

Abstract
Aims
Patients with dermatomyositis (DM) suffer from reduced aerobic metabolism contributing to impaired muscle function, which has been linked to cytochrome c oxidase (COX) deficiency in muscle tissue. This mitochondrial respiratory chain dysfunction is typically seen in perifascicular regions, which also show the most intense inflammatory reaction along with capillary loss and muscle fibre atrophy. The objective of this study was to investigate the pathobiology of the oxidative phosphorylation deficiency in DM.
Methods
Muscle biopsy specimens with perifascicular COX deficiency from five juveniles and seven adults with DM were investigated. We combined immunohistochemical analyses of subunits in the respiratory chain including complex I (subunit NDUFB8), complex II (succinate dehydrogenase, subunit SDHB) and complex IV (COX, subunit MTCO1) with in situ hybridisation, next generation deep sequencing and quantitative polymerase chain reaction (PCR).
Results
There was a profound deficiency of complexes I and IV in the perifascicular regions with enzyme histochemical COX deficiency, whereas succinate dehydrogenase activity and complex II were preserved. In situ hybridisation of mitochondrial RNA showed depletion of mitochondrial DNA (mtDNA) transcripts in the perifascicular regions. Analysis of mtDNA by next generation deep sequencing and quantitative PCR in affected muscle regions showed an overall reduction of mtDNA copy number particularly in the perifascicular regions.
Conclusion
The respiratory chain dysfunction in DM muscle is associated with mtDNA depletion causing deficiency of complexes I and IV, which are partially encoded by mtDNA, whereas complex II, which is entirely encoded by nuclear DNA, is preserved. The depletion of mtDNA indicates a perturbed replication of mtDNA explaining the muscle pathology and the disturbed aerobic metabolism.
Keywords: dermatomyositis, inflammatory myopathy, mitochondria, mitochondrial DNA, oxidative phosphorylation, respiratory chain
Cytochrome c oxidase deficiency in dermatomyositis muscle is typically seen in perifascicular regions. We demonstrate deficiency of complexes I and IV of the respiratory chain and depletion of mtDNA transcripts by in situ hybridization in these regions. Analyses of mtDNA by next‐generation deep sequencing and quantitative PCR in affected muscle regions show an overall reduction of mtDNA copy number particularly in the perifascicular regions. Our results provide evidence that respiratory chain dysfunction in dermatomyositis is associated with mtDNA depletion.
Key Points.
In dermatomyositis, muscle mitochondrial respiratory chain dysfunction is typically seen in perifascicular regions.
The respiratory chain dysfunction in dermatomyositis muscle is associated with mtDNA depletion.
The depletion in dermatomyositis of mtDNA indicates a perturbed replication of mtDNA explaining the muscle pathology and the disturbed aerobic metabolism.
INTRODUCTION
Idiopathic inflammatory myopathies (IIM) are a cause of muscle weakness in children and adults. In spite of immunosuppressive treatment, many patients suffer from a lifelong disease leading to severe disability and reduced quality of life. The classification of IIM is evolving but the main distinct subtypes include dermatomyositis (DM), inclusion body myositis (IBM), immune‐mediated necrotising myopathy and anti‐synthetase syndrome [1, 2].
DM is characterised by subacute onset of muscle weakness along with a characteristic heliotrope skin rash and Gottron's papules and occurs both in juveniles (juvenile DM) and adults (adult DM) [3]. The pathophysiology of muscle weakness in DM remains elusive. Interferon‐ß and type I interferon‐inducible proteins seem to play pivotal roles in the initiation and maintenance of the disease, which is associated with a complement mediated intramuscular microangiopathy, leading to loss of capillaries, muscle ischaemia and muscle fibre degeneration [4, 5, 6]. The histopathological changes in muscle are often highly characteristic and include perifascicular muscle fibre atrophy and capillary loss, perifascicular upregulation of major histocompatibility complex (MHC) type I and in most cases also mitochondrial abnormalities. Enzyme‐histochemical deficiency of cytochrome c oxidase (COX, complex IV of the respiratory chain) in perifascicular muscle fibres indicates a disturbance of the oxidative phosphorylation (OXPHOS) system and occurs both in juvenile and adult DM [7, 8]. There is evidence that the mitochondrial dysfunction has an important role in the pathophysiology of DM. The patients with DM have a reduced maximal aerobic capacity [9, 10], which is inversely correlated with the proportion of COX deficient muscle fibres and mitochondrial functional defects are considered a hallmark of DM [5, 11]. The exact mechanism for the mitochondrial dysfunction has not been clarified and there may be several mechanisms involved.
COX deficiency combined with preserved activity of succinate dehydrogenase (SDH, complex II of the respiratory chain) as seen in DM [7, 8] is also typical for many disorders associated with mitochondrial DNA (mtDNA) defects [12]. This combined COX deficiency and preserved SDH activity in mitochondrial diseases is because of the fact that mtDNA encodes for three important subunits of COX but no subunits of SDH, which is entirely encoded by nuclear genes [13]. Therefore, muscle diseases because of mtDNA point mutations in tRNA genes, large‐scale rearrangements (deletions and duplications) or mtDNA depletion (reduced mtDNA copy number) usually display muscle fibres with COX deficiency and preserved SDH activity [14, 15].
On this background, we aimed to explore the presence of mtDNA, mtRNA and proteins involved in the respiratory chain to investigate the role of mtDNA for the observed COX deficiency in juvenile and adult DM. The results demonstrate a clear complex I and complex IV deficiency, associated with depletion of mtDNA and mtDNA transcripts in the perifascicular regions with enzyme‐histochemical COX deficiency.
