

Abstract
Inherited peripheral neuropathy (IPN) is a heterogeneous group of disorders due to pathogenic variation in more than 100 genes. In 2012, the first cases of IPN associated with HINT1 pathogenic variations were described in 33 families sharing the same phenotype characterized by an axonal neuropathy with neuromyotonia and autosomal recessive inheritance (NMAN: OMIM #137200). Histidine Triad Nucleotide Binding Protein 1 regulates transcription, cell‐cycle control, and is possibly involved in neuropsychiatric pathophysiology. Herein, we report seven French patients with NMAN identified by Next Generation Sequencing. We conducted a literature review and compared phenotypic and genotypic features with our cohort. We identified a new HINT1 pathogenic variation involved in NMAN: c.310G>C p.(Gly104Arg). This cohort is comparable with literature data regarding age of onset (7,4yo), neuronal involvement (sensorimotor 3/7 and motor pure 4/7), and skeletal abnormalities (scoliosis 3/7, feet anomalies 6/7). We expand the phenotypic spectrum of HINT1‐related neuropathy by describing neurodevelopmental or psychiatric features in six out of seven individuals such as generalized anxiety disorder (GAD), obsessive–compulsive disorder (OCD), mood disorder and attention deficit hyperactivity disorder (ADHD). However, only 3/128 previously described patients had neuropsychiatric symptomatology or neurodevelopmental disorder. These features could be part of HINT1‐related disease, and we should further study the clinical phenotype of the patients.
Keywords: ARAN‐NM, Charcot–Marie–tooth disease, France, HINT1, Neuromyotonia, NMAN, Peripheral neuropathy
1. INTRODUCTION
Inherited peripheral neuropathy (IPN) is a highly heterogeneous group of disorders caused by pathogenic variations in more than 100 genes. 1 These pathologies are classified according to the sensory and/or motor impairment and the injury of the nerve axon (axonal neuropathy) or the myelin sheath (demyelinating neuropathy). In 2012, the first cases of hereditary peripheral neuropathy linked to eight loss‐of‐function variants of the HINT1 gene were discovered. In a heterogeneous cohort of patients affected by autosomal recessive axonal neuropathy, the authors found that 11% of cases were due to HINT1 pathogenic variants. The yield rises to 76% in individuals presenting neuropathy associated to neuromyotonia. 2
Therefore, IPN due to HINT1 pathogenic variants was defined as a novel autosomal recessive subtype characterized by the association of neuropathy and neuromyotonia and named Axonal Neuropathy with Neuromyotonia (ARAN‐NM) or Neuromyotonia and Axonal Neuropathy (NMAN) (OMIM #137200) has been described.
Neuromyotonia is a peripheral nerve hyperexcitability characterized by a persistent muscle contraction which may be associated with myokymia that occurs because of a spontaneous high frequency of motor unit discharges. Although It may be clinically detected, the continuous muscle activity is sometimes detectable only by electromyography (EMG), which reveals spontaneous, continuous, irregularly occurring doublet, triplet, or multiplet single motor unit (or partial motor unit) discharges, firing at a high intraburst frequency (30–300 Hz).
HINT1, located on chromosome 5q31.2, encodes a 126 amino acid, ubiquitously expressed homodimeric purine phosphoramidase called Histidine Triad Nucleotide Binding Protein 1, one of the 3 HINT proteins (HINT1, HINT2, and HINT3) in the human genome. HINT1 is involved in the regulation of transcription and cell‐cycle control. HINT1 modulates apoptosis in cancer cells and is a potential tumor suppressor. 3 , 4 HINT1 also interacts with transcription factors through its action on SUMO proteins. 5 Indeed, it has a sumoylase activity with a major role in sumoylation. This post‐translational modification is essential for intracellular trafficking, protein–protein, and DNA‐protein interactions, and transcriptional activities of target proteins. Sumoylase activity is altered in vitro when the HINT1 protein is mutated. 6 The role of HINT1 in the pathogenesis of ARAN‐NM may be due to the impairment of motor neuron signaling pathways in which HINT1 is implicated. 7
This protein is also possibly involved in the pathophysiology of pain, 8 addiction, 9 and brain aging. 10
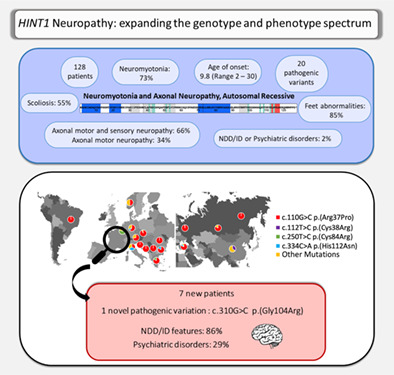
To date, 128 patients with ARAN‐NM have been described (108 families) worldwide. 11 , 12 , 13 , 14 , 15 Most of them are from eastern Europe, where the founder variant (c.110G>C, p.Arg37Pro) is mainly represented allele frequency 0.00043 in the European (Non‐finish) population (gnomAD V3.1.2), 0.002 in Russia. 15 In addition, another founder variant (c.112T>C, p.Cys38Arg) has been identified in the Chinese population. 14 Twenty sequence variations of this gene have been reported as pathogenic or probably pathogenic in a homozygous or heterozygous composite state. 11 , 15 , 16 , 17
Here, we report seven French patients with ARAN‐NM associated with homozygous pathogenic variants of HINT1. We identify a novel pathogenic variant. We conduct an exhaustive review of the literature and discuss the phenotypic spectrum of HINT1 pathogenic variants, particularly the neuropsychiatric features.
2. MATERIALS AND METHODS
2.1. Patients selection
The patients were identified during national meetings of the FILNEMUS (French rare neuromuscular diseases Healthcare Network) network. This national rare disease network brings together French molecular biology laboratories as well as clinical neurology department specialized in IPN. Six of the seven patients were identified by NGS sequencing in gene panels within the FILNEMUS network laboratories. Last patient (P1) underwent whole exome sequencing because of his particular phenotype (Appendix B).
2.2. Samples
A written and signed approval has been collected from the patients following French recommendations and in agreement with the local ethics committee rules. DNA was extracted from peripheral blood using standard procedures.
2.3. Next‐generation sequencing
NGS was performed using an in‐house designed panel containing the coding exons and flanking intronic sequences of neuromuscular disease‐associated genes. To check whether variants had already been inventoried and classified, we looked up Online Mendelian Inheritance in Man (OMIM) (https://www.ncbi.nlm.nih.gov/omim), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and HGMD (http://www.hgmd.cf.ac.uk). Finally, following the American College of Medical Genetics and Genomics (ACMG) recommendations (Richards' classification, 2015), we classified these variants into five categories: pathogenic, likely pathogenic, variant of unknown significance (VUS), likely benign, and benign.
