanayama et al., 1992), have been referenced as a major osmoregulatory protein which participate to maintain cellular homeostasis against salinity stress among marine invertebrate species (Mussels, Mytilus trossulus; Vasquez et al., 2020; Mitten crab – Eriocheir sinensis, Malik & Kim, 2021). Moreover, transcriptomic analyses in crustaceans also reported the importance of genes related to structural constituents of the cuticle under various salt conditions (Rocha et al., 2012; Seear et al., 2010). For example, high salinity stress may impact larvae development and moulting cycle with a prolonged state of inter‐moult period (Li et al., 2022). Lastly, salinity stress is also known to causes genotoxicity by DNA alteration or damage leading to downstream errors in DNA replication (Villarino et al., 2014), stress which require for DNA repair mechanisms.
Temperature is widely recognized as a critical abiotic factor driving local adaptation, especially in ectotherm (Cayuela et al., 2022; Narum et al., 2013). While candidate SNPs associated with this variable were not associated with a marked population structure within the Gulf of Maine, we detected a weak pattern of clustering (Figure 3c – PC2 axis) separating areas of rather cold waters (Newfoundland and St. Lawrence Estuary) to areas of warmer waters (i.e., southern part of the Gulf of St. Lawrence). Annotations for candidate SNPs associated to temperature allowed the identification of putative candidate genes and metabolic pathways (e.g., fatty acid metabolism, immunity or DNA repair mechanisms) that are thermo‐dependent in ectotherms. For instance, we found one SNP annotated for putative Zona pellucida‐like domain‐containing protein 3, which is tightly associated with pancreatic zymogen granules membranes (Imamura et al., 2002). In crustaceans, zymogen metabolite is a major macromolecule participating to phenoloxidase (PO) cascade activity, which plays a critical role in crustaceans during physiological responses to stress for immune reactions, cuticle mineralization and wound healing (Kuballa & Elizur, 2008; Söderhäll & Cerenius, 1998; Terwilliger, 1999). Several studies experimentally demonstrated that thermal stress (i.e., both extreme low or high temperatures) influences PO activity in decapods species such as shrimps, crabs and lobsters (Cheng et al., 2003; Gomez‐Jimenez et al., 2000; Matozzo et al., 2011). Furthermore, the candidate SNP annotated for NinaC gene also suggest a signal of thermal adaptation in lobsters. In arthropods, NinaC gene encodes for photoreceptor‐specific polypeptides which participate to mediate the transport of rhabdomeral pigments in eyes (Baumann, 1992; Matsushita & Arikawa, 1996). Although the molecular relationships between NinaC gene activity and temperature variation is still poorly understood, it has been demonstrated that both visual pigment composition and fatty acid composition of cell membranes are affected by temperature (Tsin & Beatty, 1977). Moreover, by investigating temperature‐induced response on eye‐photoreceptor molecules in three Antarctic crustacean species, Tiang (1980) found that temperature elevation (i.e. +10°C) for 7 h affects the structural integrity of rhabdomeral photoreceptor pigments in eyes.
The potential role of local chlorophyll concentration as a selective agent on lobster is similarly difficult to ascertain. Recent GEA analyses have suggested that trophic productivity may be a driver of adaptation in another marine species in the North Atlantic (Cayuela et al., 2020). However, the functional nature of this relation is unclear, and the gene–environment associations documented were based on somewhat more spatially discrete habitats (intertidal beaches used for spawning) than the “open water” sites from which most of our lobster samples were obtained. Indeed, in our study system, spatial linkages between pelagic physical conditions, primary production and the secondary production upon which lobster larvae mainly feed (large zooplankton, Juinio & Cobb, 1992) are complex and vary from year to year (De Lafontaine, 1994). Nevertheless, the associations we observed between chlorophyll concentration and adaptive genetic variation in our North and South study domains are strong and warrant further investigation. Annotations for adaptive SNP associated to SSC were mostly related to diet and growth metabolism pathways such as zinc transporter or lipid metabolism. While zinc is a micronutrient mostly taken from the diet, its deficiency has been related to growth retardation and hypoganadism in humans (Nishi, 1996). Furthermore, studies found that zinc supplementation yielded to largest weight gain in the mitten crab, E. sinensis (Li et al., 2010). Three other adaptive SNPs associated with SSC were related to annotated genes for growth and lipid metabolism proteins (i.e., fatty acid synthase‐like, galactosylceramide sulphotransferase‐like 3, trifunctional enzyme subunit alpha‐like). Overall, the associations between SNP and local chlorophyll concentration suggest that this biotic factor could be involved in microevolutionary processes affecting diet metabolism. Nevertheless, further studies are needed to assess how and at what life stage local variation in trophic conditions may leverage lobster metabolism and its development from larvae to mature stage.
Although our results are consistent with previous RADseq studies documenting an environmentally driven signature of adaptive genetic variation in marine species (Benestan et al., 2016; Cayuela et al., 2020; Stanley et al., 2018), they are also limited by the same constraints usually reported for this approach, in particular the nonexhaustive genome sampling that restrains a full assessment of candidate loci (Lowry et al., 2017) and may affect the accuracy of downstream genome scan analyses (Pavlidis et al., 2012). Additionally, note that genotype‐environment association (GEA) was only investigated through redundancy analysis. This choice was guided by several studies which concluded that RDA was generally more powerful compared to conventional univariate GEA methods (e.g., LFMM, Bayenv), especially for loci that evolved under weak selection or related to polygenic selection (Forester et al., 2018; Laporte et al., 2016). Finally, the low level of RAD tag mapping along the published reference genome (i.e., 13.3%) can be explained by three main factors. First, the published reference genome is incomplete with approximately 28% of missing assembly (note that the whole genome size for this species is expected to be between 3.06 and 4.64 Gbp; Polinski et al., 2021). Second, of the ~72% of complete assembly, 53% corresponds to repeat sequences. Third, to investigate functional annotation of DNA sequences containing our reported adaptive SNPs, we considered highly conservative filters for mapped sequences, namely a mapping score (MapQ) above 30, as well as uniquely mapped sequences (i.e., discarding any repeated sequences along the genome). This latter filtering requirement was chosen as we specifically identified and discarded putative repeated sequences using the approach for paralogy detection published by McKinney et al. (2017) and Dorant et al. (2020). As a result, approximately 33% of the lobster assembled genome, which correspond to nonduplicated DNA, could really considered for mapping and gene annotations check.
