

Abstract
Mechanophores are molecular moieties that are incorporated into polymers and respond to force with constitutional, configurational, or conformational bond rearrangements to enable functionality. Up to today, several chemically latent motifs have been activated by polymer mechanochemical methods, but the generation of secondary amines remains elusive. Here we report carbamoyloximes as mechanochemical protecting groups for secondary amines. We show that carbamoyloximes undergo force‐induced homolytic bond scission at the N−O oxime bond in polymers thus producing the free amine, as the reaction proceeds via the carbamoyloxyl and aminyl radicals, analogously to its photochemical counterpart. Eventually, we apply the carbamoyloxime motif in a force‐activated organocatalytic Knoevenagel reaction. We believe that this protecting strategy can be universally applied for many other secondary and primary amines in polymer materials.
Keywords: Amines, Mechanochemistry, Organocatalysis, Photochemistry, Polymers
Carbamoylaldoximes undergo mechanochemical homolytic bond scission of the oxime N−O bond when embedded into polymers. The subsequent radical mechanism yields mainly the nitrile, upon hydrogen dissociation, and the free amine, upon decarboxylation and hydrogen abstraction. Force‐induced organocatalysis is demonstrated for the activation of a secondary amine.

Introduction
Force is a ubiquitous stimulus that acts on polymer materials in their respective area of application and is usually associated to degradation phenomena. Structural polymers bear loads, polymers in solution are exposed to shear force, and coatings are subjected to friction and wear. Using force to activate function from latent molecular motifs (mechanophores)[ 1 , 2 ] is thus a promising method on the one hand to better understand the mechanical behavior of polymers and on the other hand to convert mechanical energy into useful chemical functionality. While the former is generally carried out by the incorporation of optical force probes,[ 3 , 4 ] the latter entails examples, such as the release of small molecules,[ 5 , 6 ] the activation of latent catalysts, [7] or the initiation of secondary reactions. [8] Therefore, reactive functional groups, such as transition metals,[ 9 , 10 , 11 , 12 , 13 ] carbenes,[ 13 , 14 , 15 ] organic acids,[ 16 , 17 , 18 ] latent nucleophiles,[ 19 , 20 ] or persistent radicals,[ 21 , 22 ] have been activated mechanochemically.
Amines are common functional groups in synthetic chemistry as organic bases, organocatalysts, or simply as nucleophiles for bond formation and polymerization reactions.[ 23 , 24 ] Thus, it is highly desirable to activate amines with an external stimulus to gain control over their reactivity. While light, redox, or enzymes have been investigated for this purpose,[ 25 , 26 ] a general method to activate latent amines by mechanical force has not yet been reported. Certainly, the release of primary amine‐bearing molecules from latent carbamates has been demonstrated both by Robb and co‐workers [27] and us, [28] and Jung and Yoon recently have shown the mechanochemical generation of imines and their subsequent hydrolysis to primary amines. [29] However, a general method for the direct activation of chemically useful secondary amines is not available.
Several latent protecting groups for the light‐induced uncaging of amines have been reported, mostly so‐called photobase generators (PBGs). [30] PBG motifs often are specific variants of carbamates which generate amines under hydrolytic conditions. However, for ordinary carbamates the nucleophilic attack of H2O to form the tetrahedral intermediate in the addition‐elimination hydrolysis mechanism is the rate‐determining step. This would also be true for a hypothetical carbamate mechanophore where the subsequent mechanochemical bond scission step would be severely rate‐limited therefore rendering carbamates unsuitable motifs for this purpose. [31]
Concurrently, oxime carbamates (or carbamoyloximes) have a relatively low bond dissociation energy (BDE) of approximately 222 kJ mol−1. [32] When these motifs are used as PBGs, [33] the light‐induced homolytic scission of the N−O bond is the key step for a subsequent radical mechanism culminating in the liberation of an amine (Scheme 1). Together, the low BDE, the initial homolytic bond scission step, and the report by Moore and co‐workers on oxime sulfonates as mechanoacid generators [17] thus plausibly support the investigation of carbamoyloximes as mechanobase generators (MBGs).
Scheme 1.
