

Abstract
In an attempt to produce a more defined, clinical-grade version of a vaccine based on Plasmodium falciparum merozoite surface protein 1 (MSP1), we evaluated the efficacy of two recombinant forms of MSP1 in an Aotus nancymai challenge model system. One recombinant vaccine, bvMSP142, based on the 42-kDa C-terminal portion of MSP1, was expressed as a secreted protein in baculovirus-infected insect cells. A highly pure baculovirus product could be reproducibly expressed and purified at yields in excess of 8 mg of pure protein per liter of culture. This protein, when tested for efficacy in the Aotus challenge model, gave significant protection, with only one of seven monkeys requiring treatment for uncontrolled parasitemia after challenge with P. falciparum. The second recombinant protein, P30P2MSP119, has been used in previous studies and is based on the smaller, C-terminal 19-kDa portion of MSP1 expressed in Saccharomyces cerevisiae. Substantial changes were made in its production process to optimize expression. The optimum form of this vaccine antigen (as judged by in vitro and in vivo indicators) was then evaluated, along with bvMSP142, for efficacy in the A. nancymai system. The new formulation of P30P3MSP119 performed significantly worse than bvMSP142 and appeared to be less efficacious than we have found in the past, with four of seven monkeys in the vaccinated group requiring treatment for uncontrolled parasitemia. With both antigens, protection was seen only when high antibody levels were obtained by formulation of the vaccines in Freund's adjuvant. Vaccine formulation in an alternate adjuvant, MF59, resulted in significantly lower antibody titers and no protection.
In the ongoing search for an asexual vaccine against malaria, merozoite surface protein 1 (MSP1) of Plasmodium falciparum remains the most advanced candidate (26). This 200-kDa molecule is expressed on the surface of the red cell invasive form of the parasite, the merozoite. On that surface, MSP1 undergoes several proteolytic processing steps to leave first the 42-kDa C terminus of MSP1 anchored to the merozoite surface by a C-terminal glycosylphosphatidyl inositol anchor and then the 19-kDa, most C-terminal part of MSP1, which remains attached to the parasite during red cell invasion (for a review, see reference 15).
While several regions of MSP1 have been identified as possible targets of protective immunity (34), we previously focused our efforts on the C-terminal 19-kDa portion, MSP119. The amino acid sequence of this region is largely conserved, with only limited point mutations having been identified (primarily at four positions, although rarer variants have been reported) (20, 30, 31). These point mutations are expressed predominantly as of all one type or all the other type. However, this expression is independent from the dimorphism present in the rest of the MSP1 molecule, in which large portions of the sequence are present in one of two major allelic families (27, 33).
MSP119 is also the target of a series of monoclonal antibodies that have the ability to inhibit the invasion of red blood cells by parasites in vitro (1, 5). Furthermore, in the rodent challenge model system of P. yoelii, MSP119 expressed in both yeast and bacteria has repeatedly been shown to protect mice against otherwise lethal infections (9, 14, 24). This protection, while involving multiple arms of the immune system, is largely antibody mediated, with high antibody titers being essential (8, 13). The conformation of MSP119 is also thought to be critical for protection, as this region of MSP1 has 12 cysteine residues and consists entirely of just two epidermal growth factor-like domains, each containing three disulfide bonds (2).
We previously produced a recombinant form of MSP119, P30P2MSP119, as a secreted protein from Saccharomyces cerevisiae. Vaccination of Aotus nancymai monkeys with this molecule has proven to protect them reproducibly from infection with the virulent FVO strain of P. falciparum (11, 22, 23). This protection also relies upon the achievement of very high antibody titers, and one of the weaknesses of the Aotus challenge model is that very few adjuvants effectively elicit high antibody titers in these monkeys; to date, only Freund's adjuvants have been used successfully to elicit protection.
Vaccines based on MSP119 have several potential problems. First, unlike the rest of MSP1, MSP119 has limited T-cell epitopes. T-cell responses to the protein are found in only 26% of naturally infected donors, and these responses may be directed to T-cell epitopes that are variant specific (10, 36). Thus, to recruit T-cell help, P30P2MSP119 has the P30 and P2 universal T-cell epitopes from tetanus toxoid linked to MSP119. Unfortunately, none of the predicted full-length P30P2MSP119 molecule can be detected when it is produced in S. cerevisiae, with both the P30 epitope and most of the P2 epitope being cleaved (23). However, it is thought that undetectable quantities of full-length protein may be present or that the cleaved P30 and P2 regions may aid protein folding during synthesis before they are cleaved, since when the equivalent unfused MSP119 protein is produced without the P30 and P2 epitopes, no protection is seen (22).
Second, more sequence variation has been found in MSP119 than was previously thought to exist (30, 31), reducing the advantages of focusing on this conserved region of MSP1. Finally, it has been found that considerable conformational variability exists in the current form of P30P2MSP119 (32a), and a way to control this variability, if not to eliminate it, needs to be found.
An alternate approach to avoid the problems associated with P30P2MSP119 would be to use a larger portion of the MSP1 molecule. In fact, the 42-kDa form of MSP1 (MSP142) is known to contain immunodominant T-cell epitopes in a region of the molecule immediately upstream from MSP119 (MSP133) (36). Despite the dimorphic nature of this region, these T-cell responses appear to be directed toward epitopes that are conserved between the two allelic families of MSP1 (10). Further, a recombinant form of the MSP142 molecule, expressed in a baculovirus system, has been shown to be protective in a monkey challenge experiment (4). The T-cell responses in that study were also directed toward T-cell epitopes in the upstream MSP133 region, rather than to T-cell epitopes in the MSP119 region. However, difficulties in making the antigen reproducibly have stymied its further development.
The purpose of the present study was to examine three issues: (i) to control the production process to produce a form of P30P2MSP119 that is more acceptable as a potential vaccine, (ii) to produce a baculovirus-expressed form of MSP142 in a reproducible manner, and (iii) to compare the relative efficacies of the two vaccines in the Aotus system.
MATERIALS AND METHODS
Saccharomyces protein production. (i) P30P2MSP119 construct.
A form of P30P2MSP119 with an amino acid sequence identical to that used previously (23) but in which codon usage was optimized for yeast expression was synthesized. The gene was cloned into the yeast episomal plasmid YEpRPEU3 (32a). Gene expression is under the control of the ADH2 promoter for ethanol-induced production, and plasmid selection is encoded by TRP1 downstream of the gene. Protein secretion is directed by the pre-pro yeast mating alpha factor signal sequence. A C-terminal six-histidine tag was added for purification.
(ii) Host cells and fermentation.
Plasmids were used to transform an S. cerevisiae VK1-derived cell line (haploid; trp1 lys2–801 pep4−::ura). Protein production was initiated using the batch-fed fermentation method previously described (12, 21). However, we attempted to optimize expression by using a Plackett-Burman matrix (29) to design eight fermentation experiments allowing us to examine variations in seven parameters simultaneously. From this selection process, based on the level of expression, the products of four fermentations were chosen for further analysis.
(iii) Protein purification.
