

Abstract
Background
Understanding complex influences on mental health problems in young people is needed to inform early prevention strategies. Both genetic and environmental factors are known to influence youth mental health, but a more comprehensive picture of their interplay, including wide‐ranging environmental exposures – that is, the exposome – is needed. We perform an integrative analysis of genomic and exposomic data in relation to internalizing and externalizing symptoms in a cohort of 4,314 unrelated youth from the Adolescent Brain and Cognitive Development (ABCD) Study.
Methods
Using novel GREML‐based approaches, we model the variance in internalizing and externalizing symptoms explained by additive and interactive influences from the genome (G) and modeled exposome (E) consisting of up to 133 variables at the family, peer, school, neighborhood, life event, and broader environmental levels, including genome‐by‐exposome (G × E) and exposome‐by‐exposome (E × E) effects.
Results
A best‐fitting integrative model with G, E, and G × E components explained 35% and 63% of variance in youth internalizing and externalizing symptoms, respectively. Youth in the top quintile of model‐predicted risk accounted for the majority of individuals with clinically elevated symptoms at follow‐up (60% for internalizing; 72% for externalizing). Of note, different domains of environmental exposures were most impactful for internalizing (life events) and externalizing (contextual including family, school, and peer‐level factors) symptoms. In addition, variance explained by G × E contributions was substantially larger for externalizing (33%) than internalizing (13%) symptoms.
Conclusions
Advanced statistical genetic methods in a longitudinal cohort of youth can be leveraged to address fundamental questions about the role of ‘nature and nurture’ in developmental psychopathology.
Keywords: Exposome, genetics, youth mental health, depression, gene–environment interaction, G × E, heritability
Introduction
Young people are at elevated risk of mental health problems, with epidemiological data suggesting about half of all mental disorders begin by mid‐adolescence (Kessler et al., 2007). Whether internalizing (e.g. depression/anxiety) or externalizing (e.g. problem behavior) in nature, early‐onset mental health problems are associated with poor lifelong outcomes including recurring psychiatric difficulties, suicidal behavior, and impaired functioning (Clayborne, Varin, & Colman, 2019; Newman et al., 1996; Woodward & Fergusson, 2001). As such, understanding complex influences on mental health problems in young people is needed to inform strategies for preventing these problems and their downstream consequences.
Broadly speaking, it is known that both genetic and environmental factors play a role in influencing mental health among youth (Kwong et al., 2019; Nikstat & Riemann, 2020). However, a complete picture remains obscured by the limited focus of prior studies. First, while psychiatric genetic research has benefited from moving to genome‐wide approaches (Sullivan & Kendler, 2021), comprehensive analyses of environmental influences have been relatively sparse. Efforts to model environmental influences have typically captured one or a handful of exposures (e.g. childhood trauma) rather than a wider range of factors that could influence mental health (Guloksuz, van Os, & Rutten, 2018). The exposome, defined as the totality of environmental exposures and their interactions (Wild, 2012), is critical for understanding human health (Manrai et al., 2017). Although our recent work has demonstrated the utility of exposure‐wide approaches in depression research (Choi et al., 2020), the cumulative exposome has remained challenging to model. Second, assessments of how environmental exposures interact with genome‐wide influences to shape mental health (Uher & Zwicker, 2017) are limited, particularly in youth; existing genome‐wide environment interaction studies of depression, for example, have been conducted primarily in adult populations (e.g. Arnau‐Soler et al., 2019; Coleman et al., 2020; Dunn et al., 2016). If an interaction exists, this suggests that genetic influences on youth mental health vary by environmental factors and/or that environmental influences differ according to genetic profiles, pointing to more precise understanding of which individuals may be most susceptible or protected. Third, potential sources of bias in gene–environment studies, such as multicollinearity between environmental exposures which is common in psychosocial data and known to bias variance estimates (Zhou & Lee, 2021), must be explicitly addressed.
Recent methods in statistical genetics can help overcome these gaps by allowing for the integrative modeling of genome‐wide (genomic) and exposure‐wide (exposomic) influences and their interactions, while accounting for potential estimation biases (Zhou & Lee, 2021). Briefly, these methods rely on characterizing genome‐wide similarity among pairs of unrelated individuals against their phenotypic similarity and can now be extended to the estimation of exposure‐wide similarities (Zhou & Lee, 2021). The Adolescent Brain and Cognitive Development (ABCD) Study – which includes genomic data, wide‐ranging environmental exposures, and rich longitudinal measurements of mental health in a cohort of more than 10,000 youth (Jernigan, Brown, & ABCD Consortium Coordinators, 2018) – provides an important opportunity to conduct these comprehensive analyses. Here, we leveraged these data to further address fundamental questions about the role of ‘nature and nurture’ in developmental psychopathology. Using a novel linear mixed model approach, we sought to decompose the main and interactive effects of the genome and exposome on internalizing and externalizing symptoms in preadolescent youth. Advancing our understanding of the complex interplay of genes and environments in youth mental health may inform for whom and how best to focus preventative efforts.
Methods
Sample
The Adolescent Brain and Cognitive Development (ABCD) Study is a multisite longitudinal cohort study following over 10,000 adolescents beginning at the ages of 9–10. ABCD participants were recruited through schools across 21 sites in the United States using multistage probability sampling to capture a nationally representative sample (Garavan et al., 2018). The study collected data from youth and parents across major domains including substance use, mental and physical health, neurocognition, culture and environment, and brain imaging (Karcher & Barch, 2021). More information regarding ABCD study recruitment, design, and measures can be found elsewhere (Karcher & Barch, 2021). Ethics approval for the parent study was obtained from all relevant institutional review boards, with written informed consent obtained from parents and verbal assent obtained from youth participants; secondary analyses of the publicly released, anonymized ABCD data were deemed by our local review board as not human subjects research. For this study, we examined the subsample with genomic data (see Appendix S1 for genomic QC and imputation details). Because estimates of whole‐genome effects can be confounded by population stratification, we focused on the largest European ancestry subgroup for primary demonstration (n = 5,369), retaining 6,834,371 variants with imputation INFO score ≥0.80 and minor allele frequency ≥1%. The top 10 genetic principal components (PCs) were used to adjust for residual stratification in subsequent analyses. We additionally retained one randomly selected individual from each related pair (pi‐hat > 0.20) resulting in a sample of genetically unrelated individuals. Our analytic sample consisted of 4,314 genetically unrelated youth with genomic data and baseline and 1‐year follow‐up symptom scores [47% female, mean age (SD; range) = 9.9 (0.6; 8.9–11.0) years] based on the ABCD 3.0 release. To leverage the diversity of the ABCD cohort, we also identified participants from non‐European groups with at least 1,000 unrelated individuals – of African (n = 1,125) and admixed American (n = 1,474) ancestry, respectively – to examine the consistency of our results.