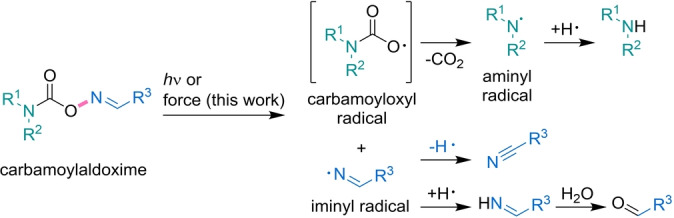
Reaction pathway of the light‐ (PBG, previous work) and force‐induced (MBG, this work) scission of carbamoyloximes. The initially cleaved bond is marked in bold pink.
Here we report the synthesis and mechanochemical activation of carbamoyloximes as a general strategy for the force‐induced generation of amines. Therefore, we rely on carbamoylaldoxime‐protected piperidine incorporated as mechanophore into linear polymer chains. We qualitatively verify the mechanochemical activity of the molecular motif by the constrained geometries simulate external forces (CoGEF) method. [34] We activate the mechanophores in solution by ultrasonication using an immersion probe sonicator (20 kHz) and analyze the reaction pathway in detail compared to its photochemical counterpart. We find that in both cases the amine is generated from the aminyl radical, which is in turn produced from the carbamoyloxyl radical, while the iminyl radical intermediate affords the related nitrile or aldehyde (Scheme 1). Eventually, we exemplarily highlight the use of this motif for force‐activated organocatalysis.
Results and Discussion
The synthesis was carried out adapting the protocol of Batey and co‐workers using carbonyldiimidazole (CDI) as safe phosgene equivalent (Scheme 2a). [35] Due to its high regioselectivity, this method did not require protecting the alkyl alcohols. First, piperidine 1 was carbamoylated to intermediate 2, which after methylation to 3 reacted neatly with aldoxime 4 affording the carbamoylaldoxime mechanophore diol 5 (see Supporting Information for details). 5 was then esterified with α‐bromoisobutyryl bromide giving bifunctional initiator 6. This was then used for Cu0‐mediated controlled radical polymerization of either methyl acrylate (MA) or hydroxyethyl acrylate (HEA) yielding hydrophobic PMA6 (various M n and Đ M, Table S1) and hydrophilic PHEA6 (M n =44 kDa, Đ M =1.30). While the carbamoylaldoxime motif was stable at temperatures used for storage (2–8 °C) and mild chemical reactions (50 °C), thermal decomposition was observed above 70 °C (Figure S1–S5).
Scheme 2.

a) Synthesis of carbamoylaldoxime mechanophore diol 5 and subsequent esterification and polymerization reactions. b) Photochemical decomposition pathway of 5.
Hereafter, the photochemical decomposition pathway of mechanophore diol 5 upon irradiation at 254 nm was investigated (Scheme 2b, Table S2). Therefore, we used nonpolar aprotic THF, polar aprotic DMF, and polar protic H2O as solvents. The proceeding photochemical reaction was monitored by UV/Vis spectroscopy (Figure S6–S8). The aromatic products derived from the iminyl radical were identified by 1H NMR spectroscopy (Figure S9–S10) while ESI‐MS and 1H NMR, after derivatization with catechol carbonate 16, were used to verify amine formation from the aminyl radical (Figure S11–S14). We found that after homolytic scission of the oxime bond, the iminyl radical was converted to nitrile 7, while aldehyde 8 was only observed in traces. Concomitantly, the carbamoyloxyl radical rapidly decarboxylated yielding the aminyl radical and thereby amine 1. The latter was detected by formation of carbamate 17 through ring‐opening of the cyclic carbonate 16, which induced a diagnostic upfield shift of the aromatic protons. Thereby, the photochemical reaction mechanism was plausibly retraced in all three solvents. The photochemical reactivity of PMA6 and PHEA6 was investigated in complete analogy to diol 5 in both THF and H2O and gave similar results (Figure S15–S17).