Fermentation culture supernatants were recovered by microfiltration (0.1-mm hollow-fiber filter; Millipore, Bedford, Mass.). The supernatant was concentrated by ultrafiltration and diafiltered with a 3-kDa spiral-fiber filter (Millipore) into 2× phosphate-buffered saline (pH 7.4; PBS). The protein was purified from the supernatant by Ni-nitrilotriacetic acid chromatography (Qiagen, Valencia, Calif.) followed by size exclusion chromatography and buffer exchange into PBS using a Superdex 75 column (Amersham Pharmacia Biotech, Piscataway, N.J.).
Baculovirus protein production.
A synthetic gene coding for the amino acid sequence of P. falciparum MSP142 (Vietnam-Oak Knoll or FVO strain; GenBank accession no. L20092) was constructed. The coding sequence of the synthetic gene was altered to a mammalian codon preference to normalize the AT content of the gene. This construct, corresponding to amino acids A-1349 to S-1723, was cloned behind the secretion signal sequence of baculovirus envelope glycoprotein gp67 into the pFastBacI baculovirus transfer vector (Life Technologies, Grand Island, N.Y.). pFastBacI-MSP1 was used to transform competent Escherichia coli DH10Bac cells for site-specific transposition of insert DNA into the baculovirus genome downstream of the polyhedrin promoter within the polyhedrin locus. Recombinant MSP142 bacmid DNA was recovered from white colonies and used to transfect Sf-9 insect cells with the cationic liposome CELLFECTIN (Life Technologies). Recombinant virus was recovered from transfected cells after 72 h, and MSP142-expressing virus clones were isolated by three rounds of virus plaque purification.
A master virus stock was established using Sf-9 insect cells, serum-free medium (Sf-900 II SFM; Life Technologies), and a multiplicity of infection of 0.1 PFU/cell. The DNA sequence of the MSP1 gene insert and flanking baculovirus polyhedrin DNA was determined to be identical to the expected input nucleotide sequence. Propagation of working virus stocks and virus plaque assays were carried out with Sf-9 insect cells.
Recombinant MSP142 protein was produced by infection of Trichoplusia ni H5 insect cells at a cell density of 1.5 × 106 cells/ml (15 liters) with bvMSP142 at a multiplicity of infection of 3 PFU/cell using serum-free medium (HyQ; HyClone). The infected cells were harvested after 3 days, and the medium was isolated by low-speed centrifugation (1,500 × g, 10 min, 4°C). The supernatant from the infected cell culture was clarified further by centrifugation (12,000 × g, 30 min, 4°C).
Baculovirus protein purification.
The clarified infected cell supernatant was concentrated and diafiltered by passage through a hollow-fiber filtration cartridge (molecular weight cutoff, 10,000; A/G Technology Corp., Needham, Mass.) into 50 mM bis-Tris-propane–10 mM sodium chloride (pH 9) using an Amicon M12 ultrafiltration system (Millipore). The concentrated crude supernatant was loaded onto a Q Sepharose FF column (Amersham Pharmacia Biotech) to capture bvMSP142, and the recombinant protein was eluted from the column using a pH gradient from pH 9 to pH 6. Fractions containing recombinant bvMSP142 were identified by Western blot analysis using rabbit antisera to P30P2MSP119 and were pooled. The Q column eluate was adjusted to 500 mM sodium chloride–5 mM imidazole, loaded directly onto an Ni-nitrilotriacetic acid column (Qiagen), washed, and eluted with 50 mM Tris–500 mM sodium chloride–300 mM imidazole (pH 8.0). The eluted protein was analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), dialyzed against 10 mM sodium phosphate (pH 6.8), and loaded directly onto a hydroxyapatite column (Bio-Rad, Hercules, Calif.). The bound bvMSP142 protein was eluted using a salt gradient from 10 mM to 1 M sodium phosphate (pH 6.8). The purified antigen was formulated with 5 mM sodium phosphate–5 mM potassium phosphate–150 mM sodium chloride (pH 7.2) and filtered aseptically through a 0.22-μm-pore-size membrane as the final bulk product.
Protein characterization.
Amino acid sequencing by automated Edman degradation and electron spray mass spectroscopy were performed with liquid samples or with samples transferred to polyvinylidene difluoride membranes after SDS-PAGE at the Biological Resources Branch, National Institute of Allergy and Infectious Diseases. Protein concentrations were determined with a bicinchoninic acid protein assay (Pierce, Rockford, Ill.). Endotoxin levels were determined with a Limulus amebocyte lysate chromogenic pyrogenicity assay performed under contract at Novavax (formerly DynCorp) Quality Control Laboratory (Rockville, Md.).
Glycosylation patterns were determined by using a five-lectin–digoxigenin–glycan detection kit (Boehringer Mannheim Biochemicals, Indianapolis, Ind.) according to the manufacturer's instructions.
Rabbit immunizations and in vitro inhibition assays.
New Zealand White rabbits were immunized with different preparations of recombinant P30P2MSP119 in complete Freund's adjuvant (CFA; Life Technologies) as previously described (19). Briefly, rabbits were immunized with 100 μg of P30P2MSP119 emulsified with an equal volume of CFA intramuscularly. Three additional booster immunizations with the same antigen dose were successively given at 28-day intervals. For each booster immunization, the mycobacterial content in the CFA was successively halved by mixing with incomplete Freund's adjuvant. Rabbits were bled for sera 7 days prior to immunization (preimmune sera) and 21 days after the tertiary and quaternary immunizations (immune sera).
In vitro parasite growth inhibition assays using rabbit sera were performed as previously described (19). Briefly, rabbit sera (preimmune and immune) were heat inactivated at 56°C for 30 min and absorbed with human erythrocytes overnight at 4°C. Parasite cultures (FVO) were synchronized by sorbitol lysis to obtain mature stages. Rabbit sera were added to parasite cultures in 96-well plates at a final concentration of 20%, and hematocrit and parasitemia were adjusted to 0.8% and approximately 0.5%, respectively. Cultures were then incubated at 37°C for 72 h with occasional mixing. At 72 h, thin blood smears were prepared and stained with Giemsa stain. Parasitemia was determined microscopically. The percentage of parasite growth inhibition by the immune sera was determined using the following formula:
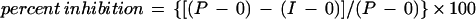 |
In this equation, P represents parasitemia with preimmune sera at 72 h; I represents parasitemia with immune sera at 72 h; and 0 represents parasitemia at 0 h.
ELISA.
Serum antibodies to recombinant proteins were assayed as described previously (12). Immulon-4 96-well plates (Dynex, Chantilly, Va.) were coated for 16 h at 4°C with 100 μl of a 1-μg/ml dilution of recombinant protein in coating buffer (15 mM Na2CO3, 35 mM NaHCO3 [pH 9.6]) per well. The plates were blocked with 5% (wt/vol) nonfat milk powder (Difco, Becton Dickinson, Sparks, Md.) in PBS (blocking buffer) for 1 h at room temperature. Serum samples were serially diluted in blocking buffer and incubated in the coated plates for 2 h at room temperature. The plates were washed extensively with PBS–0.05% Tween 20 and incubated with the appropriate secondary antibody for 1 h at room temperature. The secondary antibodies were 1:1,000 dilutions in blocking buffer of goat anti-mouse, anti-rabbit, or anti-human immunoglobulin G conjugated to alkaline phosphatase (Kirkegaard & Perry Laboratories, Gaithersburg, Md.). After the washing step was repeated, the plates were given an additional wash in Tris-buffered saline (pH 7.4). Detection was performed using 100 μl of p-nitrophenyl disodium phosphate solution (Sigma 104 phosphatase substrate [Sigma, St. Louis, Mo.]; one tablet per 5 ml of coating buffer) per well. After 20 min of incubation, the absorbance was read at 405 nm with a Dynex MR500 enzyme-linked immunosorbent assay (ELISA) plate reader. Serum dilutions that gave an absorbance value of 0.5 unit above the background were designated the endpoint of the serum ELISA titer.