2.4. Electrodiagnostic examination [EDx]: nerve conduction studies and electromyography
Results of the EDx were retrieved when available. Electrodiagnostic studies included nerve conduction studies in the upper and lower limbs and EMG using a concentric needle electrode in two muscles. Electrophysiologic studies were performed using conventional equipment and standard methods. Skin temperature was maintained in the range of 32–34°C. Patients with normal motor and sensory conduction velocities, normal or mildly decreased sensory nerve action potential amplitudes, decreased compound muscle action potential amplitudes, and neuropathic changes on ENMG, that is, an increased number of long‐duration, high‐amplitude, and polyphasic motor unit potentials (MUPs), were classified as having axonal motor peripheral neuropathy.
2.5. Statistical analysis
The characteristics of our patients were compared with patient data from a comprehensive review of the literature. A Fisher test was performed to compare the percentage of different features in these two populations.
3. RESULTS
3.1. Clinical features of HINT1 ‐positive patients
The clinical characteristics of the seven patients are summarized in Table 1 and Appendix B. Homozygous variants in HINT1 were identified in all of them. Three patients carry the variant c.110G>C (p.Arg37Pro) (P1, P3, and P4), three the variant c.334C>A (p.His112Asn) (P2, P5, and P7). P6 has a homozygous variant c.310G>C (p.Gly104Arg) never reported before. Ethnic origin and family consanguinity are reported for each patient.
TABLE 1.
Clinical characteristics of French patients carrying biallelic mutations in HINT1 compared with literature data
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | Literature review a |
|---|---|---|---|---|---|---|---|---|
| Genotype | c.110G>C p.(Arg37Pro) homozygous | c.334C>A p.(His112Asn) homozygous | c.110G>C p.(Arg37Pro) homozygous | c.110G>C p.(Arg37Pro) homozygous | c.334C>A p.(His112Asn) homozygous | c.310G>C p.(Gly104Arg) homozygous | c.334C>A p.(His112Asn) homozygous | Figure 1 |
| Sex | F | M | M | M | F | M | M | F: 58% (n = 40)–M: 42% (n = 29) |
| Origin | Austrian | Portuguese | Kosovan | French | Roma | Algerian | Roma/French | Figure 2 |
| Consanguinity | No | Yes | No | No | Yes | Yes | No | Yes: 25% (n = 14) – No: 75% (n = 42) |
| Age at examination (years) | 26 | 23 | 37 | 17 | 14 | 23 | 19 | |
| Age of onset (years) | 4 | 2 | 24 | 6 | 8 | 7 | 1 | Average: 9,8 (min 2–max 30) |
| Schooling | Normal | Specialized | Normal | Normal with LSA | Specialized | Specialized | Normal | |
| Walk | Abnormal without aids | Abnormal with orthosis | Abnormal without aids | Abnormal without aids | Abnormal without aids | Abnormal without aids | Abnormal without aids | |
| Feet abnormalities | Pes planus | Pes planus, short Achille's tendons | Pes cavus | NA | Short Achille's tendons | Pes cavus, short Achille's tendons | Pes cavus | Presence: 85% (n = 29/34) |
| Scoliosis | Presence | Presence | Absence | Presence | Absence | Absence | Absence | Presence: 55% (n = 6/11) |
| DTR | Absence | Absence | Absence | Absence | Absence | Absence | Absence | |
| Clinical motor testing | Abnormal | Abnormal | Abnormal | NA | Abnormal | Abnormal | Abnormal | |
| Symptoms suggesting myotonia | Presence | Presence | Presence | Presence | Presence | Absence | Absence | 69% (n = 58/84) |
| Additional clinical features | Muscle retractions | Lordosis, irregular tremor on the finger‐nose test, overweight, hip hyperlaxity, chronic constipation | Hypersudation, Ataxia | |||||
| Median MCV (m/s) | 52 | 56.8 | NA | NA | 46.8 | 41.8 | >40 | |
| NCS | Axonal motor neuropathy | Axonal motor and sensory neuropathy | Axonal motor and sensory neuropathy | Axonal motor and sensory neuropathy | Axonal motor neuropathy | Axonal motor neuropathy | Axonal motor neuropathy |
Axonal motor neuropathy: 34% (n = 39/114) Axonal motor and sensory neuropathy: 66% (n = 75/114) |
| Neuromyotonia | Absence | Presence | Presence | Presence | Presence | Absence | Absence | 73% (n = 63/84) |
| NDD or ID | Dyslexia | ID | Absence | NDD | NDD and ID | NDD and ID | NDD and ID | n = 1/127 |
| Psychiatric disorders | Absence | Absence | Absence | Depression, TDAH | GAD and OCD | Absence | Absence | n = 3/127 |
| Brain MRI | Normal | Normal | NA | NA | Not performed | Normal | Not performed | |
| CK (norm: 0–190 IU/L) | 339 | 620 | NA | 923 | 272 | 1636 | Normal | Inscreased 67% (n = 26/39) |
Abbreviations: DTR, deep tendon reflexes; EMG, electromyography; F, female; GAD, generalized anxiety disorder; LSA, learning support assistant; ID, intellectual deficiency; M, male; MCV, motor conduction velocity; NA, not available; NDD, neurodevelopmental disorder; NCS, nerve conduction studies; OCD, obsessive–compulsive disorder.
Exhaustive bibliography in Appendix A.
The mean age at symptoms onset was 7.4 years (range 1–24). All of them present an abnormal walking pattern with bilateral steppage gait. One of them needs orthosis (P2). Foot anomalies were found in 6/7 patients: pes planus (P1 and P2), pes cavus (P3, P6, and P7), short Achille's tendons (P2, P5, and P6). Three patients have scoliosis (P1, P2, and P4). They all have a clinical picture of peripheral neuropathy with the abolition of deep tendon reflexes, decreased muscle strength, and wasting of hands and foot muscles. Five had muscle cramps, stiffness, and slow muscle relaxation (P1‐P5), suggesting NM. Four patients had NM on EMG (P2–P5) (57%). In three patients (P1, P6, and P7) no NM was diagnosed at EMG. The NCS with reduced amplitude reflected sensory‐motor axonal neuropathy in three patients (P2–P4) and distal motor neuropathy in the four other patients (P1, P5–P7). Serum CK level was increased for 5/6 patients and was up to 8 N (N: 0–190 IU/L) in one of them (P6). This biological feature led to suspect a myopathic process, which was invalidated by a muscle biopsy.
3.2. Neuropsychiatric and neurodevelopmental features of HINT1 ‐positive patients
Six of the seven patients described have a neurodevelopmental disorder (NDD), intellectual deficiency (ID), or a psychiatric disorder. Patient 1 has dyslexia. Patients 2 and 6 have ID, but language acquisitions were made at a normal age. IQ tests have not been performed yet. Patients 4, 5, and 7 also have ID, preceded by NDD. Patient 4 also has psychiatric features such as depression and attention deficit hyperactivity disorder (ADHD). He has a treatment for these diseases. In term of schooling, he underwent normal schooling with Learning Support Assistant (LSA). Patient 5 walked at 24 months and spoke at 4 years. She has psychiatric disorders such as Generalized Anxiety Disorder (GAD) and obsessive–compulsive disorder (OCD). Patient 7 had acquisition delays, particularly in oral language. He has a mild intellectual disability that is being investigated. Because of the family context and his nomadic lifestyle, this patient did not benefit from enrolment in a specialized school. At the age of 19, he did not acquire reading skills, due to his truancy and his intellectual disability.
