

Abstract
Platelets are the “guardians” of the blood circulatory system. At sites of vessel injury, they ensure hemostasis and promote immunity and vessel repair. However, their uncontrolled activation is one of the main drivers of thrombosis. To keep circulating platelets in a quiescent state, the endothelium releases platelet antagonists including nitric oxide (NO) that acts by stimulating the intracellular receptor guanylyl cyclase (GC). The latter produces the second messenger cyclic guanosine‐3′,5′‐monophosphate (cGMP) that inhibits platelet activation by stimulating protein kinase G, which phosphorylates hundreds of intracellular targets. Intracellular cGMP pools are tightly regulated by a fine balance between GC and phosphodiesterases (PDEs) that are responsible for the hydrolysis of cyclic nucleotides. Phosphodiesterase type 5 (PDE5) is a cGMP‐specific PDE, broadly expressed in most tissues in humans and rodents. In clinical practice, PDE5 inhibitors (PDE5i) are used as first‐line therapy for erectile dysfunction, pulmonary artery hypertension, and lower urinary tract symptoms. However, several studies have shown that PDE5i may ameliorate the outcome of various other conditions, like heart failure and stroke. Interestingly, NO donors and cGMP analogs increase the capacity of anti‐platelet drugs targeting the purinergic receptor type Y, subtype 12 (P2Y12) receptor to block platelet aggregation, and preclinical studies have shown that PDE5i inhibits platelet functions. This review summarizes the molecular mechanisms underlying the effect of PDE5i on platelet activation and aggregation focusing on the therapeutic potential of PDE5i in platelet disorders, and the outcomes of a combined therapy with PDE5i and NO donors to inhibit platelet activation.
Keywords: cyclic guanosine‐3′,5′‐monophosphate; nitric oxide donors; phosphodiesterase type 5; phosphodiesterase type 5 inhibitors; platelet disorders; thrombosis
1. INTRODUCTION
Platelets are small anucleated blood cells that ensure hemostasis at sites of blood vessel injury by aggregating and releasing a plethora of bioactive molecules, thereby limiting blood loss and pathogen entry, and promoting vessel repair. 1
When the endothelial lining is intact and healthy, platelets are maintained in a resting non‐adhesive state by short‐lived molecules constitutively released by the endothelium, namely nitric oxide (NO) and prostacyclin (prostaglandin I2 [PGI2]). NO is an apolar gas produced from L‐arginine by nitric oxide synthase (NOS) with an intravascular half‐life of 2 ms that can enter platelets by simple diffusion and stimulate the intracellular soluble receptor guanylyl cyclase (sGC), which in turn catalyzes the conversion of guanosine‐5′‐triphosphate (GTP) to cyclic guanosine 3′,5′‐monophosphate (cGMP). 2 Prostacyclin is a lipid mediator of the eicosanoid family (half‐life of 42 s) that binds the platelet G protein‐coupled prostacyclin receptor (IP), which stimulates adenylyl cyclase to convert adenosine‐5′‐triphosphate (ATP) to adenosine 3′,5′‐monophosphate (cAMP). 3 cGMP and cAMP activate protein kinase G (PKG) and A (PKA), respectively, that phosphorylate hundreds of intracellular targets to ultimately inhibit the activation of platelets flowing along the endothelium. 4 The NO and PGI2 paracrine effect also extends to nearby leukocytes and smooth muscle cells. 5 Thus, their overall outcome is to inhibit/dampen thrombosis and inflammation and promote vasodilation.
When the endothelial lining is injured or inflamed, the local concentration of these key inhibitory mediators is reduced locally and the molecular breaks are released and thereby smooth muscle cells contract and platelets and leukocytes are more prone to become active. 6 At the sites of injury platelets sense and respond to exposed components of the extracellular matrix (ECM) such as collagen, and locally generated soluble agonists, such as thrombin; that is, the product of the coagulation cascade. 7 These agonists trigger intracellular signaling cascades that promote the conversion of integrin receptors from a low‐ to a high‐affinity state for their ligands (integrin inside‐out activation). 7 The β1 integrins (α2β1, α6β1, α5β1, αIIbβ3, αvβ3) support adhesion and spreading of the first layer of platelets to the ECM. 8 αIIbβ3, the most abundant integrin expressed in platelets, in its active state binds to plasmatic fibrinogen or von Willebrand factor (VWF) released from the injured endothelium, supporting the formation of a three‐dimensional platelet aggregate (hemostatic plug) at the injury site. 9 The controlled amplification of activation that is necessary to recruit more platelets and to ensure stability of the growing aggregate is mediated by co‐stimulatory signaling provided by the short‐lived autocrine/paracrine agonists thromboxane (Tx)A2 and adenosine diphosphate (ADP), which are released from activated platelets and can only act locally like NO and prostacyclin. 10 , 11
Uncontrolled activation of the platelet pro‐adhesive and secretory functions is one of the main drivers of thrombosis and can also exacerbate other disease states such as inflammation, infections, 12 diabetes, and cancer. 13 , 14 For instance, COVID‐19 patients display hyperactive platelets and a higher risk of thrombotic complications, 12 which could be explained, at least in part, by the endothelial dysfunction and the reduced NO bioavailability documented in these patients.
In clinical practice, the strategy most widely used to counteract pathological platelet activation is to inhibit the amplificatory pathways mediated by TxA2 and ADP by treatment of patients with aspirin (which prevents TxA2 synthesis) and/or ADP receptor, purinergic receptor type Y, subtype 12 (P2Y12) blockers such as clopidogrel. 15 In principle, one could lower platelet activation not only by inhibiting the activation pathways but also by boosting the inhibitory pathways.
One way to do this is by increasing the intracellular levels of cGMP. Intracellular cGMP pools are tightly regulated by a fine balance between the NO receptor guanylyl cyclase (GC) and phosphodiesterases (PDE) that are responsible for cyclic nucleotides hydrolysis. 16 In mammals, the PDE superfamily includes 60 different isoforms with different specificity for cAMP and/or cGMP. 17 Phosphodiesterase type 5 (PDE5) is a cGMP‐specific phosphodiesterase broadly expressed in most tissues in humans and mice, 18 , 19 , 20 which jumped to the headlines thanks to the development of its pharmacological inhibitor, sildenafil (Viagra). In clinical practice PDE5 inhibitors (PDE5i) are used as first‐line therapy for erectile dysfunction (ED), pulmonary arterial hypertension (PAH), and lower urinary tract symptoms (LUTS). 21 However, several studies have shown that PDE5 may ameliorate the outcome in various other conditions, including heart failure, 22 stroke, 23 diabetic nephropathy, 24 , 25 peripheral artery disease, and premature ejaculation, 26 and its clinical applications are increasing in several fields. 27 , 28 , 29 , 30
As expected, NO donors and cGMP analogs greatly increase the capacity of P2Y12 inhibitors to block platelet aggregation 31 and PDE5i in particular have been shown to inhibit platelet function. 32 However, the pathological context in which these therapeutic strategies could be most effective and safe remains to be established.
