

Abstract
Aromatic NH2 groups are essential as hydrogen-bonding donors in secondary structures of DNA and RNA. Although rapid rotations of NH2 groups of adenine and guanine bases were previously characterized, there has been a lack of quantitative information about slow rotations of cytosine NH2 groups in Watson-Crick base pairs. In this study, using an NMR method we had recently developed, we determined the kinetic rate constants and energy barriers for cytosine NH2 rotations in a 15-base-pair DNA duplex. Our data show that the rotational dynamics of cytosine NH2 groups depend on local environments. Qualitative correlation between the ranges of 15N chemical shifts and rotational timescales for various NH2 groups of nucleic acids and proteins illuminates a relationship between the partial double-bond character of the C-N bond and the timescale for NH2 rotations.
Graphical Abstract
Base pairing of nucleic acids is crucial for gene replication, recombination, transcription, and translation in life. The dynamic nature of base pairs has been revealed by nuclear magnetic resonance (NMR) spectroscopy.1,2 Nucleic acids undergo conformational transitions between Watson-Crick base pair and Hoogsteen base pair.3–6 Transient breakage of a base pair (‘base-opening’) can lead to an extrahelical conformation where a base is flipped out of double helix and recognized by enzymes for base excision repair.7,8 Therefore, the dynamic properties of base pairs are important for our understanding of the functions of DNA and RNA as well as their interactions with proteins.
NH2 groups of adenine (A), cytosine (C), and guanine (G) bases serve as hydrogen-bonding donors in base pairing. NH2 rotations of A/G bases in DNA were studied through NMR line-shape analysis.9,10 Despite their hydrogen-bonding with base-pair partners, adenine/guanine NH2 groups rapidly rotate on a timescale of 10−2–10−5 s, depending on temperature and hydrogen bonds. Due to the rapid exchange caused by the rotations, the two 1H resonances of adenine/guanine NH2 groups are severely broadened or averaged into a single resonance at physiological temperature.9–11 By contrast, cytosine NH2 groups typically exhibit two distinct 1H resonances, clearly indicating that their rotations are slower.11 It was proposed that cytosine NH2 groups do not even rotate within Watson-Crick GC base pairs.12
In this communication, we report the kinetics and the energy barriers for rotations of cytosine NH2 groups in double-stranded DNA. Nuclear magnetization transfer via the exchange between the two 1H resonances of each NH2 group is difficult to distinguish from transfer via cross-relaxation that occurs simultaneously. Recently, to investigate NH2 rotations, we developed a new NMR method named “ODDS-zz-EXSY” (off-diagonal–diagonal swapping zz-exchange spectroscopy). This method cancels cross-relaxation, controls the positions of auto and exchange cross peaks, and allows us to accurately measure the rate constants for NH2 rotations in the slow exchange regime.13 In our prior work, we used ODDS-zz-EXSY to investigate slow rotations of protein Asn/Gln side-chain NH2 groups. In our current study, we apply it to the rotational dynamics of cytosine NH2 group in double-stranded DNA.
A 13C,15N-labeled 15 base-pair (bp) DNA duplex (Figure 1A) was used in this study. Details of the sample preparation are described in the Supporting Information (SI). This 15-bp DNA contains 8 cytosine bases that form hydrogen bonds with guanine bases. 1H, 13C, 15N resonances were assigned as described in the SI. As shown in Figure 1B, the cytosine NH2 groups exhibit well-resolved signals in the 1H-15N heteronuclear single-quantum coherence (HSQC) spectra. NMR signals from the NH2 groups of the terminal cytosine (C0 and C15) were broadened, presumably due to the rapid hydrogen exchange, and therefore were excluded from further analysis. We investigated the behavior of the other 6 cytosine NH2 groups in the 15-bp DNA.
Figure 1.