MATERIAL AND METHODS
Muscle samples
Archival muscle biopsy specimens from 12 patients with DM (5 juveniles, juvenile DM1‐5; 7 adults, adult DM1‐7) were selected based on early DM with no or short treatment and the presence of marked deficiency of COX in the perifascicular regions. Summary of patient data is given in Table 1. All patients had muscle weakness, seven displayed a typical skin rash and three had joint symptoms. Cancer was not reported in any patient. No data on myositis‐specific and myositis‐associated autoantibodies were available. Control skeletal muscle specimens included muscle biopsy specimens from 14 individuals (C1–14, age range 3–60 years) who had been investigated for a possible muscle disorder, but in whom the clinical and pathological investigations excluded muscle disease (Table 2). Four disease control skeletal muscle specimens (DC) were included (Table 2, DC1–4). Three were from patients with mitochondrial myopathy, two of these due to the “common” 4977 bp mtDNA deletion (m.8470–13,447) and one caused by a mtDNA point mutation (m.5669G > A) in the tRNA (Asn) gene (MT‐TN). The fourth disease control was a biopsy from a patient with IBM with typical multiple mtDNA deletions in muscle. Open skeletal muscle biopsies were performed in all individuals. Specimens were snap‐frozen in isopentane cooled by liquid nitrogen and stored at −80°C until analysed.
TABLE 1.
Summary of patients with dermatomyositis in this study at the time of diagnosis
| Cases | Sex | Age at biopsy (y) | Duration of muscle symptoms | CK | Muscle | Extramuscular manifestations | |
|---|---|---|---|---|---|---|---|
| Juvenile DM | 1 | M | 4 | 6 mo | Normal | n/a | Skin |
| 2 | M | 6 | n/a | Normal | Tibialis anterior | Skin, joints | |
| 3 | F | 9 | 4 mo | Elevated | n/a | Skin | |
| 4 | M | 12 | 3 mo | Elevated | Vastus lateralis | Skin, fever, myocarditis | |
| 5 | F | 13 | 3 mo | Elevated | n/a | Skin | |
| Adult DM | 1 | F | 36 | 1 mo | n/a | Tibialis anterior | Skin, dysphagia, joints, Raynaud's syndrome |
| 2 | M | 37 | n/a | n/a | Vastus lateralis | Lung | |
| 3 | F | 40 | 3 mo | Normal | Biceps | Joints, fatigue | |
| 4 | M | 47 | 2 mo | Normal | Tibialis anterior | Skin | |
| 5 | F | 56 | n/a | n/a | Deltoid | Skin | |
| 6 | F | 60 | 1.5 y | Normal | Deltoid | Skin, joints, fever | |
| 7 | M | 88 | n/a | Normal | Deltoid | Skin | |
M, male; F, female; y, year; mo, months; n/a, not available.
TABLE 2.
Summary of controls used in this study
| C | Sex | Age at biopsy (y) | C | Sex | Age at biopsy (y) |
|---|---|---|---|---|---|
| 1 | F | 3 | 8 | F | 41 |
| 2 | M | 5 | 9 | M | 49 |
| 3 | M | 11 | 10 | M | 51 |
| 4 | F | 13 | 11 | F | 54 |
| 5 | M | 17 | 12 | F | 54 |
| 6 | F | 34 | 13 | M | 56 |
| 7 | M | 36 | 14 | F | 60 |
| DC | Sex | Age at biopsy (y) | Disease | ||
| 1 | F | 29 | mtDNA, single deletion | ||
| 2 | M | 50 | mtDNA, tRNA point mutation | ||
| 3 | M | 69 | Inclusion body myositis | ||
| 4 | F | 30 | mtDNA single deletion | ||
C, control; F, female; M, Male; y, year; DC, disease control.
Enzyme and immunohistochemical analyses
Cryosections, 8‐μm thick, were cut and mounted on Superfrost Plus slides (J1800AMNZ, Thermo Scientific). Standard techniques were applied for enzyme histochemical analyses of COX, SDH and combined COX‐SDH [16]. For immunohistochemical studies of the mitochondrial respiratory chain complex, cryosections were fixed in 4% formaldehyde at 4°C for 10 min, washed in Tris‐buffered saline‐Tween 20 (TBS‐T) for 10 min, permeabilised in a graded methanol series (70% 10 min, 95% 10 min, 100% 20 min, 95% 10 min and 70% 10 min) and washed in TBS‐T for 5 min. For the study of MHC‐class I (HLA‐ABC), the cryosections were fixed with acetone. The tissue sections were processed in a Dako Autostainer using the EnVision FLEX DAB+ Substrate Chromogen System kit and incubated with the following primary antibodies for 1 h: anti‐NDUFB8 (complex I, ab110242, Abcam, UK, 1:100), anti‐SDHB (complex II, ab14714, Abcam, 1:500), anti‐MTCO1 (complex IV, ab14705, Abcam, 1:2000), anti‐VDAC1 (porin, a mitochondrial marker, ab14734, Abcam, 1:2000) and anti‐human HLA‐ABC (M0736, clone W6/32, Dako, 1:1000). These antibodies to subunits of complex I, II and IV of the respiratory chain have been well characterised and demonstrated to be reliable tools in studies on mitochondrial myopathies [17, 18, 19].