2. SIGNALING PATHWAYS REGULATING CGMP IN PLATELETS
2.1. Nitric oxide
Cytosolic levels of cGMP are regulated by a fine balance between the activity of the NO‐stimulated sGC and PDEs. The harmonized activity of these players converges in a complex signaling pathway whose initiator is NO, a free radical, naturally produced through oxidation of the amino acid L‐arginine by the NOS. The main source of NO in the blood is endothelial NOS (eNOS), but there is also a NOS resident in platelets that contributes to the vascular pool of NO. 33 NO regulation of vascular tone occurs through different pathways according to cell needs and environmental stimuli. Under physiological conditions, NO mainly synthesized by endothelial eNOS induces cGMP production via stimulation of sGC in vascular smooth muscle cells. 34 The increase of cGMP activates PKG thus preventing calcium influx and promoting cytosolic calcium reuptake into the sarcoplasmic reticulum. These coupled events, acting on myosin–actin bridges, finally induce muscle relaxation. Under hypoxic conditions sGC‐mediated cGMP production is inhibited and Rho‐associated protein kinase (ROCK) activation mediated by inosine cyclic monophosphate (cIMP) triggers muscle contraction. 35 These mechanisms ensure vascular homeostasis and prevent vascular damage that occurs in several pathological conditions such as atherosclerosis in which NO concentration declines promoting smooth muscle cell proliferation and ECM deposition. 5 NO is best known for its stimulatory role on sGC. However, NO can downregulate platelet function, independently of sGC, through nitration or S‐nytrosilation of intracellular signaling proteins. 36 , 37 S‐nytrosilation of N‐ethylmaleimide–sensitive factor (NSF) was shown to inhibit platelet granule release, indispensable for platelet activation, by impairing its ability to disassemble the soluble N‐ethylmaleimide NSF attachment receptor (SNARE) complex. 36 Even though it is well established that NO is a strong negative regulator of platelet activation and adhesion, 38 , 39 under oxidative stress conditions, there is an impairment of NO signaling in platelets called “NO resistance” leading to dysfunction of the classical NO/cGMP/PKG pathway. Superoxide anion (O2−) radicals can act as NO scavengers and at the same time inactivate sGC. 40
Platelet sensitivity to NO as well as cGMP and sGC decreases with age, and among young people, women are the ones to show a higher expression of cGMP and sGC. 41 Homocysteine, an eNOS inhibitor, induces formation of the peroxynitrite biomarker nitrotyrosine, and leads to decreased NO bioavailability, NO impairment, endothelial damage, and stimulation of platelet aggregation. 42 Enhanced intraplatelet reactive oxygen species production decreases NO bioavailability, and potential defects in the heme group of sGC are probably implicated in the platelet hyperaggregability associated with a high‐fat diet. 43 The platelet inhibitor aspirin can also exert its function via NOS acetylation increasing NO production and activating its downstream signaling. 44
2.2. Soluble guanylate cyclase
NO acts as the initiator of a cascade of signals that converge in the activity of its cytosolic receptor, namely sGC, a member of the nucleotide cyclase family. 45 sGC activity increases several fold when binding to NO, 46 leading to cGMP generation. sGC is a heterodimer consisting of an alpha bound to a beta subunit, and the alpha subunit is present in two isoforms. 45 Strategies for the genomic deletion of individual subunits have been developed in recent years. Removal of either one of the alpha isoforms has no effect on sGC, while depletion of the beta subunit coding gene led to total absence of the sGC, and consequent interruption of the NO/sGC/cGMP/PKG signaling transduction pathway. 47 As a heme‐containing α/β– heterodimer, the redox state of the heme moiety is a crucial regulator for binding of NO to sGC, for the reason that sGC can bind NO and become active only when the heme iron is in its reduced state (Fe2+), while in presence of oxidized state iron (Fe3+), sGC is insensitive to NO and unable to bind to it. 48 Even though, as mentioned in the previous section, there are NO‐dependent sGC‐independent inhibitory mechanisms, several mouse models confirmed the inhibitory role of the NO/GC/cGMP axis in platelets. 47 , 49 Consistently, NO‐independent pharmacological sGC agonists inhibit platelets. 50 They can be distinguished in sGC stimulators and sGC activators that are effective on Fe2+ or Fe3+ iron, respectively. A known sGC stimulator, YC‐1 (3‐[5‐hydroxymethyl‐2furyl]‐1‐benzyl indazole), acts on sGC by sensitizing it to CO, stimulating sGC in a NO‐similar manner. 51 sGC stimulators also increase sGC activity acting on specific regions in the structure of the enzyme, such as BAY 41‐2272, which acts on the regulatory region on the sGC α1 subunit, increasing the enzyme activity. 52 Activation of the sGC can be induced in a heme‐dependent or ‐independent manner. The heme‐dependent activation of sGC is observed during NO binding to Fe2+, 53 or during treatment with nitro vasodilators such as glyceryl trinitrate, that result in activation of sGC by modulating the heme moiety, forming a nitrosyl heme of the enzyme. 54 YC‐1, on the other hand, activates sGC in both NO‐dependent and NO‐independent manners. 55 The sGC activator HMR1766 reduces platelet activation in vivo after chronic treatment of diabetic rats. 56 The sGC activator BAY 60‐2770 was shown to overstimulate platelet sGC and to inhibit platelet aggregation, adhesion, intracellular Ca2+ levels, and integrin aIIbβ3 activation particularly in heme‐oxidizing conditions. 57 The GC stimulator riociguat is effective in the treatment of pulmonary hypertension but it was shown to inhibit platelet activation in whole blood only at concentrations above 50 μM, which is much higher than the concentrations reached in the plasma of patients (150–500 nM), suggesting that platelets may not be the target of this drug. 58 On the other hand, the phosphodiesterase 3 (PDE3) inhibitor dipyridamole, a widely used antiplatelet drug, has been shown to augment NO production and bioavailability under diabetic conditions 59 and on experimental models of ischemic limbs. 60 In comparison to riociguat, dipyridamole is more effective because it increases NO bioavailability; inhibits the cellular reuptake of adenosine, thus inhibiting platelet aggregation; and has been shown to play an effective role in platelets, where it inhibited shear‐induced platelet aggregation with a larger efficacy in whole blood compared to platelet rich plasma, 61 making it a more reliable treatment for platelet‐related disorders. Thrombospondin‐1 (TSP‐1) is a universal inhibitor of sGC that blocks both heme‐dependent and ‐independent activation. 62 TSP‐1 increases with age, and in chronic diseases such as diabetes and atherosclerosis, thus the therapeutic potential of drugs that target sGC could be compromised in these pathological conditions in which TSP‐1 signaling is elevated. 62 While the inhibitory effect of sGC in NO‐stimulated circulating platelets is unquestionable, its role during thrombus formation seems to be more complex. Increasing evidence suggests that sGC can be activated downstream of the VWF receptor GP‐Ib‐IX‐V 63 , 64 or the Toll‐like receptor (TLR)4 stimulated by pathogen‐associated molecular patterns (PAMPS) like lipopolysaccharide or damage‐associated molecular patterns (DAMPS) and can have a stimulatory effect on platelet activation 65 in synergy with other platelet agonists. Moreover, a recent study has shown that sGC has mechanosensitive properties and its inhibitory function is potentiated by shear stress. 66 These apparently contradictory observations are conciliated by a proposed model in which, during thrombus formation, sGC might display a biphasic role in the modulation of platelet function. 67 In the early stages of thrombus formation, platelet sGC/cGMP axis is activated by VWF or PAMPs and DAMPs situated at the site of injury and promotes thrombus formation. 68 During the late stages of thrombus growth, the effect of high shear stress stimulates the sGC/cGMP inhibitory signaling axis and results in the limitation of platelet activation and of thrombus growth. 68 NO‐dependent or ‐independent activation of sGC leads to increase of cGMP, whose levels are finely regulated by the action of cGMP‐hydrolyzing phosphodiesterases.