Brain MRI are available for three of our patients (P1–P2, P6), and there were no abnormalities.
We describe below the clinical history of Patient 6, for whom we identified a new pathogenic variants in HINT1.
3.3. Patient 6
This young Algerian man, the 7th child of a consanguineous couple (consanguinity coefficient of 1/32), has been followed in the neuro‐pediatrics department since the age of 14. He has ID. One of his sisters also has ID and motor neuropathy. As she lives in Algeria, neither the clinical examination nor the genetic study could be done. The patient was able to walk and run at a normal age. However, from the age of 7 years, deformities of both feet and difficulties in running appeared with falls occurring during middle school.
Difficulties in learning were noticed since elementary school: he repeated several classes with the acquisition of reading and writing in Arabic (his native language) and significant difficulties in French. He has acquired the ability to count but not to add or subtract. He had to leave the regular school curriculum to enter a specialized school. Brain MRI was normal. The first clinical examination at 14 years reported pes cavus, clawed toes, impossible walking on heels, difficulty on toes, and the abolition of tendon reflexes in lower limbs. He has no scoliosis. The patient had a slightly elongated face with prognathism. At 16 years of age, he underwent bilateral Achilles tenotomy with bilateral triple arthrodesis. Hyperhydrosis of the hands was noted, which required botulinum toxin injections without success. At the last examination, at 22 years of age, he could not stand on heels and toes. He has a steppage gait with a reduced balance. Deep tendon reflexes were absent in the lower limbs and present in the upper limbs. Examination of sensation was normal except for proprioceptive ataxia.
The upper limbs muscular strength was fairly preserved (proximal 5/5, Flexors of the fingers 3/5, and Interosseous of the short abductors of the thumb 4/5). In the lower limbs, it is rated as proximal 4+/5, sural level 3/5, anterior leg 2/5, and big toes 0/5.
No neuromyotonia was present, either clinically or by EMG exploration.
Sensory nerve conduction velocities (SNCV) were normal in all examined nerves. Motor nerve conduction velocities (MNCV) and amplitudes of compound muscle action potential (CMAP) were unremarkable in the right median nerve, but reduced amplitudes of CMAP were observed in both ulnar (left: 4.2mv; right: 2.1 mv) and left median nerves (5.3mv). The distal motor latencies (DML) were prolonged in both median (left: 5.4 ms; right: 5.5 ms) and right ulnar nerves (4.08 ms). CMAPs of lower limbs nerves were not recordable. Electromyography (EMG) shows chronic denervation in the tibialis anterior and rectus femoris but did not reveal any complex repetitive discharges (CRD). The ENMG was classified as axonal motor peripheral neuropathy.
Muscle MRI showed a severe and symmetrical fatty infiltration of the anterolateral compartment of legs and tibialis posterior while soleus, gastrocnemius lateralis, and gastrocnemius medialis were relatively spared. Pelvic and thighs muscles were of normal appearance. There was no significant hypersignal on T2/STIR sequences. Serum CK levels were elevated at 1696 IU/L (N: 0–190 IU/L).
Next Generation Sequencing of a panel of 116 genes involved in hereditary sensory‐motor neuropathies allowed to identify a new c.310G>C p. (Gly104Arg) homozygous missense variant in HINT1. According to ACMG guidelines, we classified this variant as likely pathogenic. This variant is absent from the general population database gnomAD v3.1.2 [PM2]. It impacts a highly conserved amino acid which is near a functional site of the protein (AMP binding site at amino acid 105–108) (https://www.ebi.ac.uk/pdbe/pdbe-kb/proteins/P49773) (Figure 1) [PM1]. Bioinformatics prediction scores support a deleterious impact of the variant: REVEL (0,888) Pathogenic, SIFT (0), Damaging, CADD (26.6) [PP3]. Furthermore, HINT1 is a gene with a low rate of benign missense variants. False‐sense variants are a common mechanism of ARAN‐NM disease. [PP2]. Finally, our patient's history and clinical presentation suggested the diagnosis of axonal neuropathy associated with the HINT1 gene. [PP4].
FIGURE 1.
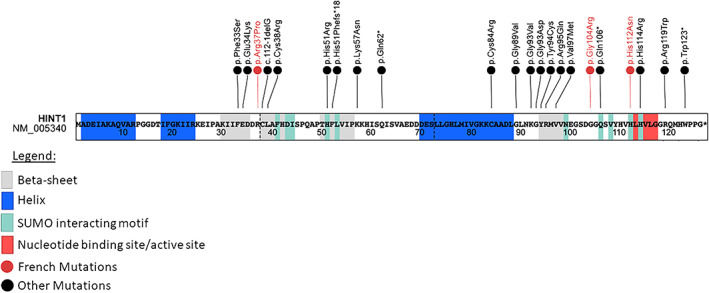
Schematic illustration of HINT1 protein and mutations [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
We hereby report seven patients with ARAN‐NM. Patient 6 is the first north‐African patient published. He is homozygous for a new missense pathogenic variant of the HINT1 gene: c.310G>C (p.Gly104Arg). The other patients were homozygous for known pathogenic variants: c.110G>C (p.Arg37Pro) and c.334C>A (p.His112Asn).
Our results show, in contrast to other Western‐European countries such as UK and Spain 18 , that although very rare, ARAN‐NM is present in France.
To date, 21 variants of HINT1 have been described in 18 publications. It represents 128 patients from 108 families (Appendix A). More than 80% were identified in South‐ or Eastern‐Europe (Figures 1 and 2). HINT1 pathogenic variants have also been described in other populations: Western/Northern‐European (n = 12; 9.4%), Chinese (n = 9; 7.1%) or Brazilian (n = 1; 0.8%). Among the 21 already published variants, we focus on the most frequent variants. First, the founder variant c.110G>C p.(Arg37Pro) is described in ARAN‐NM in 89 families, at a homozygous state for 77 of them, from all around the world (Eastern Europe, Asia, and South America). It is the most frequent variant in every studied population, except for Chinese and Belgian cohorts. 12 , 16 Herein, we report three homozygous patients (43%) carriers of this variant originating from Austria, Kosovo, and France (P1‐P3‐P4). The second most frequently encountered variant is c.250T>C p.(Cys84Arg) was identified in six Belgian and Greek families. 2 , 19 We did not find such variant in our French cohort patients. The third, c.334C>A p.(His112Asn) was found in five families from Eastern Europe. 2 It is the cause of ARAN‐NM in 3/7 of our patients originating from Portugal (P2) and French Roma families (P5, P7).
FIGURE 2.