Competitive ELISA.
Inhibition-competition ELISAs were performed as described above, except that prior to use, serum was preincubated for 2 h at room temperature in blocking buffer containing various concentrations of one of the recombinant proteins described above.
Indirect immunofluorescence assays (IFAs).
Thin films were made on Toxoplasma slides (Bellco, Vineland, N.J.) from cultured parasites, fixed in 90% (vol/vol) acetone–10% (vol/vol) methanol for 10 min at −20°C, and then air dried. Polyclonal antibodies were diluted with 5% skim milk powder in PBS-Tween 20, and 5 μl was spotted on the slides. The slides were incubated in a sealed, moist box for 2 h at room temperature. The slides were washed three times for 5 min each time with PBS-Tween 20 and air dried. A secondary antibody, fluorescein isothiocyanate-conjugated goat anti-human immunoglobulin (Kirkegaard & Perry Laboratories), was diluted appropriately in 5% skim milk powder in PBS-Tween 20, spotted on the slides, and incubated for 1 h. The slides were rewashed, air dried, and mounted with an antifade solution to retard photobleaching (SlowFade; Molecular Probes, Eugene, Oreg.). Fluorescence was examined under an Olympus BH2 UV microscope with a 100× oil immersion objective.
Vaccination and challenge infection of malaria-naive Aotus monkeys.
Monkeys were housed at the Primate Research Facility, National Institutes of Health, in compliance with a National Institutes of Health Animal Care and Use Committee-approved protocol (LPD-8E). Monkeys were stratified by weight and sex and randomly assigned to vaccine groups by card draw. Group assignment was masked to the primary investigators who cared for or vaccinated the animals, read smears, or determined when a monkey should be drug cured. Only when all monkeys had been treated were the codes revealed to these investigators.
Thirty-six A. nancymai monkeys were used in the study. They were divided into four vaccine groups of seven monkeys per group and two control groups of four monkeys each. The four vaccine groups received the following: bvMSP142 formulated in Freund's adjuvant; bvMSP142 formulated in MF59 adjuvant; P30P2MSP119 formulated in Freund's adjuvant; and P30P2MSP119 formulated in MF59 adjuvant. The two control groups received adjuvant alone (either Freund's or MF59).
Monkeys received 250 μg of the respective recombinant protein per vaccination. Monkeys received three vaccinations, each 3 weeks apart. For the animals receiving vaccinations formulated in Freund's adjuvant, the first vaccination was an emulsion of 250 μl of antigen (in PBS) with 250 μl of CFA (Sigma) given subcutaneously at four sites on the back; the next two vaccinations were emulsions with incomplete Freund's adjuvant (Sigma) given in the same manner. The control group for this adjuvant received the same regimen, with PBS replacing the antigen.
For the animals receiving vaccinations formulated in MF59, all vaccinations were a mixture of 250 μl of antigen (in PBS) with 250 μl of MF59 (a generous gift from John Donnelly, Chiron Corporation, Los Angeles, Calif.) given intramuscularly at two sites in the thigh. As before, the control group for this adjuvant received the same regimen, with PBS replacing the antigen.
Seven days after the third vaccination, an A. nancymai donor monkey (2767) was infected intravenously with approximately 106 freshly thawed P. falciparum parasites of the FVO strain from a frozen sample from monkey T774 (A. nancymai). Previously, a large infection bank of parasites had been prepared by infecting monkey T774 from a frozen sample derived originally from monkey A1–936, kindly provided by W. E. Collins, Centers for Disease Control and Prevention (monkey A1–936 sample passaged through monkeys A11, 1588, and 2544 prior to T774). Five days later, when 4% parasitemia had been reached in the donor monkey, blood was collected, washed, and diluted in RPMI medium to 104 parasitized red blood cells/ml. The donor monkey then was drug-cured with 25 mg of mefloquine/kg of body weight. Monkeys in the experimental groups were each challenged with 1 ml of 104 parasitized red blood cells/ml by intravenous infusion. The challenge infection was administered 12 days after the third vaccination. One investigator during the challenge was aware of the group code and used that information to ensure that a control monkey was challenged first and last.
Hematocrit and Giemsa-stained thin smears were made from blood collected by puncture of superficial veins in the dorsum of the calf. Hematocrit values were determined daily; the plasma portions from hematocrit samples were retained for antibody analysis, and the blood portions were archived for later parasite analysis. Blood smears were prepared for each monkey on challenge day 0 and then daily from day 4 until final treatment on day 30. After chemotherapy, blood smears were prepared daily until there was no detectable parasitemia for two consecutive days. Monkeys were drug cured with 25 mg of mefloquine/kg given orally at 4% or greater parasitemia or at a hematocrit below 25%. All untreated monkeys were given chemotherapy on day 30. Parasitemia was calculated based on an examination of approximately 2,000 red blood cells (equivalent to 10 high-power fields); if no parasites were seen, then 40 more high-power fields were examined.
Statistical methods.
Trial outcomes were measured with a primary statistical endpoint and several secondary endpoints. In the past, we have found that Aotus monkeys that control their parasitemia either self-cure or suffer anemia. A treatment criterion for the trial was a drop in the hematocrit below 25%. Thus, monkeys that control their parasitemia but suffer anemia will, at some stage, require treatment for anemia. At this point, it is impossible to say what would have occurred to such a monkey's parasite burden; the monkey may have self-cured or continued to control the parasite burden, or it may have lost control and suffered acute parasitemia. Thus, for the primary endpoint, we included data up until the first monkey was treated for hematocrit rather than parasitemia. On that day, all monkeys were ranked in order of cumulative parasitemia. Monkeys that were treated for parasitemia prior to the day of data collection were ranked first, in order of their cumulative parasitemia until treatment. Then, monkeys that required treatment for hematocrit were ranked in the same fashion. The lowest ranked monkeys were those that did not require treatment up until that point, and they also were ranked in order of cumulative parasitemia. A nonparametric Wilcoxon rank sum analysis was performed to compare test groups to control groups.
Secondary statistical comparisons were also made. Student's t tests were used to compare antibody responses elicited to the vaccines, and linear regression analysis was performed to correlate antibody responses to protection from challenge. Nonparametric tests were also performed using a Mann-Whitney analysis to compare discontinuous data between vaccine groups (e.g., days to peak parasitemia, days to treatment, peak parasitemia, and parasitemia at the time of treatment).
RESULTS
Optimization of P30P2MSP119 production.
A matrix of S. cerevisiae fermentations were performed with P30P2MSP119 to evaluate parameters critical to the fermentation process. From this selection process, on the basis of expression levels, four conditions were chosen. Each condition gave rise to a slightly different purified product, as judged by the relative amounts of the various protein species derived from P30P2MSP119. The different resulting SDS-PAGE banding patterns are shown in Fig. 1, where the “standard” conditions are those used in past studies. The variables included both chemical medium components and physical conditions (temperature and pH) in an attempt to define both the critical medium components (removing undefined variable components, such as yeast extract) and the critical physical parameters (Table 1). The banding patterns shown in Fig. 1 were reproducible from fermentation to fermentation. The differences between the banding patterns were only slight but were consistently obtained and reflected the degree to which yeast proteases had removed various lengths of the N-terminal region of the protein. The differences in processing may reflect the presence or absence of inhibitory components (e.g., amino acids) in the more complex medium formulations.