2.3. Phosphodiesterases and platelets
Phosphodiesterases belong to a large family of enzymes virtually expressed in all tissues and responsible for cyclic nucleotide hydrolysis. 18 Eleven PDE families have been identified so far that, following multiple splicing processes, give rise to more than 100 PDE isoforms in humans and rodents. 69 PDEs differ in their structure, properties, location, cellular expression, and targets. 17 PDE5 is the principal cGMP hydrolyzer, for which three isoforms have been identified in humans and mice. 18 , 70 Given the broad expression and the ability of PDE5 to control cGMP levels in the cell, it has been proposed as a pivotal effector in many biological processes such as platelet activation and aggregation, smooth muscle relaxation, immune response, and heart muscle contraction. 71
Three PDE isoenzymes are expressed in platelets, PDE2, PDE3, and PDE5, and while PDE2 hydrolyzes cAMP, and PDE3 both cAMP and cGMP, PDE5 is a cGMP‐specific enzyme. 32 The high expression of PDE5 in platelets leads to rapid cGMP hydrolysis, allowing platelets to promptly activate and aggregate when the endothelium is injured. 21
2.4. cGMP effectors
The basal cGMP level in platelets is reported to be around 0.5 μM. NO‐mediated activation of sGC results in a rapid 10‐fold increase in cGMP levels. 72 This transient elevation is mirrored by activation and subsequent phosphorylation of PDE5. 73 Phosphorylation of PDE5 has been suggested to enhance cGMP hydrolysis in vitro. 74 , 75
Since its discovery in rat urine about six decades ago, 76 various studies have been conducted to better understand cGMP's effect on platelet function. cGMP intracellular levels in platelets are tightly controlled by the activity of sGC and cGMP‐hydrolyzing PDEs. The main effector of cGMP is PKG that inhibits platelet activation by phosphorylating multiple substrates. One of its targets is IP3R‐associated PKG substrate (IRAG) that results in the inhibition of IP3‐dependent Ca2+ release from the intracellular stores. 77 By blocking Ca2+ mobilization, cGMP negatively regulates all aspects of platelet activation, from integrin activation to cytoskeleton remodeling and granule release. In addition, phosphorylation of regulatory proteins of Ras‐like guanine‐nucleotide‐binding protein (Rap1GAP2) 78 and of RhoA (Myo9b and GEF‐H1) contributes to the inhibition of platelet aggregation and of cytoskeletal dynamics. Additionally, PKG was shown to inhibit the platelet procoagulant response (PS exposure, mitochondrial membrane depolarization) 79 but its impact on platelet lifespan is yet to be determined. A cross‐talk between cGMP and cAMP has been reported in many tissue and cell types including platelets. Most notably, cGMP inhibits PDE3, which degrades cAMP, 80 and stimulates PDE2, which degrades both cGMP and cAMP, with the final effect of increasing cAMP. Thus, cGMP inhibits platelet activation in a PKG‐dependent (NO/cGMP/PKG) and PKG‐independent (NO/cGMP/PDE3A/cAMP/PKA) manner (Figure 1). 77 , 81 Moreover, PKA is able to phosphorylate and activate PDE5 on the PKG site (Ser92), but with 10‐fold lower efficiency than PKG. 82 Only a few substrates are known to be selectively phosphorylated by PKA (i.e., PDE3, IP3R, TRPC, MLCK) and PKG (i.e., PDE5 and IRAG), 83 while PKA and PKG can phosphorylate both the same substrates including the vasodilator‐stimulated phosphoprotein (VASP), 84 LIM and SH3 domain protein (LASP), 85 and heat shock protein 27 (HSP27). 86 The NO/sGC/cGMP pathway plays an important role in the activity of different cell types, organs, and tissues, most importantly in the brain, the corpus cavernosum, and the cardiovascular system. 87 In platelets, studies in humans and genetically modified animal models have demonstrated that upregulating the cGMP/PKG pathway is critical for reducing platelet aggregation and the risk of myocardial infarction. 88
FIGURE 1.
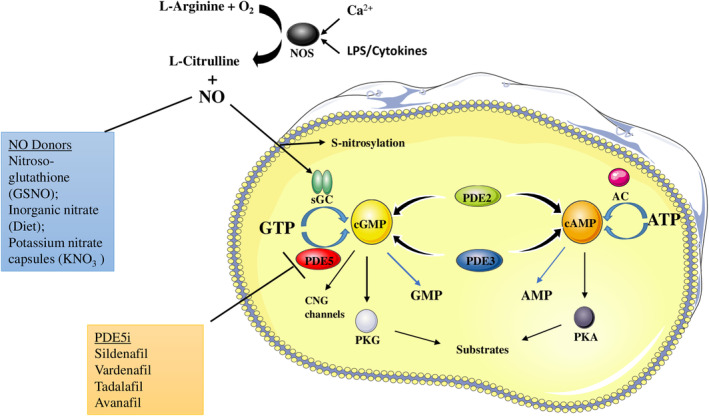
Nitric oxide (NO)/cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG) transduction signaling pathway in platelets. NO is produced by oxidation of L‐arginine by the NO synthase (NOS) enzymes, and acts as an activator for soluble guanylyl cyclase (sGC), which induces cGMP production through guanosine‐5′‐triphosphate (GTP) phosphorylation. Many NO donors, endogenous and exogenous ones, augment NO bioavailability, thus leading to a major activation of the cGMP pathway. cGMP can both inhibit and enhance cyclic adenosine monophosphate (cAMP) production in platelets, by allosterically inhibiting phosphodiesterase 3 (PDE3)—a cAMP inhibitor, and by stimulating phosphodiesterase 2 (PDE2) that degrades cAMP, so NO/cGMP can inhibit platelet activation either in a cGMP‐dependent PKG–dependent (NO‐cGMP‐PKG) or –independent (NO‐cGMP‐PDE3A‐cAMP‐PKA) pathway. cGMP binds to the three different effector proteins, PKGs, PDEs, and the cyclic nucleotide‐gated cation channels (CNG channels) that mediate sensory transduction in cells. Phosphodiesterase 5 (PDE5) is a cGMP‐specific phosphodiesterase that targets it and inhibits the crucial NO/cGMP/PKG signaling pathway. In presence of PDE5 inhibitors (PDE5i), PDE5 cannot exert its hydrolyzing function, by allowing cGMP to continue its platelet inhibitory function through PKG. AMP, adenosine monophosphate; ATP, adenosine triphosphate; PS, lipopolysaccharide; PKA, protein kinase A.