Analysis of mtDNA transcripts by in situ hybridisation
Because the mtDNA is transcribed as a single polycistronic precursor RNA, we chose to investigate the overall presence of transcripts by combining a probe for MT‐ND1 transcripts (encoding NADH–ubiquinone oxidoreductase chain 1) in the minor arc of mtDNA with a probe for MT‐ND4 transcripts (encoding NADH–ubiquinone oxidoreductase chain 4) in the major arc of mtDNA. In this way, we reduced the influence of possible large‐scale deletions on the results because such deletions are located in either the minor or the major arc of mtDNA. Cryosections, 10‐μm thick, were mounted on Superfrost Plus slides and included muscle biopsy specimens from two patients with DM (one adult and one juvenile), one normal control and one mitochondrial disease control (with the “common” 4977 bp mtDNA deletion). The analysis was performed by RNAscope technology (Advanced Cell Diagnostics, Inc., Hayward, CA, USA) according to the manufacturer's protocol, including a chromogenic RNAscope 2.5 HD Assay Brown‐Hs (cat no. 322370), and RNAscope probes for MT‐ND1 (RNAscope® Probe ‐ Hs‐MT‐ND1; 7 ZZ‐probe set targeting 3310–4049 of NC_012920.1) and MT‐ND4 (RNAscope® Probe ‐ Hs‐MT‐ND4; 20 ZZ‐probe set targeting 10,953–12,135 of NC_012920.1).
Quantification of mtDNA by qPCR in COX‐deficient and COX‐normal muscle fibres
To study if the relative amount of mtDNA was lower in COX‐deficient (COX−) regions compared to COX‐normal (COX+) in the same muscle sections, four groups of approximately 30 muscle fibres in each section, either COX+ or COX−, were isolated with a tungsten needle from 16‐μm thick cryosections from four patients with DM after SDH staining. We used sequential cryosections with combined COX‐SDH staining as a reference to select the regions of interest to be isolated; the brown fibres were regarded as COX+, the blue fibres were regarded as COX−. Quantitative PCR analysis was performed as previously described [15, 20]. Briefly, the nuclear gene CCR5 was selected to serve as the nuclear genome control, and the Hs99999149_s1 primer/probe set was used (TaqMan Gene Expression Assays; Life Technologies, Carlsbad, CA, USA). mtDNA primers (forward nt 3782–3806 and reverse nt 3846–3826) and a FAM‐labelled probe (nt 3808–3824) were chosen from the ND1 region, a region that is rarely deleted. The mtDNA copy number was determined by the mtDNA/nDNA ratio. The relative mtDNA levels in COX− and COX+ regions were then compared.
Analysis of mtDNA by deep sequencing
To investigate the overall mtDNA copy number, presence of mtDNA rearrangements and mtDNA single‐nucleotide polymorphisms (SNPs), total genomic DNA was isolated from the muscle biopsy specimens from two patients with juvenile DM, two patients with adult DM (Table 1) and 14 controls (C1–14) (Table 2) using standard protocols. DNA was subjected to whole genome sequencing (WGS) using the TruSeq™ PCR‐Free library preparation kit (Illumina, San Diego, CA, USA), and the Illumina's HiSeq X platform was used for sequencing (Illumina, San Diego, CA, USA).
To identify mtDNA copy number, large mtDNA deletions and duplications, we applied the MitoSAlt pipeline, and the paired‐end reads from the WGS were aligned as previously described [21]. Gapped alignments, indicative of deletions/duplications, were clustered and visualised as described [22]. A deletion or duplication was considered only if it was supported by five sequencing reads with a minimum heteroplasmy level of 0.01%. Heteroplasmy levels for individual deletions and duplications were estimated by comparing the number of reads supporting the corresponding breakpoints to the total number of reads overlapping the breakpoints (including wild type).
Analysis of mtDNA copy number in relation to nuclear DNA was estimated as previously described: mtDNA copy number = mitochondrial genome coverage × 2/nuclear genome coverage [23]. To identify somatic mtDNA SNPs, the paired‐end reads from the WGS were aligned to the reference genome (hg19) using the CLC Biomedical Genomics workbench (Qiagen). The data were analysed using Ingenuity Variant Analysis (www.ingenuity.com/products/variant-analysis) (Qiagen). A level of at least 1% heteroplasmy was set to exclude sequencing errors and below 20% to exclude most of the inherited variants, which are frequently homoplasmic or show a high heteroplasmy level.
RESULTS
In order to get insights into the molecular pathobiology explaining the COX deficiency in the peripheral parts of the muscle fascicles in DM, we combined DNA, RNA and protein analyses.
Combined complex I and IV OXPHOS deficiency in DM
As part of the inclusion criteria, all twelve cases of DM showed profound enzyme histochemical COX deficiency in the perifascicular regions of the muscle biopsies (Figure 1). These regions also showed an upregulation of MHC‐class I, indicating an association between inflammation and COX deficiency (Figure 1). Immunohistochemical analysis of complex I and IV subunits showed markedly reduced expression in the perifascicular regions with low COX enzymatic activity (Figure 2, left panel). In line with the preserved SDH activity, no deficiency of complex II was identified (Figure 2, left panel). There was no loss of mitochondria in the perifascicular regions, as shown by normal presence of the mitochondrial marker VDAC1. A combined complex I and IV (COX) deficiency with preserved complex II (SDH) activity in muscle fibres was also seen in mitochondrial myopathy disease controls, such as the tRNA (Asn) m.5669G > A point mutation and the “common” large‐scale mtDNA deletion involving several tRNA genes (Figure 2, right panel).