Further developments, especially in lobster genome research combined with functional validation and experimental studies will be required to provide a more detailed interpretation pertaining to the functional role of the putatively adaptive candidate loci we identified. For instance, the use of transcriptomic approaches to assess physiological and metabolic responses locally adapted populations to various environmental conditions may be helpful to biologically measure how gene–environment associations modulate individual fitness under contrasted environmental conditions in terms of salinity, temperature or trophic factors.
5. CONCLUSION
While delineating biologically meaningful population units in species showing high levels of gene flow most often represents a very challenging endeavour, our study indicates that weak population structure in high gene flow systems can be resolved at various spatial scales. Toward this goal, we showed that putative adaptive markers represent an untapped source of genetic variation capable of informing about fine‐scale population structure despite weak global genetic differentiation, as previously proposed by Gagnaire et al. (2015). As such, with the increasing availability of extended genomic data, our study, along with others, demonstrates the value of including adaptive loci, whether alone or in conjunction with neutral loci, for population genetic studies.
AUTHOR CONTRIBUTIONS
Louis Bernatchez and Rémy Rochette designed and planned the project. Yann Dorant, Martin Laporte, Quentin Rougemont, and Hugo Cayuela performed research. Yann Dorant analysed the data. Quentin Rougemont performed demographic analyses. Yann Dorant, Martin Laporte, Quentin Rougemont, Hugo Cayuela, Rémy Rochette and Louis Bernatchez wrote the manuscript.
CONFLICT OF INTEREST
The authors declare no competing interest.
Supporting information
Appendix S1
Table S1
ACKNOWLEDGEMENTS
This research is part of the ‘Lobster Node’ of the NSERC Canadian Fisheries Research Network (CFRN). We aim to foremost thank lobster fishers and their association leads for their implication in various axes of this work, such as project design, field work and sampling. We also thank other external collaborators for their contribution to our sampling efforts in the USA: W. Watson, J. Carloni, T. Pugh and L. Dellinger. Project design and work plan were developed in collaboration with scientists from the Department of Fisheries and Oceans (B. Sainte‐Marie, J. Chassé, M. Comeau, J. Tremblay) and the PEI Department of Fisheries and Communities (R. MacMillan), representatives of fishermen associations and the facilitator of the Lobster Node, M. Allain. We would like to thank A. Boudreau, V. Brzeski, Y. Carignan, Clearwater, B. Comeau, M. Comeau, J. P. Allard, M. Deraspe, N. Davis, S. Delorey, C. Denton, R. Doucelle, J. Grignon, M. Haarr, R. MacMillan, G. Paulin and M. Thériault who helped to collect the samples. We are grateful to A. Devault and the Arbor Biosciences team for DNA probes synthesis and methodological advice. We also thank the personal of the IBIS sequencing platform for their assistance in developing the Rapture assay for highly multiplexed configuration. Finally, we warmly thank B Ste‐Marie and C. Mérot for their constructive comments and advice to improve the manuscript. We are also grateful to L. Rieseberg and two anonymous reviewers for their constructive comments on a previous version of this manuscript. This research was funded by a Strategic Partnership Grants for Projects from Natural Sciences and Engineering Research Council of Canada to LB and RR. This research is a contribution to the research programme of Resources Aquatiques Québec (RAQ).
Dorant, Y. , Laporte, M. , Rougemont, Q. , Cayuela, H. , Rochette, R. , & Bernatchez, L. (2022). Landscape genomics of the American lobster (Homarus americanus). Molecular Ecology, 31, 5182–5200. 10.1111/mec.16653
Handling Editor: Loren Rieseberg
Contributor Information
Yann Dorant, Email: y.dorant@gmail.com.
Rémy Rochette, Email: rochette@unb.ca.
Louis Bernatchez, Email: louis.bernatchez@bio.ulaval.ca.
DATA AVAILABILITY STATEMENT
Demultiplexed raw reads (FASTQ format) generated for this study have been deposited in the National Center for Biotechnology Information Sequence Read Archive database (NCBI) under BioProject accession no. PRJNA862675, PRJNA645159 and PRJNA645211. Sampling data information are available in Table S1.