FIG. 1.

Different forms of P30P2MSP119 produced by various fermentation conditions (standard, 5b, 6b, and 8b). The products of the four different fermentations were purified identically, and equal amounts were run in nonreducing SDS-PAGE. A, B, and C indicate the migration positions of the three known major polypeptides derived from the sequence of P30P2MSP119 (A, NISQ…; B, FIGITEVENISQ…; and C, EVENISQ… ). The band migrating above band A is a longer protein with an inconsistent starting point. The material migrating below band C is known to be misfolded P30P2MSP119 (32a). The sizes of the molecular mass standards are shown on the left in kilodaltons.
TABLE 1.
Various parameters used to produce different forms of P30P2MSP119a
| Condition | MgSO4 added | Yeast extract added | Rich mediumb supplement during:
|
Alternate carbon source (glycerol) during expression | Temp (°C) | |
|---|---|---|---|---|---|---|
| Growth | Expression | |||||
| Standardc | + | + | + | + | + | 25 |
| 5b | − | + | − | − | + | 30 |
| 6b | + | − | + | − | − | 30 |
| 8b | − | − | + | + | + | 25 |
+, yes; −, no.
Rich medium is a mixture of 25% glucose, 1% yeast nitrogen base, 1% yeast extract, and 2% acid-hydrolyzed Casamino Acids.
Standard conditions are those used to produce P30P2MSP119 in previously published studies.
To evaluate which of the four products would be most effective as a vaccine, each was used to immunize five rabbits in a four-immunization schedule using CFA and incomplete Freund's adjuvant. The antibodies elicited were then evaluated for titers against recombinant MSP119 by an ELISA and for biological efficacy by in vitro inhibition of parasite invasion of red blood cells (Table 2).
TABLE 2.
Immune responses in rabbits to variant forms of P30P2MSP119
| P30P2MSP119 variant | Mean ELISA titera (% SE) | % Inhibition of invasion in vitro (% SE) | Correlation coefficient (r2) of ELISA titers versus % inhibition (P) |
|---|---|---|---|
| Standardb | 760,000 (56) | 60.2 (55.2) | 0.6974 (0.44) |
| 5b | 796,000 (69) | 53.2 (54.0) | 0.5705 (0.06) |
| 6b | 936,000 (43) | 80.9 (31.4) | 0.8258 (0.02) |
| 8b | 770,000 (56) | 70.9 (33.1) | 0.6790 (0.06) |
Five rabbits were immunized in each group.
Standard conditions are those used to produce P30P2MSP119 in previously published studies.
From this process, condition 6b was chosen as producing the best product—it elicited the highest antibody titers, elicited the highest percentage of invasion inhibition, and produced the best (and only significant) correlation between antibody titers and percentage of invasion inhibition (Table 2). Although none of the differences between conditions 5b, 6b, 8b, and standard achieved significance, condition 6b also produced the most uniform responses among the five animals for both titers and percentage of inhibition (i.e., the lowest standard error). To evaluate the reproducibility of the production process, these studies were reproduced for three independent runs using only condition 6b; no significant differences were found among the three products in terms of ELISA titers (data not shown). Thus, all subsequent studies were performed with this product (from condition 6b).
MSP119 sequence variation.
As part of the above evaluation of each P30P2MSP119 product, competitive ELISAs were performed. The ability of each of the forms of P30P2MSP119 (standard, 5b, 6b, and 8b) to compete with standard P30P2MSP119 for the binding of monoclonal antibody 5.2 was assayed. No significant differences were observed (data not shown). However, during the course of these studies, it was noted that the specificities of the antibody responses of different rabbits to immunization with P30P2MSP119 varied dramatically (Fig. 2). Within MSP119, two major allelic forms, which reflect variation at four amino acid positions, can be seen. The four amino acids are dimorphic, and here the allelic variation is described as either Q-KNG or E-TSR (i.e., there is either a Q or an E at one dimorphic site, etc.) (20). Some rabbits (e.g., 8075, immunized with 5b P30P2MSP119) showed a response that was totally allele independent. That is, after immunization with P30P2MSP119 (which is of the Q-KNG allele), standard molecules of either the Q-KNG or the E-TSR allele could successfully inhibit binding of the rabbit antisera to the Q-KNG coating antigen during a competitive ELISA. Alternately, with rabbit 8089 (immunized with 8b P30P2MSP119), the E-TSR allele of MSP119 was not as effective as the Q-KNG allele of MSP119 at outcompeting the antibody response.
FIG. 2.

Examples of competitive ELISAs performed with sera from rabbits immunized with P30P2MSP119. Sera from four rabbits immunized with P30P2MSP119 (Q-KNG allele) were mixed with various concentrations of a competitor MSP119 of either the Q-KNG allele (open circles) or the E-TSR allele (closed circles) prior to an ELISA with the Q-KNG allele as the coating antigen. Rabbits 8075 and 8077 were immunized with P30P2MSP119 produced under condition 5b, rabbit 8089 was immunized with P30P2MSP119 from condition 8b, and rabbit 8076 was immunized with P30P2MSP119 from standard conditions.
However, after immunization of rabbit 8077 with P30P2MSP119 (5b), a significant proportion of the response was allele specific, as the inhibition of binding reached a plateau and increasing concentrations of the alternate E-TSR form of MSP119 were unable to completely inhibit binding to the Q-KNG coating antigen; this result indicated that one or more B-cell epitopes recognized by the sera were specific for the Q-KNG allele of P30P2MSP119. With rabbit 8076 (immunized with standard P30P2MSP119), the response elicited was almost totally allele specific for the Q-KNG allele of the immunizing P30P2MSP119. In all, of 16 rabbit polyclonal sera to P30P2MSP119 tested, 2 showed no allele specificity in response, 4 showed a reduced recognition of the alternate E-TSR allele, 7 showed a clear lack of some B-cell epitopes, and 3 showed a complete lack of recognition of the alternate allelic form of MSP119.
Production of recombinant bvMSP142.
We found that the critical factor required for the successful purification of a soluble full-length form of MSP142 from baculovirus (bvMSP142) was initial production levels. That is, bvMSP142 is a difficult molecule to purify, possessing hydrophobic properties that lead to strong associations with heterologous proteins. If initial production levels are not sufficiently high, then the purification process becomes too difficult to give a useful product. In practical terms, we found that if the bvMSP142 product after infection was not easily distinguishable as a discrete band on a Coomassie blue-stained SDS-polyacrylamide gel of a crude culture supernatant, then purification of the supernatant would not be successful. Ultimately, the use of Hi-5 cells, a defined serum-free medium, and a 3-day infection period allowed us to achieve reproducibly useful yields of bvMSP142 (a final yield of 7.3 mg of purified bvMSP142 per liter of culture was achieved). Furthermore, a robust and reproducible purification strategy was also achieved; we believe that this strategy will prove to be scalable (V. Cioce et al., unpublished).