3. THE THERAPEUTIC POTENTIAL OF PDE5 INHIBITORS IN PLATELET DISORDERS
PDE5 inhibitors reached their breakthrough at the end of the last century and are currently used as first‐line therapy in patients with ED, PAH, and LUTS. 21 The most widely used PDE5i, sildenafil, commonly known as Viagra, was discovered in 1989. 89 Its ameliorating outcomes in patients with ED depend on its vasodilating properties, which are beneficial also in other clinical settings including cardiovascular disease (CVD). 90 , 91 , 92 , 93 In preclinical studies, PDE5 inhibitors’ effects on thrombosis have been examined in an experimental rat model of thrombotic suture suggesting a benefit in applying sildenafil in the anastomosis with already present thrombogenic disease. 94 The same effect is shown in clinical studies in which sildenafil reduced thrombosis, thromboembolic events, and the risk of thrombotic strokes in patients during low‐level hemolysis (LLH) on HeartMate II support. 95 Moreover, a retrospective analysis of clinical studies assessing the potential effect of PDE5i in ED suggests that use of sildenafil improves the outcome of CVD patients 96 and that there should be no safety concerns in the use of this drug in CVD patients. 97
An analysis of a Dutch registry (INTERMACS) of patients that used PDE5i after left ventricular assist device placement demonstrates that PDE5 inhibition improves survival and reduces thrombotic events. 98 Thus, the reduction in CVD risk is not only due to the vasodilating effect of PDE5i but also to their ability to inhibit platelet function. 99 Even though sildenafil shows several beneficial effects, its short half‐life 100 has led to the development of other PDE5 inhibitors, such as tadalafil, vardenafil, and avanafil with longer half‐lives. 21 Tadalafil has shown promising results in ex vivo studies and clinical trials, where its administration inhibits platelet activation, increases cGMP levels, 101 and reduces inflammation after treatment with 5 mg/day tadalafil. 102
Vardenafil is not only more effective than sildenafil in extending NO‐induced relaxation of trabecular smooth muscle cells, but can also inhibit Ca2+ flux, thereby inhibiting platelet activation. 103
4. PDE5 INHIBITORS COMBINED WITH NO DONORS COULD HAVE A SYNERGIC EFFECT ON PLATELET INHIBITION
PDE5 inhibitors alone have displayed limited effect as platelet aggregation inhibitors. 32 It has been demonstrated that in platelets, the presence of NO is able to induce an increase of cGMP content promoting the intracellular accumulation of PDE5i raising their affinity for PDE5. 104 Moreover, a synergic effect of PDE5i in combination with NO for the treatment of COVID‐19 has been proposed 28 and sildenafil has been shown to prompt the anti‐aggregatory effect of NO donors that inhibit platelet activation and aggregation through cGMP‐dependent and ‐independent pathways. 105 , 106
A more recent study 107 has shown very promising outcomes on ED patients by combining low‐dose tadalafil with dietary nutritional supplements that can boost the endothelial NO production, such as Panax ginseng, traditionally used for its properties as a vascular endothelial cell‐derived NO secretion promoter; moringa oleifera, known for its anticoagulant properties; and rutin, which possesses anticoagulant and antithrombotic properties. 108
Another study has shown that the use of compounds that augment NO levels and eNOS activity, such as endocannabinoid anandamide, shows an effect on platelet NO/cGMP pathway, by increasing in a dose‐ and time‐dependent manner NO and cGMP levels in human platelets and stimulating eNOS activity, contributing to an extended platelet survival. 109
All together, these data suggest a potential beneficial effect of PDE5 inhibitors combined with NO donors in platelets. However, dedicated studies deeply assessing this concern are missing.
5. WHO SHOULD TAKE PDE5 INHIBITORS AS ANTI‐PLATELET THERAPY?
The inhibitory effect of PDE5i on platelet aggregation suggests that these drugs alone or in combination with other therapies could be employed in the setting of CVD to reduce the risk of thrombosis and minimize the infarct size. 110 However, randomized clinical trials examining the safety and efficacy of PDE5i in CVD are still lacking.
The TARDIS trial 111 showed that combining aspirin, clopidogrel, and dipyridamole, a pan‐PDE inhibitor, did not reduce the incidence and severity of recurrent stroke compared to clopidogrel alone or aspirin and dipyridamole combined, but significantly increased the chance of bleeding. Thus, further trials are needed to find the clinical setting, modalities, and timing of administration in which these drugs could be most beneficial. Another recent study shows how the differential reactivity of platelets in males and females is at least in part due to differences in the NO–sGC signaling pathway, and that women are indeed prone to benefit from larger doses of antiplatelet drugs that block the P2Y12 receptor. 112
The L‐arginine/NO/cGMP pathway is impaired in platelets from obese adults, associated with reduced superoxide dismutase (SOD) activity, resulting in reduced NO bioavailability that supports platelet hyper aggregation. 113 Thus, among obese patients, combination therapy with NO donors and PDE5 could be preferable. In addition to their ability to form thrombi, platelets can also foster pathological conditions through their capacity to sense the activated endothelium, release chemokines, and bind leukocytes. In the setting of the arteries, platelets participate in the initial stages of atherosclerosis by arresting leukocytes 114 and facilitating their transmigration at sites of endothelial activation. 115 In veins where the endothelium is activated by flow stagnation and hypoxia platelets activate neutrophils to release neutrophil extracellular traps, which in turn promote thrombosis. 116 After stroke, platelets contribute to ischemia reperfusion injury and stroke progression through the recruitment of T cells. The common feature of these pathological states is endothelial dysfunction that results in reduced bioavailability of the platelet antagonists NO and PGI2 and decrease of the platelet activation threshold.
Notably, a recent study 117 demonstrated that tadalafil does not affect platelet adhesion to collagen and thrombus formation (platelet functions required for hemostasis) but significantly reduces platelet adhesion to inflamed endothelial cells and release of chemokines from activated platelets (platelet functions that foster inflammation). Because tadalafil also reduces inflammation 30 one could envision the use of this drug to prevent the progression of thrombo‐inflammatory disease without undermining hemostasis. However, these studies need confirmation among human subjects.
6. CONCLUSIONS
This review dissects the NO/cGMP/PKG/PDE5 pathway and its relevance in platelet physiopathology, and summarizes recent findings coming from preclinical studies that might shed light on the therapeutic potential of PDE5i as anti‐platelet treatment. From the analysis of available studies, the increase of cGMP levels through PDE5i or sGC stimulation emerge as a promising therapeutic tool in platelet disorders. However, randomized controlled trials are needed to assess efficacy and safety of these treatments.
AUTHOR CONTRIBUTIONS
AD and FC did the literature research and wrote the first draft of the review. AD and FC designed the figure. LS and MAV performed critical revision for important intellectual content. All the authors reviewed and approved the final version of the manuscript for consideration of publication.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
CONSENT FOR PUBLICATION
All authors have read and approved the submission of the manuscript.
ACKNOWLEDGMENTS
We would like to thank Dr. Francesca Sciarra for assisting with the references of our manuscript. Open Access Funding provided by Universita degli Studi di Roma La Sapienza within the CRUI‐CARE Agreement.