REFERENCES
- Alexander, D. H. , Novembre, J. , & Lange, K. (2009). Fast model‐based estimation of ancestry in unrelated individuals. Genome Research, 19(9), 1655–1664. 10.1101/gr.094052.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali, O. A. , O'Rourke, S. M. , Amish, S. J. , Meek, M. H. , Luikart, G. , Jeffres, C. , & Miller, M. R. (2016). RAD capture (Rapture): Flexible and efficient sequence‐based genotyping. Genetics, 202(2), 389–400. 10.1534/genetics.115.183665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annis, E. R. (2005). Temperature effects on the vertical distribution of lobster postlarvae (Homarus americanus). Limnology and Oceanography, 50(6), 1972–1982. 10.4319/lo.2005.50.6.1972 [DOI] [Google Scholar]
- Assis, J. , Tyberghein, L. , Bosch, S. , Verbruggen, H. , Serrão, E. A. , & Clerck, O. D. (2017). Bio‐ORACLE v2.0: Extending marine data layers for bioclimatic modelling. Global Ecology and Biogeography, 27(3), 277–284. 10.1111/geb.12693 [DOI] [Google Scholar]
- Attard, C. R. M. , Beheregaray, L. B. , Sandoval‐Castillo, J. , Jenner, K. C. S. , Gill, P. C. , Jenner, M.‐N. M. , Morrice, M. G. , & Möller, L. M. (2018). From conservation genetics to conservation genomics: A genome‐wide assessment of blue whales (Balaenoptera musculus) in Australian feeding aggregations. Royal Society Open Science, 5(1), 170925. 10.1098/rsos.170925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, N. , & Bengtsson, B. O. (1986). The barrier to genetic exchange between hybridising populations. Heredity, 57(3), 357–376. 10.1038/hdy.1986.135 [DOI] [PubMed] [Google Scholar]
- Barton, N. H. (1979). The dynamics of hybrid zones. Heredity, 43(3), 341–359. 10.1038/hdy.1979.87 [DOI] [Google Scholar]
- Batista, P. D. , Janes, J. K. , Boone, C. K. , Murray, B. W. , & Sperling, F. A. H. (2016). Adaptive and neutral markers both show continent‐wide population structure of mountain pine beetle (Dendroctonus ponderosae). Ecology and Evolution, 6(17), 6292–6300. 10.1002/ece3.2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann, O. (1992). Structural interactions of actin filaments and endoplasmic reticulum in honeybee photoreceptor cells. Cell and Tissue Research, 268(1), 71–79. 10.1007/BF00338055 [DOI] [PubMed] [Google Scholar]
- Béné, C. , Arthur, R. , Norbury, H. , Allison, E. H. , Beveridge, M. , Bush, S. , Campling, L. , Leschen, W. , Little, D. , Squires, D. , Thilsted, S. H. , Troell, M. , & Williams, M. (2016). Contribution of fisheries and aquaculture to food security and poverty reduction: Assessing the current evidence. World Development, 79, 177–196. 10.1016/j.worlddev.2015.11.007 [DOI] [Google Scholar]
- Benestan, L. , Gosselin, T. , Perrier, C. , Sainte‐Marie, B. , Rochette, R. , & Bernatchez, L. (2015). RAD genotyping reveals fine‐scale genetic structuring and provides powerful population assignment in a widely distributed marine species, the American lobster (Homarus americanus). Molecular Ecology, 24(13), 3299–3315. 10.1111/mec.13245 [DOI] [PubMed] [Google Scholar]
- Benestan, L. , Quinn, B. K. , Maaroufi, H. , Laporte, M. , Clark, F. K. , Greenwood, S. J. , Rochette, R. , & Bernatchez, L. (2016). Seascape genomics provides evidence for thermal adaptation and current‐mediated population structure in American lobster (Homarus americanus). Molecular Ecology, 25(20), 5073–5092. 10.1111/mec.13811 [DOI] [PubMed] [Google Scholar]
- Bernatchez, L. (2016). On the maintenance of genetic variation and adaptation to environmental change: Considerations from population genomics in fishes. Journal of Fish Biology, 89(6), 2519–2556. 10.1111/jfb.13145 [DOI] [PubMed] [Google Scholar]
- Bernatchez, L. , Wellenreuther, M. , Araneda, C. , Ashton, D. T. , Barth, J. M. I. , Beacham, T. D. , Maes, G. E. , Martinsohn, J. T. , Miller, K. M. , Naish, K. A. , Ovenden, J. R. , Primmer, C. R. , Suk, H. Y. , Therkildsen, N. O. , & Withler, R. E. (2017). Harnessing the power of genomics to secure the future of seafood. Trends in Ecology & Evolution, 32(9), 665–680. 10.1016/j.tree.2017.06.010 [DOI] [PubMed] [Google Scholar]
- Bierne, N. , Welch, J. , Loire, E. , Bonhomme, F. , & David, P. (2011). The coupling hypothesis: Why genome scans may fail to map local adaptation genes. Molecular Ecology, 20(10), 2044–2072. 10.1111/j.1365-294X.2011.05080.x [DOI] [PubMed] [Google Scholar]
- Blais, M. , Galbraith, P. S. , Plourde, S. , Scarratt, M. , Devine, L. , & Lehoux, C. (2019). Chemical and biological oceanographic conditions in the Estuary and Gulf of St. Lawrence during 2017 . 61.
- Bonardelli, J. C. , Himmelman, J. H. , & Drinkwater, K. (1993). Current variability and upwelling along the north shore of Baie des Chaleurs. Atmosphere‐Ocean, 31(4), 541–565. 10.1080/07055900.1993.9649485 [DOI] [Google Scholar]
- Campbell, A. , & Stasko, A. B. (1986). Movements of lobsters (Homarus americanus) tagged in the Bay of Fundy, Canada. Marine Biology, 92(3), 393–404. 10.1007/BF00392680 [DOI] [Google Scholar]
- Carr, S. M. , & Marshall, H. D. (2008). Intraspecific phylogeographic genomics from multiple complete mtDNA genomes in Atlantic cod (Gadus morhua): Origins of the “Codmother,” transatlantic vicariance and midglacial population expansion. Genetics, 180(1), 381–389. 10.1534/genetics.108.089730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. , Hohenlohe, P. A. , Bassham, S. , Amores, A. , & Cresko, W. A. (2013). Stacks: An analysis tool set for population genomics. Molecular Ecology, 22(11), 3124–3140. 10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayuela, H. , Dorant, Y. , Forester, B. R. , Jeffries, D. L. , Mccaffery, R. M. , Eby, L. A. , Hossack, B. R. , Gippet, J. M. W. , Pilliod, D. S. , & Chris Funk, W. (2022). Genomic signatures of thermal adaptation are associated with clinal shifts of life history in a broadly distributed frog. Journal of Animal Ecology, 91, 1222–1238. 10.1111/1365-2656.13545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayuela, H. , Rougemont, Q. , Laporte, M. , Mérot, C. , Normandeau, E. , Dorant, Y. , Tørresen, O. K. , Hoff, S. N. K. , Jentoft, S. , Sirois, P. , Castonguay, M. , Jansen, T. , Praebel, K. , Clément, M. , & Bernatchez, L. (2020). Shared ancestral polymorphisms and chromosomal rearrangements as potential drivers of local adaptation in a marine fish. Molecular Ecology, 29(13), 2379–2398. 10.1111/mec.15499 [DOI] [PubMed] [Google Scholar]