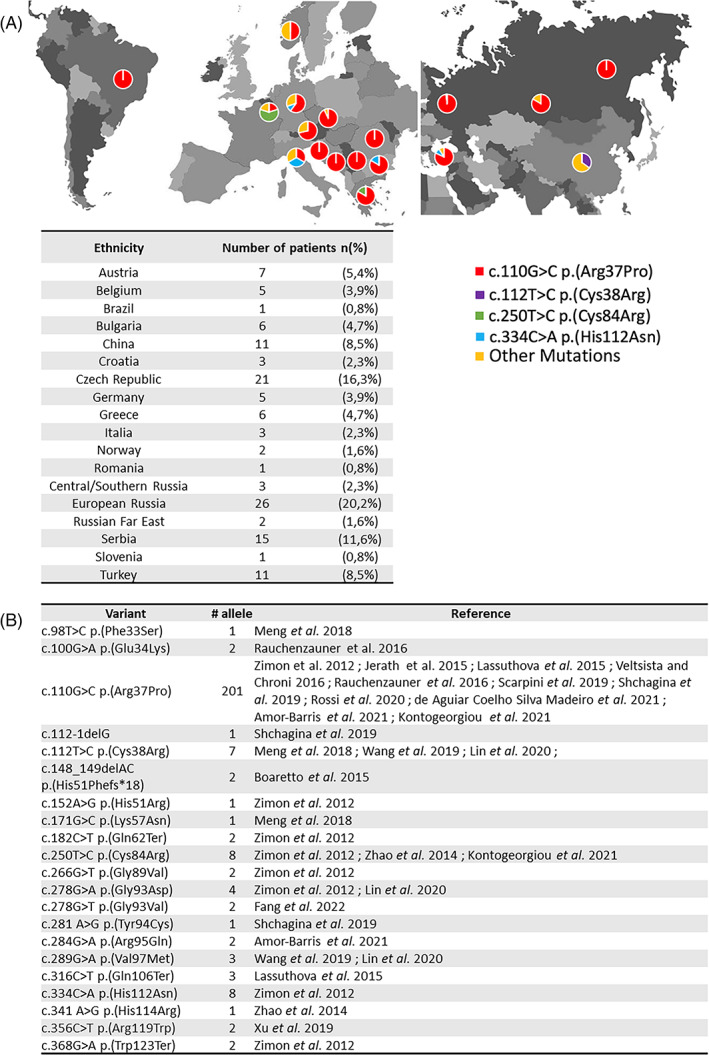
(A) Geographic distribution of published HINT1 pathogenic mutations. (B) HINT1 pathogenic mutations and their publications [Colour figure can be viewed at wileyonlinelibrary.com]
The description of our patients is consistent with data from the literature. The age of onset of the disease is in childhood with an average of 7.4 years (range: 1–24 years) compared to 9.8yo (range: 2–30 years) in scientific literature. Axonal damage is constant and affects motor nerves in all our patients as previously described (114/114 patients) and sensory nerves in 3/7 (43%) of our patients compared to 75/114 (66%) of previously published patients.
From a clinical point of view, we observed scoliosis in 3/7 (43%) of the patients, which is consistent with the data in the literature 6/11 (55%). 20 , 21 We notice foot abnormalities in 6/7 patients (86%) as described in the literature 29/34 (85%). 12 , 19 , 20 , 21 , 22 , 23 , 24 , 25
Serum CK level was increased for 26/39 (67%) published patients 2 , 12 , 14 , 20 , 24 , 25 , 26 , 27 and for 5/6 (83%) of ours (data unavailable for patient P3). In average, CK serum levels was up to 3.5 N which might led to suspect a myopathic process.
Neuromyotonia confirmed by electromyography was observed in 75% of patients published in the literature (63/84) 2 , 14 , 25 , 26 , 27 , 28 and in 4/7 (57%) of our patients. The rate of patients with neuromyotonia is not significantly different in these two populations, although the incidence in ours appears lower (p = 0.38). Neuromyotonia seems to be less significantly associated to ARAN‐NM than previously thought. Thus, the clinical picture of our patients with HINT1‐related neuropathy might be less typical and may induce diagnostic delays, as noticed in Patient 1 (Appendix B).
An important point to underline is the presence of neuropsychiatric signs or neurodevelopmental disorders. Six out of the seven patients we described here had NDD or ID. Two of them (P4 and P5) also had psychiatric features such as depression, ADHD (P4), or GAD, and OCD (P5).
To our knowledge, pathogenic variants in the HINT gene have mainly been described in patients with disorders of the peripheral nervous system of the ARAN‐NM type. Some authors 24 , 29 reported neuropsychiatric symptomatology without NM in patients with a homozygous c.110G>C variant and moderate ID. Recently, a Norvegian patient has been described with late language development. Furthermore, he was diagnosed with ADHD, with reported mood and conduct problems. 11 Other authors did not give any information about the mental health of their patients.
Concerning ID, one of the limitations of our study is that these patients did not benefit from other genetic analyses. Context of consanguinity (P2, P5, and P6) may increase the risk of other autosomal recessive diseases that might explain ID.
However, recent findings have shown that HINT1 pathogenic variants can be associated with neuropsychiatric disorders. Indeed, association studies have identified a higher proportion of variants in the HINT1 chromosomal region in schizophrenic patients or drug‐addicted patients. 9 , 30 Furthermore, animal studies show that Hint1‐KO mice exhibit reduced social interaction behaviors, aggressive behavior, sensorimotor gating deficits, apathetic and self‐neglect behaviors. 31 Hint1‐KO mice exhibited schizophrenia‐like behaviors, and dysfunction in the dopaminergic and glutamatergic systems may be involved in the abnormalities in Hint1‐KO mice. 32 Interestingly, HINT1 deficiency in aged mice is also described with reduced anxiety‐like and depression‐like behaviors and enhanced cognitive performances. 33
Human post‐mortem studies reveal a differential expression of HINT1 in schizophrenia brains versus controls. 34 HINT1 is also involved in the behavioral abnormalities induced by social isolation. 35
This work allowed us to identify 7 French patients with ARAN‐NM, which raises to 22 the number of HINT1 pathogenic variants described in the literature and involved in ARAN‐NM. Furthermore, we extend the geographical scope of this disorder with the identification of the first affected patient of Algerian origin. The description of neurodevelopmental and psychiatric disorders in our patients is consistent with data from animal and functional studies. These disorders could be an integral part of the phenotypic spectrum of this disease, and it would be interesting to further study the clinical picture of the ARAN‐NM patients.
CONFLICT OF INTEREST
The authors declare that they have no competing interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/cge.14198.
ACKNOWLEDGEMENT
We thank all the participants and the FILNEMUS—French rare neuromuscular diseases Healthcare Network.