Measures
Youth internalizing and externalizing symptoms
The parent‐completed Child Behavior Checklist (CBCL; Achenbach, 2001) was used to index current youth internalizing and externalizing symptoms. The internalizing composite scale of the CBCL comprises the following subscale domains: Anxious/Depressed (e.g. cries a lot, feels worthless or inferior), Withdrawn/Depressed (e.g. very little he/she enjoys, underactive, sad, depressed), and Somatic Complaints (e.g. headaches, nausea, feels sick, stomachaches). The externalizing composite scale of the CBCL comprises the following subscale domains: Rule‐Breaking Behavior (e.g. lying or cheating, swearing or obscene language) and Aggressive Behavior (e.g. argues a lot, destroys his/her own things). For our main analyses, we used continuous internalizing (INT) and externalizing (EXT) T‐scores (typically scaled at mean 50 and SD 10) at the 1‐year follow‐up assessment. For sensitivity analyses, we also created a dichotomous indicator of clinically elevated symptoms at baseline, that is, T‐scores ≥70 (Pandolfi, Magyar, & Dill, 2012).
Environmental exposures
Environmental exposures in our modeled exposome consisted of 133 survey‐based and/or derived variables (Tables S1 and S2) including proximal contextual variables (which may influence youth’s day‐to‐day experiences directly, for example, peer, family, and school factors; k = 36), broader contextual variables (which reflect broader environments surrounding youth, for example, neighborhood and geocoded environmental factors; k = 49), and life events variables (which include potential stressors, for example, injury, illness, loss, and family conflict; k = 48). All exposures were measured at baseline except life events, which were reported at 1‐year follow‐up but pertained to discrete events occurring prior to follow‐up, so were also included. We included variables reported by parents and/or youth where available, which may tap into different risk and protective experiences. Following data preprocessing (Appendix S2), we observed low missingness (0%–4%) among exposures and imputed these values for sample size consistency using the missForest package in R (Stekhoven & Bühlmann, 2011), a nonparametric method that efficiently handles mixed variable types and performs imputation by fitting iterative random forests (k = 100) on observed data to predict missing data.
Statistical analyses
Integrative analysis of genomic and exposomic data (IGE)
Using linear mixed models with genome‐based restricted maximum likelihood (GREML), we performed a comprehensive decomposition of genomic and exposomic influences on our two outcomes of interest: internalizing (INT) and externalizing (EXT) symptom scores at the 1‐year follow‐up. Scores were preadjusted (i.e. taking residuals of a regression model adjusted for the following variables) for youth age at baseline, sex, study site, and top 10 ancestry‐specific genetic PCs, then standardized to mean 0 and SD 1. To estimate the proportion of outcome variance attributable to additive and nonadditive (statistical) effects of the whole genome and modeled exposome – including interactions between exposome and genome, and between exposomic variables – we constructed a set of relationship matrices (Appendix S3 for matrix equations).
First, a relatedness matrix capturing genome‐wide variation (G) across unrelated individuals was generated in PLINK 1.9 (Chang et al., 2015). Second, the exposome matrix (E) was generated using MTG2 v2.22 (Lee & van der Werf, 2016) after transformation of the exposure data using principal components analysis (PCA). Specifically, we extracted the eigenvectors of the k × k covariance matrix of exposures (equivalent to the right singular vectors of the n × k exposure matrix) via eigen decomposition. We then multiplied the exposure matrix by the eigenvectors to produce a column‐orthogonal, and subsequently column‐standardized, matrix for fitting the linear mixed models. For greater detail, see Zhou and Lee (2021). For the E matrix, we used k = 133 environmental factors that included proximal contextual variables, broader contextual variables, and prior life events described above. Third, the genome‐by‐exposome interactions matrix (G × E) was computed as the Hadamard product of the transformed exposome matrix and genomic relatedness matrix. Fourth, an exposome‐by‐exposome interactions matrix (E × E) was generated from the pairwise products between all environmental factors, again pretransformed to account for potential multicollinearity. Coding procedures, including steps to pretransform exposures and generate relationship matrices, are outlined in detail in the MTG2 manual (https://sites.google.com/view/s‐hong‐lee‐homepage/mtg2).
Variance in INT or EXT symptoms explained by these components was estimated in the following nested models: (a) G only; (b) G and E; (c) G, E, and G × E; and (d) G, E, G × E, and E × E. Each model was statistically evaluated against the previous model using likelihood ratio tests, with a significant result (p < .0083 = .05/6 based on three model comparisons for two outcomes), indicating model fit improvement due to the additional variance component. The model after which no significant improvement was detected was considered the best‐fitting model. Variance component estimates whose 95% confidence intervals did not contain zero were interpreted as significant. We then used a fivefold cross‐validation approach to index the predictive accuracy of the best‐fitting model for each outcome by generating model‐predicted values in 80% of the sample (training) and calculating Pearson correlations with observed outcome values in the remaining 20% holdout sample (testing), averaging these correlations across five nonoverlapping random splits, as in prior work (Zhou & Lee, 2021). For further interpretation, we divided individuals in each testing set based on quintiles of model‐predicted values (based on best linear unbiased predictions summed across all random effect components) and, using the metafor R package, meta‐analyzed the beta increase in standardized symptom scores associated with membership in each prediction quintile compared to the bottom reference quintile. We also calculated the average risk concentration (i.e. proportion of all youth with clinically elevated symptoms at follow‐up; n = 154 for INT and n = 81 for EXT) represented across model‐predicted quintiles. We then performed sensitivity analyses to evaluate the best‐fitting model and its variance components for consistency: (i) after removing individuals with clinically elevated symptoms at baseline (n = 140 for INT and n = 75 for EXT), (ii) when adjusting for parental educational attainment, (iii) including perinatal exposures, or (iv) without pretransforming exposures for independence. We also assessed potential differences by reporter source by modeling the exposome component based on child‐reported (k = 61) versus parent‐reported (k = 39) exposures. Though underpowered, we also evaluated the best‐fitting model in the admixed American and African ancestry groups.