- Charmantier, G. , Haond, C. , Lignot, J. , & Charmantier‐Daures, M. (2001). Ecophysiological adaptation to salinity throughout a life cycle: A review in homarid lobsters. Journal of Experimental Biology, 204(5), 967–977. [DOI] [PubMed] [Google Scholar]
- Cheng, W. , Juang, F.‐M. , Li, J.‐T. , Lin, M.‐C. , Liu, C.‐H. , & Chen, J.‐C. (2003). The immune response of the giant freshwater prawn Macrobrachium rosenbergii and its susceptibility to Lactococcus garvieae in relation to the moult stage. Aquaculture, 218(1), 33–45. 10.1016/S0044-8486(02)00415-5 [DOI] [Google Scholar]
- D'Aloia, C. C. , Andrés, J. A. , Bogdanowicz, S. M. , McCune, A. R. , Harrison, R. G. , & Buston, P. M. (2020). Unraveling hierarchical genetic structure in a marine metapopulation: A comparison of three high‐throughput genotyping approaches. Molecular Ecology, 29(12), 2189–2203. 10.1111/mec.15405 [DOI] [PubMed] [Google Scholar]
- De Lafontaine, Y. D. (1994). Zooplankton biomass in the Southern Gulf of St. Lawrence: Spatial patterns and the influence of freshwater runoff. Canadian Journal of Fisheries and Aquatic Sciences, 51, 617–635. 10.1139/f94-063 [DOI] [Google Scholar]
- DeFaveri, J. , & Merilä, J. (2014). Local adaptation to salinity in the three‐spined stickleback? Journal of Evolutionary Biology, 27(2), 290–302. 10.1111/jeb.12289 [DOI] [PubMed] [Google Scholar]
- Dorant, Y. , Benestan, L. , Rougemont, Q. , Normandeau, E. , Boyle, B. , Rochette, R. , & Bernatchez, L. (2019). Comparing Pool‐seq, Rapture, and GBS genotyping for inferring weak population structure: The American lobster (Homarus americanus) as a case study. Ecology and Evolution, 9(11), 6606–6623. 10.1002/ece3.5240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorant, Y. , Cayuela, H. , Wellband, K. , Laporte, M. , Rougemont, Q. , Mérot, C. , Normandeau, E. , Rochette, R. , & Bernatchez, L. (2020). Copy number variants outperform SNPs to reveal genotype–temperature association in a marine species. Molecular Ecology, 29, 4765–4782. 10.1111/mec.15565 [DOI] [PubMed] [Google Scholar]
- Dupont, C. L. , Larsson, J. , Yooseph, S. , Ininbergs, K. , Goll, J. , Asplund‐Samuelsson, J. , McCrow, J. P. , Celepli, N. , Allen, L. Z. , Ekman, M. , Lucas, A. J. , Hagström, Å. , Thiagarajan, M. , Brindefalk, B. , Richter, A. R. , Andersson, A. F. , Tenney, A. , Lundin, D. , Tovchigrechko, A. , … Bergman, B. (2014). Functional tradeoffs underpin salinity‐driven divergence in microbial community composition. PLoS One, 9(2), e89549. 10.1371/journal.pone.0089549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forester, B. R. , Lasky, J. R. , Wagner, H. H. , & Urban, D. L. (2018). Comparing methods for detecting multilocus adaptation with multivariate genotype–environment associations. Molecular Ecology, 27(9), 2215–2233. 10.1111/mec.14584 [DOI] [PubMed] [Google Scholar]
- Funk, W. C. , McKay, J. K. , Hohenlohe, P. A. , & Allendorf, F. W. (2012). Harnessing genomics for delineating conservation units. Trends in Ecology & Evolution, 27(9), 489–496. 10.1016/j.tree.2012.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnaire, P.‐A. , Broquet, T. , Aurelle, D. , Viard, F. , Souissi, A. , Bonhomme, F. , Arnaud‐Haond, S. , & Bierne, N. (2015). Using neutral, selected, and hitchhiker loci to assess connectivity of marine populations in the genomic era. Evolutionary Applications, 8(8), 769–786. 10.1111/eva.12288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Jimenez, S. , Uglow, R. F. , & Gollas‐Galvan, T. (2000). The effects of cooling and emersion on total haemocyte count and phenoloxidase activity of the spiny lobster Panulirus interruptus . Fish & Shellfish Immunology, 10(7), 631–635. 10.1006/fsim.2000.0277 [DOI] [PubMed] [Google Scholar]
- Guillen, J. , Natale, F. , Carvalho, N. , Casey, J. , Hofherr, J. , Druon, J.‐N. , Fiore, G. , Gibin, M. , Zanzi, A. , & Martinsohn, J. T. (2019). Global seafood consumption footprint. Ambio, 48(2), 111–122. 10.1007/s13280-018-1060-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst, R. N. , Hernandez, R. D. , Williamson, S. H. , & Bustamante, C. D. (2009). Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genetics, 5(10), e1000695. 10.1371/journal.pgen.1000695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings, A. (1993). Complex interactions between dispersal and dynamics: Lessons from coupled logistic equations. Ecology, 74(5), 1362–1372. 10.2307/1940066 [DOI] [Google Scholar]
- Hawkins, S. J. , Bohn, K. , Sims, D. W. , Ribeiro, P. , Faria, J. , Presa, P. , Pita, A. , Martins, G. M. , Neto, A. I. , Burrows, M. T. , & Genner, M. J. (2016). Fisheries stocks from an ecological perspective: Disentangling ecological connectivity from genetic interchange. Fisheries Research, 179, 333–341. 10.1016/j.fishres.2016.01.015 [DOI] [Google Scholar]
- Hewitt, G. (2000). The genetic legacy of the Quaternary ice ages. Nature, 405(6789), 907–913. 10.1038/35016000 [DOI] [PubMed] [Google Scholar]
- Hill, B. T. , Ruffman, A. , & Drinkwater, K. (2002). Historical record of the incidence of sea ice on the Scotian Shelf and the Gulf of St. Lawrence . 10.