The identity and purity of the final product were assayed by biochemical and immunological methods (Table 3). The final product runs as two closely migrating bands in nonreducing SDS-PAGE (Fig. 3). We have not yet determined the difference between the two bands, because they have identical N termini (as determined by direct sequencing) and presumably identical C termini (as they are purified with a C-terminal six-histidine tag). The two mass peaks observed by mass spectroscopy suggest that they may have differences in glycosylation patterns. Interestingly, about half of the protein produced by the infected insect cells is not secreted and remains intracellular. This protein is also recoverable and appears indistinguishable from secreted bvMSP142. However, to date, we have used only the secreted protein in vaccine trials.
TABLE 3.
Characterization of bvMSP142
| bvMSP142 | % Final puritya | Endotoxin level (U/mg)b | Mass (kDa) determined by:
|
N-terminal sequenced | Glycosylation pattern | Monoclonal antibody recognitione | |
|---|---|---|---|---|---|---|---|
| Gel filtration chromatography | Electron spray mass spectroscopyc | ||||||
| Secreted | 97.4 | 0.78 | 81.5f | 43,320.0; 44,404.0f | NH2 ADPAVTPSVIDNIL | High mannose | 5.2, 2D634, 13E353, 4H919, 12.1, 1E1, 12.8, 2F10 |
| Intracellular | ND | ND | 100.0 | 41,320; 42,276; 43,660 | NH2 ADPAVTPSVIDNIL | High mannose | Same as for secreted bvMSP142 |
Determined by scanning densitometry. ND, not determined.
Determined by Limulus amebocyte lysate assay.
Major mass peaks are listed.
ADP is a vector-derived cloning sequence; underlining indicates the start of the MSP142 sequence. The predicted sequence is NH2 ADPAVT.
Monoclonal antibodies recognizing conformational epitopes in MSP119 with different specificities were used in Western blot analyses: 5.2 (32), 2D634, 13E353, 4H919 (7), 12.1 (25), 1E1, 12.8, and 2F10 (16). Antibodies 5.2 and 12.8 recognize the first EGF domain; 4H919 recognizes the second EGF domain; and 2D634, 13E353, 12.1, and 12.8 recognize both EGF domains.
Predicted: 41,924.4.
FIG. 3.

Purity of the recombinant bvMSP142 protein. Coomassie blue-stained SDS-PAGE analysis of purified bvMSP142. Lane 1, molecular weight markers; lane 2, purified bvMSP142 run under nonreducing conditions; lane 3, purified bvMSP142 run under reducing conditions. Purity estimates obtained by laser scanning densitometry were 97.4 and 94.4% for lanes 2 and 3, respectively.
Aotus challenge trial. (i) Antibodies.
Thirty-six A. nancymai monkeys were immunized, three times each at 3-week intervals. Some animals received secreted recombinant bvMSP142, formulated in either CFA or incomplete Freund's adjuvant (seven animals) or MF59 adjuvant (seven animals). Other animals received recombinant P30P2MSP119, formulated in either CFA or incomplete Freund's adjuvant (seven animals) or MF59 adjuvant (seven animals). A control group of four animals was also immunized for each of the two adjuvant groups. Two weeks after the final vaccination, all monkeys were challenged with 104 P. falciparum parasites of the virulent FVO strain, freshly passaged through a donor monkey. Animals were treated either when parasitemia increased to greater than 4%, as determined with thin blood smears, or when the hematocrit dropped to less than 25%.
Prechallenge antibody titers for each animal, as measured by ELISAs and IFAs, are shown in Fig. 4. For both antigens, the antibody titers determined by ELISAs were significantly lower in the animals receiving MF59 than in those receiving Freund's adjuvant. Animals immunized with bvMSP142 in Freund's adjuvant had 16 times more antibody than animals immunized with bvMSP142 in MF59. There was an eightfold difference between the corresponding groups immunized with P30P2MSP119. Thus, in the MF59 groups, all animals required treatment for parasitemia, while in the Freund's groups, one of seven animals vaccinated with bvMSP142 and four of seven animals vaccinated with P30P2MSP119 required treatment for parasitemia.
FIG. 4.

Antibody titers of individual monkeys prior to challenge. Shown on the x axis is the immunogen-adjuvant combination for that group of monkeys (42/CFA, bvMSP142 in CFA; 19/MF59, P30P2MSP119 in MF59, etc). (A and B) ELISA titers are recorded as the inverse of the serum dilution corresponding to an optical density at 405 nm of 0.5. (C) IFA titers are recorded as the inverse of the serum dilution corresponding to a reading above the background. ELISA titers were measured against two capture antigens, bvMSP142 (A) and P30P2MSP119 (B). Open circles represent animals which required treatment for parasitemia during challenge; closed circles represent animals which controlled their parasitemia but required treatment for anemia; closed triangles indicate animals that self-cured without treatment. Significant differences determined by Student's t test between animals immunized with the same antigen but with either Freund's adjuvant or MF59 are shown.
There was no significant difference in the antibody titers elicited by immunization with bvMSP142 compared to those elicited by immunization with P30P2MSP119 when the same adjuvant was used. This was true regardless of the antigen used for capture in the ELISA (P30P2MSP119 or bvMSP142). This was also true despite the fact that the animals receiving P30P2MSP119 received 3.5 times the molar dosage of antigen (all animals received 250 μg of antigen per dose, but P30P2MSP119 has a mass of 12 kDa and the mass of bvMSP142 is 42 kDa).
(ii) Parasitemia.
The course of parasitemia for each animal is shown in Fig. 5. The primary statistical endpoint analysis for the trial—a Wilcoxon rank sum test to compare cumulative levels of parasitemia for all of the groups up to day 14 (the first day when a monkey required treatment for anemia rather than parasitemia)—showed no significant differences between any of the groups.
FIG. 5.

Course of the daily parasitemia in individual monkeys. Monkeys were challenged on day 0 with 104 P. falciparum FVO parasites. Parasitemia was determined by counting 2,000 red blood cells on Giemsa-stained thin smears. The broken line is for ease of reference between graphs. Also indicated are the treatment times for uncontrolled parasitemia greater than 4% (P), for hematocrits below 25% (H), and for self-curing animals (∗).
By secondary measures, however, the course of parasitemia for animals vaccinated with either bvMSP142 or P30P2MSP119 in Freund's adjuvants did differ significantly from the course of parasitemia for control animals, as measured by several parameters (Table 4). Animals immunized with either antigen in MF59 showed no significant difference in the course of parasitemia compared to the results for the relevant control animals. However, one animal (T740) immunized with bvMSP142 in MF59 did appear to briefly control its parasitemia before succumbing. The mechanism of this control (and its loss) is unclear, as this animal did not have the highest antibody titers prior to challenge (it was fourth highest in its group), and the titer in this animal was well below those in the animals receiving Freund's adjuvants.
TABLE 4.