Sonication of PMA6 in THF and PHEA6 in H2O then verified the mechanochemical activity of the carbamoyloxime (Table S3). Upon sonication of PMA6 with M n=107 kDa (PMA6107 ) a new peak emerged in the gel permeation chromatography (GPC) elugram at ca. half the initial molar mass suggesting bond scission in the center of the polymer chain (Figure 1a). Sonication of a control PMA chain with M n =94 kDa and Đ M=1.13 (PMA94 ) under identical conditions showed a considerably reduced mechanochemical conversion (Figure 1b). This underlined that the carbamoyloxime moiety was cleaved at a significantly increased rate compared to random scission of the PMA backbone. The sonication of PHEA6 in H2O and subsequent UV/Vis spectroscopy confirmed this result (Figure S18) against PMA and PHEA control polymers as well as control 5 (Figure S32–S33).
Figure 1.
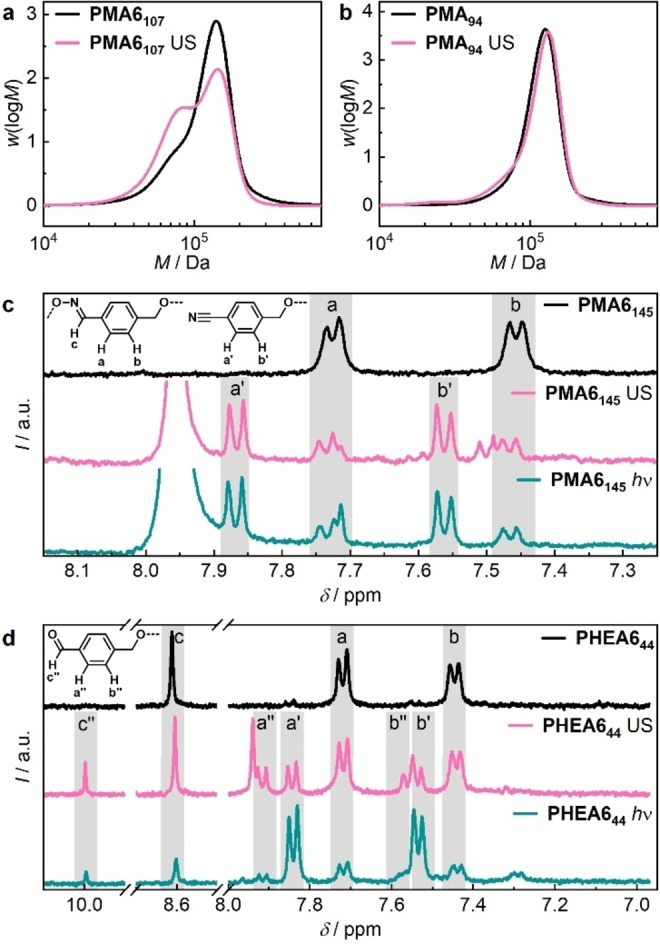
Mechanochemical activation of the carbamoyloxime mechanophore. Molar mass distributions obtained by GPC in THF via RI detector before and after 1 h sonication in THF of a) PMA6107 and b) PMA94 control polymer. Comparison of 1H NMR spectra in DMSO‐d6 : c) PMA6145 before (top), after sonication for 5 h in DMF (middle), and after irradiation for 1 h with light at 254 nm in DMF (bottom). d) PHEA644 before (top), after sonication for 1 h in H2O (middle), and after irradiation for 1 h with light at 254 nm in H2O (bottom). See Figure S30–S31 for a more detailed 1H NMR comparison.
Since accelerated mechanochemical bond scission is a necessary but not a sufficient criterion for the identification of a mechanophore, we subsequently investigated the mechanochemical reaction products by 1H NMR spectroscopy. Therefore, PMA6145 was both cleaved photochemically and mechanochemically in DMF. The 1H NMR spectra revealed identical reaction products through the diagnostic signals at δ=7.86 and 7.57 ppm corresponding to the nitrile PMA7 (Figure 1c). This was confirmed for a variety of reaction conditions under variable sonication times, molar masses, different solvents, and by dedicated synthesis of a nitrile control polymer (Figure S19–S21). A comparison between photochemical and mechanochemical reactivity was then also carried out in H2O using PHEA644 (Figure 1d). While irradiation with light yielded mostly nitrile PHEA7, sonication gave a considerably increased fraction of aldehyde PHEA8 as product. This implies that under mechanochemical conditions in H2O the iminyl radical is likely to either abstract or lose an H‐atom while all other investigated conditions appear to favor solely the detachment of an H‐atom to form the nitrile (cf. Scheme 1 and 2).