- Hohenlohe, P. A. , Funk, W. C. , & Rajora, O. P. (2021). Population genomics for wildlife conservation and management. Molecular Ecology, 30(1), 62–82. 10.1111/mec.15720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudon, C. (1994). Large‐scale analysis of Atlantic Nova Scotia American lobster (Homarus americanus) landings with respect to habitat, temperature, and wind conditions. Canadian Journal of Fisheries and Aquatic Sciences, 51(6), 1308–1321. 10.1139/f94-130 [DOI] [Google Scholar]
- Imamura, T. , Asada, M. , Vogt, S. K. , Rudnick, D. A. , Lowe, M. E. , & Muglia, L. J. (2002). Protection from pancreatitis by the zymogen granule membrane protein integral membrane‐associated protein‐1. Journal of Biological Chemistry, 277(52), 50725–50733. 10.1074/jbc.M204159200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incze, L. , Xue, H. , Wolff, N. , Xu, D. , Wilson, C. , Steneck, R. , Wahle, R. , Lawton, P. , Pettigrew, N. , & Chen, Y. (2010). Connectivity of lobster (Homarus americanus) populations in the coastal Gulf of Maine: Part II. Coupled biophysical dynamics. Fisheries Oceanography, 19(1), 1–20. 10.1111/j.1365-2419.2009.00522.x [DOI] [Google Scholar]
- Juinio, M. , & Cobb, J. (1992). Natural diet and feeding habits of the postlarval lobster Homarus americanus . Marine Ecology Progress Series, 85, 83–91. 10.3354/meps085083 [DOI] [Google Scholar]
- Jury, S. H. , Pugh, T. L. , Henninger, H. , Carloni, J. T. , & Watson, W. H. (2019). Patterns and possible causes of skewed sex ratios in American lobster (Homarus americanus) populations. Invertebrate Reproduction & Development, 63(3), 189–199. 10.1080/07924259.2019.1595184 [DOI] [Google Scholar]
- Kanayama, H. , Tamura, T. , Ugai, S. , Kagawa, S. , Tanahashi, N. , Yoshimura, T. , Tanaka, K. , & Ichihara, A. (1992). Demonstration that a human 26S proteolytic complex consists of a proteasome and multiple associated protein components and hydrolyzes ATP and ubiquitin‐ligated proteins by closely linked mechanisms. European Journal of Biochemistry, 206(2), 567–578. 10.1111/j.1432-1033.1992.tb16961.x [DOI] [PubMed] [Google Scholar]
- Katavouta, A. , Thompson, K. R. , Lu, Y. , & Loder, J. W. (2016). Interaction between the tidal and seasonal variability of the Gulf of Maine and Scotian Shelf Region. Journal of Physical Oceanography, 46(11), 3279–3298. 10.1175/JPO-D-15-0091.1 [DOI] [Google Scholar]
- Keller, S. R. , Chhatre, V. E. , & Fitzpatrick, M. C. (2018). Influence of range position on locally adaptive gene–environment associations in populus flowering time genes. Journal of Heredity, 109(1), 47–58. 10.1093/jhered/esx098 [DOI] [PubMed] [Google Scholar]
- Kenchington, E. L. , Harding, G. C. , Jones, M. W. , & Prodöhl, P. A. (2009). Pleistocene glaciation events shape genetic structure across the range of the American lobster, Homarus americanus . Molecular Ecology, 18(8), 1654–1667. 10.1111/j.1365-294X.2009.04118.x [DOI] [PubMed] [Google Scholar]
- Koutitonsky, V. G. , & Bugden, G. L. (1991). The physical oceanography of the Gulf of St. Lawrence: A review with emphasis on the synoptic variability of the motion. In The Gulf of St. Lawrence: Small ocean or big estuary? (Vol. 113, pp. 57–90). Canadian Special Publication of Fisheries and Aquatic Sciences. [Google Scholar]
- Kuballa, A. V. , & Elizur, A. (2008). Differential expression profiling of components associated with exoskeletal hardening in crustaceans. BMC Genomics, 9(1), 575. 10.1186/1471-2164-9-575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landguth, E. L. , & Balkenhol, N. (2012). Relative sensitivity of neutral versus adaptive genetic data for assessing population differentiation. Conservation Genetics, 13(5), 1421–1426. 10.1007/s10592-012-0354-x [DOI] [Google Scholar]
- Laporte, M. , Pavey, S. A. , Rougeux, C. , Pierron, F. , Lauzent, M. , Budzinski, H. , Labadie, P. , Geneste, E. , Couture, P. , Baudrimont, M. , & Bernatchez, L. (2016). RAD sequencing reveals within‐generation polygenic selection in response to anthropogenic organic and metal contamination in North Atlantic eels. Molecular Ecology, 25, 219–237. 10.1111/mec.13466 [DOI] [PubMed] [Google Scholar]
- Le Fouest, V. , Zakardjian, B. , Saucier, F. J. , & Çizmeli, S. A. (2006). Application of SeaWIFS‐ and AVHRR‐derived data for mesoscale and regional validation of a 3‐D high‐resolution physical–biological model of the Gulf of St. Lawrence (Canada). Journal of Marine Systems, 60(1), 30–50. 10.1016/j.jmarsys.2005.11.008 [DOI] [Google Scholar]
- Legendre, L. (1973). Phytoplankton organization in Baie des Chaleurs (Gulf of St Lawrence). Journal of Ecology, 61(1), 135–149. 10.2307/2258923 [DOI] [Google Scholar]
- Lehnert, S. J. , DiBacco, C. , Wyngaarden, M. V. , Jeffery, N. W. , Lowen, J. B. , Sylvester, E. V. A. , Wringe, B. F. , Stanley, R. R. E. , Hamilton, L. C. , & Bradbury, I. R. (2019). Fine‐scale temperature‐associated genetic structure between inshore and offshore populations of sea scallop (Placopecten magellanicus). Heredity, 122(1), 69–80. 10.1038/s41437-018-0087-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenormand, T. (2002). Gene flow and the limits to natural selection. Trends in Ecology & Evolution, 17(4), 183–189. 10.1016/S0169-5347(02)02497-7 [DOI] [Google Scholar]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. ArXiv:1303.3997 [q‐Bio]. http://arxiv.org/abs/1303.3997