Finally, where a significant interaction (e.g. G × E) variance component was observed, we further decomposed the E matrix into major subdomains – proximal contextual variables (k = 36), broader contextual variables (k = 49), and life events variables (k = 48) – to separately examine the influence of each subdomain and assess its potential role in the interaction. Lastly, we fitted the best‐fitting model including all subdomain exposome (and genome‐exposome) matrices simultaneously.
Results
Variance decomposition results for INT and EXT, estimating joint contributions of genomic and exposomic influences on mental health symptoms in ABCD participants, are reported in Table 1. First, when examined alone (G model), the G variance component () was modest and nonsignificant for INT symptoms (6%), but sizeable and significant for EXT symptoms (19%). Second, including additive effects of the exposome (G and E model) improved the genomics‐only model. Here, remained modest for INT (6%) but was markedly attenuated for EXT (8%), while the E variance component () was estimated at 16% for INT and 23% for EXT. Third, including interactive effects between the exposome and genome (G, E, and G × E model) showed further improvement over the previous model. Here, and remained virtually unchanged in magnitude, while the G × E variance component () was estimated to be 13% for INT and 33% for EXT, indicating sizeable unique variance explained by genome‐exposome interactions. Fourth, including nonadditive effects of the exposome (G, E, G × E, and E × E model) did not result in significant improvements. Here, the E × E variance component () accounted for <1% of phenotypic variance in INT and EXT symptoms, while other component estimates remained unchanged. Thus, the more parsimonious G, E, and G × E model was considered to be the best‐fitting model for both outcomes. The proportion of phenotypic variance in INT and EXT symptoms explained by each component in the best‐fitting model is visualized in Figure 1.
Table 1.
Genomic and exposomic variance components for youth internalizing and externalizing symptoms
| Model | G only | G and E | G, E, and G × E | G, E, G × E, and E × E | |
|---|---|---|---|---|---|
| INT | |||||
|
|
0.058 (−0.092, 0.208) | 0.06 (−0.069, 0.19) | 0.063 (−0.067, 0.191) | 0.063 (−0.066, 0.192) | |
|
|
0.161 (0.116, 0.206) a | 0.161 (0.115, 0.205) a | 0.161 (0.115, 0.205) a | ||
|
|
0.129 (0.042, 0.215) a | 0.126 (0.037, 0.215) a | |||
|
|
−0.002 (−0.024, 0.019) | ||||
| LKH | −2,160.3820 | −1,914.1290, p = 4.06e‐109 b | −1,909.7914, p = .0032 b | −1,909.7730, p = .848 | |
| EXT | |||||
|
|
0.192 (0.036, 0.347) a | 0.084 (−0.036, 0.203) | 0.074 (−0.042, 0.189) | 0.072 (−0.044, 0.187) | |
|
|
0.233 (0.171, 0.294) a | 0.226 (0.165, 0.286) a | 0.226 (0.165, 0.286) a | ||
|
|
0.327 (0.239, 0.414) a | 0.334 (0.242, 0.424) a | |||
|
|
0.005 (−0.014, 0.024) | ||||
| LKH | −2,157.5381 | −1,745.7653, p = 4.10e‐181 b | −1,715.3967, p = 6.52e‐15 b | −1,715.2634, p = .606 | |
σ 2 = variance components reflect proportions of total variance estimated by the model; residual (e) components not shown.
Nonzero overlapping 95% confidence interval.
Significant model likelihood ratio test compared to previous nested model.
Figure 1.
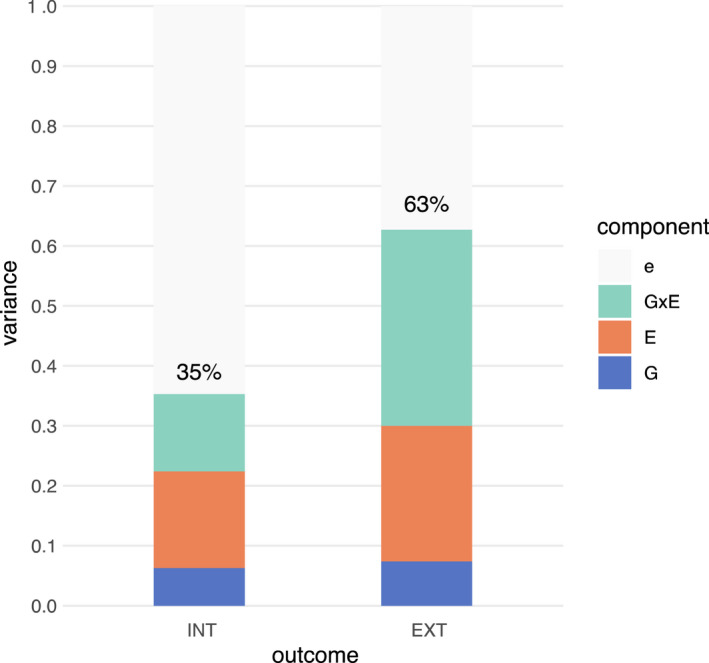
Estimated total proportion of variance in INT and EXT symptoms explained by G, E, and G × E components. e, residual variance; E, modeled exposome effects based on 133 exposure variables; G, genomic effects; G × E, genome‐by‐exposome effects [Colour figure can be viewed at wileyonlinelibrary.com]
In fivefold cross‐validation, we found that predicted values by the best model (G, E, and G × E) were significantly correlated with observed values of INT [fivefold average r = .36 (range = .32–.43), p < 2.2e‐16] and EXT [fivefold average r = .45 (range = .40–.51), p < 2.2e‐16] in the holdout sets. Individuals in the top quintile of model‐predicted values had symptom scores at least 1 SD or higher compared to those in the bottom quintile (B = 1.00 and 1.25 for INT and EXT, respectively; Figure 2A) in the holdout sets, translating to roughly 10–12 points higher on their CBCL T‐score. The upper quintiles of model‐predicted values also accounted for the large majority of youth with clinically elevated symptoms at follow‐up (81% for INT and 91% for EXT in the top two quintiles; 60% and 72% in the top quintile alone; Figure 2B).