FIGURE 1.
Perifascicular atrophy and COX deficiency (A and B) and upregulation of MHC‐class I (D and E) in juvenile (juvenile DM5) and adult (adult DM4) DM. Serial sections demonstrate COX deficiency (blue fibres) in the same regions as upregulation of MHC‐class I is seen. In the control, there is no perifascicular atrophy or COX deficiency (C) and no expression of MHC‐class I in muscle fibres (F). Bar = 250 μm
FIGURE 2.
Enzyme and immunohistochemical assays of skeletal muscle biopsy in patients with DM (left panels, juvenile DM5 and adult DM3), a normal control (middle panel), and two patients with mitochondrial myopathy as disease controls (right panels: mtDNA deletion (m.8470–13,447) and mtDNA point mutation (MT‐TN, m.5669G > A). Each case panel consists of enzyme‐histochemistry of COX and immunohistochemistry of mitochondrial respiratory chain complex I subunit NDUFB8, complex II subunit SDHB, complex IV subunit MTCO1 and the mitochondrial marker VDAC1. In patients with DM, there is a combined deficiency of complexes I and IV in the perifascicular regions with COX deficiency. Due to mtDNA mutations, there is combined complex I and complex IV deficiency in the COX deficient fibres in mitochondrial myopathy. Bar = 250 μm
Depletion of mtDNA transcripts in COX‐deficient perifascicular regions in DM
Because there was a reduced expression of the partly mtDNA encoded enzyme complexes I and IV in the respiratory chain, we analysed the level of mtDNA transcripts in the same perifascicular regions in muscle biopsies by in situ hybridisation. The analysis, which included a combination of probes targeting MT‐ND1 and MT‐ND4, demonstrated reduced numbers of mitochondrial transcripts in perifascicular regions corresponding to the regions that showed low expression of complex IV but preserved expression of complex II (Figure 3). To validate the in situ hybridisation assay, we analysed muscle tissue from a normal control and a patient with a single mtDNA deletion (Figure 4). The signal from combined ND1/ND4 probes corresponded as expected to the overall amount of mitochondria in each fibre as seen in normal muscle (Figure 4D) and mitochondrial myopathy muscle (Figure 4E). When using only the MT‐ND4 probe (ND4) corresponding to the deleted part of mtDNA, the signal was profoundly reduced in COX deficient fibres confirming that the in situ hybridisation assay reflects the number of mtDNA transcripts (Figure 4F).
FIGURE 3.
Serial sections of a muscle biopsy from a juvenile patient with DM (juvenile DM5) showing deficiency of complex IV but not complex II in the perifascicular regions where in situ hybridisation demonstrates the depletion of mtDNA transcripts. (A) Enzyme histochemistry of complex IV. (B) Immunohistochemistry of complex II subunit SDHB. (c) In situ hybridisation of mtDNA transcripts with probes ND1 and ND4. (D) A schematic drawing of the mtDNA genome with the locations of the ND1 and ND4 probes marked in red. Bar = 200 μm
FIGURE 4.
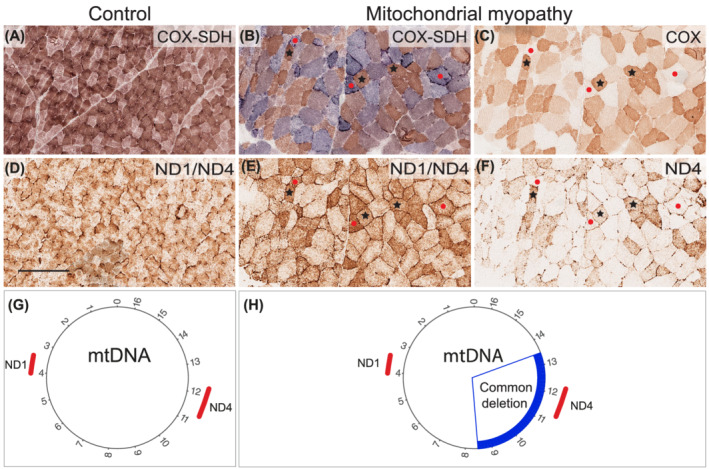
Enzyme histochemical and RNA in situ hybridisation assays of skeletal muscle biopsy. Enzyme histochemistry of COX/SDH in the normal control (A) and the disease control (B) and COX in the disease control (C). In COX/SDH staining, the blue fibres indicate COX deficiency. Figures 4D–H show RNA in situ hybridisation assay with either a combination of probes targeting MT‐ND1 (ND1, m.3310–4049) and MT‐ND4 (ND4, m.10953–12,135), or only the ND4 probe. The combination of both probes (ND1/ND4) shows that the number of transcripts corresponds to the number of mitochondria both in control (A and D) and a disease control (B and E). The mtDNA deletion (m.8470–13,447) causes depletion of mtDNA corresponding to the ND4 probe and therefore, depletion of the corresponding mtDNA transcripts (F). Control = Normal control. Mitochondrial myopathy = disease control (DC1) with the single 4977 bp “common” deletion in mtDNA. Black stars indicate COX‐positive muscle fibres and red dots indicate COX‐deficient fibres. Bar = 250 μm
Depletion of mitochondrial DNA in DM
To investigate if the reduced levels of transcripts, shown by in situ hybridisation, are associated with mtDNA depletion in the same regions, we isolated skeletal muscle fibres and estimated the amount of mtDNA by applying qPCR. We then compared COX− with COX+ fibres in two adult and two juvenile cases of DM. The analysis showed a reduced amount of mtDNA in COX− compared to COX+ fibres (p < 0.05) (Figure 5A). To investigate whether there was an overall reduction of mtDNA, we applied deep sequencing of skeletal muscle DNA, followed by bioinformatics analysis. Detailed characterisation of mtDNA in muscle samples from four patients (two juveniles and two adults) with DM and 14 age‐matched controls (five juveniles and nine adults) was performed. A mean coverage of 43,000× (range 17,000–63,000) in the patients and 127,000× (range 75,000–230,000) in controls was obtained for mtDNA. Estimations of mtDNA copy number revealed a significantly reduced amount of mtDNA in the DM muscles (p < 0.001) with a mean of 2169 mtDNA copies per 1 nuclear DNA copy (range 889–3032) compared to controls with a mean of 6590 (range 3672–9706) (Figure 5B).