Significance of protection obtained through vaccination
| Vaccine | No. of animals:
|
P value fora:
|
|||||
|---|---|---|---|---|---|---|---|
| Self-curing | Treated for:
|
Days to treatment | Parasitemia at treatment | Days to peak parasitemia | Peak parasitemia | ||
| Anemia | Parasitemia | ||||||
| bvMSP142 in Freund's adjuvant | 2 | 4 | 1 | 0.019 | 0.004 | NS | 0.004 |
| P30P2MSP119 in Freund's adjuvant | 0 | 3 | 4 | 0.029 | 0.029 | 0.015 | 0.029 |
| bvMSP142 in MF59 | 0 | 0 | 7 | NS | NS | NS | NS |
| P30P2MSP119 in MF59 | 0 | 0 | 7 | NS | NS | NS | NS |
| Both antigens in Freund's adjuvant | NS | 0.009 | NS | 0.009 | |||
P values are for comparisons of groups receiving the individual vaccines to groups receiving the adjuvant only, except that P values for groups receiving both antigens represent comparisons of the immunized groups to each other. P values were determined with a nonparametric Mann-Whitney test. NS, not significant.
There was a clear difference in the level of protection obtained by animals receiving bvMSP142 in Freund's adjuvants compared to that obtained by animals receiving P30P2MSP119 in Freund's adjuvants. However, of the parameters in Table 4, this difference in the level of protection between the two antigens reached significance only for peak parasitemia and parasitemia at the time of treatment.
Regression analysis did show some correlations between antibody titers and the measures of protection shown in Table 4. For bvMSP142-immunized monkeys, ELISA titers correlated positively with days to treatment (r2 = 0.54) and negatively with peak parasitemia and parasitemia at the time of treatment (r2 = 0.52 and 0.54, respectively). For P30P2MSP119-immunized monkeys, the correlation coefficients were weaker, although titers still correlated positively with days to treatment and days to peak parasitemia (r2 = 0.46 and 0.56, respectively) and negatively with peak parasitemia and parasitemia at the time of treatment (r2 = 0.19 and 0.23, respectively).
DISCUSSION
Baculovirus-expressed bvMSP142 performed exceptionally well in the current trial, a result consistent with a previous report (4). Six of the seven animals immunized with Freund's adjuvant-bvMSP142 were able to control a virulent infection by P. falciparum, two of seven completely clearing their parasites. Since we believe that we have solved many of the production problems associated with expressing bvMSP142 reproducibly in a robust process, we find these results to be intriguing; we believe that further testing of this molecule is of the highest priority. One of the most interesting questions to examine in future studies is whether this form of bvMSP142 will protect against a challenge with a heterologous allele of MSP1.
In contrast, the P30P2MSP119 molecule used here performed less effectively than bvMSP142. This form of P30P2MSP119 is the end product resulting from an extensive effort to define and control the critical parameters in the production process in preparation for further clinical testing of this molecule. However, despite the results of rabbit immunizations that gave strong indications of optimal immunogenicity, this batch of P30P2MSP119 performed less effectively than it has in previous trials with A. nancymai. Table 5 presents a summary of the protection seen in the present and previous trials. The lesser efficacy seen in the present study may be due to the alterations made to the production process or may simply reflect normal variance, hidden by the small numbers of animals used by necessity for all these experiments. However, further evaluation of this form of P30P2MSP119 is clearly warranted before clinical manufacture.
TABLE 5.
Protection obtained with P30P2MSP119 in previous Aotus trials after challenge with P. falciparum FVO parasites
| Study | Species | No. of vaccinations | Dose (μg) | No. (%) of animals:
|
|||
|---|---|---|---|---|---|---|---|
| Total | Self-cured | Cured for anemia | With virulent parasitemia | ||||
| 23 | A. nancymai | 3 | 250d | 2a | 2 | 0 | 0 |
| A. vociferans | 2a | 0 | 0 | 2c | |||
| 11 | A. vociferans | 4 | 200e | 4a | 3 | 0 | 1 |
| 4b | 1 | 2 | 1 | ||||
| 22 | A. nancymai | 3 | 250d | 10a | 7 | 2 | 1c |
| A. vociferans | 2a | 0 | 1 | 1 | |||
| A. Egan et al., submitted for publication | A. nancymai | 4 | 200e | 3b | 1 | 1 | 1 |
| C. A. Long et al., unpublished data | A. vociferans | 4 | 200e | 7b | 2 | 0 | 5 |
| Total | A. nancymai | 15 | 10 (67) | 3 (20) | 2 (13) | ||
| A. vociferans | 19 | 6 (32) | 3 (4) | 10 (53) | |||
| This study | A. nancymai | 3 | 250d | 7a | 0 (0) | 3 (43) | 4 (57) |
Monkeys were challenged with 104 FVO P. falciparum parasites.
Monkeys were challenged with 105 FVO P. falciparum parasites.
Onset of parasitemia was delayed for one animal.
Vaccinations were given subcutaneously with CFA, followed by IFA, and followed by IFA.
The fourth vaccination was given intramuscularly without adjuvant.
It is difficult to say which form of antigen is overall most efficacious in the Aotus model. Table 5 shows that in A. nancymai, including in the present trial, P30P2MSP119 enables 45.5% of monkeys to self-cure (10 of 22) and 27.3% of monkeys to control parasitemia at low levels until they require treatment for anemia (6 of 22) but leaves 27.3% unprotected (6 of 22). With much smaller animal numbers, the equivalent results for bvMSP142 are 28.6% (two of seven), 57.1% (four of seven), and 14.3% (one of seven) respectively. Thus, historically, 73% of monkeys vaccinated with P30P2MSP119 gain some measure of protection, compared to 86% for bvMSP142.
The C-terminal 19-kDa region of MSP119 has long been regarded as an attractive vaccine candidate. One of its principal advantages was thought to be the conserved nature of the amino acid sequence in this region. Although this region is known to possess four point mutations (described by Q-KNG and E-TSR) (20), which are largely linked in character, it was hoped that these details would not present the serious problems associated with the dimorphism exhibited in the rest of the MSP1 molecule. Indeed, previous studies have found a consistent serological cross-reactivity to the two different allelic forms in both infected naive Aotus monkeys (18) and actively immunized animals (17, 21). However, our finding that rabbits immunized with one allelic form (Q-KNG) mount an antibody response with various degrees of specificity, which in some cases can be entirely allele specific, may negate this advantage. That such specific responses can be mounted should come as no surprise now that the three-dimensional structure of MSP119 is known (6, 28). The most surface-exposed portion of the molecule is the KN-TS loop. So, it is easy to imagine this being a prominent and entirely specific B-cell epitope. Indeed, although the studies mentioned above using polyclonal antiserum demonstrated no allele specificity, it has been previously noted that monoclonal antibodies do differentiate epitopes defined by both the E-to-Q and the TSR-to-KNG variations (35).
Thus, one of the perceived disadvantages of using a larger portion of MSP1, such as MSP142, which contains some of the large dimorphic regions of MSP1, may not be such a disadvantage when compared to the use of P30P2MSP119. A vaccine based on either may require two allelic forms to be produced to adequately cover the repertoire of sequence diversity, a diversity that, for MSP119, is rapidly expanding beyond the originally described four positions (30, 31).
Formulation of either P30P2MSP119 or bvMSP142 in MF59 instead of Freund's adjuvants abrogated any protective response. Rodent models have shown that a critical requirement for induced protection with vaccines based on MSP1 is a high antibody titer (8, 13). Titers with MF59 were between 8 and 15 times lower than those with Freund's adjuvants, and this result almost certainly explains the lack of protection. Our findings are also consistent with those of other studies using P30P2MSP119 or MSP119 and a series of six other adjuvants, all of which resulted in low antibody titers and poor protection (3, 22).