APPENDIX A. Clinical characteristics of published patients carrying biallelic mutations in HINT1
| Publications | Variant | State | Number of Families (subject) | Sex ratio (M‐F) | Age at onset (min–max) | Feet abnormalies | Scoliosis | CPK | Clinical sugg. NM | Electrical NM | Neuropathy type | Consanguinity family (subject) | Geographic Origin |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zimon et al. 2 | c.110G>C (p.Arg37Pro) | hom | 23 (37) | 8–25 | 10 (3–25) | NA | NA | +(10/21) | 25/37 | 27/37 | HMSN (20), HMN (17) | Yes 2(5); No 17(25) | Serbia (15), Bulgaria (4), Turkey (9), Croatia (3), Austria (4), Roma (1), Europe (1) |
| c.110G>C (p.Arg37Pro) + c.250T>C (p.Cys84Arg) | comp het | 1 (2) | NA | 10 | NA | NA | NA | 2/2 | 2/2 | HMSN (2) | No 1(2) | Belgium (2) | |
| c.110G>C (p.Arg37Pro) + c.266G>T (p.Gly89Val) | comp het | 2 (2) | 1–1 | 9.5 (7–12) | NA | NA | + (1), NA (1) | 1/2 | 1/2 | HMSN (2) | No 2(2) | Austria (2) | |
| c.110G>C (p.Arg37Pro) + c.334C>A (p.His112Asn) | comp het | 1 (2) | 1–1 | 8 (6–10) | NA | NA | + (1) | 2/2 | 2/2 | HMN (2) | No 1(2) | Bulgaria (2) | |
| c.152A>G (p.His51Arg) + c.250T>C (p.Cys84Arg) | comp het | 1 (1) | 0–1 | 12 | NA | NA | + (1) | 1/1 | 1/1 | HMN (1) | No 1 (1) | Belgium (1) | |
| c.182C>T (p.Gln62Ter) + c.278G>A (p.Gly93Asp) | comp het | 1 (2) | 1–1 | 10 | NA | NA | + (2) | 2/2 | 2/2 | HMN (2) | No 1(2) | China (2) | |
| c.334C>A (p.His112Asn) | hom | 3 (3) | 2–1 | 6 (4–8) | NA | NA | + (2/3) | 2/3 | 3/3 | HMSN (2), HMN (1) | Yes 2(2) No 1(1) | Italia (1), roma (1), Turkey (1) | |
| c.368G>A (p.Trp123Ter) | hom | 1 (1) | 0–1 | 12 | NA | NA | NA | 1/1 | 1/1 | HMSN (1) | Yes 1(1) | Turkey (1) | |
| Zhao et al. 19 | c.250T>C (p.Cys84Arg) | hom | 1 (1) | 0–1 | 17 | pes cavus | NA | NA | 0/1 | 0/1 | HMN (1) | No 1(1) | Belgium (1) |
| c.250T>C (p.Cys84Arg) + c.341 A>G (p.His114Arg) | comp het | 1 (1) | 1–0 | 28 | pes cavus | NA | NA | 0/1 | 0/1 | HMN (1) | No 1(1) | Belgium (1) | |
| Jerath et al. 20 | c.110G>C (p.Arg37Pro) | hom | 1(1) | 1–0 | 13 | inverted feet 1/1 | 1/1 | + (1) | 0/1 | 1/1 | HMN (1) | No 1(1) | Slovenia (1) |
| Boaretto et al. 22 | c.148_149delAC (p.His51Phefs*18) | hom | 1 (1) | 1–0 | 12 | bilateral tendon transfer and calcaneal osteotomy | NA | NA | 0/1 | 0/1 | HMN (1) | No 1(1) | Italia (1) |
| Lassuthova et al. 21 | c.110G>C (p.Arg37Pro) | hom | 17 (18) | NA | 7.5 (1–12) | 13/16 | 5/10 (8 NA) | NA | 9/11 | NA | HMSN (9), HMN (2), NA (7) | NA | Czech Republic (18) |
| c.110G>C (p.Arg37Pro) + c.316C>T (p.Gln106Ter) | comp het | 2 (3) | NA | 7 (4–10) (1 NA) | 2/2 (1 NA) | NA | NA | NA | NA | NA (3) | NA | Czech Republic (3) | |
| Veltsista and Chroni 27 | c.110G>C (p.Arg37Pro) | hom | 1(2) | 2–0 | 10.5 (4–17) | NA | NA | + (2) | 2/2 | 2/2 | HMN (2) | No 1(2) | Greece (2) |
| Rauchenzauner et al. 26 | c.100G>A (p.Glu34Lys) | hom | 1 (1) | 0–1 | 3 | NA | NA | + (1) | 1/1 | 1/1 | HMSN (1) | No 1(1) | Austria (1) |
| Meng et al. 14 | c.112 T>C (p.Cys38Arg) + c.171G>C (p.Lys57Asn) | comp het | 1 (1) | 0–1 | 2 | NA | NA | + (1) | 1/1 | 1/1 | HMN (1) | No 1(1) | China (1) |
| c.112 T>C (p.Cys38Arg) | hom | 1 (1) | 1–0 | 10 | NA | NA | NA | 0/1 | 0/1 | HMSN (1) | No 1(1) | China (1) | |
| c.112 T>C (p.Cys38Arg) + c.98T>C (p.Phe33Ser) | comp het | 1 (1) | 1–0 | 16 | NA | NA | + (1) | 1/1 | 1/1 | HMSN (1) | No 1(1) | China (1) | |
| Wang et al. 25 | c.112 T>C (p.Cys38Arg) | hom | 1 (1) | 0–1 | 15 | third brachymetatarsus | NA | + (1) | 1/1 | 1/1 | HMN (1) | Yes 1(1) | China (1) |
| c.289G>A (p.Val97Met) | hom | 1 (1) | 1–0 | 13 | pes cavus | NA | + (1) | 1/1 | 1/1 | HMN (1) | Yes 1(1) | China (1) | |
| Scarpini et al. 24 | c.110G>C (p.Arg37Pro) | hom | 1 (1) | 1–0 | 1 | pes cavus | NA | + (1) | 0/1 | 0/1 | HMSN (1) | Yes 1(1) | Italia (1) |
| Shchagina et al. 15 | c.110G>C (p.Arg37Pro) | hom | 29 (29) | NA | NA | NA | NA | NA | NA | NA | HMSN (28) NA (1) | NA | Western Russia (25), Russian Far East (2), Middle‐south Russia (2) |
| c.110G>C (p.Arg37Pro) + c.112‐1delG | comp het | 1 (1) | NA | NA | NA | NA | NA | NA | NA | HMSN (1) | NA | Western Russia (1) | |
| c.110G>C (p.Arg37Pro) + c. 281 A>G (p.Tyr94Cys) | comp het | 1 (1) | NA | NA | NA | NA | NA | NA | NA | HMSN (1) | NA | Middle‐south Russia (1) | |
| Xu et al. 17 | c.356C>T (p.Arg119Trp) | hom | 1(1) | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Rossi et al. 29 | c.110G>C (p.Arg37Pro) | hom | 1 (1) | 1–0 | 14 | NA | NA | NA | NA | NA | HMSN (1) | NA | Romania (1) |
| Lin et al. 16 | c.278G>A (p.Gly93Asp) + c.289G>A (p.Val97Met) | comp het | 1(1) | 0–1 | 5 | NA | NA | NA | 1/1 | NA | NA | NA | China (1) |
| c.112 T>C (p.Cys38Arg) + c.278G>A (p.Gly93Asp) | comp het | 1(1) | 1–0 | 7 | NA | NA | NA | 1/1 | NA | NA | NA | China (1) | |
| de Aguiar Coelho Silva Madeiro et al. 12 | c.110G>C (p.Arg37Pro) | hom | 1 (1) | 1–0 | 8 | pes cavus | NA | + (1) | 0/1 | 1/1 | HMN (1) | No 1(1) | Brazil (1) |
| Amor‐Barris et al. 11 | c.110G>C (p.Arg37Pro) + c.284G>A (p.Arg95Gln) | comp het | 2 (2) | 20 | 13.5 (6–20) | 1/2 | NA | NA | 0/2 | 0/2 | HMSN (2) | No 2(2) | Norway (2) |
| Kontogeorgiou et al. 28 | c.110G>C (p.Arg37Pro) | hom | 2 (2) | 1–1 | 14.5 (14–15) | 1/2 | NA | NA | 2/2 | 2/2 | HMN (1) ‐ HMSN (1) | No 2(2) | Greece (2) |