- Li, W. , Li, S. , Wang, X. , Chen, H. , Hao, H. , & Wang, K.‐J. (2022). Internal carbohydrates and lipids as reserved energy supply in the pubertal molt of Scylla paramamosain . Aquaculture, 549, 737736. 10.1016/j.aquaculture.2021.737736 [DOI] [Google Scholar]
- Li, W.‐W. , Jin, X.‐K. , He, L. , Jiang, H. , Gong, Y.‐N. , Xie, Y.‐N. , & Wang, Q. (2010). Molecular cloning, characterization, expression and activity analysis of cathepsin L in Chinese mitten crab, Eriocheir sinensis . Fish & Shellfish Immunology, 29(6), 1010–1018. 10.1016/j.fsi.2010.08.007 [DOI] [PubMed] [Google Scholar]
- Linck, E. , & Battey, C. J. (2019). Minor allele frequency thresholds strongly affect population structure inference with genomic data sets. Molecular Ecology Resources, 19(3), 639–647. 10.1111/1755-0998.12995 [DOI] [PubMed] [Google Scholar]
- Lou, R. N. , Fletcher, N. K. , Wilder, A. P. , Conover, D. O. , Therkildsen, N. O. , & Searle, J. B. (2018). Full mitochondrial genome sequences reveal new insights about post‐glacial expansion and regional phylogeographic structure in the Atlantic silverside (Menidia menidia). Marine Biology, 165(8), 124. 10.1007/s00227-018-3380-5 [DOI] [Google Scholar]
- Lowry, D. B. , Hoban, S. , Kelley, J. L. , Lotterhos, K. E. , Reed, L. K. , Antolin, M. F. , & Storfer, A. (2017). Responsible RAD: Striving for best practices in population genomic studies of adaptation. Molecular Ecology Resources, 17(3), 366–369. 10.1111/1755-0998.12677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart, G. , England, P. R. , Tallmon, D. , Jordan, S. , & Taberlet, P. (2003). The power and promise of population genomics: From genotyping to genome typing. Nature Reviews Genetics, 4(12), 981–994. 10.1038/nrg1226 [DOI] [PubMed] [Google Scholar]
- Malik, A. , & Kim, C.‐B. (2021). Role of transportome in the gills of Chinese mitten crabs in response to salinity change: A meta‐analysis of RNA‐seq datasets. Biology, 10(1), 39. 10.3390/biology10010039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.Journal, 17(1), 10–12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Matozzo, V. , Gallo, C. , & Marin, M. G. (2011). Effects of temperature on cellular and biochemical parameters in the crab Carcinus aestuarii (Crustacea, Decapoda). Marine Environmental Research, 71(5), 351–356. 10.1016/j.marenvres.2011.04.001 [DOI] [PubMed] [Google Scholar]
- Matsushita, A. , & Arikawa, K. (1996). Disruption of actin filament organization by cytochalasin D inhibits rhabdom synthesis in the compound eye of the crab Hemigrapsus sanguineus . Cell and Tissue Research, 286(1), 167–174. 10.1007/s004410050685 [DOI] [Google Scholar]
- McCauley, D. J. , Pinsky, M. L. , Palumbi, S. R. , Estes, J. A. , Joyce, F. H. , & Warner, R. R. (2015). Marine defaunation: Animal loss in the global ocean. Science, 347(6219), 1255641. 10.1126/science.1255641 [DOI] [PubMed] [Google Scholar]
- McCraken, F. D. (1997). Canso marine environment workshop part 1 of 4 parts, executive summary (No. 834; p. vii + 17).
- McCusker, M. R. , & Bentzen, P. (2010). Historical influences dominate the population genetic structure of a sedentary marine fish, Atlantic wolffish (Anarhichas lupus), across the North Atlantic Ocean. Molecular Ecology, 19(19), 4228–4241. 10.1111/j.1365-294X.2010.04806.x [DOI] [PubMed] [Google Scholar]
- McIntyre, P. B. , Liermann, C. A. R. , & Revenga, C. (2016). Linking freshwater fishery management to global food security and biodiversity conservation. Proceedings of the National Academy of Sciences of the United States of America, 113(45), 12880–12885. 10.1073/pnas.1521540113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney, G. J. , Waples, R. K. , Seeb, L. W. , & Seeb, J. E. (2017). Paralogs are revealed by proportion of heterozygotes and deviations in read ratios in genotyping‐by‐sequencing data from natural populations. Molecular Ecology Resources, 17(4), 656–669. 10.1111/1755-0998.12613 [DOI] [PubMed] [Google Scholar]
- Milano, I. , Babbucci, M. , Cariani, A. , Atanassova, M. , Bekkevold, D. , Carvalho, G. R. , Espiñeira, M. , Fiorentino, F. , Garofalo, G. , Geffen, A. J. , Hansen, J. H. , Helyar, S. J. , Nielsen, E. E. , Ogden, R. , Patarnello, T. , Stagioni, M. , Tinti, F. , & Bargelloni, L. (2014). Outlier SNP markers reveal fine‐scale genetic structuring across European hake populations (Merluccius merluccius). Molecular Ecology, 23(1), 118–135. 10.1111/mec.12568 [DOI] [PubMed] [Google Scholar]
- Narum, S. R. , Campbell, N. R. , Meyer, K. A. , Miller, M. R. , & Hardy, R. W. (2013). Thermal adaptation and acclimation of ectotherms from differing aquatic climates. Molecular Ecology, 22(11), 3090–3097. 10.1111/mec.12240 [DOI] [PubMed] [Google Scholar]
- Nishi, Y. (1996). Zinc and growth. Journal of the American College of Nutrition, 15(4), 340–344. 10.1080/07315724.1996.10718608 [DOI] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , & Wagner, H. (2018). Vegan: Ecological diversity R package, version 2.5‐4.