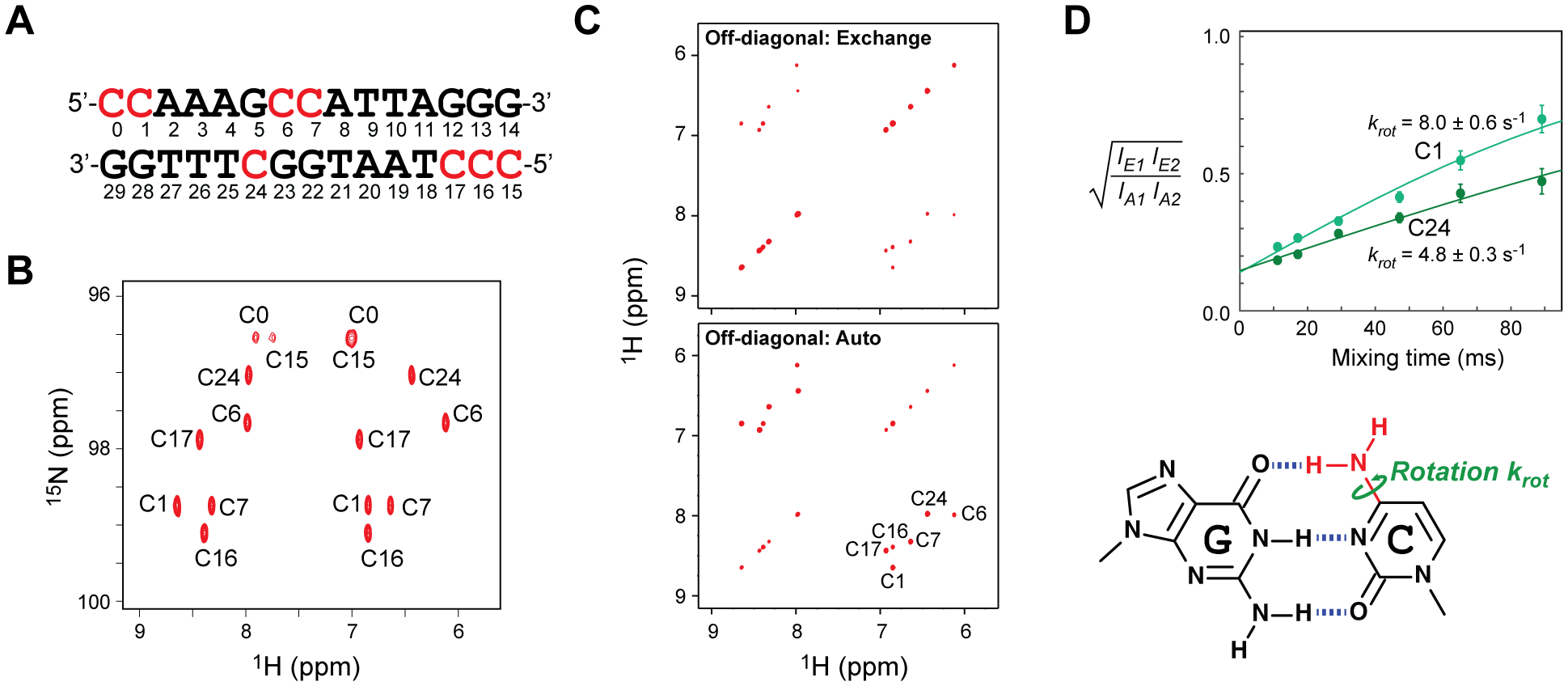
NMR investigations of cytosine NH2 rotations in double-stranded DNA. (A) 15-bp DNA used in this study. The residue numbering is based on that of Fernandez et al.14 Cytosine and guanine nucleotides in this DNA were labeled by 13C and 15N isotopes. The sample solution contained 0.3 mM DNA, 20 mM potassium succinate (pH 5.8), 100 mM KCl, and 0.4 mM NaF. (B) 13C-decoupled 1H-15N HSQC spectrum for cytosine NH2 groups in the 15-bp DNA duplex at 25°C. The resonances were assigned as described in the Supporting Information. (C)Examples of ODDS-zz-EXSY sub-spectra for cytosine NH2 groups of the 13C/15N-labeled 15-bp DNA at 25°C (with a mixing time of 47 ms). In the second sub-spectrum, off-diagonal and diagonal cross peaks are swapped through coherence transfer between 2HzaNz and 2HzbNz terms via two 1JNH couplings.13 (D) Ratios of intensities of off-diagonal exchange cross peaks and off-diagonal auto cross peaks for the NH2 groups of C1 and C24 at 25°C. Solid curves represent the best-fit curves obtained through the fitting with Eq. 1. The krot constant represents a rate constant for a 180° rotation and is a half of the exchange rate constant kex.