The challenge system combination of A. nancymai monkeys and the virulent parasite strain FVO thus requires a powerful adjuvant to produce high antibody titers. Thus, the model may be used to determine whether a powerful immune response generated against a particular antigen will be protective. That is, it can identify antigens of possible use in a vaccine but currently cannot be used to evaluate the formulation of an antigen in an adjuvant suitable for human use.
One weakness of the Aotus model is that a significant number of animals require treatment for anemia rather than parasitemia. These are animals that have managed to control their virulent parasites but are unable to clear them. We regard these animals as protected, but they remain a cause for concern. The vaccine-induced immune response has had an antiparasite effect, allowing the animals to control an otherwise lethal infection, but the cause of the drop in the hematocrit remains unclear.
The form of bvMSP142 used the present study can be produced reproducibly at the 15-liter scale. The purification procedure is also reproducible and appears to be robust enough for scaling-up purposes. The resultant recombinant protein is recognized by a panel of conformationally restricted monoclonal antibodies, and high titers generated by vaccination with the antigen provide protection from a lethal P. falciparum challenge. In a clinical situation, these high titers may be obtained by continual boosting of blood-stage infections. Alternatively, multiple components may act synergistically to be more efficacious and lower the absolute antibody requirement for any one antigen. For these reasons, we are encouraged by the results of this study and intend to take this antigen into a clinical development program.
ACKNOWLEDGMENTS
We gratefully acknowledge the contribution of Joseph Tropea for the cloning of the synthetic yeast codon-optimized form of P30P2MSP119, Anthony Holder for Western blots of baculovirus-expressed MSP142 using various monoclonal antibodies against MSP1, and John Donnelly of Chiron Corporation for the generous gift of adjuvant MF59.
REFERENCES
- 1.Blackman J M, Heidrich H G, Donachie S, McBride J S, Holder A A. A single fragment of a malaria merozoite surface protein remains on the parasite during red cell invasion and is the target of invasion-inhibiting antibodies. J Exp Med. 1990;172:379–382. doi: 10.1084/jem.172.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blackman J M, Ling I T, Nicholls S C, Holder A A. Proteolytic processing of the Plasmodium falciparum merozoite surface protein-1 produces a membrane-bound fragment containing two epidermal growth factor-like domains. Mol Biochem Parasitol. 1991;49:29–33. doi: 10.1016/0166-6851(91)90127-r. [DOI] [PubMed] [Google Scholar]
- 3.Burghaus P A, Wellde B T, Hall T, Richards R L, Egan A F, Riley E M, Ballou W R, Holder A A. Immunization of Aotus nancymai with recombinant C terminus of Plasmodium falciparum merozoite surface protein 1 in liposomes and alum adjuvant does not induce protection against a challenge infection. Infect Immun. 1996;64:3614–3619. doi: 10.1128/iai.64.9.3614-3619.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang S P, Case S E, Gosnell W L, Hashimoto A, Kramer K J, Tam L Q, Hashiro C Q, Nikaido C M, Gibson H L, Lee-Ng C T, Barr P J, Yokota B T, Hui G S. A recombinant baculovirus 42-kilodalton C-terminal fragment of Plasmodium falciparum merozoite surface protein 1 protects Aotus monkeys against malaria. Infect Immun. 1996;64:253–261. doi: 10.1128/iai.64.1.253-261.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chappel J A, Holder A A. Monoclonal antibodies that inhibit Plasmodium falciparum invasion in vitro recognise the first growth factor-like domain of merozoite surface protein-1. Mol Biochem Parasitol. 1993;60:303–311. doi: 10.1016/0166-6851(93)90141-j. [DOI] [PubMed] [Google Scholar]
- 6.Chitarra V, Holm I, Bentley G A, Petres S, Longacre S. The crystal structure of C-terminal merozoite surface protein 1 at 1.8 A resolution, a highly protective malaria vaccine candidate. Mol Cell. 1999;3:457–464. doi: 10.1016/s1097-2765(00)80473-6. [DOI] [PubMed] [Google Scholar]
- 7.Cooper J A, Cooper L T, Saul A J. Mapping of the region predominantly recognized by antibodies to the Plasmodium falciparum merozoite surface antigen MSA 1. Mol Biochem Parasitol. 1992;51:301–312. doi: 10.1016/0166-6851(92)90080-4. [DOI] [PubMed] [Google Scholar]
- 8.Daly T M, Long C A. Humoral response to a carboxyl-terminal region of the merozoite surface protein-1 plays a predominant role in controlling blood-stage infection in rodent malaria. J Immunol. 1995;155:236–243. [PubMed] [Google Scholar]
- 9.Daly T M, Long C A. A recombinant 15-kilodalton carboxyl-terminal fragment of Plasmodium yoelii yoelii 17XL merozoite surface protein 1 induces a protective immune response in mice. Infect Immun. 1993;61:2462–2467. doi: 10.1128/iai.61.6.2462-2467.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egan A, Waterfall M, Pinder M, Holder A, Riley E. Characterization of human T- and B-cell epitopes in the C terminus of Plasmodium falciparum merozoite surface protein 1: evidence for poor T-cell recognition of polypeptides with numerous disulfide bonds. Infect Immun. 1997;65:3024–3031. doi: 10.1128/iai.65.8.3024-3031.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Egan A F, Blackman M J, Kaslow D C. Vaccine efficacy of recombinant Plasmodium falciparum merozoite surface protein 1 in malaria-naive, -exposed, and/or -rechallenged Aotus vociferans monkeys. Infect Immun. 2000;68:1418–1427. doi: 10.1128/iai.68.3.1418-1427.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gozar M M, Price V L, Kaslow D C. Saccharomyces cerevisiae-secreted fusion proteins Pfs25 and Pfs28 elicit potent Plasmodium falciparum transmission-blocking antibodies in mice. Infect Immun. 1998;66:59–64. doi: 10.1128/iai.66.1.59-64.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirunpetcharat C, Good M F. Deletion of Plasmodium berghei-specific CD4+ T cells adoptively transferred into recipient mice after challenge with homologous parasite. Proc Natl Acad Sci USA. 1998;95:1715–1720. doi: 10.1073/pnas.95.4.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirunpetcharat C, Tian J H, Kaslow D C, van Rooijen N, Kumar S, Berzofsky J A, Miller L H, Good M F. Complete protective immunity induced in mice by immunization with the 19-kilodalton carboxyl-terminal fragment of the merozoite surface protein-1 (MSP1[19]) of Plasmodium yoelii expressed in Saccharomyces cerevisiae: correlation of protection with antigen-specific antibody titer, but not with effector CD4+ T cells. J Immunol. 1997;159:3400–3411. [PubMed] [Google Scholar]