| c.110G>C (p.Arg37Pro) + c.250T>C (p.Cys84Arg) | comp het | 2 (2) | 1–1 | 10.5 (8–13) | 2/2 | NA | NA | 2/2 | 2/2 | HMN (2) | No 2(2) | Greece (2) | |
| Gentile et al. 23 | c.110G>C (p.Arg37Pro) | hom | 1 (1) | 0–1 | 10 | shortening of Achille's tendons | NA | NA | NA | NA | HMSN | No 1(1) | Eastern Europe origin (1) |
| Fang et al. 13 | c.278G>T (p.Gly93Val) | hom | 1 (1) | 0–1 | 30 | Pes cavus | NA | NA | 0/1 | 0/1 | HMN | Yes 1(1) | China (1) |
| Our patients | c.110G>C (p.Arg37Pro) | hom | 3(3) | 2–1 | 11.3 (4–24) | Pes planus (1), Pes cavus(1), NA (1) | 1/3 | + (1), NA (2) | 3/3 | 2/2, 1 (NA) | HMSN (2), HMN (1) | Yes 2(2) No 1(1) | Austria (1), Kosovo (1), France (1) |
| c.334C>A (p.His112Asn) | hom | 2(2) | 1–1 | 5 (2–8) | Pes planus (1), short Achille's tendons (2) | 1/2 | + (2) | 2/2 | 1/1 1 (NA) | HMSN (1), HMN (1) | Yes 2(2) | Portugal (1), French roma (2) | |
| c.310G>C (p.Gly104Arg) | hom | 1(1) | 1–0 | 7 | Pes cavus, short Achille's tendons | 0/1 | + (1) | 0/1 | 0/1 | HMN (1) | Yes 1(1) | Algeria (1) |
Abbreviations: Comp. het., compound heterozygote; F, female; Hom, homozygous; HMSN, hereditary motor sensory neuropathy; HMN, hereditary motor neuropathy; M, male; NA, not available; NM, neuromyotonia; +, increased; −, normal.
APPENDIX B. Exhaustive description of P1‐P5, P7
B.1. PATIENT 1
First patient was a 26‐year‐old Austrian female. She acquired walking at a normal age [12 months]. Her symptoms started at the age of 4 years old with internally rotated feet and difficulties in stretching her knees and elbows. She has stiffness, cramps, muscle retractions. She also has flat feet, scoliosis, and distal muscular atrophy of all 4 limbs.
She is impaired in fine motor skills and complains of pain in her arms and hands.
At 15 years of age, the patient noticed that walking on her heels was impossible. At 16 years old, she was fatigued during exercise and fell a few times a month. She is followed by a physiotherapist once a week.
We can notice an asymmetrical disorder predominating on the lower left part of her face, present since childhood.
Achilles deep tendon reflexes (DTR) are bilaterally abolished, the patellas DTR are diminished, and they are normal in the upper limbs. The patient presents a picture of pure axonal motor neuropathy. However, this patient has numerous retractions (knees, elbows, digital flexors) as well as distal amyotrophy of all 4 limbs. The EMNG is in favor of a chronic distal motor neuropathy.
The patient underwent a muscle biopsy, which was in favor of a muscle denervation/reinnervation formula, probably corresponding to neuropathic or even motoneuron damage.
We did not report any psychiatric disorder or psychomotor development, although we did note dyslexia which required follow‐up by a speech therapist. Brain MRI was normal and serum CK level was increased to 339 IU.
This clinical picture first made us think of a distal spinal muscular atrophy but the various genetic examinations did not allow us to find a genetic origin to her pathology. Then, after a few years of diagnostic delay, an exome study allowed to find a homozygous mutation c.110G>C p.(Arg37Pro) in the HINT1 gene. The diagnosis of neuropathy linked to HINT1 could thus be made, even in the absence of neuromyotonia.
B.2. PATIENT 2
Patient 2 is a 23‐year‐old Portuguese male, from a consanguineous relationship. Walking was acquired at 13 months of age but first symptoms appeared around 2 years of age. Psychomotor development was not normal, and this patient had an intellectual disability that was not assessed. Because of his difficulties at school, this patient followed a specialized schooling.
He presents a bilateral steppage when walking but does not need help. He also has distal amyotrophy in leading to a decrease in motor strength and no DTR is seen in all 4 limbs. There are flat feet with short Achilles tendons, muscle stiffness and scoliosis. He is followed twice a week by a physiotherapist.
Serum CK level is increased (620 IU/L), brain MRI is normal.
The presence of clinical neuromyotonia was confirmed by ENMG. This examination reveals an axonal sensitivomotor neuropathy.
Molecular analysis show a homozygous mutation of the HINT1 gene c.334C>A p.(His112Asn).
Two of her cousins, also from a consanguineous union, would also have a similar clinical picture, but we have no further information.
B.3. PATIENT 3
He is a 37‐year‐old man from Kosovo, with no consanguinity in the family. The psychomotor development was normal and the first symptoms appeared at the age of 24. Clinically, the patient presented with distal involvement of the lower limbs characterized by progressive weakness, severe muscle amyotrophy (anterior and lateral compartment of the legs) and imbalance (positive Romberg sign). The patient walks with bilateral steppage but does not require assistance and there is no limitation of walking. He works in the building trade and requires weekly follow‐up by a physiotherapist. He has localized cramps in his thighs, and sensations of stiffness and muscular blocking. This patient has pes cavus with moderate achilleic retraction. The DTR are abolished in all 4 limbs.
Sensory impairment was also present with bilateral hypoesthesia and hypopallesthesia from the knee to the feet.
Genetic analysis identified the c.110G>C mutation p.(Arg37Pro) in the homozygous state.