Using the ODDS-zz-EXSY method,13 we measured the kinetics of NH2 rotations for these cytosine bases. Each ODDS-zz-EXSY experiment provides two 2D 1H-1H sub-spectra for NH2 groups selectively. Examples of these sub-spectra are shown in Figure 1C. In one of the sub-spectra, the exchange cross peaks arising from the NH2 rotations (which cause interconversion of the product operator terms 2HzaNz and 2HzbNz during the mixing time) are observed at off-diagonal positions whereas the auto cross peaks are observed at diagonal positions. Because the ODDS-zz-EXSY method cancels cross-relaxation, the off-diagonal cross peaks arise from exchange. These data clearly show that cytosine NH2 groups undergo rotations in a slow exchange regime. In the other sub-spectrum, for which the 2HzaNz and 2HzbNz terms are interconverted through coherence transfer via two scalar 1JNH couplings,13 the exchange cross peaks are located at the diagonal positions and the auto cross peaks are at the off-diagonal positions. This feature of the ODDS-zz-EXSY method greatly facilitate quantitative analysis of auto cross peaks.13
We measured ODDS-zz-EXSY sub-spectra using 6 different mixing times. From the signal intensity ratios, we determined the rate constant krot for rotations of the cytosine NH2 groups through nonlinear least-squares fitting with:13
| [1], |
where IE1 and IE2 are the intensities of two off-diagonal exchange cross peaks; IA1 and IA2 are those of two off-diagonal auto cross peaks; q is a parameter representing the incompleteness of the ODDS filter;13 and Tm is the mixing time. Figure 1D shows examples of time courses and best-fit curves. At 25°C, the rate constants krot for cytosine NH2 rotations ranged from 4.8 s−1 to 24 s−1 in the 15-bp DNA. Interestingly, even among cytosine bases in the middle region of the 15-bp DNA, significant variation in krot rate constants was observed. For example, the krot constant for C6 (15 ± 1 s−1) was three times as large as that for C24 (4.8 ± 0.3 s−1).
We measured the rate constants krot for all non-terminal cytosine NH2 groups at 15, 20, 25, and 30°C. These rate constants are shown in Figure 2A. The rotation kinetics were faster at higher temperatures. Through fitting to the temperature dependence data using the Eyring equation, we determined the energy barriers for NH2 rotations. The activation free energy (ΔG‡) and its enthalpic (ΔH‡) and entropic (–TΔS‡) components obtained for each cytosine NH2 group are shown in Figure 2B. For the majority of cytosine NH2 groups, the energy barrier for rotations about the C-N bond is largely enthalpic with only little or no entropic contribution.
Figure 2.