FIGURE 5.
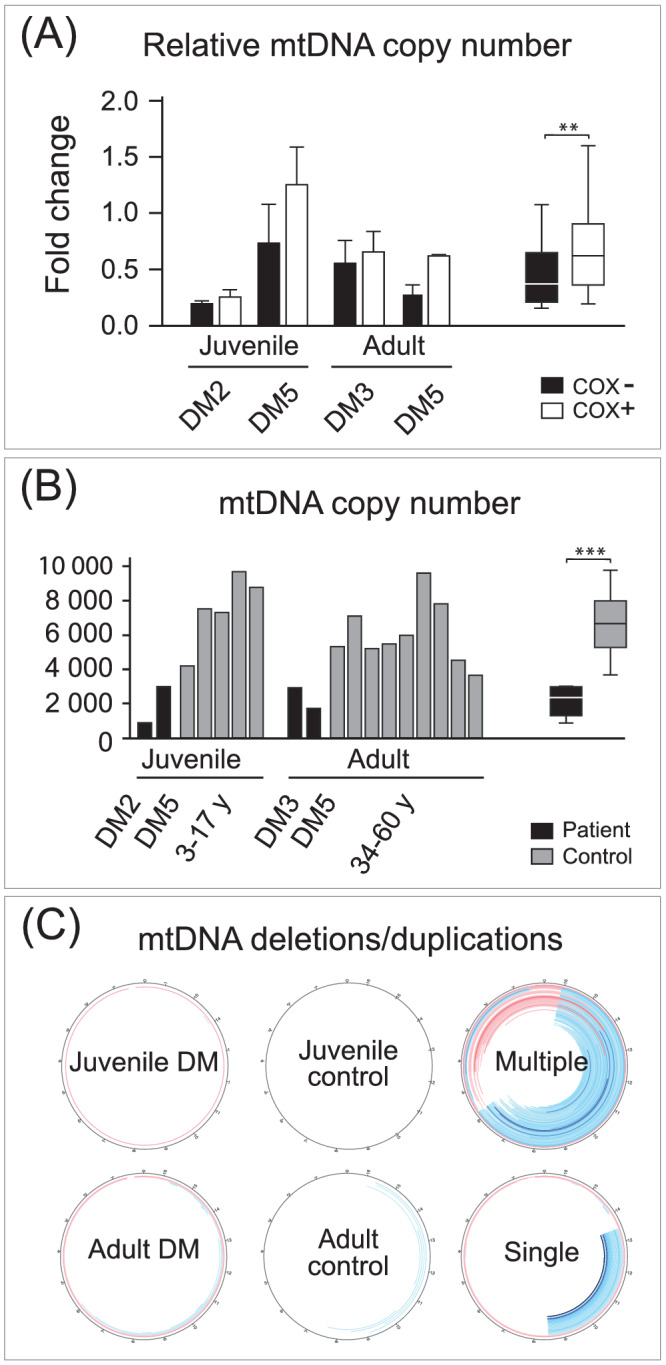
Analysis of mtDNA in muscle biopsy specimens from patients with juvenile and adult DM and controls. (A) Quantitative PCR analysis showing a relatively lower amount of mtDNA in COX‐deficient (COX−) compared to COX‐normal (COX+) muscle fibres in two juvenile (juvenile DM2 and juvenile DM5) and two adult (adult DM3 and adult DM5) patients with DM. Columns show mean fold change and standard deviation. The difference between COX− and COX+ samples was verified by two‐sample t test, **p < 0.05. Columns show median, quartiles and range. (B) mtDNA copy number in relation to nuclear DNA in two juvenile patients with DM (juvenile DM2 and juvenile DM5) and two adult patients with DM (adult DM3 and adult DM5), and age‐matched controls (C1–14) analysed by deep sequencing showing lower amount of mtDNA copy number in patients with DM compared to controls. The difference between DM and control samples was verified by two‐sample t test, ***p < 0.001. Columns show median, quartiles and range. (C) Analysis of mtDNA rearrangements (deletions blue and duplications red) in two patients with DM, one juvenile (juvenile DM5) and one adult (adult DM3), in normal controls, and in two disease controls (one with multiple mtDNA deletions [DC3] and one with a single mtDNA deletion [DC4])
Very low numbers of large‐scale mtDNA deletions or duplications (<0.5%) were identified in DM muscles and not different from controls at a stringent clustering threshold of a minimum five reads supporting a breakpoint (Figure 5C). This may be compared to the results from mtDNA analysis of muscle tissue from a patient with IBM showing the typical presence of multiple mtDNA deletions and a patient with a single mtDNA deletion (Figure 5C). Analysis of small somatic mtDNA variants such as single‐nucleotide variants and small insertions or deletions in coding regions showed a similar pattern with no or few variants as in controls (data not shown).