- Ormerod, S. J. (2003). Current issues with fish and fisheries: Editor's overview and introduction. Journal of Applied Ecology, 40(2), 204–213. 10.1046/j.1365-2664.2003.00824.x [DOI] [Google Scholar]
- Palumbi, S. R. (2003). Population genetics, demographic connectivity, and the design of marine reserves. Ecological Applications, 13(sp1), 146–158. 10.1890/1051-0761(2003)013[0146:PGDCAT]2.0.CO;2 [DOI] [Google Scholar]
- Pauly, D. , Christensen, V. , Guénette, S. , Pitcher, T. J. , Sumaila, U. R. , Walters, C. J. , Watson, R. , & Zeller, D. (2002). Towards sustainability in world fisheries. Nature, 418(6898), 689–695. 10.1038/nature01017 [DOI] [PubMed] [Google Scholar]
- Pavlidis, P. , Jensen, J. D. , Stephan, W. , & Stamatakis, A. (2012). A critical assessment of storytelling: Gene ontology categories and the importance of validating genomic scans. Molecular Biology and Evolution, 29(10), 3237–3248. 10.1093/molbev/mss136 [DOI] [PubMed] [Google Scholar]
- Pecuchet, L. , Törnroos, A. , & Lindegren, M. (2016). Patterns and drivers of fish community assembly in a large marine ecosystem. Marine Ecology Progress Series, 546, 239–248. 10.3354/meps11613 [DOI] [Google Scholar]
- Pembleton, L. W. , Cogan, N. O. I. , & Forster, J. W. (2013). StAMPP: An R package for calculation of genetic differentiation and structure of mixed‐ploidy level populations. Molecular Ecology Resources, 13(5), 946–952. 10.1111/1755-0998.12129 [DOI] [PubMed] [Google Scholar]
- Pita, A. , Casey, J. , Hawkins, S. J. , Villarreal, M. R. , Gutiérrez, M.‐J. , Cabral, H. , Carocci, F. , Abaunza, P. , Pascual, S. , & Presa, P. (2016). Conceptual and practical advances in fish stock delineation. Fisheries Research, 173, 185–193. 10.1016/j.fishres.2015.10.029 [DOI] [Google Scholar]
- Polinski, J. M. , Zimin, A. V. , Clark, K. F. , Kohn, A. B. , Sadowski, N. , Timp, W. , Ptitsyn, A. , Khanna, P. , Romanova, D. Y. , Williams, P. , Greenwood, S. J. , Moroz, L. L. , Walt, D. R. , & Bodnar, A. G. (2021). The American lobster genome reveals insights on longevity, neural, and immune adaptations. Science Advances, 7(26), eabe8290. 10.1126/sciadv.abe8290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn, B. K. , Chassé, J. , & Rochette, R. (2017). Potential connectivity among American lobster fisheries as a result of larval drift across the species' range in eastern North America. Canadian Journal of Fisheries and Aquatic Sciences, 74(10), 1549–1563. 10.1139/cjfas-2016-0416 [DOI] [Google Scholar]
- Rocha, J. , Garcia‐Carreño, F. L. , Muhlia‐Almazán, A. , Peregrino‐Uriarte, A. B. , Yépiz‐Plascencia, G. , & Córdova‐Murueta, J. H. (2012). Cuticular chitin synthase and chitinase mRNA of whiteleg shrimp Litopenaeus vannamei during the molting cycle. Aquaculture, 330–333, 111–115. 10.1016/j.aquaculture.2011.12.024 [DOI] [Google Scholar]
- Seear, P. J. , Tarling, G. A. , Burns, G. , Goodall‐Copestake, W. P. , Gaten, E. , Özkaya, Ö. , & Rosato, E. (2010). Differential gene expression during the moult cycle of Antarctic krill (Euphausia superba). BMC Genomics, 11(1), 582. 10.1186/1471-2164-11-582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafer, A. B. A. , & Wolf, J. B. W. (2013). Widespread evidence for incipient ecological speciation: A meta‐analysis of isolation‐by‐ecology. Ecology Letters, 16(7), 940–950. 10.1111/ele.12120 [DOI] [PubMed] [Google Scholar]
- Smit, A. J. , Roberts, M. , Anderson, R. J. , Dufois, F. , Dudley, S. F. J. , Bornman, T. G. , Olbers, J. , & Bolton, J. J. (2013). A coastal seawater temperature dataset for biogeographical studies: Large biases between in situ and remotely‐sensed data sets around the coast of South Africa. PLoS One, 8(12), e81944. 10.1371/journal.pone.0081944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderhäll, K. , & Cerenius, L. (1998). Role of the prophenoloxidase‐activating system in invertebrate immunity. Current Opinion in Immunology, 10(1), 23–28. 10.1016/S0952-7915(98)80026-5 [DOI] [PubMed] [Google Scholar]
- Stanley, R. R. E. , DiBacco, C. , Lowen, B. , Beiko, R. G. , Jeffery, N. W. , Wyngaarden, M. V. , Bentzen, P. , Brickman, D. , Benestan, L. , Bernatchez, L. , Johnson, C. , Snelgrove, P. V. R. , Wang, Z. , Wringe, B. F. , & Bradbury, I. R. (2018). A climate‐associated multispecies cryptic cline in the northwest Atlantic. Science Advances, 4(3), eaaq0929. 10.1126/sciadv.aaq0929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshima, H. , Iguchi, K. , Hashiguchi, Y. , & Nishida, M. (2016). Using dense locality sampling resolves the subtle genetic population structure of the dispersive fish species Plecoglossus altivelis . Molecular Ecology, 25(13), 3048–3064. 10.1111/mec.13650 [DOI] [PubMed] [Google Scholar]
- Terwilliger, N. B. (1999). Hemolymph proteins and molting in crustaceans and insects. American Zoologist, 39(3), 589–599. 10.1093/icb/39.3.589 [DOI] [Google Scholar]
- Thomas, M. L. H. (Eds.) (1983). Marine and coastal systems of the Quoddy region, New Brunswick. Department of Fisheries and Oceans. [Google Scholar]
- Thurston, H. (1990). Tidal life, a natural history of the Bay of Fundy. Camden House Publishing. [Google Scholar]
- Tiang, K. M. (1980). Structure, light & temperature induced changes in the compound eyes of Antarctic crustaceans [Thesis, Master of Science (MSc), The University of Waikato, Hamilton, New Zealand]. https://hdl.handle.net/10289/12864