Degjoni A, Campolo F, Stefanini L, Venneri MA. The NO/cGMP/PKG pathway in platelets: The therapeutic potential of PDE5 inhibitors in platelet disorders. J Thromb Haemost. 2022;20:2465‐2474. doi: 10.1111/jth.15844
Manuscript handled by: Katsue Suzuki‐Inoue
Final decision: Katsue Suzuki‐Inoue, 08 August 2022
REFERENCES
- 1. Martinod K, Deppermann C. Immunothrombosis and thromboinflammation in host defense and disease. Platelets. 2021;32:314‐324. doi: 10.1080/09537104.2020.1817360 [DOI] [PubMed] [Google Scholar]
- 2. Thomas DD, Liu X, Kantrow SP, Lancaster JR Jr. The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2 . Proc Natl Acad Sci USA. 2001;98:355‐360. doi: 10.1073/pnas.98.1.355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yan K, Gao LN, Cui YL, Zhang Y, Zhou X. The cyclic AMP signaling pathway: exploring targets for successful drug discovery (Review). Mol Med Rep. 2016;13:3715‐3723. doi: 10.3892/mmr.2016.5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smolenski A. Novel roles of cAMP/cGMP‐dependent signaling in platelets. J Thromb Haemost. 2012;10:167‐176. doi: 10.1111/j.1538-7836.2011.04576.x [DOI] [PubMed] [Google Scholar]
- 5. Gimbrone MA Jr, García‐Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620‐636. doi: 10.1161/circresaha.115.306301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Favero G, Paganelli C, Buffoli B, Rodella LF, Rezzani R. Endothelium and its alterations in cardiovascular diseases: life style intervention. Biomed Res Int. 2014;2014:801896. doi: 10.1155/2014/801896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stefanini L, Bergmeier W. RAP GTPases and platelet integrin signaling. Platelets. 2019;30:41‐47. doi: 10.1080/09537104.2018.1476681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Varga‐Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler Thromb Vasc Biol. 2008;28:403‐412. doi: 10.1161/atvbaha.107.150474 [DOI] [PubMed] [Google Scholar]
- 9. Durrant TN, van den Bosch MT, Hers I. Integrin α(IIb)β(3) outside‐in signaling. Blood. 2017;130:1607‐1619. doi: 10.1182/blood-2017-03-773614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jackson SP, Nesbitt WS, Westein E. Dynamics of platelet thrombus formation. J Thromb Haemost. 2009;7(Suppl 1):17‐20. doi: 10.1111/j.1538-7836.2009.03401.x [DOI] [PubMed] [Google Scholar]
- 11. Bergmeier W, Stefanini L. Platelets at the vascular interface. Res Pract Thromb Haemost. 2018;2:27‐33. doi: 10.1002/rth2.12061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Portier I, Campbell RA. Role of platelets in detection and regulation of infection. Arterioscler Thromb Vasc Biol. 2021;41:70‐78. doi: 10.1161/atvbaha.120.314645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thon JN, Italiano JE. Platelets: production, morphology and ultrastructure. Handb Exp Pharmacol. 2012;(210):3‐22. doi: 10.1007/978-3-642-29423-5_1 [DOI] [PubMed] [Google Scholar]
- 14. Palacios‐Acedo AL, Mège D, Crescence L, Dignat‐George F, Dubois C, Panicot‐Dubois L. Platelets, thrombo‐inflammation, and cancer: collaborating with the enemy. Front Immunol. 2019;10:1805. doi: 10.3389/fimmu.2019.01805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wallentin L. P2Y(12) inhibitors: differences in properties and mechanisms of action and potential consequences for clinical use. Eur Heart J. 2009;30:1964‐1977. doi: 10.1093/eurheartj/ehp296 [DOI] [PubMed] [Google Scholar]
- 16. Francis SH, Busch JL, Corbin JD, Sibley D. cGMP‐dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525‐563. doi: 10.1124/pr.110.002907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Keravis T, Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br J Pharmacol. 2012;165:1288‐1305. doi: 10.1111/j.1476-5381.2011.01729.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Campolo F, Zevini A, Cardarelli S, et al. Identification of murine phosphodiesterase 5A isoforms and their functional characterization in HL‐1 cardiac cell line. J Cell Physiol. 2018;233:325‐337. doi: 10.1002/jcp.25880 [DOI] [PubMed] [Google Scholar]
- 19. Cesarini V, Guida E, Campolo F, et al. Type 5 phosphodiesterase (PDE5) and the vascular tree: from embryogenesis to aging and disease. Mech Ageing Dev. 2020;190:111311. doi: 10.1016/j.mad.2020.111311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin CS. Tissue expression, distribution, and regulation of PDE5. Int J Impot Res. 2004;16(Suppl 1):S8‐s10. doi: 10.1038/sj.ijir.3901207 [DOI] [PubMed] [Google Scholar]
- 21. Andersson KE. PDE5 inhibitors – pharmacology and clinical applications 20 years after sildenafil discovery. Br J Pharmacol. 2018;175:2554‐2565. doi: 10.1111/bph.14205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Redfield MM, Chen HH, Borlaug BA, et al. Effect of phosphodiesterase‐5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268‐1277. doi: 10.1001/jama.2013.2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang RL, Zhang ZG, Chopp M. Targeting nitric oxide in the subacute restorative treatment of ischemic stroke. Expert Opin Investig Drugs. 2013;22:843‐851. doi: 10.1517/13543784.2013.793672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hong JH, Kwon YS, Kim IY. Pharmacodynamics, pharmacokinetics and clinical efficacy of phosphodiesterase‐5 inhibitors. Expert Opin Drug Metab Toxicol. 2017;13:183‐192. doi: 10.1080/17425255.2017.1244265 [DOI] [PubMed] [Google Scholar]
- 25. Pofi R, Fiore D, De Gaetano R, et al. Phosphodiesterase‐5 inhibition preserves renal hemodynamics and function in mice with diabetic kidney disease by modulating miR‐22 and BMP7. Sci Rep. 2017;7:44584. doi: 10.1038/srep44584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krishnappa P, Fernandez‐Pascual E, Carballido J, Martinez‐Salamanca JI. Sildenafil/Viagra in the treatment of premature ejaculation. Int J Impot Res. 2019;31:65‐70. doi: 10.1038/s41443-018-0099-2 [DOI] [PubMed] [Google Scholar]
- 27. Pofi R, Gianfrilli D, Badagliacca R, Di Dato C, Venneri MA, Giannetta E. Everything you ever wanted to know about phosphodiesterase 5 inhibitors and the heart (but never dared ask): how do they work? J Endocrinol Invest. 2016;39:131‐142. doi: 10.1007/s40618-015-0339-y [DOI] [PubMed] [Google Scholar]
- 28. Isidori AM, Giannetta E, Pofi R, et al. Targeting the NO‐cGMP‐PDE5 pathway in COVID‐19 infection. The DEDALO project. Andrology. 2021;9:33‐38. doi: 10.1111/andr.12837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Campolo F, Pofi R, Venneri MA, Isidori AM. Priming metabolism with the type 5 phosphodiesterase: the role of cGMP‐hydrolyzing enzymes. Curr Opin Pharmacol. 2021;60:298‐305. doi: 10.1016/j.coph.2021.08.007 [DOI] [PubMed] [Google Scholar]
- 30. Pofi R, Giannetta E, Feola T, et al. Sex‐specific effects of daily tadalafil on diabetic heart kinetics in RECOGITO, a randomized, double‐blind, placebo‐controlled trial. Sci Transl Med. 2022;14:eabl8503. doi: 10.1126/scitranslmed.abl8503 [DOI] [PubMed] [Google Scholar]