DISCUSSION
Mitochondrial dysfunction plays an important role in muscle weakness and fatigue in DM, because patients show a reduced aerobic capacity and there is histopathological and biochemical evidence of OXPHOS dysfunction mainly affecting perifascicular regions [5, 7, 8, 9, 24]. Although the mitochondrial impairment is well established, the reason for the respiratory chain dysfunction has remained unclear. To study the role of mtDNA in the pathobiology of OXPHOS deficiency in DM, we investigated the respiratory chain enzyme complexes in muscle biopsy specimens from patients with juvenile and adult DM. We found that the respiratory chain complexes I and IV, which contain mtDNA encoded subunits, were deficient in perifascicular regions, whereas the entirely nuclear encoded complex II was unaltered in parallel with an unchanged mitochondrial density. This finding strongly indicates defective mtDNA since this pattern of combined complex I and complex IV deficiency with preserved complex II activity is typical for myopathies due to mtDNA deletions, mitochondrial tRNA gene mutations or mtDNA depletion, because these conditions cause an overall perturbed protein synthesis of mtDNA encoded proteins [17, 18]. To explore the hypothesis that mtDNA depletion is the cause of the complex I and complex IV deficiency, we analysed mtDNA transcripts by in situ hybridisation. This technique allows for a comparison between different single muscle fibres regarding the number of transcripts, enzyme activity and protein expression in the same fibres in consecutive sections. We found that mtDNA transcripts were reduced in the perifascicular regions that showed decreased COX activity and reduced complex I and complex IV proteins, indicating mtDNA depletion in these perifascicular regions. To further prove the mtDNA depletion hypothesis, we performed two different mtDNA analyses. First, we analysed the mtDNA copy number by comparing COX deficient fibres with COX positive fibres by qPCR analysis and found a lower copy number of mtDNA in the COX deficient fibres indicating mtDNA depletion. Second, we analysed, by deep sequencing, the overall amount of mtDNA and presence of point mutations or large‐scale rearrangements of mtDNA in the muscle specimens from patients with DM and age matched controls. We found a significant reduction of mtDNA copy number in patients with DM. No increase in somatic point mutations or large‐scale rearrangements were found. These different investigations clearly demonstrate that respiratory chain deficiency in patients with DM is associated with mtDNA and mtRNA depletion.
The mtDNA and mtRNA depletion in muscle is apparently part of the pathobiology of mitochondrial dysfunction in both juvenile and adult DM. Depletion of mtDNA is associated with a variety of genetic disorders collectively known as mitochondrial DNA depletion syndrome (MDS) [25]. These disorders are caused by defective mtDNA maintenance implying defects in mtDNA replication [26]. Common examples are disease‐causing variants in mitochondrial polymerase gamma, the mtDNA helicase twinkle or deficiency of mtDNA building blocks, for example, secondary to thymidine kinase 2 (TK2) deficiency. The roles of these proteins and others that are involved in mtDNA replication for the mtDNA depletion observed in muscle biopsies from patients with DM remain to be determined [27]. Depletion of mtDNA is also seen in another type of inflammatory myopathy, IBM, supporting the concept that muscle inflammation may affect mtDNA replication [22]. However, the explanation for OXPHOS deficiency in IBM muscle, in addition to mtDNA depletion, is multiple mtDNA deletions that are clonally expanded in muscle fibre segments [28, 29]. Such multiple mtDNA deletions do not explain the respiratory chain deficiency in DM because the amount of large‐scale mtDNA rearrangements in muscle from patients with DM did not differ from controls.
The reduced mtDNA copy number in muscle from patients with DM shows a similar perifascicular distribution as the MHC‐class I upregulation, indicating a link between the inflammation in DM muscle and an impaired mtDNA replication. The accentuated inflammation in perifascicular regions in DM is associated with high levels of different cytokines, deposition of complement complex C5b‐9, loss of capillaries and presumably regional ischaemia [6, 30]. By combining in vivo and in vitro studies on humans with DM and experimental myositis in mice, Meyer et al. [5] suggested that INF‐ß‐induced reactive oxygen species (ROS) are major players in the OXPHOS deficiency. They showed that mitochondrial respiration was reduced in human myotubes in vitro after exposure to INF‐ß, which was reversible after the addition of the ROS scavenger N‐acetyl cysteine. However, these experiments do not explain the mtDNA depletion that is associated with and at least partly causing the respiratory chain dysfunction in humans with DM. In a murine model of inflammation‐induced colon tumourigenesis with mtDNA depletion, the suggested mechanism was reduced replication of mtDNA by inflammation‐induced DNA methylation and silencing of the mtDNA polymerase (PolγA) [31]. Further research on the biological mechanisms that explain the link between inflammation and mtDNA depletion will be important for understanding the pathobiology of muscle weakness in patients with DM and development of novel treatment strategies.
This study was designed to investigate the pathobiology of the COX deficiency, which is frequently present in patients with DM and usually shows a perifascicular distribution. Therefore, we selected patients with this specific change in their muscle biopsies. Because we used archival material, we did not have the possibility to correlate our findings with subtypes of DM as defined by different auto‐antibody profiles [3]. However, in a recent study, several different subtypes of DM were associated with mitochondrial dysfunction indicating that it is not auto‐antibody specific [5]. These questions may be further analysed in a prospective study.