- Tine, M. , Kuhl, H. , Gagnaire, P.‐A. , Louro, B. , Desmarais, E. , Martins, R. S. T. , Hecht, J. , Knaust, F. , Belkhir, K. , Klages, S. , Dieterich, R. , Stueber, K. , Piferrer, F. , Guinand, B. , Bierne, N. , Volckaert, F. A. M. , Bargelloni, L. , Power, D. M. , Bonhomme, F. , … Reinhardt, R. (2014). European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nature Communications, 5(1), 5770. 10.1038/ncomms6770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres, G. , Anger, K. , & Giménez, L. (2021). Effects of short‐term and continuous exposure to reduced salinities on the biochemical composition of larval lobster, Homarus gammarus . Zoology, 144, 125885. 10.1016/j.zool.2020.125885 [DOI] [PubMed] [Google Scholar]
- Tran Lu Y, A. , Ruault, S. , Daguin‐Thiébaut, C. , Castel, J. , Bierne, N. , Broquet, T. , Wincker, P. , Perdereau, A. , Arnaud‐Haond, S. , Gagnaire, P.‐. A. , Jollivet, D. , Hourdez, S. , & Bonhomme, F. (2022). Subtle limits to connectivity revealed by outlier loci within two divergent metapopulations of the deep‐sea hydrothermal gastropod Ifremeria nautilei . Molecular Ecology, 31(10), 2796–2813. 10.1111/mec.16430 [DOI] [PubMed] [Google Scholar]
- Tsin, A. T. C. , & Beatty, D. D. (1977). Visual pigment changes in rainbow trout in response to temperature. Science, 195(4284), 1358–1360. 10.1126/science.841335 [DOI] [PubMed] [Google Scholar]
- Tusso, S. , Nieuwenhuis, B. P. S. , Weissensteiner, B. , Immler, S. , & Wolf, J. B. W. (2021). Experimental evolution of adaptive divergence under varying degrees of gene flow. Nature Ecology and Evolution, 5, 338–349. 10.1038/s41559-020-01363-2 [DOI] [PubMed] [Google Scholar]
- Tyrmi, J. S. , Vuosku, J. , Acosta, J. J. , Li, Z. , Sterck, L. , Cervera, M. T. , Savolainen, O. , & Pyhäjärvi, T. (2020). Genomics of clinal local adaptation in Pinus sylvestris under continuous environmental and spatial genetic setting. G3: Genes, Genomes, Genetics, 10(8), 2683–2696. 10.1534/g3.120.401285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez, M. C. , Martinez, D. A. , & Tomanek, L. (2020). Multiple stressor responses are regulated by sirtuins in Mytilus congeners . Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology, 246, 110719. 10.1016/j.cbpa.2020.110719 [DOI] [PubMed] [Google Scholar]
- Villarino, G. H. , Bombarely, A. , Giovannoni, J. J. , Scanlon, M. J. , & Mattson, N. S. (2014). Transcriptomic analysis of Petunia hybrida in response to salt stress using high throughput RNA sequencing. PLoS One, 9(4), e94651. 10.1371/journal.pone.0094651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahle, R. A. , Bergeron, C. , Tremblay, J. , Wilson, C. , Burdett‐Coutts, V. , Comeau, M. , Rochette, R. , Lawton, P. , Glenn, R. , & Gibson, M. (2013). The geography and bathymetry of American lobster benthic recruitment as measured by diver‐based suction sampling and passive collectors. Marine Biology Research, 9(1), 42–58. 10.1080/17451000.2012.727428 [DOI] [Google Scholar]
- Waples, R. S. (1998). Separating the wheat from the chaff: Patterns of genetic differentiation in high gene flow species. Journal of Heredity, 89(5), 438–450. 10.1093/jhered/89.5.438 [DOI] [Google Scholar]
- Waples, R. S. , & Gaggiotti, O. (2006). What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Molecular Ecology, 15(6), 1419–1439. 10.1111/j.1365-294X.2006.02890.x [DOI] [PubMed] [Google Scholar]
- Waples, R. S. , Punt, A. E. , & Cope, J. M. (2008). Integrating genetic data into management of marine resources: How can we do it better? Fish and Fisheries, 9(4), 423–449. 10.1111/j.1467-2979.2008.00303.x [DOI] [Google Scholar]
- Wares, J. P. (2002). Community genetics in the Northwestern Atlantic intertidal. Molecular Ecology, 11(7), 1131–1144. 10.1046/j.1365-294X.2002.01510.x [DOI] [PubMed] [Google Scholar]
- Weir, B. S. , & Cockerham, C. C. (1984). Estimating F‐statistics for the analysis of population structure. Evolution, 38(6), 1358–1370. 10.1111/j.1558-5646.1984.tb05657.x [DOI] [PubMed] [Google Scholar]
- Wenne, R. , Bernaś, R. , Kijewska, A. , Poćwierz‐Kotus, A. , Strand, J. , Petereit, C. , Plauška, K. , Sics, I. , Árnyasi, M. , & Kent, M. P. (2020). SNP genotyping reveals substructuring in weakly differentiated populations of Atlantic cod (Gadus morhua) from diverse environments in the Baltic Sea. Scientific Reports, 10(1), 9738. 10.1038/s41598-020-66518-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock, M. C. , & Lotterhos, K. E. (2015). Reliable detection of loci responsible for local adaptation: Inference of a null model through trimming the distribution of FST. The American Naturalist, 186(Suppl 1), S24–S36. 10.1086/682949 [DOI] [PubMed] [Google Scholar]
- Wu, T. D. , & Watanabe, C. K. (2005). GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics, 21(9), 1859–1875. 10.1093/bioinformatics/bti310 [DOI] [PubMed] [Google Scholar]
- Xue, H. , Incze, L. , Xu, D. , Wolff, N. , & Pettigrew, N. (2008). Connectivity of lobster populations in the coastal Gulf of Maine: Part I: Circulation and larval transport potential. Ecological Modelling, 210(1–2), 193–211. 10.1016/j.ecolmodel.2007.07.024 [DOI] [Google Scholar]
- Xuereb, A. , D'Aloia, C. C. , Andrello, M. , Bernatchez, L. , & Fortin, M.‐J. (2020). Incorporating putatively neutral and adaptive genomic data into marine conservation planning. Conservation Biology, 35, 909–920. 10.1111/cobi.13609 [DOI] [PubMed] [Google Scholar]
- Yi, X. , & Latch, E. K. (2022). Nonrandom missing data can bias principal component analysis inference of population genetic structure. Molecular Ecology Resources, 22(2), 602–611. 10.1111/1755-0998.13498 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Table S1
Data Availability Statement
Demultiplexed raw reads (FASTQ format) generated for this study have been deposited in the National Center for Biotechnology Information Sequence Read Archive database (NCBI) under BioProject accession no. PRJNA862675, PRJNA645159 and PRJNA645211. Sampling data information are available in Table S1.