- 31. Kirkby NS, Lundberg MH, Chan MV, et al. Blockade of the purinergic P2Y12 receptor greatly increases the platelet inhibitory actions of nitric oxide. Proc Natl Acad Sci USA. 2013;110:15782‐15787. doi: 10.1073/pnas.1218880110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gresele P, Momi S, Falcinelli E. Anti‐platelet therapy: phosphodiesterase inhibitors. Br J Clin Pharmacol. 2011;72:634‐646. doi: 10.1111/j.1365-2125.2011.04034.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Naseem KM, Riba R. Unresolved roles of platelet nitric oxide synthase. J Thromb Haemost. 2008;6:10‐19. doi: 10.1111/j.1538-7836.2007.02802.x [DOI] [PubMed] [Google Scholar]
- 34. Furchgott RF, Vanhoutte PM. Endothelium‐derived relaxing and contracting factors. FASEB J. 1989;3:2007‐2018. [PubMed] [Google Scholar]
- 35. Mizuno Y, Isotani E, Huang J, Ding H, Stull JT, Kamm KE. Myosin light chain kinase activation and calcium sensitization in smooth muscle in vivo. Am J Physiol Cell Physiol. 2008;295:C358‐C364. doi: 10.1152/ajpcell.90645.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morrell CN, Matsushita K, Chiles K, et al. Regulation of platelet granule exocytosis by S‐nitrosylation. Proc Natl Acad Sci USA. 2005;102:3782‐3787. doi: 10.1073/pnas.0408310102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Walsh GM, Leane D, Moran N, et al. S‐Nitrosylation of platelet alphaIIbbeta3 as revealed by Raman spectroscopy. Biochemistry. 2007;46:6429‐6436. doi: 10.1021/bi0620712 [DOI] [PubMed] [Google Scholar]
- 38. Lucas KA, Pitari GM, Kazerounian S, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev. 2000;52:375‐414. [PubMed] [Google Scholar]
- 39. Kobsar A, Koessler J, Kehrer L, Gambaryan S, Walter U. The thrombin inhibitors hirudin and Refludan(®) activate the soluble guanylyl cyclase and the cGMP pathway in washed human platelets. Thromb Haemost. 2012;107:521‐529. doi: 10.1160/th11-07-0461 [DOI] [PubMed] [Google Scholar]
- 40. Rajendran S, Chirkov YY. Platelet hyperaggregability: impaired responsiveness to nitric oxide ("platelet NO resistance") as a therapeutic target. Cardiovasc Drugs Ther. 2008;22:193‐203. doi: 10.1007/s10557-008-6098-7 [DOI] [PubMed] [Google Scholar]
- 41. Goubareva I, Gkaliagkousi E, Shah A, Queen L, Ritter J, Ferro A. Age decreases nitric oxide synthesis and responsiveness in human platelets and increases formation of monocyte‐platelet aggregates. Cardiovasc Res. 2007;75:793‐802. doi: 10.1016/j.cardiores.2007.05.021 [DOI] [PubMed] [Google Scholar]
- 42. Signorello MG, Segantin A, Passalacqua M, Leoncini G. Homocysteine decreases platelet NO level via protein kinase C activation. Nitric Oxide. 2009;20:104‐113. doi: 10.1016/j.niox.2008.11.005 [DOI] [PubMed] [Google Scholar]
- 43. Monteiro PF, Morganti RP, Delbin MA, et al. Platelet hyperaggregability in high‐fat fed rats: a role for intraplatelet reactive‐oxygen species production. Cardiovasc Diabetol. 2012;11:5. doi: 10.1186/1475-2840-11-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Awtry EH, Loscalzo J. Aspirin. Circulation. 2000;101:1206‐1218. doi: 10.1161/01.cir.101.10.1206 [DOI] [PubMed] [Google Scholar]
- 45. Denninger JW, Marletta MA. Guanylate cyclase and the .NO/cGMP signaling pathway. Biochim Biophys Acta. 1999;1411:334‐350. doi: 10.1016/s0005-2728(99)00024-9 [DOI] [PubMed] [Google Scholar]
- 46. Kosarikov DN, Young P, Uversky VN, Gerber NC. Human soluble guanylate cyclase: functional expression, purification and structural characterization. Arch Biochem Biophys. 2001;388:185‐197. doi: 10.1006/abbi.2001.2284 [DOI] [PubMed] [Google Scholar]
- 47. Friebe A, Koesling D. The function of NO‐sensitive guanylyl cyclase: what we can learn from genetic mouse models. Nitric Oxide. 2009;21:149‐156. doi: 10.1016/j.niox.2009.07.004 [DOI] [PubMed] [Google Scholar]
- 48. Stasch JP, Schmidt PM, Nedvetsky PI, et al. Targeting the heme‐oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest. 2006;116:2552‐2561. doi: 10.1172/jci28371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Friebe A, Mergia E, Dangel O, Lange A, Koesling D. Fatal gastrointestinal obstruction and hypertension in mice lacking nitric oxide‐sensitive guanylyl cyclase. Proc Natl Acad Sci USA. 2007;104:7699‐7704. doi: 10.1073/pnas.0609778104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stasch JP, Pacher P, Evgenov OV. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation. 2011;123:2263‐2273. doi: 10.1161/circulationaha.110.981738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Friebe A, Schultz G, Koesling D. Sensitizing soluble guanylyl cyclase to become a highly CO‐sensitive enzyme. EMBO J. 1996;15:6863‐6868. [PMC free article] [PubMed] [Google Scholar]
- 52. Stasch JP, Dembowsky K, Perzborn E, Stahl E, Schramm M. Cardiovascular actions of a novel NO‐independent guanylyl cyclase stimulator, BAY 41‐8543: in vivo studies. Br J Pharmacol. 2002;135:344‐355. doi: 10.1038/sj.bjp.0704483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent and selective inhibition of nitric oxide‐sensitive guanylyl cyclase by 1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one. Mol Pharmacol. 1995;48:184‐188. [PubMed] [Google Scholar]
- 54. Kosarikov DN, Lee JM, Uversky VN, Counts GN. Role of conformational changes in the heme‐dependent regulation of human soluble guanylate cyclase. J Inorg Biochem. 2001;87:267‐276. doi: 10.1016/s0162-0134(01)00387-7 [DOI] [PubMed] [Google Scholar]
- 55. Martin E, Lee YC, Murad F. YC‐1 activation of human soluble guanylyl cyclase has both heme‐dependent and heme‐independent components. Proc Natl Acad Sci USA. 2001;98:12938‐12942. doi: 10.1073/pnas.231486198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schäfer A, Flierl U, Kobsar A, Eigenthaler M, Ertl G, Bauersachs J. Soluble guanylyl cyclase activation with HMR1766 attenuates platelet activation in diabetic rats. Arterioscler Thromb Vasc Biol. 2006;26:2813‐2818. doi: 10.1161/01.Atv.0000249407.92147.12 [DOI] [PubMed] [Google Scholar]
- 57. Mendes‐Silverio CB, Leiria LO, Morganti RP, et al. Activation of haem‐oxidized soluble guanylyl cyclase with BAY 60‐2770 in human platelets lead to overstimulation of the cyclic GMP signaling pathway. PLoS One. 2012;7:e47223. doi: 10.1371/journal.pone.0047223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Reiss C, Mindukshev I, Bischoff V, et al. The sGC stimulator riociguat inhibits platelet function in washed platelets but not in whole blood. Br J Pharmacol. 2015;172:5199‐5210. doi: 10.1111/bph.13286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pattillo CB, Bir SC, Branch BG, et al. Dipyridamole reverses peripheral ischemia and induces angiogenesis in the Db/Db diabetic mouse hind‐limb model by decreasing oxidative stress. Free Radic Biol Med. 2011;50:262‐269. doi: 10.1016/j.freeradbiomed.2010.10.714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Venkatesh PK, Pattillo CB, Branch B, et al. Dipyridamole enhances ischaemia‐induced arteriogenesis through an endocrine nitrite/nitric oxide‐dependent pathway. Cardiovasc Res. 2010;85:661‐670. doi: 10.1093/cvr/cvq002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu Y, Cone J, Le SN, et al. Cilostazol and dipyridamole synergistically inhibit human platelet aggregation. J Cardiovasc Pharmacol. 2004;44:266‐273. doi: 10.1097/00005344-200408000-00017 [DOI] [PubMed] [Google Scholar]