CONCLUSION
We have investigated the pathobiology of the COX deficiency that is seen in many cases of juvenile and adult DM. It is characteristically seen in the perifascicular regions in the muscle tissue that also show other typical alterations such as MHC‐class I upregulation, muscle fibre atrophy and capillary loss. We found a reduced mtDNA copy number corresponding to reduced levels of mitochondrial transcripts and respiratory chain proteins involved in the partly mtDNA encoded complexes I and IV, but not those involved in the entirely nuclear DNA encoded complex II. The mtDNA depletion may be explained by perturbed mtDNA replication, and therefore, mechanisms leading to impaired mtDNA replication in patients with DM warrant further investigations.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
CH‐O, UL and AO designed the study and experiments. CH‐O, UL, KV, DL, SR, CT and AO performed and interpreted the data. CH‐O, UL and AO wrote the manuscript. All authors commented and approved the manuscript.
ETHICAL STATEMENT
The study was approved by the Swedish Ethical Review Authority and conducted according to the Declaration of Helsinki of 1975 (Dnr 2022‐00026‐01).
FUNDING STATEMENT
This work was supported by grants from the Swedish Research Council (No 2018‐02821 to AO) and the Swedish state under the agreement between the Swedish government and the county councils, the ALF‐agreement (ALFGBG‐716821 to AO, ALFGBG‐79230 to CH‐O and ALFGBG‐872571 to UL) and the Western Sweden Muscle Foundation (to AO and CH‐O).
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/nan.12841.
ACKNOWLEDGEMENTS
We thank Brith Leidvik for technical assistance. The authors would like to acknowledge the Clinical Genomics Stockholm facility at Science for Life Laboratory for providing assistance in next generation sequencing.
Hedberg‐Oldfors C, Lindgren U, Visuttijai K, et al. Respiratory chain dysfunction in perifascicular muscle fibres in patients with dermatomyositis is associated with mitochondrial DNA depletion. Neuropathol Appl Neurobiol. 2022;48(7):e12841. doi: 10.1111/nan.12841
Carola Hedberg‐Oldfors and Ulrika Lindgren contributed equally to this study.
Funding information Vetenskapsrådet
DATA AVAILABILITY STATEMENT
The data are available upon request.
REFERENCES
- 1. Lundberg IE, de Visser M, Werth VP. Classification of myositis. Nat Rev Rheumatol. 2018;14(5):269‐278. doi: 10.1038/nrrheum.2018.41 [DOI] [PubMed] [Google Scholar]
- 2. Mariampillai K, Granger B, Amelin D, et al. Development of a new classification system for idiopathic inflammatory myopathies based on clinical manifestations and myositis‐specific autoantibodies. JAMA Neurol. 2018;75(12):1528‐1537. doi: 10.1001/jamaneurol.2018.2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mammen AL, Allenbach Y, Stenzel W, et al. 239th ENMC international workshop: classification of dermatomyositis, Amsterdam, the Netherlands, 14‐16 December 2018. Neuromuscul Disord. 2020;30(1):70‐92. doi: 10.1016/j.nmd.2019.10.005 [DOI] [PubMed] [Google Scholar]
- 4. Dalakas MC. Inflammatory muscle diseases. N Engl J Med. 2015;372(18):1734‐1747. doi: 10.1056/NEJMra1402225 [DOI] [PubMed] [Google Scholar]
- 5. Meyer A, Laverny G, Allenbach Y, et al. IFN‐beta‐induced reactive oxygen species and mitochondrial damage contribute to muscle impairment and inflammation maintenance in dermatomyositis. Acta Neuropathol. 2017;134(4):655‐666. doi: 10.1007/s00401-017-1731-9 [DOI] [PubMed] [Google Scholar]
- 6. Lahoria R, Selcen D, Engel AG. Microvascular alterations and the role of complement in dermatomyositis. Brain. 2016;139(7):1891‐1903. doi: 10.1093/brain/aww122 [DOI] [PubMed] [Google Scholar]
- 7. Woo M, Chung SJ, Nonaka I. Perifascicular atrophic fibers in childhood dermatomyositis with particular reference to mitochondrial changes. J Neurol Sci. 1988;88(1‐3):133‐143. doi: 10.1016/0022-510X(88)90211-0 [DOI] [PubMed] [Google Scholar]
- 8. Alhatou MI, Sladky JT, Bagasra O, Glass JD. Mitochondrial abnormalities in dermatomyositis: characteristic pattern of neuropathology. J Mol Histol. 2004;35(6):615‐619. doi: 10.1007/s10735-004-2194-6 [DOI] [PubMed] [Google Scholar]
- 9. Cea G, Bendahan D, Manners D, et al. Reduced oxidative phosphorylation and proton efflux suggest reduced capillary blood supply in skeletal muscle of patients with dermatomyositis and polymyositis: a quantitative 31P‐magnetic resonance spectroscopy and MRI study. Brain. 2002;125(7):1635‐1645. doi: 10.1093/brain/awf163 [DOI] [PubMed] [Google Scholar]
- 10. Wiesinger GF, Quittan M, Nuhr M, et al. Aerobic capacity in adult dermatomyositis/polymyositis patients and healthy controls. Arch Phys Med Rehabil. 2000;81(1):1‐5. doi: 10.1016/S0003-9993(00)90212-0 [DOI] [PubMed] [Google Scholar]