- 15.Holder A A, Blackman M J, Burghaus P A, Chappel J A, Ling I T, McCallum-Deighton N, Shai S. A malaria merozoite surface protein (MSP1)—structure, processing and function. Mem Inst Oswaldo Cruz. 1992;87:37–42. doi: 10.1590/s0074-02761992000700004. [DOI] [PubMed] [Google Scholar]
- 16.Holder A A, Lockyer M J, Odink K G, Sandhu J S, Riveros-Moreno V, Nicholls S C, Hillman Y, Davey L S, Tizard M L, Schwarz R T, et al. Primary structure of the precursor to the three major surface antigens of Plasmodium falciparum merozoites. Nature. 1985;317:270–273. doi: 10.1038/317270a0. [DOI] [PubMed] [Google Scholar]
- 17.Hui G S, Gosnell W L, Case S E, Hashiro C, Nikaido C, Hashimoto A, Kaslow D C. Immunogenicity of the C-terminal 19-kDa fragment of the Plasmodium falciparum merozoite surface protein 1 (MSP1), YMSP1(19) expressed in S. cerevisiae. J Immunol. 1994;153:2544–2553. [PubMed] [Google Scholar]
- 18.Hui G S, Nikaido C, Hashiro C, Kaslow D C, Collins W E. Dominance of conserved B-cell epitopes of the Plasmodium falciparum merozoite surface protein, MSP1, in blood-stage infections of naive Aotus monkeys. Infect Immun. 1996;64:1502–1509. doi: 10.1128/iai.64.5.1502-1509.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hui G S, Tam L Q, Chang S P, Case S E, Hashiro C, Siddiqui W A, Shiba T, Kusumoto S, Kotani S. Synthetic low-toxicity muramyl dipeptide and monophosphoryl lipid A replace Freund complete adjuvant in inducing growth-inhibitory antibodies to the Plasmodium falciparum major merozoite surface protein, gp195. Infect Immun. 1991;59:1585–1591. doi: 10.1128/iai.59.5.1585-1591.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jongwutiwes S, Tanabe K, Kanbara H. Sequence conservation in the C-terminal part of the precursor to the major merozoite surface proteins (MSP1) of Plasmodium falciparum from field isolates. Mol Biochem Parasitol. 1993;59:95–100. doi: 10.1016/0166-6851(93)90010-u. [DOI] [PubMed] [Google Scholar]
- 21.Kaslow D C, Hui G, Kumar S. Expression and antigenicity of Plasmodium falciparum major merozoite surface protein (MSP1(19)) variants secreted from Saccharomyces cerevisiae. Mol Biochem Parasitol. 1994;63:283–289. doi: 10.1016/0166-6851(94)90064-7. [DOI] [PubMed] [Google Scholar]
- 22.Kumar S, Collins W, Egan A, Yadava A, Garraud O, Blackman M J, Guevara Patino J A, Diggs C, Kaslow D C. Immunogenicity and efficacy in Aotus monkeys of four recombinant Plasmodium falciparum vaccines in multiple adjuvant formulations based on the 19-kilodalton C terminus of merozoite surface protein 1. Infect Immun. 2000;68:2215–2223. doi: 10.1128/iai.68.4.2215-2223.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Yadava A, Keister D B, Tian J H, Ohl M, Perdue-Greenfield K A, Miller L H, Kaslow D C. Immunogenicity and in vivo efficacy of recombinant Plasmodium falciparum merozoite surface protein-1 in Aotus monkeys. Mol Med. 1995;1:325–332. [PMC free article] [PubMed] [Google Scholar]
- 24.Ling I T, Ogun S A, Holder A A. Immunization against malaria with a recombinant protein. Parasite Immunol. 1994;16:63–67. doi: 10.1111/j.1365-3024.1994.tb00324.x. [DOI] [PubMed] [Google Scholar]
- 25.McBride J S, Newbold C I, Anand R. Polymorphism of a high molecular weight schizont antigen of the human malaria parasite Plasmodium falciparum. J Exp Med. 1985;161:160–180. doi: 10.1084/jem.161.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller L H, Hoffman S L. Research toward vaccines against malaria. Nat Med. 1998;4:520–524. doi: 10.1038/nm0598supp-520. [DOI] [PubMed] [Google Scholar]
- 27.Miller L H, Roberts T, Shahabuddin M, McCutchan T F. Analysis of sequence diversity in the Plasmodium falciparum merozoite surface protein-1 (MSP-1) Mol Biochem Parasitol. 1993;59:1–14. doi: 10.1016/0166-6851(93)90002-f. [DOI] [PubMed] [Google Scholar]
- 28.Morgan W D, Birdsall B, Frenkiel T A, Gradwell M G, Burghaus P A, Syed S E, Uthaipibull C, Holder A A, Feeney J. Solution structure of an EGF module pair from the Plasmodium falciparum merozoite surface protein 1. J Mol Biol. 1999;289:113–122. doi: 10.1006/jmbi.1999.2753. [DOI] [PubMed] [Google Scholar]
- 29.Plackett R L, Burman J P. The design of optimum multifactor experiments. Biometrika. 1946;33:305. [Google Scholar]
- 30.Qari S H, Shi Y P, Goldman I F, Nahlen B L, Tibayrenc M, Lal A A. Predicted and observed alleles of Plasmodium falciparum merozoite surface protein-1 (MSP-1), a potential malaria vaccine antigen. Mol Biochem Parasitol. 1998;92:241–252. doi: 10.1016/s0166-6851(98)00010-3. [DOI] [PubMed] [Google Scholar]
- 31.Sakihama N, Kimura M, Hirayama K, Kanda T, Na-Bangchang K, Jongwutiwes S, Conway D, Tanabe K. Allelic recombination and linkage disequilibrium within Msp-1 of Plasmodium falciparum, the malignant human malaria parasite. Gene. 1999;230:47–54. doi: 10.1016/s0378-1119(99)00069-4. [DOI] [PubMed] [Google Scholar]
- 32.Siddiqui W A, Tam L Q, Kan S C, Kramer K J, Case S E, Palmer K L, Yamaga K M, Hui G S. Induction of protective immunity to monoclonal-antibody-defined Plasmodium falciparum antigens requires strong adjuvant in Aotus monkeys. Infect Immun. 1986;52:314–318. doi: 10.1128/iai.52.1.314-318.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32a.Stowers A W, Zhang Y, Shimp R L, Kaslow D C. Structural conformers produced during material vaccine production in yeast. Yeast. 2001;18:137–150. doi: 10.1002/1097-0061(20010130)18:2<137::AID-YEA657>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 33.Tanabe K, Mackay M, Goman M, Scaife J G. Allelic dimorphism in a surface antigen gene of the malaria parasite Plasmodium falciparum. J Mol Biol. 1987;195:273–287. doi: 10.1016/0022-2836(87)90649-8. [DOI] [PubMed] [Google Scholar]
- 34.Tian J H, Kumar S, Kaslow D C, Miller L H. Comparison of protection induced by immunization with recombinant proteins from different regions of merozoite surface protein 1 of Plasmodium yoelii. Infect Immun. 1997;65:3032–3036. doi: 10.1128/iai.65.8.3032-3036.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tolle R, Bujard H, Cooper J A. Plasmodium falciparum: variations within the C-terminal region of merozoite surface antigen-1. Exp Parasitol. 1995;81:47–54. doi: 10.1006/expr.1995.1091. [DOI] [PubMed] [Google Scholar]
- 36.Udhayakumar V, Anyona D, Kariuki S, Shi Y P, Bloland P B, Branch O H, Weiss W, Nahlen B L, Kaslow D C, Lal A A. Identification of T and B cell epitopes recognized by humans in the C-terminal 42-kDa domain of the Plasmodium falciparum merozoite surface protein (MSP)-1. J Immunol. 1995;154:6022–6030. [PubMed] [Google Scholar]