Figure 2.
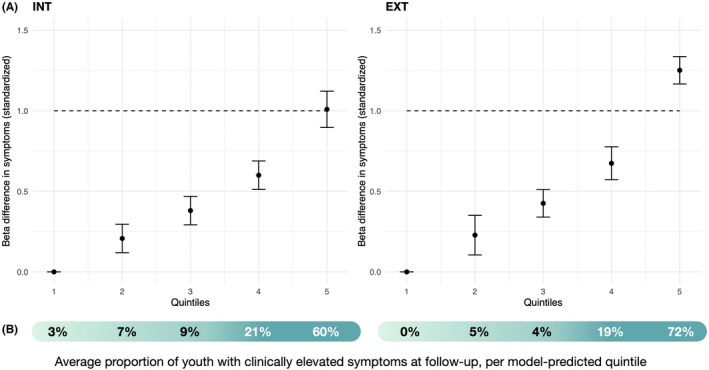
Integrative (G, E, and G × E) model predictions of elevated internalizing and externalizing symptoms at follow‐up. (A) Beta values associated with each model‐predicted quintile (meta‐analyzed across the fivefold holdout splits) are shown with 95% CI; individuals in quintile 1 of model‐predicted values are considered as the reference group. Dashed line shows 1 SD increase in symptoms. (B) Risk concentrations (i.e. proportions of all youth with clinically elevated symptoms for each follow‐up outcome) are shown in the shaded bar below for each quintile [Colour figure can be viewed at wileyonlinelibrary.com]
In sensitivity analyses, and were minimally attenuated when further adjusting for parental educational attainment (Table S3), did not increase substantially when including perinatal exposures (Table S4), and remained significant albeit slightly attenuated, by about 2%–4%, after excluding participants with elevated baseline symptoms (Table S5). When exposures were not initially transformed for independence, and were reduced but remained significant (Table S6). When separating child‐ and parent‐reported exposures (Table S7), significant and were observed for both outcomes regardless of child‐ or parent‐reported exposures, with slightly higher based on parent‐reported exposures and slightly higher based on child‐reported exposures. For the admixed American and African ancestry groups (Table S8), for both outcomes was similar to the European ancestry group; was qualitatively larger in the African ancestry group across both outcomes; and was significant for EXT but not INT in the admixed American ancestry group.
When decomposing the full E matrix and re‐estimating the G, E, and G × E model where E comprised only variables pertaining to each subdomain – proximal contextual factors, broader contextual factors, or life event factors – a significant persisted for all three subdomains (Table 2). Comparing across the three subdomains, the largest was observed when E comprised life events for INT (11%) and when E comprised proximal contextual variables for EXT (16%). For both outcomes, was smallest in magnitude when E comprised broader contextual variables (3% for INT; 5% for EXT), though still nonzero. Notably, was substantially reduced for EXT when jointly modeled with based on proximal contextual or life events factors, but remained relatively intact (i.e. significant) when jointly modeled with based on broader contextual variables. On the other hand, was only significant when E comprised either proximal contextual variables or life event variables, but not when E comprised broader contextual variables – with the largest based on E comprising proximal contextual variables (14% and 26% for INT and EXT, respectively). These results were consistent with simultaneous models (Tables S9 and S10) where, even considered together, all three exposome subdomains contributed significant across both outcomes, while proximal contextual and life event subdomains both contributed significant , but the broad contextual subdomain did not.
Table 2.
Decomposition of best‐model components based on exposomic subdomains
| Proximal contextual (k = 36) | Broader contextual (k = 49) | Life events (k = 48) | ||
|---|---|---|---|---|
| INT | ||||
|
|
0.053 (−0.085, 0.191) | 0.060 (−0.087, 0.206) b | 0.055 (−0.08, 0.189) | |
|
|
0.079 (0.039, 0.119) a | 0.029 (0.013, 0.045) a | 0.108 (0.06, 0.155) a , b | |
|
|
0.129 (0.058, 0.198) a , b | −0.012 (−0.04, 0.016) | 0.119 (0.061, 0.176) a | |
| EXT | ||||
|
|
0.079 (−0.044, 0.201) | 0.167 (0.019, 0.316) a , b | 0.134 (0, 0.268) | |
|
|
0.163 (0.084, 0.242) a , b | 0.045 (0.023, 0.067) a | 0.112 (0.062, 0.161) a | |
|
|
0.280 (0.21, 0.35) a , b | 0.038 (−0.026, 0.102) | 0.199 (0.138, 0.258) a | |
σ 2 = variance components reflect proportions of total variance estimated by the model; residual (e) components not shown.
Nonzero overlapping 95% confidence interval.
Largest variance explained by that component (row‐wise) across the three exposomic subdomains: Proximal contextual = E comprising 36 friends/caregiver/family/school variables; Broader contextual = E comprising 49 neighborhood/area‐level variables; Life events = E comprising 48 life event variables.
Discussion
In this study, we applied advanced statistical genetics methods in a longitudinal cohort of preadolescent youth to address fundamental questions about the role of ‘nature and nurture’ in developmental psychopathology. Because of our ability to explicitly model joint genomic and exposomic effects as well as their potential interactions, this represents to our knowledge the most comprehensive decomposition to date of genetic and environmental influences on mental health in youth using molecular genetic data.