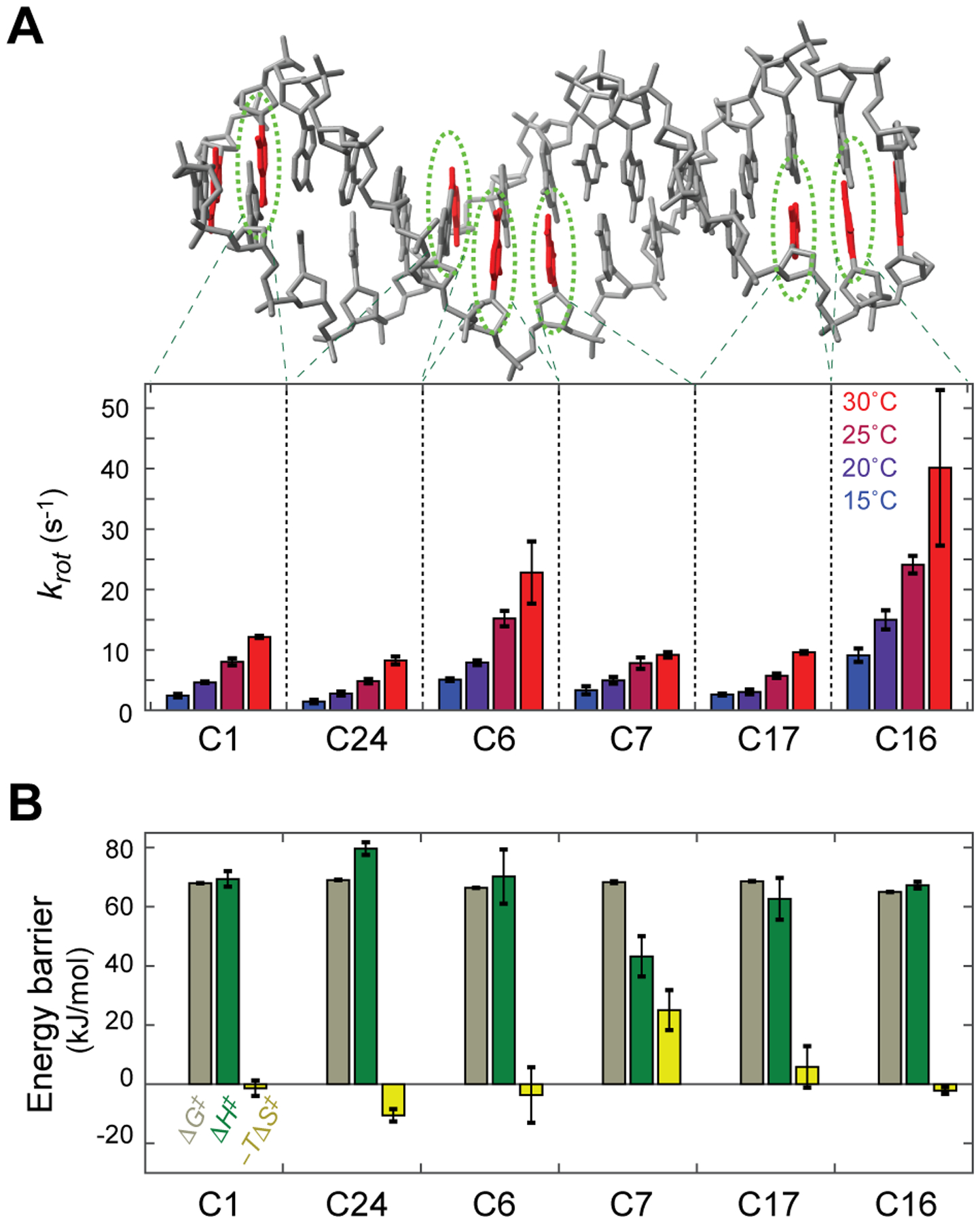
Temperature dependence of cytosine NH2 rotations in the 15-bp DNA duplex. (A) Rate constants (krot) for cytosine NH2 rotations measured at 15, 20, 25, and 30°C. (B) Energy barriers for cytosine NH2 rotations. The activation free energy and its enthalpic and entropic components were determined from the temperature dependence of krot constants.