Hereafter, we evaluated the mechanochemical selectivity. Therefore, we sonicated PMA6 and PHEA6 in different solvents and compared the desired mechanochemical conversion determined by 1H NMR to the overall degree of bond scission determined by GPC (Table S4, Figure S22–S29). Depending on the sonication conditions, we found that 42–66 % of the overall bond scission events were productive and occurred at the desired N−O bond of the carbamoylaldoxime. These values were moderate and we recognize that precise computations, in addition to kinetic analyses, will be necessary in the future to obtain a quantitative mechanochemical understanding regarding rate constant, F max, and E max. Nevertheless, we performed CoGEF simulations (Figures S36) for a qualitative indication that the N−O bond is the most prone one to undergo scission when the mechanophore is subjected to mechanical force. Furthermore, the mechanochemical origin of the observed reaction was unequivocally verified by the sonication of control polymer chains with terminally substituted mechanophores yielding no productive conversion (Figure S32–S34).
Though it appeared plausible to assume that the desired amine is formed under mechanochemical conditions in complete analogy to the photochemical pathway after the above results, we carried out a qualitative labelling experiment on sonicated PMA6. Therefore, we used Rhodamine B isothiocyanate RhBITC which formed a stable thiourea upon reaction with the amine allowing its observation by UV/Vis spectroscopy (Scheme S1). After optimization of the labelling conditions using piperidine as small molecule (Figure S37–S39), PMA680 was labelled using 10 equiv of RhBITC. GPC was then performed using a UV/Vis detector at 351 nm where only the dye absorbed. PMA680 before sonication did not react with the dye and showed only a very small residual absorbance in the UV channel coinciding with the RI channel of the polymer (Figure 2a). This signal did not significantly stand out from the overall background noise and might be caused by coelution of small amounts of unreacted dye that remained adsorbed non‐covalently to the polymer. On the contrary, PMA680 after sonication exhibited a clear peak in the UV channel that coincided with the peak of the RI channel at half the initial molar mass (Figure 2b). These results were comparable to dye‐labelled photochemically cleaved PMA680 (Figure 2c) thus confirming the successful mechanochemical generation of the amine.
Figure 2.
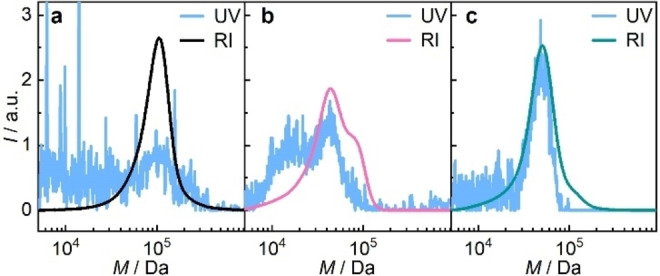
Derivatization of PMA680 before and after carbamoylaldoxime bond scission with RhBITC. GPC elugrams in THF containing both RI and UV (351 nm) detector traces of a) pristine PMA680 , b) PMA680 sonicated for 2 h in THF : H2O=7 : 3 (v:v), and c) PMA680 irradiated for 20 min with light at 254 nm in THF.
Hereafter, we aimed to apply this method in a proof‐of‐concept experiment where the mechanochemical bond scission of PMA6 would yield a secondary amine PMA1 that would then catalyze a Knoevenagel reaction (Scheme 3). Therefore, we carried out ex situ experiments where the catalyst generated by sonication or irradiation with light was mixed with the reagents in CDCl3 after its activation (Table S5 and S6). We used 4‐nitrobenzaldehyde 18 in combination with diethyl malonate 19 in 50‐fold excess to achieve a pseudo first‐order reaction and monitored the conversion to product 20 by 1H NMR (Table S7–S9, Figure S40–S45).
Scheme 3.