Key findings
Several notable findings emerged. First, an integrative model including additive and interactive influences of the genome and exposome explained over 30% and 60% of variance in youth internalizing and externalizing symptoms at follow‐up, respectively. By contrast, a single variable with known impacts on externalizing behavior – parental separation/divorce (Garriga & Pennoni, 2020), which was associated with externalizing symptoms in our sample (p < 1e‐10 in adjusted linear regression) – accounted for only 1.1% of its variance. Moreover, joint models of polygenic risk and a small selection of environmental exposures in a recent study of 1,154 preadolescent youth in the Netherlands accounted for no more than 11%–12% variance of internalizing and externalizing symptoms (Ensink et al., 2020), highlighting the enhanced explanatory power of our integrative genomic and exposomic modeling. In fivefold cross‐validation, youth in the top quintile of predicted values from the integrative model had symptom T‐scores that were on average 10–12 points (at least 1 SD) higher than those in the bottom quintile. For context, an increase of 15 points above the mean T‐score reflects ‘borderline’ clinical risk (Achenbach, 2001). Moreover, among all youth with clinically elevated internalizing and externalizing symptoms at follow‐up, the majority (80%–90%) were captured in the upper distribution of model‐predicted values.
Second, exposomic influences generally overshadowed genomic contributions to mental health symptoms in preadolescent youth, perhaps unsurprisingly. The additive modeled exposome explained a substantial proportion of variation in observed symptoms (16% for internalizing and 23% for externalizing). Among exposomic subdomains, life events such as experiences of injury, illness, or loss appeared most influential for internalizing symptoms in youth, substantiating known relationships between stressful experiences and depression (Hammen, 2005; Kessler, 1997; Mazure, 1998), while proximal contextual variables including family, school, and peer‐level factors were most influential for externalizing symptoms, suggesting that everyday contexts where a child is embedded have some of the most powerful influences on problem behavior (Bank, Burraston, & Snyder, 2004; Tomé, Matos, Simões, Diniz, & Camacho, 2012). Broader contextual factors including neighborhood crime and area‐level deprivation, while non‐negligible in their influences, contributed less to overall symptom variance than proximal contextual variables or life events.
We initially found that genome‐wide influences explained more than twice the variance in externalizing symptoms (19%) compared to internalizing symptoms (6%) at this age. This aligns with twin‐based literature, indicating externalizing symptoms are more heritable than internalizing symptoms in early adolescence (Bergen, Gardner, & Kendler, 2007; Nikstat & Riemann, 2020) – potentially due to measurement, as externalizing behaviors are more easily observed and potentially captured with reduced noise (Bartels et al., 2004), or life course timing, since depression and other internalizing conditions may onset later in adolescence and young adulthood (Nivard et al., 2017). Unlike twin studies that extrapolate genetic and environmental influences based on inherited similarities/differences, we examined common genomic variation in unrelated individuals. While not significant, our G estimate for internalizing symptoms is similar to reported SNP‐based heritability of self‐reported internalizing symptoms (6%) from a recent meta‐analytic Genome‐Wide Association Study (GWAS) of 20 childhood and adolescent cohorts spanning a broader age range (Jami et al., 2021).
When modeled alongside the exposome, genomic effects on internalizing symptoms remained unchanged in magnitude, suggesting relative independence from exposomic effects, whereas genomic effects on externalizing symptoms were markedly attenuated. Our findings suggest that the modeled exposome may account for more than half of the estimated genome‐wide effects on externalizing symptoms. It could be that genomic effects on externalizing symptoms are mediated through environmental pathways; for example, genome‐wide variation may increase the likelihood of certain exposures (e.g. negative life events, Johnson, Rhee, Whisman, Corley, & Hewitt, 2013), which then influence externalizing risk. Alternatively, genetic factors that underlie endophenotypes of externalizing behavior (e.g. impulsivity) may also shape environmental factors (e.g. family conflict), reflecting gene–environment correlation (Lau & Eley, 2008), though our results remained largely consistent when excluding youth with clinically elevated externalizing symptoms at baseline. Genome‐wide effects were most attenuated when the modeled exposome consisted of proximal contextual or life events variables, indicating these factors may be particularly intertwined with the genetic basis of externalizing behavior. If so, genetic risk for youth problem behavior could potentially be modified by interventions targeting these environmental factors.
Third, genome–exposome interactions appear to contribute substantially to youth mental health symptoms. Modeling these interactions explained an additional 13% of variation in internalizing and an additional 33% in externalizing symptoms. This suggests a key role for ‘nurture’ since exposomic influences on youth mental health were not only more profound on their own, but appear to also modulate genomic influences as well. Interestingly, genome‐wide variation appeared to interact mostly with proximal contextual factors or life events, rather than broader contextual variables, in shaping youth symptoms. On the other hand, exposome‐by‐exposome interaction effects were modest in boosting explanatory power, suggesting the environmental factors we considered in this sample are more likely to operate additively. However, accounting for their potential intercorrelations appeared to increase exposomic signal. In sensitivity analyses where exposures were not initially transformed for independence, the estimated exposome effect was qualitatively reduced albeit still significant. One potential interpretation is that we may generally underestimate exposomic effects due to multicollinearity. Simulation work has shown that correlation structure among exposures can produce biased estimates (Zhou & Lee, 2021) as the effective number of exposures becomes smaller than the actual number of modeled exposures.
Strengths and limitations
This study offers numerous strengths, including the explicit modeling of an ‘exposome’ based on 133 different variables (at family, peer, school, neighborhood, life event, and broader environmental levels), and longitudinal design with temporal separation of modeled exposures from outcomes at follow‐up. We were able to further dissect the exposome into conceptual subdomains to examine relative contributions of multilevel factors from additive and interactive perspectives. Furthermore, we modeled genome‐wide similarity and differences across individuals, rather than relying on polygenic scores that largely capture additive effects from GWAS for specific traits only and to date explain limited phenotypic variance in psychiatric outcomes (Murray et al., 2021). For example, a recent study from the ABCD cohort reported even the most predictive polygenic scores (e.g. ADHD PRS) explained no more than 1.4% of variance in CBCL‐related outcomes (Waszczuk et al., 2021).