- 62. Miller TW, Isenberg JS, Roberts DD. Thrombospondin‐1 is an inhibitor of pharmacological activation of soluble guanylate cyclase. Br J Pharmacol. 2010;159:1542‐1547. doi: 10.1111/j.1476-5381.2009.00631.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li Z, Ajdic J, Eigenthaler M, Du X. A predominant role for cAMP‐dependent protein kinase in the cGMP‐induced phosphorylation of vasodilator‐stimulated phosphoprotein and platelet inhibition in humans. Blood. 2003;101:4423‐4429. doi: 10.1182/blood-2002-10-3210 [DOI] [PubMed] [Google Scholar]
- 64. Yin H, Liu J, Li Z, Berndt MC, Lowell CA, Du X. Src family tyrosine kinase Lyn mediates VWF/GPIb‐IX‐induced platelet activation via the cGMP signaling pathway. Blood. 2008;112:1139‐1146. doi: 10.1182/blood-2008-02-140970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Beaulieu LM, Freedman JE. Inflammation & the platelet histone trap. Blood. 2011;118:1714‐1715. doi: 10.1182/blood-2011-06-362764 [DOI] [PubMed] [Google Scholar]
- 66. Wen L, Feil S, Wolters M, et al. A shear‐dependent NO‐cGMP‐cGKI cascade in platelets acts as an auto‐regulatory brake of thrombosis. Nat Commun. 2018;9:4301. doi: 10.1038/s41467-018-06638-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li Z, Delaney MK, O'Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341‐2349. doi: 10.1161/atvbaha.110.207522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang G, Xiang B, Dong A, et al. Biphasic roles for soluble guanylyl cyclase (sGC) in platelet activation. Blood. 2011;118:3670‐3679. doi: 10.1182/blood-2011-03-341107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309‐327. doi: 10.1161/01.RES.0000256354.95791.f1 [DOI] [PubMed] [Google Scholar]
- 70. Lin CS, Chow S, Lau A, Tu R, Lue TF. Human PDE5A gene encodes three PDE5 isoforms from two alternate promoters. Int J Impot Res. 2002;14:15‐24. doi: 10.1038/sj.ijir.3900802 [DOI] [PubMed] [Google Scholar]
- 71. Zhu B, Strada SJ. The novel functions of cGMP‐specific phosphodiesterase 5 and its inhibitors in carcinoma cells and pulmonary/cardiovascular vessels. Curr Top Med Chem. 2007;7:437‐454. doi: 10.2174/156802607779941198 [DOI] [PubMed] [Google Scholar]
- 72. Schwarz UR, Walter U, Eigenthaler M. Taming platelets with cyclic nucleotides. Biochem Pharmacol. 2001;62:1153‐1161. doi: 10.1016/s0006-2952(01)00760-2 [DOI] [PubMed] [Google Scholar]
- 73. Mullershausen F, Koesling D, Friebe A. NO‐sensitive guanylyl cyclase and NO‐induced feedback inhibition in cGMP signaling. Front Biosci. 2005;10:1269‐1278. doi: 10.2741/1617 [DOI] [PubMed] [Google Scholar]
- 74. Wyatt TA, Naftilan AJ, Francis SH, Corbin JD. ANF elicits phosphorylation of the cGMP phosphodiesterase in vascular smooth muscle cells. Am J Physiol. 1998;274:H448‐H455. doi: 10.1152/ajpheart.1998.274.2.H448 [DOI] [PubMed] [Google Scholar]
- 75. Rybalkin SD, Rybalkina IG, Feil R, Hofmann F, Beavo JA. Regulation of cGMP‐specific phosphodiesterase (PDE5) phosphorylation in smooth muscle cells. J Biol Chem. 2002;277:3310‐3317. doi: 10.1074/jbc.M106562200 [DOI] [PubMed] [Google Scholar]
- 76. Ashman DF, Lipton R, Melicow MM, Price TD. Isolation of adenosine 3′, 5′‐monophosphate and guanosine 3′, 5′‐monophosphate from rat urine. Biochem Biophys Res Commun. 1963;11:330‐334. doi: 10.1016/0006-291x(63)90566-7 [DOI] [PubMed] [Google Scholar]
- 77. Antl M, von Brühl ML, Eiglsperger C, et al. IRAG mediates NO/cGMP‐dependent inhibition of platelet aggregation and thrombus formation. Blood. 2007;109:552‐559. doi: 10.1182/blood-2005-10-026294 [DOI] [PubMed] [Google Scholar]
- 78. Schultess J, Danielewski O, Smolenski AP. Rap1GAP2 is a new GTPase‐activating protein of Rap1 expressed in human platelets. Blood. 2005;105:3185‐3192. doi: 10.1182/blood-2004-09-3605 [DOI] [PubMed] [Google Scholar]
- 79. Rukoyatkina N, Walter U, Friebe A, Gambaryan S. Differentiation of cGMP‐dependent and ‐independent nitric oxide effects on platelet apoptosis and reactive oxygen species production using platelets lacking soluble guanylyl cyclase. Thromb Haemost. 2011;106:922‐933. doi: 10.1160/th11-05-0319 [DOI] [PubMed] [Google Scholar]
- 80. Maurice DH, Haslam RJ. Molecular basis of the synergistic inhibition of platelet function by nitrovasodilators and activators of adenylate cyclase: inhibition of cyclic AMP breakdown by cyclic GMP. Mol Pharmacol. 1990;37:671‐681. [PubMed] [Google Scholar]
- 81. Haslam RJ, Dickinson NT, Jang EK. Cyclic nucleotides and phosphodiesterases in platelets. Thromb Haemost. 1999;82:412‐423. [PubMed] [Google Scholar]
- 82. Thomas MK, Francis SH, Corbin JD. Characterization of a purified bovine lung cGMP‐binding cGMP phosphodiesterase. J Biol Chem. 1990;265:14964‐14970. [PubMed] [Google Scholar]
- 83. Kleppe R, Jonassen I, Døskeland SO, Selheim F. Mathematical modelling of nitric oxide/cyclic GMP/cyclic AMP signalling in platelets. Int J Mol Sci. 2018;19:612. doi: 10.3390/ijms19020612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Aszódi A, Pfeifer A, Ahmad M, et al. The vasodilator‐stimulated phosphoprotein (VASP) is involved in cGMP‐ and cAMP‐mediated inhibition of agonist‐induced platelet aggregation, but is dispensable for smooth muscle function. EMBO J. 1999;18:37‐48. doi: 10.1093/emboj/18.1.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Keicher C, Gambaryan S, Schulze E, Marcus K, Meyer HE, Butt E. Phosphorylation of mouse LASP‐1 on threonine 156 by cAMP‐ and cGMP‐dependent protein kinase. Biochem Biophys Res Commun. 2004;324:308‐316. doi: 10.1016/j.bbrc.2004.08.235 [DOI] [PubMed] [Google Scholar]
- 86. Walter U, Gambaryan S. cGMP and cGMP‐dependent protein kinase in platelets and blood cells. Handb Exp Pharmacol. 2009;(191):533‐548. doi: 10.1007/978-3-540-68964-5_23 [DOI] [PubMed] [Google Scholar]
- 87. Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F. Cyclic GMP‐dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ Res. 2003;93:907‐916. doi: 10.1161/01.Res.0000100390.68771.Cc [DOI] [PubMed] [Google Scholar]
- 88. Erdmann J, Stark K, Esslinger UB, et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature. 2013;504:432‐436. doi: 10.1038/nature12722 [DOI] [PubMed] [Google Scholar]
- 89. Goldstein I, Burnett AL, Rosen RC, Park PW, Stecher VJ. The serendipitous story of sildenafil: an unexpected oral therapy for erectile dysfunction. Sex Med Rev. 2019;7:115‐128. doi: 10.1016/j.sxmr.2018.06.005 [DOI] [PubMed] [Google Scholar]
- 90. Halcox JP, Nour KR, Zalos G, et al. The effect of sildenafil on human vascular function, platelet activation, and myocardial ischemia. J Am Coll Cardiol. 2002;40:1232‐1240. doi: 10.1016/s0735-1097(02)02139-3 [DOI] [PubMed] [Google Scholar]
- 91. Venneri MA, Barbagallo F, Fiore D, et al. PDE5 inhibition stimulates Tie2‐expressing monocytes and angiopoietin‐1 restoring angiogenic homeostasis in diabetes. J Clin Endocrinol Metab. 2019;104:2623‐2636. doi: 10.1210/jc.2018-02525 [DOI] [PubMed] [Google Scholar]