- 11. Zhong D, Wu C, Bai J, Xu D, Zeng X, Wang Q. Co‐expression network analysis reveals the pivotal role of mitochondrial dysfunction and interferon signature in juvenile dermatomyositis. PeerJ. 2020;8:e8611. doi: 10.7717/peerj.8611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Larsson NG, Oldfors A. Mitochondrial myopathies. Acta Physiol Scand. 2001;171(3):385‐393. doi: 10.1046/j.1365-201x.2001.00842.x [DOI] [PubMed] [Google Scholar]
- 13. Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13(12):878‐890. doi: 10.1038/nrg3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pfeffer G, Chinnery PF. Diagnosis and treatment of mitochondrial myopathies. Ann Med. 2013;45(1):4‐16. doi: 10.3109/07853890.2011.605389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roos S, Lindgren U, Ehrstedt C, Moslemi AR, Oldfors A. Mitochondrial DNA depletion in single fibers in a patient with novel TK2 mutations. Neuromuscul Disord. 2014;24(8):713‐720. doi: 10.1016/j.nmd.2014.05.009 [DOI] [PubMed] [Google Scholar]
- 16. Dubowitz V, Sewry C, Oldfors A. Muscle biopsy. A practical approach. 5th ed. Amsterdam: Elsevier; 2021. [Google Scholar]
- 17. Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017;241(2):236‐250. doi: 10.1002/path.4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rocha MC, Grady JP, Grunewald A, et al. A novel immunofluorescent assay to investigate oxidative phosphorylation deficiency in mitochondrial myopathy: understanding mechanisms and improving diagnosis. Sci Rep. 2015;5(1):15037. doi: 10.1038/srep15037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roos S, Hedberg‐Oldfors C, Visuttijai K, et al. Expression pattern of mitochondrial respiratory chain enzymes in skeletal muscle of patients with mitochondrial myopathy associated with the homoplasmic m.14674T>C variant. Brain Pathol. 2021;32(4):e13038. doi: 10.1111/bpa.13038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roos S, Macao B, Fuste JM, et al. Subnormal levels of POLgammaA cause inefficient initiation of light‐strand DNA synthesis and lead to mitochondrial DNA deletions and autosomal dominant progressive external ophthalmoplegia. Hum Mol Genet. 2013;22(12):2411‐2422. doi: 10.1093/hmg/ddt094 [DOI] [PubMed] [Google Scholar]
- 21. Basu S, Xie X, Uhler JP, et al. Accurate mapping of mitochondrial DNA deletions and duplications using deep sequencing. PLoS Genet. 2020;16(12):e1009242. doi: 10.1371/journal.pgen.1009242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hedberg‐Oldfors C, Lindgren U, Basu S, et al. Mitochondrial DNA variants in inclusion body myositis characterized by deep sequencing. Brain Pathol. 2021;31(3):e12931. doi: 10.1111/bpa.12931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ding J, Sidore C, Butler TJ, et al. Assessing mitochondrial DNA variation and copy number in lymphocytes of ~2,000 Sardinians using tailored sequencing analysis tools. PLoS Genet. 2015;11(7):e1005306. doi: 10.1371/journal.pgen.1005306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Varadhachary AS, Weihl CC, Pestronk A. Mitochondrial pathology in immune and inflammatory myopathies. Curr Opin Rheumatol. 2010;22(6):651‐657. doi: 10.1097/BOR.0b013e32833f108a [DOI] [PubMed] [Google Scholar]
- 25. Suomalainen A, Isohanni P. Mitochondrial DNA depletion syndromes‐‐many genes, common mechanisms. Neuromuscul Disord. 2010;20(7):429‐437. doi: 10.1016/j.nmd.2010.03.017 [DOI] [PubMed] [Google Scholar]
- 26. Viscomi C, Zeviani M. MtDNA‐maintenance defects: syndromes and genes. J Inherit Metab Dis. 2017;40(4):587‐599. doi: 10.1007/s10545-017-0027-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gustafsson CM, Falkenberg M, Larsson NG. Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem. 2016;85(1):133‐160. doi: 10.1146/annurev-biochem-060815-014402 [DOI] [PubMed] [Google Scholar]
- 28. Oldfors A, Moslemi AR, Jonasson L, Ohlsson M, Kollberg G, Lindberg C. Mitochondrial abnormalities in inclusion‐body myositis. Neurology. 2006;66(Issue 1, Supplement 1):S49‐S55. doi: 10.1212/01.wnl.0000192127.63013.8d [DOI] [PubMed] [Google Scholar]
- 29. Oldfors A, Larsson NG, Lindberg C, Holme E. Mitochondrial DNA deletions in inclusion body myositis. Brain. 1993;116(Pt 2):325‐336. doi: 10.1093/brain/116.2.325 [DOI] [PubMed] [Google Scholar]
- 30. De Paepe B. Vascular changes and perifascicular muscle fiber damage in dermatomyositis: another question of the chicken or the egg that is on our mind. Ann Transl Med. 2017;5(1):22. doi: 10.21037/atm.2016.12.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maiuri AR, Li H, Stein BD, Tennessen JM, O'Hagan HM. Inflammation‐induced DNA methylation of DNA polymerase gamma alters the metabolic profile of colon tumors. Cancer Metab. 2018;6(1):9. doi: 10.1186/s40170-018-0182-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data are available upon request.