Several limitations, however, should be noted. First, it is worth considering that despite modeling a set of genome, exposome, and genome‐by‐exposome influences, a notable proportion of variance in youth mental health symptoms remained ‘missing’, or unexplained. This might be explained by the fact that our modeled exposome, while wide‐ranging in scope, was limited to assessed variables rather than all possible environmental exposures that could shape youth mental health, including those occurring earlier in life, though we found including several common perinatal exposures contributed minimally to additional variance. We also did not examine the role of rare or X‐linked genomic variation. It may be that no set of genomic and/or exposomic factors is deterministic of youth mental health symptoms, raising the importance of modeling idiographic variation. Second, this was a study of mental health symptoms in preadolescent youth, whereas the heritability – and epidemiological risk – of internalizing and externalizing phenotypes tends to increase over time in young people (Bergen et al., 2007). However, early identification of at‐risk individuals is arguably timely for life course prevention. Third, despite leveraging the largest genetic ancestry group for demonstration, our sample size was relatively modest for detecting significant genome‐wide effects for complex psychiatric traits, though the magnitudes of heritability estimates were not dissimilar from other recent studies. Fourth, there may be reverse influences of internalizing/externalizing risk on the modeled exposome; directionality is not established, although we sought to temporally separate outcome measures from the environmental exposures at baseline or prior to follow‐up. Finally, symptom outcomes were parent‐reported, which may differ from other informants such as teachers or youth themselves. Shared method variance where outcome and exposures are reported by the same informant (Xerxa et al., 2021) may upward bias the exposome estimates, although we found that exposome and genome‐exposome influences persisted even when relying solely on child‐reported exposures. Because some exposures were only reported by child versus parent (e.g. school vs. neighborhood factors), we cannot fully untangle shared method bias from substantive differences in assessed exposures; this may require studies where environmental exposures have been consistently measured across multiple informants. For generalizability, our further replications in various age brackets of youth from this and other large‐scale developmental cohorts, with different modes of measurement and across diverse settings, are warranted and currently ongoing. Certain environmental exposures (e.g. neighborhood contextual factors) may influence mental health more substantially as older youth develop greater awareness of contexts outside of home and school and may suggest the need for dynamic prevention approaches that shift in their targets over time.
Conclusion
In conclusion, the relative contributions and interplay between genes and environment has been a central focus of developmental psychopathology. Applying recent advances in statistical genetic modeling to a longitudinal cohort comprising genomic, exposomic, and clinical data, we demonstrate the influence of multilevel contexts and their interaction with genome‐wide variation on emotional and behavioral problems in young people. Identifying exposomic components of mental health risk may inform the design of prevention strategies.
Supporting information
Appendix S1. Genomic data processing.
Appendix S2. Phenotypic data pre‐processing.
Appendix S3. Relationship matrix specifications.
Table S1. Assessment of exposures in the study cohort.
Table S2. More information on exposures.
Table S3. Estimated variance components for G, E, G × E model when further pre‐adjusting outcomes for parental educational attainment.
Table S4. Estimated variance components for G, E, G × E model when including perinatal exposures.
Table S5. Estimated variance components for G, E, G × E model when excluding individuals with clinically elevated baseline symptoms.
Table S6. Estimated variance components for G, E, G × E model where E components were not initially PCA‐transformed.
Table S7. Estimated variance components for G, E, G × E model based on only parent‐ versus child‐reported exposures.
Table S8. Estimated variance components for G, E, G × E model in the African and admixed American ancestry groups.
Table S9. Estimated variance components for G, E, G × E model of INT where multiple E subdomains were simultaneously modeled.
Table S10. Estimated variance components for G, E, G × E model of EXT where multiple E subdomains were simultaneously modeled.
Acknowledgements
The authors would like to thank Xuan Zhou, PhD, and Zhaowen Liu, PhD, for methodological support and guidance, and Yingzhe Zhang, MS, for assistance with quality control and processing of cross‐ancestry genomic data. K.W.C. was supported in part by the National Institute of Mental Health (K08MH127413), Kaplen Fellowship on Depression from the Harvard Medical School, and NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation. J.W.S. is supported in part by a gift from the Demarest Lloyd Jr. Foundation and the Tommy Fuss Fund.
Data used in the preparation of this article were obtained from the Adolescent Brain Cognitive Development (ABCD) Study (https://abcdstudy.org), held in the NIMH Data Archive (NDA). This is a multisite, longitudinal study designed to recruit more than 10,000 children aged 9–10 and follow them over 10 years into early adulthood. The ABCD Study® is supported by the National Institutes of Health and additional federal partners under award numbers U01DA041048, U01DA050989, U01DA051016, U01DA041022, U01DA051018, U01DA051037, U01DA050987, U01DA041174, U01DA041106, U01DA041117, U01DA041028, U01DA041134, U01DA050988, U01DA051039, U01DA041156, U01DA041025, U01DA041120, U01DA051038, U01DA041148, U01DA041093, U01DA041089, U24DA041123, U24DA041147. A full list of supporters is available at https://abcdstudy.org/federal‐partners.html. A listing of participating sites and a complete listing of the study investigators can be found at https://abcdstudy.org/consortium_members. ABCD consortium investigators designed and implemented the study and/or provided data but did not necessarily participate in the analysis or writing of this report. This manuscript reflects the views of the authors and may not reflect the opinions or views of the NIH or ABCD consortium investigators. The ABCD data repository grows and changes over time. The ABCD data used in this report came from DOI https://doi.org/10.15154/1524700.
J.W.S. is a member of the Leon Levy Foundation Neuroscience Advisory Board, the Scientific Advisory Board of Sensorium Therapeutics and has received honoraria for internal seminars at Biogen, Inc and Tempus Labs. He is PI of a collaborative study of the genetics of depression and bipolar disorder sponsored by 23andMe for which 23andMe provides analysis time as in‐kind support but no payments. The remaining authors have declared that they have no competing or potential conflicts of interest.
Key points.