- 92. Venneri MA, Giannetta E, Panio G, et al. Chronic inhibition of PDE5 limits pro‐inflammatory monocyte‐macrophage polarization in streptozotocin‐induced diabetic mice. PLoS One. 2015;10:e0126580. doi: 10.1371/journal.pone.0126580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yang HM, Jin S, Jang H, et al. Sildenafil reduces neointimal hyperplasia after angioplasty and inhibits platelet aggregation via activation of cGMP‐dependent protein kinase. Sci Rep. 2019;9:7769. doi: 10.1038/s41598-019-44190-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pingarrón‐Martín L, Arias‐Gallo LJ. Sildenafil effect on prevention of thrombosis after microsurgical anastomosis: experimental rat model of thrombotic suture. Oral Maxillofac Surg. 2014;18:53‐58. doi: 10.1007/s10006-012-0387-9 [DOI] [PubMed] [Google Scholar]
- 95. Saeed O, Rangasamy S, Selevany I, et al. Sildenafil is associated with reduced device thrombosis and ischemic stroke despite low‐level hemolysis on Heart Mate II support. Circ Heart Fail. 2017;10:e004222. doi: 10.1161/circheartfailure.117.004222 [DOI] [PubMed] [Google Scholar]
- 96. Andersson DP, Trolle Lagerros Y, Grotta A, Bellocco R, Lehtihet M, Holzmann MJ. Association between treatment for erectile dysfunction and death or cardiovascular outcomes after myocardial infarction. Heart (British Cardiac Society). 2017;103:1264‐1270. doi: 10.1136/heartjnl-2016-310746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Herrmann HC, Chang G, Klugherz BD, Mahoney PD. Hemodynamic effects of sildenafil in men with severe coronary artery disease. N Engl J Med. 2000;342:1622‐1626. doi: 10.1056/nejm200006013422201 [DOI] [PubMed] [Google Scholar]
- 98. Xanthopoulos A, Wolski K, Wang Q, et al. Postimplant phosphodiesterase‐5 inhibitor use in centrifugal flow left ventricular assist devices. JACC Heart failure. 2022;10:89‐100. doi: 10.1016/j.jchf.2021.09.008 [DOI] [PubMed] [Google Scholar]
- 99. Lewis GD, Witzke C, Colon‐Hernandez P, Guerrero JL, Bloch KD, Semigran MJ. Sildenafil improves coronary artery patency in a canine model of platelet‐mediated cyclic coronary occlusion after thrombolysis. J Am Coll Cardiol. 2006;47:1471‐1477. doi: 10.1016/j.jacc.2005.11.060 [DOI] [PubMed] [Google Scholar]
- 100. Eardley I, Ellis P, Boolell M, Wulff M. Onset and duration of action of sildenafil for the treatment of erectile dysfunction. Br J Clin Pharmacol. 2002;53(Suppl 1):61s‐65s. doi: 10.1046/j.0306-5251.2001.00034.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. De Bon E, Bonanni G, Saggiorato G, Bassi P, Cella G. Effects of tadalafil on platelets and endothelium in patients with erectile dysfunction and cardiovascular risk factors: a pilot study. Angiology. 2010;61:602‐606. doi: 10.1177/0003319710362977 [DOI] [PubMed] [Google Scholar]
- 102. Demirci A, Ozgur BC. The effect of using tadalafil 5 mg/day on neutrophil‐lymphocyte and platelet‐lymphocyte ratios in mild‐medium and severe erectile dysfunction patients; and comparison of clinical response. Andrologia. 2019;51:e13347. doi: 10.1111/and.13347 [DOI] [PubMed] [Google Scholar]
- 103. Toque HA, Teixeira CE, Priviero FB, Morganti RP, Antunes E, De Nucci G. Vardenafil, but not sildenafil or tadalafil, has calcium‐channel blocking activity in rabbit isolated pulmonary artery and human washed platelets. Br J Pharmacol. 2008;154:787‐796. doi: 10.1038/bjp.2008.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bajraktari G, Burhenne J, Bugert P, Haefeli WE, Weiss J. Cyclic guanosine monophosphate modulates accumulation of phosphodiesterase 5 inhibitors in human platelets. Biochem Pharmacol. 2017;145:54‐63. doi: 10.1016/j.bcp.2017.08.026 [DOI] [PubMed] [Google Scholar]
- 105. Gudmundsdóttir IJ, McRobbie SJ, Robinson SD, Newby DE, Megson IL. Sildenafil potentiates nitric oxide mediated inhibition of human platelet aggregation. Biochem Biophys Res Commun. 2005;337:382‐385. doi: 10.1016/j.bbrc.2005.09.060 [DOI] [PubMed] [Google Scholar]
- 106. Dunkern TR, Hatzelmann A. The effect of sildenafil on human platelet secretory function is controlled by a complex interplay between phosphodiesterases 2, 3 and 5. Cell Signal. 2005;17:331‐339. doi: 10.1016/j.cellsig.2004.07.007 [DOI] [PubMed] [Google Scholar]
- 107. Mirone V, Napolitano L, di Villa D'E, et al. A new original nutraceutical formulation ameliorates the effect of Tadalafil on clinical score and cGMP accumulation. Arch Ital Urol Androl. 2021;93:221‐226. doi: 10.4081/aiua.2021.2.221 [DOI] [PubMed] [Google Scholar]
- 108. Satish A, Sairam S, Ahmed F, Urooj A. Moringa oleifera Lam.: protease activity against blood coagulation cascade. Pharm Res. 2012;4:44‐49. doi: 10.4103/0974-8490.91034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Signorello MG, Giacobbe E, Passalacqua M, Leoncini G. The anandamide effect on NO/cGMP pathway in human platelets. J Cell Biochem. 2011;112:924‐932. doi: 10.1002/jcb.23008 [DOI] [PubMed] [Google Scholar]
- 110. Tzoumas N, Farrah TE, Dhaun N, Webb DJ. Established and emerging therapeutic uses of PDE type 5 inhibitors in cardiovascular disease. Br J Pharmacol. 2020;177:5467‐5488. doi: 10.1111/bph.14920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Bath PM, Woodhouse LJ, Appleton JP, et al. Antiplatelet therapy with aspirin, clopidogrel, and dipyridamole versus clopidogrel alone or aspirin and dipyridamole in patients with acute cerebral ischaemia (TARDIS): a randomised, open‐label, phase 3 superiority trial. Lancet (London, England). 2018;391:850‐859. doi: 10.1016/s0140-6736(17)32849-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ranucci M, Aloisio T, Di Dedda U, Menicanti L, de Vincentiis C, Baryshnikova E. Gender‐based differences in platelet function and platelet reactivity to P2Y12 inhibitors. PLoS One. 2019;14:e0225771. doi: 10.1371/journal.pone.0225771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Leite NR, Siqueira de Medeiros M, Mury WV, et al. Platelet hyperaggregability in obesity: is there a role for nitric oxide impairment and oxidative stress? Clin Exp Pharmacol Physiol. 2016;43:738‐744. doi: 10.1111/1440-1681.12589 [DOI] [PubMed] [Google Scholar]
- 114. Huo Y, Schober A, Forlow SB, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61‐67. doi: 10.1038/nm810 [DOI] [PubMed] [Google Scholar]
- 115. Momi S, Falcinelli E, Petito E, Ciarrocca Taranta G, Ossoli A, Gresele P. Matrix metalloproteinase‐2 on activated platelets triggers endothelial PAR‐1 initiating atherosclerosis. Eur Heart J. 2022;43:504‐514. doi: 10.1093/eurheartj/ehab631 [DOI] [PubMed] [Google Scholar]
- 116. Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107:15880‐15885. doi: 10.1073/pnas.1005743107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Coenen DM, Heinzmann ACA, Oggero S, et al. Inhibition of phosphodiesterase 3A by cilostazol dampens proinflammatory platelet functions. Cell. 2021;10:1998. doi: 10.3390/cells10081998 [DOI] [PMC free article] [PubMed] [Google Scholar]