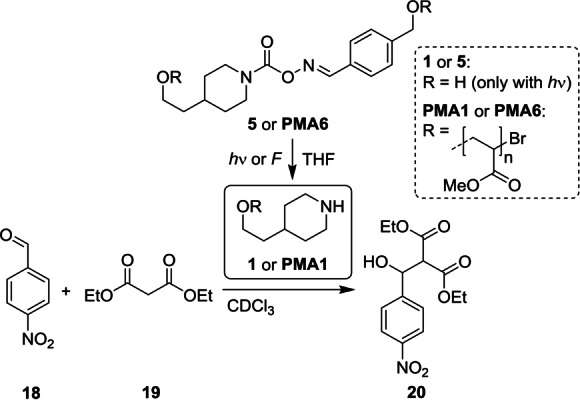
Photo‐ and mechanochemical reactions of 5 and PMA6, respectively, yielding amines 1 (by light) or PMA1 (by ultrasound) to catalyze the Knoevenagel reaction of 4‐nitrobenzaldehyde 18 with diethyl malonate 19 to product 20.
First, we verified the catalytic activity of the generated piperidine 1 by irradiation of carbamoyloxime 5 with light and then combining it with the reactants for 2 h. While a 0.49 mm solution of irradiated 5 gave a conversion of 23 % 20, pristine 1.00 mm 1 yielded 60 % 20, and pristine 5.50 mm 5 (as a negative control) only 7 % 20 (Table S7 and S9). Analogously, the catalysis experiments performed with photochemically generated PMA1 from PMA6 were also successful affording a conversion of 34 % (2 h reaction duration, 0.44 mm, Table S7). This suggested that the PMA chain was fully swollen with CDCl3 and reagents permeated the hydrodynamic coil efficiently to the reactive chain‐terminal amine moiety.
Subsequently, a PMA6116 solution was sonicated to generate PMA1 mechanochemically. Note that we found that the sonication conditions produced acids, electrophiles, and/or other species as minor byproducts that deactivated piperidine at low concentrations. Therefore, n‐butylamine (200 μL) was added to the sonication solution as scavenger for these byproducts. This primary amine, however, was removed by dialysis before the reactants were combined and in addition was verified to not catalyze the reaction of 18 with 19. The sonication of PMA6116 was performed in triplicate for 3 h giving mechanophore conversions between 52–58 % (Table S8). This corresponded to a final mechanophore concentration of 2.35–2.61 mm in the catalysis solution. Thereby, catalytic conversions from 26–63 % 20 were obtained after 2 h reaction, which is significantly higher than the conversions of 1–2 % with sonicated PMA42 as negative control (Figure 3, Table S8). These data clearly underline that the amine PMA1 was efficiently formed upon sonication and catalyzed the Knoevenagel reaction, therefore demonstrating that the carbamoyloxime motif is suitable for force‐induced organocatalysis applications.
Figure 3.
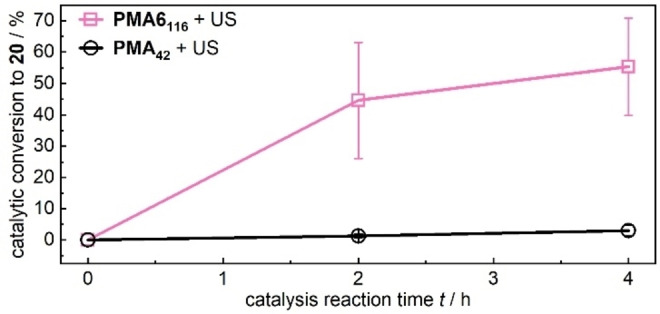
Knoevenagel reaction of 4‐nitrobenzaldehyde 18 with diethyl malonate 19 to product 20 over 2 and 4 h catalysis reaction time in the presence of sonicated PMA6116 or control PMA42 . Mean values±SD from the mean. N=3 independent sonication and subsequent catalysis runs of the same polymer sample. All data are reported in Table S8.