Interestingly, the C7 NH2 group exhibited a significant entropic contribution and a relatively small activation enthalpy (ΔH‡). The smaller ΔH‡ for this NH2 group was obvious in a weaker dependence of its krot rate constant on temperature (see also Eyring plots in Figure S1 in the SI). The negative ΔS‡ for this NH2 group suggests that the transition state is entropically unfavorable, which might be caused by a larger degree of exposure of hydrophobic bases to solvent via a kink of DNA. Because a kink of DNA occurs more easily at C-A steps15 and C7 is located at a C-A step in the middle of DNA, the unique ΔH‡ and –TΔS‡ of this residue might be related to the deformability.
Although both C1 and C16 are located at a position second to a 5’-terminus, the krot rate constants of the C16 NH2 group were larger than those of the C1 NH2 group by a factor of 3. The activation free energy (ΔG‡) for C1 NH2 rotations was larger by 2.7 kJ/mol than that for C16 NH2 rotations. The 3’-neighboring nucleotides (A2 and C17) are different for C1 and C16. A recent study shows that the base-stacking energies for CpA (TpG) steps are significantly more favorable than those for CpC (GpG) steps.16 More stable base-stacking of CpA might impede rotations of the C1 NH2 group.
Now, with our data on cytosine NH2 rotations in double-stranded DNA, more comprehensive information is available about hindered rotations about sp2 C-N bonds in biomolecules. NMR spectroscopy has been used to study rotations about sp2 C-N bonds of arginine (Arg), asparagine (Asn), and glutamine (Gln) in proteins.13,17–24 Arg side-chain sp2 C-N bond rotations are far faster than Asn/Gln side-chain NH2 rotations. This difference can be ascribed to the different chemical structures between the guanidinium (Arg) and carbamoyl (Asn/Gln) moieties. However, it is less clear why NH2 rotation timescales are different between A/G and C bases despite the similarity in local chemical structures. Since solvent-accessible surface areas of cytosine NH2 groups are not significantly smaller than those of adenine/guanine NH2 groups (Figure S2 in the SI), the slower NH2 rotations for C bases are not due to a smaller degree of exposure to solvent.
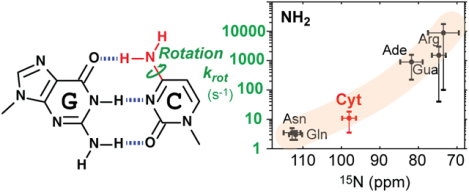
The difference in the rotational dynamics may stem from different degrees of the partial double-bond character in the C-N bonds. Due to strong shielding effects of π electrons, 15N chemical shifts qualitatively reflect the double-bond characters. For example, Lys side-chain amino groups single-bonded to sp3 carbon exhibit 15N chemical shifts around ~24 ppm in the NH2 state (~33 ppm in the NH3+ state),22 whereas 15N chemical shifts of nitrogen atoms double-bonded to an sp2 carbon atom are far larger (e.g., adenine N1, ~225 ppm; guanine N7, ~237 ppm). 15N chemical shifts of cytosine NH2 (~98 ppm) are larger than 15N chemical shifts of adenine NH2 (~80 ppm) and guanine NH2 (~76 ppm). These 15N chemical shifts suggest that the partial double-bond character in the C-N bond is stronger for cytosine NH2 groups than for adenine and guanine NH2 groups, which is consistent with the slower rotations of cytosine NH2 groups. The same rule can also explain the difference between protein side-chain NH2 groups: Arg NH2 (15N ~72 ppm) rotates far faster than Asn NH2 (15N ~113 ppm) and Gln NH2 (15N ~112 ppm). The qualitative correlation between the typical ranges of NH2 rotation rates and 15N chemical shifts is shown in Figure 3. These data suggest that the timescale of NH2 rotations about a C-N bond is related to the degree of partial double-bond character in the C-N bond.
Figure 3.