B.4. PATIENT 4
This 17‐year‐old male, second child of an unrelated couple, presents a mixed length‐dependent sensitivomotor polyneuropathy associated with neuromyotonia. The first symptoms appeared very early at the time of walking acquisition around 20 months. Falls were frequent and are still present today. Motor difficulties became more pronounced around the age of 6 years with the appearance of excess weight. There is a bowing gait with hip limp and motor deficits in the lower limbs. The patient also presents a hyperlordosis and a hyperlaxity of the hip. The unipodal station is unstable.
In the upper limbs, there is an irregular tremor on the finger‐nose test and a bilateral hollow hand.
DTR are absent in all 4 limbs.
Clinical symptoms suggesting myotonia are noted. ENMG reveals axonal motor and sensory neuropathy associated with neuromyotonia. Serum CK level is high (923 IU/L).
Some clinical signs are to be noted in this patient. He presents a deformation of the toes, as well as chronic constipation. In addition, this young man presented difficulties in school learning, and he needs a Learning Support Assistant. Concerning his mild intellectual deficiency, no IQ test had been performed nor genetic testing. There is no cerebral MRI available for him.
We can note psychiatric features. Since he was 13 year, he is treated for depression.
Genetic analysis identified the c.110G>C mutation p.(Arg37Pro) in the homozygous state.
B.5. PATIENT 5
This 14‐year‐old is the only daughter of cousin couple of rom origin. She initially presented a psychomotor developmental disorder with a delayed walking age (24 months) and language acquired at 4 years of age. She has an intellectual deficiency with an IQ of 50.
Clinically, she has muscular amyotrophy of the lower limbs with a decrease in motor strength rated at 4/5. She also has clinical neuromyotonia and short Achille's tendons. Serum CK level is slightly increased at 272 IU/L. DTR are abolished in all four limbs. The ENMG is in favor of a pure axonal motor neuropathy. Symptom suggesting myotonia and neuromyotonia are observed.
This patient also presents psychiatric disorders such as generalized anxiety disorders and obsessive–compulsive disorders. No cerebral MRI is available for her because she is claustrophobic.
Molecular analysis revealed a homozygous mutation of the HINT1 gene c.334C>A p.(His112Asn).
B.6. PATIENT 7
He is a young man from French roma family. There is no family consanguinity. This patient had a delayed language acquisition. Ability to walk was acquired at a normal age. However, the first motor symptoms appeared shortly afterwards with falls.
At 19 year, he presents an abolition of DTR in the 4 limbs with a decreased motor strength in the lower limbs. It is quoted at 3/5 in distality. The patient complains of cramps. Pes cavus are noted. There is a mild ID but no IQ test had been performed. No other clinical particularity is reported.
Serum CK level is within the norms. The ENMG shows a motor polyneuropathy of the 4 limbs, predominantly in the lower limbs. There is no neuromyotonia on ENMG.
A genetic exploration found the c.334C>A p.(His112Asn) variant in HINT1.
Morel V, Campana‐Salort E, Boyer A, et al. HINT1 neuropathy: Expanding the genotype and phenotype spectrum. Clinical Genetics. 2022;102(5):379‐390. doi: 10.1111/cge.14198
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. Benquey T, Pion E, Cossée M, et al. A National French Consensus on gene list for the diagnosis of Charcot‐Marie‐tooth disease and related disorders using next‐generation sequencing. Genes. 2022;13(2). doi: 10.3390/genes13020318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zimoń M, Baets J, Almeida‐Souza L, et al. Loss‐of‐function mutations in HINT1 cause axonal neuropathy with neuromyotonia. Nat Genet. 2012;44(10):1080‐1083. doi: 10.1038/ng.2406 [DOI] [PubMed] [Google Scholar]
- 3. Weiske J, Huber O. The histidine triad protein Hint1 triggers apoptosis independent of its enzymatic activity. J Biol Chem. 2006;281(37):27356‐27366. doi: 10.1074/jbc.M513452200 [DOI] [PubMed] [Google Scholar]
- 4. Zambelli D, Zuntini M, Nardi F, et al. Biological indicators of prognosis in Ewing's sarcoma: an emerging role for lectin galactoside‐binding soluble 3 binding protein (LGALS3BP). Int J Cancer. 2010;126(1):41‐52. doi: 10.1002/ijc.24670 [DOI] [PubMed] [Google Scholar]
- 5. Schöler J, Ferralli J, Thiry S, Chiquet‐Ehrismann R. The intracellular domain of teneurin‐1 induces the activity of microphthalmia‐associated transcription factor (MITF) by binding to transcriptional repressor HINT1. J Biol Chem. 2015;290(13):8154‐8165. doi: 10.1074/jbc.M114.615922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cortés‐Montero E, Rodríguez‐Muñoz M, Sánchez‐Blázquez P, Garzón J. The axonal motor neuropathy‐related HINT1 protein is a zinc‐ and calmodulin‐regulated cysteine SUMO protease. Antioxid Redox Signal. 2019;31(7):503‐520. doi: 10.1089/ars.2019.7724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cortés‐Montero E, Rodríguez‐Muñoz M, Sánchez‐Blázquez P, Garzón‐Niño J. Human HINT1 mutant proteins that cause axonal motor neuropathy exhibit anomalous interactions with partner proteins. Mol Neurobiol. 2021;58(4):1834‐1845. doi: 10.1007/s12035-020-02265-x [DOI] [PubMed] [Google Scholar]
- 8. Rodríguez‐Muñoz M, Cortés‐Montero E, Onetti Y, Sánchez‐Blázquez P, Garzón‐Niño J. The σ1 receptor and the HINT1 protein control α2δ1 binding to glutamate NMDA receptors: implications in neuropathic pain. Biomolecules. 2021;11(11):1681. doi: 10.3390/biom11111681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jackson KJ, Chen Q, Chen J, Aggen SH, Kendler KS, Chen X. Association of the histidine‐triad nucleotide‐binding protein‐1 (HINT1) gene variants with nicotine dependence. Pharmacogenomics J. 2011;11(4):251‐257. doi: 10.1038/tpj.2010.