While genetic and environmental factors likely both influence youth mental health, the relative contributions and interplay between genome‐wide influences and wide‐ranging environmental exposures (i.e. the exposome) have not been well characterized.
Applying recent developments in the field of statistical genetics, we perform an integrative analysis of genomic and exposomic effects as well as their interactive influences on internalizing and externalizing symptoms in a longitudinal cohort of youth.
Best‐fitting integrative models explained 35% and 63% of variance in youth internalizing and externalizing symptoms, with genome‐exposome interactions contributing significant explanatory variance (13% and 33%, respectively).
Different domains of environmental exposures were identified as most relevant for youth symptoms of internalizing (life events) and externalizing (proximal contextual factors including family, peer, and school).
Findings suggest that future research would benefit from combined approaches integrating multiple exposures and that multilevel strategies will be important for early preventive interventions in youth mental health.
Conflict of interest statement: See Acknowledgements for full disclosures.
References
- Achenbach, T.M. , & Rescorla, L.A. (2001). Manual for the ASEBA school‐age forms & profiles: An integrated system of multi‐informant assessment. Burlington, VT: University of Vermont, Research Center for Children, Youth, & Families. [Google Scholar]
- Arnau‐Soler, A. , Macdonald‐Dunlop, E. , Adams, M.J. , Clarke, T.‐K. , MacIntyre, D.J. , Milburn, K. , … & Thomson, P.A. (2019). Genome‐wide by environment interaction studies of depressive symptoms and psychosocial stress in UKBiobank and Generation Scotland. Translational Psychiatry, 9, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bank, L. , Burraston, B. , & Snyder, J. (2004). Sibling conflict and ineffective parenting as predictors of adolescent boys' antisocial behavior and peer difficulties: Additive and interactional effects. Journal of Research on Adolescence, 14, 99–125. [Google Scholar]
- Bartels, M. , Boomsma, D.I. , Hudziak, J.J. , Rietveld, M.J.H. , van Beijsterveldt, T.C.E.M. , & van den Oord, E.J.C.G. (2004). Disentangling genetic, environmental, and rater effects on internalizing and externalizing problem behavior in 10‐year‐old twins. Twin Research, 7, 162–175. [DOI] [PubMed] [Google Scholar]
- Bergen, S.E. , Gardner, C.O. , & Kendler, K.S. (2007). Age‐related changes in heritability of behavioral phenotypes over adolescence and young adulthood: A meta‐analysis. Twin Research and Human Genetics, 10, 423–433. [DOI] [PubMed] [Google Scholar]
- Chang, C.C. , Chow, C.C. , Tellier, L.C. , Vattikuti, S. , Purcell, S.M. , & Lee, J.J. (2015). Second‐generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K.W. , Stein, M.B. , Nishimi, K.M. , Ge, T. , Coleman, J.R.I. , Chen, C.‐Y. , … & Smoller, J.W. (2020). An exposure‐wide and Mendelian randomization approach to identifying modifiable factors for the prevention of depression. The American Journal of Psychiatry, 177, 944–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayborne, Z.M. , Varin, M. , & Colman, I. (2019). Systematic review and meta‐analysis: Adolescent depression and long‐term psychosocial outcomes. Journal of the American Academy of Child and Adolescent Psychiatry, 58, 72–79. [DOI] [PubMed] [Google Scholar]
- Coleman, J.R.I. , Peyrot, W.J. , Purves, K.L. , Davis, K.A.S. , Rayner, C. , Choi, S.W. , … & Breen, G. (2020). Genome‐wide gene‐environment analyses of major depressive disorder and reported lifetime traumatic experiences in UKBiobank. Molecular Psychiatry, 25, 1430–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, E.C. , Wiste, A. , Radmanesh, F. , Almli, L.M. , Gogarten, S.M. , Sofer, T. , … & Smoller, J.W. (2016). Genome‐wide association study (GWAS) and genome‐wide by environment interaction study (GWEIS) of depressive symptoms in African American and Latina/Hispanic Women. Depression and Anxiety, 33, 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensink, J.B.M. , de Moor, M.H.M. , Zafarmand, M.H. , de Laat, S. , Uitterlinden, A. , Vrijkotte, T.G.M. , … & Middeldorp, C.M. (2020). Maternal environmental risk factors and the development of internalizing and externalizing problems in childhood: The complex role of genetic factors. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics, 183, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavan, H. , Bartsch, H. , Conway, K. , Decastro, A. , Goldstein, R.Z. , Heeringa, S. , … & Zahs, D. (2018). Recruiting the ABCD sample: Design considerations and procedures. Developmental Cognitive Neuroscience, 32, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga, A. , & Pennoni, F. (2020). The causal effects of parental divorce and parental temporary separation on children's cognitive abilities and psychological well‐being according to parental relationship quality. Social Indicators Research, 161, 963–987. [Google Scholar]
- Guloksuz, S. , van Os, J. , & Rutten, B.P.F. (2018). The exposome paradigm and the complexities of environmental research in psychiatry. JAMA Psychiatry, 75, 985–986. [DOI] [PubMed] [Google Scholar]
- Hammen, C. (2005). Stress and depression. Annual Review of Clinical Psychology, 1, 293–319. [DOI] [PubMed] [Google Scholar]
- Jami, E.S. , Hammerschlag, A.R. , Ip, H.F. , Allegrini, A.G. , Benyamin, B. , Border, R. , … & Middeldorp, C.M. (2022). Genome‐wide association meta‐analysis of childhood and adolescent internalising symptoms. Journal of the American Academy of Child and Adolescent Psychiatry, 61, 934–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernigan, T.L. , Brown, S.A. , & ABCD Consortium Coordinators . (2018). Introduction. Developmental Cognitive Neuroscience, 32, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, D.P. , Rhee, S.H. , Whisman, M.A. , Corley, R.P. , & Hewitt, J.K. (2013). Genetic and environmental influences on negative life events from late childhood to adolescence. Child Development, 84, 1823–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karcher, N.R. , & Barch, D.M. (2021). The ABCD study: Understanding the development of risk for mental and physical health outcomes. Neuropsychopharmacology, 46, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler, R.C. (1997). The effects of stressful life events on depression. Annual Review of Psychology, 48, 191–214. [DOI] [PubMed] [Google Scholar]