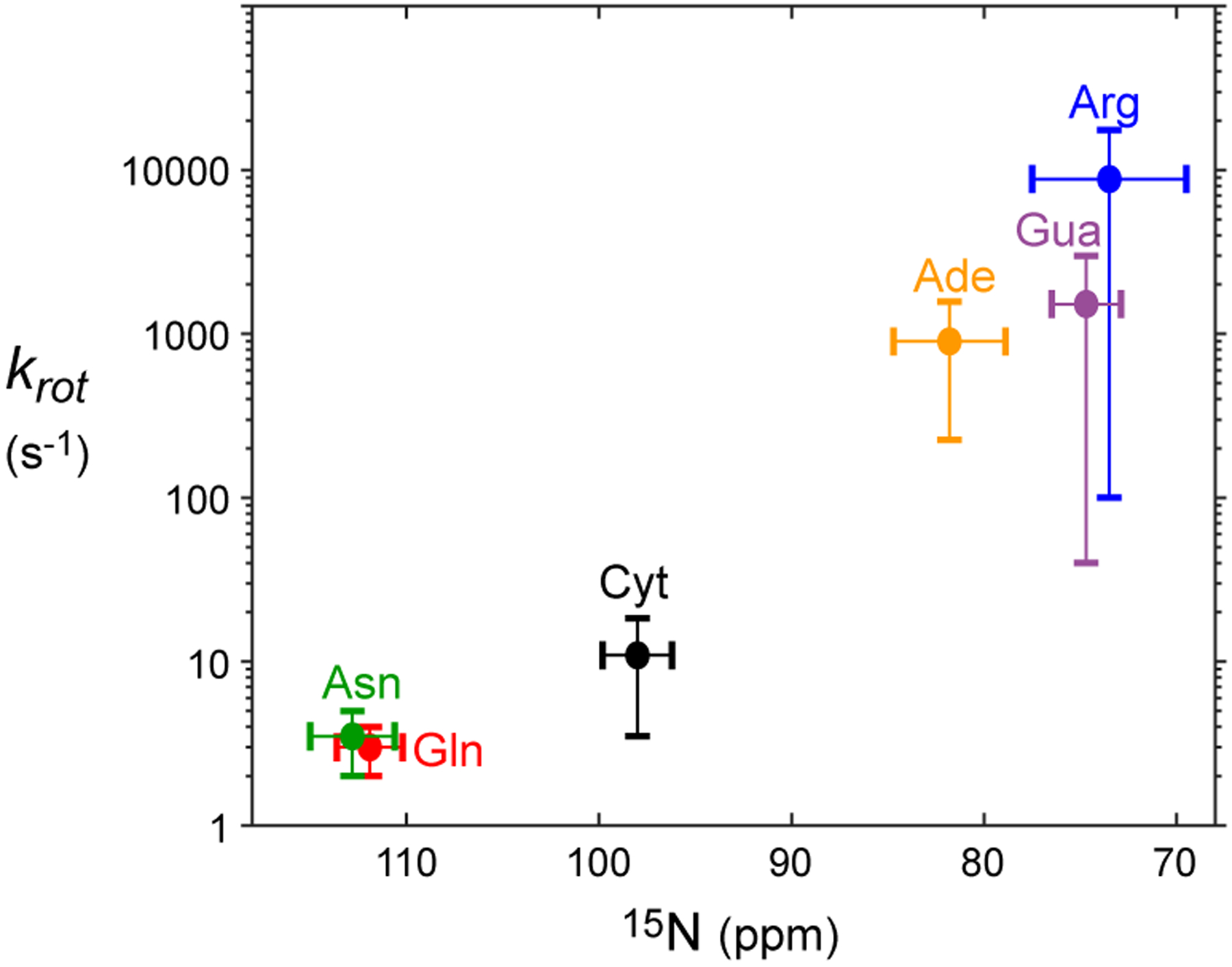
Qualitative correlation between the ranges of 15N chemical shifts and rotation rate constants krot for biomolecular NH2 groups. The horizontal position and width of a cross represent the average and the standard deviation of 15N chemical shifts of each NH2 type in the Biological Magnetic Resonance Bank (BMRB) database. The vertical position and width represent a range of krot rate constants for Asp/Gln,13 Arg,23 cytosine (Cyt; current work), adenine (Ade),10 or guanine (Gua)9 NH2 groups at 25°C. Because of the limited information and factors that influence NH2 rotations (e.g., hydrogen bonds), the shown ranges of krot rate constants provide only qualitative information.