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abdel Rassoul R, Alves S, Pantesco V, et al. Distinct transcriptome expression of the temporal cortex of the primate Microcebus murinus during brain aging versus Alzheimer's disease‐like pathology. PloS One. 2010;5(9):e12770. doi: 10.1371/journal.pone.0012770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amor‐Barris S, Høyer H, Brauteset LV, et al. HINT1 neuropathy in Norway: clinical, genetic and functional profiling. Orphanet J Rare Dis. 2021;16(1):116. doi: 10.1186/s13023-021-01746-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Aguiar Coelho Silva Madeiro B, Peeters K, Santos de Lima EL, et al. HINT1 founder mutation causing axonal neuropathy with neuromyotonia in South America: A case report. Mol Genet Genomic Med. 2021;9(10):e1783. doi: 10.1002/mgg3.1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fang J, Huang H, Lei Q, et al. Myasthenia gravis coexisting with HINT1‐related motor axonal neuropathy without neuromyotonia: A case report. BMC Neurol. 2022;22(1):168. doi: 10.1186/s12883-022-02690-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meng L, Fu J, Lv H, Zhang W, Wang Z, Yuan Y. Novel mutations in HINT1 gene cause autosomal recessive axonal neuropathy with neuromyotonia in two cases of sensorimotor neuropathy and one case of motor neuropathy. Neuromuscul Disord NMD. 2018;28(8):646‐651. doi: 10.1016/j.nmd.2018.05.003 [DOI] [PubMed] [Google Scholar]
- 15. Shchagina OA, Milovidova TB, Murtazina AF, et al. HINT1 gene pathogenic variants: the most common cause of recessive hereditary motor and sensory neuropathies in Russian patients. Mol Biol Rep. 2020;47(2):1331‐1337. doi: 10.1007/s11033-019-05238-z [DOI] [PubMed] [Google Scholar]
- 16. Lin S, Xu L‐Q, Xu G‐R, et al. Whole exome sequencing reveals a broader variant spectrum of Charcot‐Marie‐tooth disease type 2. Neurogenetics. 2020;21(2):79‐86. doi: 10.1007/s10048-019-00591-4 [DOI] [PubMed] [Google Scholar]
- 17. Xu J, Yang Y, Liu Y. Analysis of HINT1 gene variant in a case with neuromyotonia and axonal neuropathy. J Med Genet. 2019;36(8):817‐820. doi: 10.3760/cma.j.issn.1003-9406.2019.08.016 [DOI] [PubMed] [Google Scholar]
- 18. Horga A, Cottenie E, Tomaselli PJ, et al. Absence of HINT1 mutations in a UKand Spanish cohort of patients with inherited neuropathies. J Neurol. 2015;262(8):1984‐1986. doi: 10.1007/s00415-015-7851-z [DOI] [PubMed] [Google Scholar]
- 19. Zhao H, Race V, Matthijs G, et al. Exome sequencing reveals HINT1 mutations as a cause of distal hereditary motor neuropathy. Eur J Human Genet EJHG. 2014;22(6):847‐850. doi: 10.1038/ejhg.2013.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jerath NU, Shy ME, Grider T, Gutmann L. A case of neuromyotonia and axonal motor neuropathy: A report of a HINT1 mutation in the United States. Muscle Nerve. 2015;52(6):1110‐1113. doi: 10.1002/mus.24774 [DOI] [PubMed] [Google Scholar]
- 21. Laššuthová P, Brožková DŠ, Krůtová M, et al. Mutations in HINT1 are one of the most frequent causes of hereditary neuropathy among Czech patients and neuromyotonia is rather an underdiagnosed symptom. Neurogenetics. 2015;16(1):43‐54. doi: 10.1007/s10048-014-0427-8 [DOI] [PubMed] [Google Scholar]
- 22. Boaretto F, Cacciavillani M, Mostacciuolo ML, et al. Novel loss‐of‐function mutation of the HINT1 gene in a patient with distal motor axonal neuropathy without neuromyotonia. Muscle Nerve. 2015;52(4):688‐689. doi: 10.1002/mus.24720 [DOI] [PubMed] [Google Scholar]
- 23. Gentile L, Russo M, Taioli F, et al. Rare among rare: phenotypes of uncommon CMT genotypes. Brain Sci. 2021;11(12):1616. doi: 10.3390/brainsci11121616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scarpini G, Spagnoli C, Salerno GG, Rizzi S, Frattini D, Fusco C. Autosomal recessive axonal neuropathy caused by HINT1 mutation: new association of a psychiatric disorder to the neurologic phenotype. Neuromuscul Disorders NMD. 2019;29(12):979. doi: 10.1016/j.nmd.2019.05.001 [DOI] [PubMed] [Google Scholar]
- 25. Wang Z, Lin J, Qiao K, et al. Novel mutations in HINT1 gene cause the autosomal recessive axonal neuropathy with neuromyotonia. Eur J Med Genet. 2019;62(3):190‐194. doi: 10.1016/j.ejmg.2018.07.009 [DOI] [PubMed] [Google Scholar]
- 26. Rauchenzauner M, Frühwirth M, Hecht M, Kofler M, Witsch‐Baumgartner M, Fauth C. A novel variant in the HINT1 gene in a girl with autosomal recessive axonal neuropathy with neuromyotonia: thorough neurological examination gives the clue. Neuropediatrics. 2016;47(2):119‐122. doi: 10.1055/s-0035-1570493 [DOI] [PubMed] [Google Scholar]
- 27. Veltsista D, Chroni E. A first case report of HINT1‐related axonal neuropathy with neuromyotonia in a Greek family. Clin Neurol Neurosurg. 2016;148:85‐87. doi: 10.1016/j.clineuro.2016.07.012 [DOI] [PubMed] [Google Scholar]
- 28. Kontogeorgiou Z, Voudommatis C, Kartanou C, et al. HINT1‐related neuropathy in Greek patients with Charcot‐Marie‐tooth disease. J Peripheral Nervous Syst JPNS. 2021;26(4):444‐448. doi: 10.1111/jns.12473 [DOI] [PubMed] [Google Scholar]
- 29. Rossi S, Perna A, Modoni A, et al. Response to “Autosomal recessive axonal neuropathy caused by HINT1 mutation: new association of a psychiatric disorder to the neurological phenotype.”. Neuromuscul Disord NMD. 2020;30(3):265‐266. doi: 10.1016/j.nmd.2020.01.003 [DOI] [PubMed] [Google Scholar]
- 30. Baron M. Genetics of schizophrenia and the new millennium: progress and pitfalls. Am J Hum Genet. 2001;68(2):299‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Varadarajulu J, Lebar M, Krishnamoorthy G, et al. Increased anxiety‐related behaviour in Hint1 knockout mice. Behav Brain Res. 2011;220(2):305‐311. doi: 10.1016/j.bbr.2011.02.012 [DOI] [PubMed] [Google Scholar]
- 32. Lei G, Liu F, Liu P, et al. Does genetic mouse model of constitutive Hint1 deficiency exhibit schizophrenia‐like behaviors? Schizophr Res. 2020;222:304‐318. doi: 10.1016/j.schres.2020.05.018 [DOI] [PubMed] [Google Scholar]
- 33. Zhou Y, Li S, Deng L, Ma Y, Lei G, Dang Y. HINT1 deficiency in aged mice reduces anxiety‐like and depression‐like behaviours and enhances cognitive performances. Exp Gerontol. 2022;159:111683. doi: 10.1016/j.exger.2021.111683 [DOI] [PubMed] [Google Scholar]
- 34. Chen Q, Wang X, O'Neill FA, Walsh D, Kendler KS, Chen X. Is the histidine triad nucleotide‐binding protein 1 (HINT1) gene a candidate for schizophrenia? Schizophr Res. 2008;106(2–3):200‐207. doi: 10.1016/j.schres.2008.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dang Y‐H, Liu P, Ma R, et al. HINT1 is involved in the behavioral abnormalities induced by social isolation rearing. Neurosci Lett. 2015;607:40‐45. doi: 10.1016/j.neulet.2015.08.026 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Research data are not shared.