Conclusion
We here demonstrated a carbamoyloxime motif that acted as latent protecting group for the secondary amine piperidine. The synthesis of the mechanophore is straightforward (only three steps are required), efficient, and versatile, since the involved reactions are essentially not affected by the nature of the amine and the aromatic oxime precursors. We then transformed these carbamoyloximes into the free amine both photochemically by irradiation with light and mechanochemically by ultrasonication. With extensive control experiments, analyses, and qualitative computational investigations, we traced the reaction mechanism and identified all major intermediates and products of the reaction—both in the small molecular variants and when incorporated into polymer chains. Subsequently, we applied this motif for a force‐activated organocatalytic Knoevenagel reaction. Thus we have, for the first time, demonstrated a force‐responsive molecular protecting group for secondary amines—a mechanobase generator. Compared to previous examples of mechanochemical base activation (carbenes or primary amines), this system offers certain advantages. First, the stability of amines is significantly higher than carbenes, hence their applicability is less limited by degradation. Second, the reported activation of primary amines relies on the formation of imines which are then hydrolyzed to primary amines, thus the system requires a second input and the presence of water. The carbamoyloxime mechanophore here reported shows good efficiency for the direct formation of secondary amines with the sole input of mechanical force. Moreover, the mechanoresponsive oxime bond could in principle be exploited more broadly by varying the original structure of the mechanophore on different molecular moieties. Due to this potentially universal character of the latent carbamoyloxime strategy, we believe that this method will be applicable to other secondary and primary amines as well thus opening a new avenue for the mechanochemical control over functional groups in polymer systems.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
D. C. and R. G. are grateful for support by a Freigeist‐Fellowship of the Volkswagen Foundation (No. 92888) and by the Competence Center Bio4MatPro of the German Federal Ministry of Education and Research BMBF (No. 031B1148A). Parts of the analytical investigations were performed at the Center for Chemical Polymer Technology CPT, which was supported by the European Commission and the federal state of North Rhine‐Westphalia (No. 300088302). We thank Nils Münsterman and Lea Zimmer for their synthetic contributions. Open Access funding enabled and organized by Projekt DEAL.
D. Campagna, R. Göstl, Angew. Chem. Int. Ed. 2022, 61, e202207557; Angew. Chem. 2022, 134, e202207557.
Data Availability Statement
The data that support the findings of this study are documented in the Supporting Information and are openly available in Zenodo at https://doi.org/10.5281/zenodo.6849938, reference number [36].
References
- 1. Chen Y., Mellot G., van Luijk D., Creton C., Sijbesma R. P., Chem. Soc. Rev. 2021, 50, 4100–4140. [DOI] [PubMed] [Google Scholar]
- 2. O'Neill R. T., Boulatov R., Nat. Chem. Rev. 2021, 5, 148–167. [DOI] [PubMed] [Google Scholar]
- 3. He S., Stratigaki M., Centeno S. P., Dreuw A., Göstl R., Chem. Eur. J. 2021, 27, 15889–15897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Traeger H., Kiebala D. J., Weder C., Schrettl S., Macromol. Rapid Commun. 2021, 42, 2000573. [DOI] [PubMed] [Google Scholar]
- 5. Versaw B. A., Zeng T., Hu X., Robb M. J., J. Am. Chem. Soc. 2021, 143, 21461–21473. [DOI] [PubMed] [Google Scholar]
- 6. Küng R., Göstl R., Schmidt B. M., Chem. Eur. J. 2022, 28, e202103860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Groote R., Jakobs R. T. M., Sijbesma R. P., Polym. Chem. 2013, 4, 4846–4859. [Google Scholar]
- 8. Hu H., Ma Z., Jia X., Mater. Chem. Front. 2020, 4, 3115–3129. [Google Scholar]