However, the observed variation in rates and barriers for rotations among cytosine NH2 groups (Figure 2) should reflect local environments around individual NH2 groups in the same DNA. Local conformational dynamics such as the base-opening process and transitions between Watson-Crick and Hoogsteen base pairs depend on sequence context.3,25 DNA bendability also depends on sequence context.15 Since the hydrogen bond of each cytosine NH2 group remains in both Watson-Crick and Hoogsteen base pairs, base-opening dynamics that break the hydrogen bond may be more relevant to the cytosine NH2 rotations in double-stranded DNA. The behavior of cytosine NH2 groups may be even more diverse in RNA due to its structural diversity. The ODDS-zz-EXSY method is readily applicable to 15N- or 13C/15N-labeled RNA as well.
Supplementary Material
ACKNOWLEDGEMENT
This work was supported by Grant R35-GM130326 from the National Institutes of Health and Grant H-2104-20220331 from the Welch Foundation. We thank Karina Bien for language editing.
ABBREVIATIONS
- HSQC
heteronuclear single-quantum coherence
- ODDS-zz-EXSY
off-diagonal–diagonal swapping zz-exchange spectroscopy
Footnotes
Supporting Information. Materials and methods; the Eyring plot of temperature dependence of krot rate constants; and Solvent-accessible surface areas of NH2 groups in the 15-bp DNA.
REFERENCES
- 1.Kimsey I, and Al-Hashimi HM (2014) Increasing occurrences and functional roles for high energy purine-pyrimidine base-pairs in nucleic acids, Curr Opin Struct Biol 24, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao B, and Zhang Q (2015) Characterizing excited conformational states of RNA by NMR spectroscopy, Curr Opin Struct Biol 30, 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvey HS, Gottardo FL, Nikolova EN, and Al-Hashimi HM (2014) Widespread transient Hoogsteen base pairs in canonical duplex DNA with variable energetics, Nat Commun 5, 4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nikolova EN, Goh GB, Brooks CL, and Al-Hashimi HM (2013) Characterizing the Protonation State of Cytosine in Transient G·C Hoogsteen Base Pairs in Duplex DNA, J Am Chem Soc 135, 6766–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nikolova EN, Gottardo FL, and Al-Hashimi HM (2012) Probing Transient Hoogsteen Hydrogen Bonds in Canonical Duplex DNA Using NMR Relaxation Dispersion and Single-Atom Substitution, J Am Chem Soc 134, 3667–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nikolova EN, Kim E, Wise AA, O’Brien PJ, Andricioaei I, and Al-Hashimi HM (2011) Transient Hoogsteen base pairs in canonical duplex DNA, Nature 470, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, and Stivers JT (2007) Enzymatic capture of an extrahelical thymine in the search for uracil in DNA, Nature 449, 433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stivers JT (2008) Extrahelical Damaged Base Recognition by DNA Glycosylase Enzymes, Chemistry 14, 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adrian M, Winnerdy FR, Heddi B, and Phan AT (2017) Rotation of Guanine Amino Groups in G-Quadruplexes: A Probe for Local Structure and Ligand Binding, Biophys J 113, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michalczyk R, and Russu IM (1999) Rotational Dynamics of Adenine Amino Groups in a DNA Double Helix, Biophys J 76, 2679–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mueller L, Legault P, and Pardi A (1995) Improved RNA Structure Determination by Detection of NOE Contacts to Exchange-Broadened Amino Protons, J Am Chem Soc 117, 11043–11048. [Google Scholar]