- Kessler, R.C. , Amminger, G.P. , Aguilar‐Gaxiola, S. , Alonso, J. , Lee, S. , & Ustün, T.B. (2007). Age of onset of mental disorders: A review of recent literature. Current Opinion in Psychiatry, 20, 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong, A.S.F. , López‐López, J.A. , Hammerton, G. , Manley, D. , Timpson, N.J. , Leckie, G. , & Pearson, R.M. (2019). Genetic and environmental risk factors associated with trajectories of depression symptoms from adolescence to young adulthood. JAMA Network Open, 2, e196587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau, J.Y.F. , & Eley, T.C. (2008). Disentangling gene‐environment correlations and interactions on adolescent depressive symptoms. Journal of Child Psychology and Psychiatry, 49, 142–150. [DOI] [PubMed] [Google Scholar]
- Lee, S.H. , & van der Werf, J.H.J. (2016). MTG2: An efficient algorithm for multivariate linear mixed model analysis based on genomic information. Bioinformatics, 32, 1420–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrai, A.K. , Cui, Y. , Bushel, P.R. , Hall, M. , Karakitsios, S. , Mattingly, C.J. , … & Patel, C.J. (2017). Informatics and data analytics to support exposome‐based discovery for public health. Annual Review of Public Health, 38, 279–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazure, C.M. (1998). Life stressors as risk factors in depression. Clinical Psychology: Science and Practice, 5, 291–313. [Google Scholar]
- Murray, G.K. , Lin, T. , Austin, J. , McGrath, J.J. , Hickie, I.B. , & Wray, N.R. (2021). Could polygenic risk scores be useful in psychiatry?: A review. JAMA Psychiatry, 78, 210–219. [DOI] [PubMed] [Google Scholar]
- Newman, D.L. , Moffitt, T.E. , Caspi, A. , Magdol, L. , Silva, P.A. , & Stanton, W.R. (1996). Psychiatric disorder in a birth cohort of young adults: Prevalence, comorbidity, clinical significance, and new case incidence from ages 11 to 21. Journal of Consulting and Clinical Psychology, 64, 552–562. [PubMed] [Google Scholar]
- Nikstat, A. , & Riemann, R. (2020). On the etiology of internalizing and externalizing problem behavior: A twin‐family study. PLoS One, 15, e0230626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivard, M.G. , Lubke, G.H. , Dolan, C.V. , Evans, D.M. , St Pourcain, B. , Munafò, M.R. , & Middeldorp, C.M. (2017). Joint developmental trajectories of internalizing and externalizing disorders between childhood and adolescence. Development and Psychopathology, 29, 919–928. [DOI] [PubMed] [Google Scholar]
- Pandolfi, V. , Magyar, C.I. , & Dill, C.A. (2012). An initial psychometric evaluation of the CBCL 6‐18 in a sample of youth with autism spectrum disorders. Research in Autism Spectrum Disorders, 6, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stekhoven, D.J. , & Bühlmann, P. (2011). MissForest—Non‐parametric missing value imputation for mixed‐type data. Bioinformatics, 28, 112–118. [DOI] [PubMed] [Google Scholar]
- Sullivan, P.F. , & Kendler, K.S. (2021). The state of the science in psychiatric genomics. Psychological Medicine, 51, 2145–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomé, G. , Matos, M. , Simões, C. , Diniz, J.A. , & Camacho, I. (2012). How can peer group influence the behavior of adolescents: Explanatory model. Global Journal of Health Science, 4, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uher, R. , & Zwicker, A. (2017). Etiology in psychiatry: Embracing the reality of poly‐gene‐environmental causation of mental illness. World Psychiatry, 16, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waszczuk, M.A. , Miao, J. , Docherty, A.R. , Shabalin, A.A. , Jonas, K.G. , Michelini, G. , & Kotov, R. (2021). General v. specific vulnerabilities: Polygenic risk scores and higher‐order psychopathology dimensions in the Adolescent Brain Cognitive Development (ABCD) Study. Psychological Medicine, 1–10. 10.1017/s0033291721003639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild, C.P. (2012). The exposome: From concept to utility. International Journal of Epidemiology, 41, 24–32. [DOI] [PubMed] [Google Scholar]
- Woodward, L.J. , & Fergusson, D.M. (2001). Life course outcomes of young people with anxiety disorders in adolescence. Journal of the American Academy of Child and Adolescent Psychiatry, 40, 1086–1093. [DOI] [PubMed] [Google Scholar]
- Xerxa, Y. , Rescorla, L.A. , van der Ende, J. , Hillegers, M.H.J. , Verhulst, F.C. , & Tiemeier, H. (2021). From parent to child to parent: Associations between parent and offspring psychopathology. Child Development, 92, 291–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X. , & Lee, S.H. (2021). An integrative analysis of genomic and exposomic data for complex traits and phenotypic prediction. Scientific Reports, 11, 21495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Genomic data processing.
Appendix S2. Phenotypic data pre‐processing.
Appendix S3. Relationship matrix specifications.
Table S1. Assessment of exposures in the study cohort.
Table S2. More information on exposures.
Table S3. Estimated variance components for G, E, G × E model when further pre‐adjusting outcomes for parental educational attainment.
Table S4. Estimated variance components for G, E, G × E model when including perinatal exposures.
Table S5. Estimated variance components for G, E, G × E model when excluding individuals with clinically elevated baseline symptoms.
Table S6. Estimated variance components for G, E, G × E model where E components were not initially PCA‐transformed.
Table S7. Estimated variance components for G, E, G × E model based on only parent‐ versus child‐reported exposures.
Table S8. Estimated variance components for G, E, G × E model in the African and admixed American ancestry groups.
Table S9. Estimated variance components for G, E, G × E model of INT where multiple E subdomains were simultaneously modeled.
Table S10. Estimated variance components for G, E, G × E model of EXT where multiple E subdomains were simultaneously modeled.