- 9. Di Giannantonio M., Ayer M. A., Verde-Sesto E., Lattuada M., Weder C., Fromm K. M., Angew. Chem. Int. Ed. 2018, 57, 11445–11450; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11616–11621. [Google Scholar]
- 10. Sha Y., Zhang Y., Xu E., Wayne McAlister C., Zhu T., Craig S. L., Tang C., Chem. Sci. 2019, 10, 4959–4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levy A., Feinstein R., Diesendruck C. E., J. Am. Chem. Soc. 2019, 141, 7256–7260. [DOI] [PubMed] [Google Scholar]
- 12. Michael P., Binder W. H., Angew. Chem. Int. Ed. 2015, 54, 13918–13922; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14124–14128. [Google Scholar]
- 13. Piermattei A., Karthikeyan S., Sijbesma R. P., Nat. Chem. 2009, 1, 133–137. [DOI] [PubMed] [Google Scholar]
- 14. Nixon R., De Bo G., Nat. Chem. 2020, 12, 826–831. [DOI] [PubMed] [Google Scholar]
- 15. Shen H., Larsen M. B., Roessler A. G., Zimmerman P. M., Boydston A. J., Angew. Chem. Int. Ed. 2021, 60, 13559–13563; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 13671–13675. [Google Scholar]
- 16. Diesendruck C. E., Steinberg B. D., Sugai N., Silberstein M. N., Sottos N. R., White S. R., Braun P. V., Moore J. S., J. Am. Chem. Soc. 2012, 134, 12446–12449. [DOI] [PubMed] [Google Scholar]
- 17. Nagamani C., Liu H., Moore J. S., J. Am. Chem. Soc. 2016, 138, 2540–2543. [DOI] [PubMed] [Google Scholar]
- 18. Lin Y., Kouznetsova T. B., Craig S. L., J. Am. Chem. Soc. 2020, 142, 99–103. [DOI] [PubMed] [Google Scholar]
- 19. Zhang H., Gao F., Cao X., Li Y., Xu Y., Weng W., Boulatov R., Angew. Chem. Int. Ed. 2016, 55, 3040–3044; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3092–3096. [Google Scholar]
- 20. Pan Y., Zhang H., Xu P., Tian Y., Wang C., Xiang S., Boulatov R., Weng W., Angew. Chem. Int. Ed. 2020, 59, 21980–21985; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 22164–22169. [Google Scholar]
- 21. Imato K., Irie A., Kosuge T., Ohishi T., Nishihara M., Takahara A., Otsuka H., Angew. Chem. Int. Ed. 2015, 54, 6168–6172; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6266–6270. [Google Scholar]
- 22. Seshimo K., Sakai H., Watabe T., Aoki D., Sugita H., Mikami K., Mao Y., Ishigami A., Nishitsuji S., Kurose T., Ito H., Otsuka H., Angew. Chem. Int. Ed. 2021, 60, 8406–8409; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 8487–8490. [Google Scholar]
- 23. Nielsen M., Worgull D., Zweifel T., Gschwend B., Bertelsen S., Jørgensen K. A., Chem. Commun. 2011, 47, 632–649. [DOI] [PubMed] [Google Scholar]
- 24. Jin F.-L., Li X., Park S.-J., J. Ind. Eng. Chem. 2015, 29, 1–11. [Google Scholar]
- 25. Klán P., Šolomek T., Bochet C. G., Blanc A., Givens R., Rubina M., Popik V., Kostikov A., Wirz J., Chem. Rev. 2013, 113, 119–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maity C., Trausel F., Eelkema R., Chem. Sci. 2018, 9, 5999–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zeng T., Hu X., Robb M. J., Chem. Commun. 2021, 57, 11173–11176. [DOI] [PubMed] [Google Scholar]
- 28. Shi Z., Song Q., Göstl R., Herrmann A., CCS Chem. 2021, 3, 2333–2344. [Google Scholar]
- 29. Jung S., Yoon H. J., Angew. Chem. Int. Ed. 2021, 60, 23564–23568; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 23756–23760. [Google Scholar]
- 30. Suyama K., Shirai M., Prog. Polym. Sci. 2009, 34, 194–209. [Google Scholar]
- 31. Akbulatov S., Tian Y., Kapustin E., Boulatov R., Angew. Chem. Int. Ed. 2013, 52, 6992–6995; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7130–7133. [Google Scholar]
- 32. Lide D. R., CRC Handbook of Chemistry and Physics: A Ready-Reference Book of Chemical and Physical Data, CRC Press, Boca Raton, 2003. [Google Scholar]
- 33. Walton J. C., Molecules 2016, 21, 63.26751437 [Google Scholar]
- 34. Beyer M. K., J. Chem. Phys. 2000, 112, 7307–7312. [Google Scholar]
- 35. Grzyb J. A., Shen M., Yoshina-Ishii C., Chi W., Brown R. S., Batey R. A., Tetrahedron 2005, 61, 7153–7175. [Google Scholar]
- 36.D. Campagna, R. Göstl, Zenodo 2022 [dataset], doi: 10.5281/zenodo.6849938. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are documented in the Supporting Information and are openly available in Zenodo at https://doi.org/10.5281/zenodo.6849938, reference number [36].