- 12.Williams LD, Williams NG, and Shaw BR (1990) In a model cytosine:guanine base pair, one amino group rotates and the other does not, J Am Chem Soc 112, 829–833. [Google Scholar]
- 13.Wang X, Yu B, and Iwahara J (2021) Hindered Rotations of Protein Asparagine/Glutamine Side-Chain NH2 Groups: Impact of Hydrogen Bonding with DNA, J Phys Chem Lett 12, 11378–11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez C, Szyperski T, Billeter M, Ono A, Iwai H, Kainosho M, and Wuthrich K (1999) Conformational changes of the BS2 operator DNA upon complex formation with the Antennapedia homeodomain studied by NMR with 13C/15N-labeled DNA, J Mol Biol 292, 609–617. [DOI] [PubMed] [Google Scholar]
- 15.Dickerson RE (1998) DNA bending: The prevalence of kinkiness and the virtues of normality, Nucleic Acids Res 26, 1906–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruse H, Banáš P, and Šponer J (2019) Investigations of Stacked DNA Base-Pair Steps: Highly Accurate Stacking Interaction Energies, Energy Decomposition, and Many-Body Stacking Effects, J Chem Theory Comput 15, 95–115. [DOI] [PubMed] [Google Scholar]
- 17.Birdsall B, Polshakov VI, and Feeney J (2000) NMR studies of ligand carboxylate group interactions with arginine residues in complexes of Lactobacillus casei dihydrofolate reductase with substrates and substrate analogues, Biochemistry 39, 9819–9825. [DOI] [PubMed] [Google Scholar]
- 18.Gerecht K, Figueiredo AM, and Hansen DF (2017) Determining rotational dynamics of the guanidino group of arginine side chains in proteins by carbon-detected NMR, Chem Commun 53, 10062–10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guenneugues M, Drevet P, Pinkasfeld S, Gilquin B, Ménez A, and Zinn-Justin S (1997) Picosecond to hour time scale dynamics of a “three finger” toxin: correlation with its toxic and antigenic properties, Biochemistry 36, 16097–16108. [DOI] [PubMed] [Google Scholar]
- 20.Karunanithy G, Reinstein J, and Hansen DF (2020) Multiquantum Chemical Exchange Saturation Transfer NMR to Quantify Symmetrical Exchange: Application to Rotational Dynamics of the Guanidinium Group in Arginine Side Chains, J Phys Chem Lett 11, 5649–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan WD, Birdsall B, Nieto PM, Gargaro AR, and Feeney J (1999) 1H/15N HSQC NMR studies of ligand carboxylate group interactions with arginine residues in complexes of brodimoprim analogues and Lactobacillus casei dihydrofolate reductase, Biochemistry 38, 2127–2134. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen D, Chen C, Pettitt BM, and Iwahara J (2019) NMR methods for characterizing the basic side chains of proteins: electrostatic interactions, hydrogen bonds, and conformational dynamics, Methods Enzymol 615, 285–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nieto PM, Birdsall B, Morgan WD, Frenkiel TA, Gargaro AR, and Feeney J (1997) Correlated bond rotations in interactions of arginine residues with ligand carboxylate groups in protein ligand complexes, FEBS Lett 405, 16–20. [DOI] [PubMed] [Google Scholar]
- 24.Yamazaki T, Pascal SM, Singer AU, Formankay JD, and Kay LE (1995) NMR Pulse Schemes for the Sequence-Specific Assignment of Arginine Guanidino 15N and 1H Chemical-Shifts in Proteins, J Am Chem Soc 117, 3556–3564. [Google Scholar]
- 25.Coman D, and Russu IM (2005) A Nuclear Magnetic Resonance Investigation of the Energetics of Basepair Opening Pathways in DNA, Biophys J 89, 3285–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.