

Abstract
Human umbilical cord-derived mesenchymal stem cells (UC-MSCs) are considered an attractive cell source for tissue regeneration. However, environmental oxidative stress can trigger premature senescence in MSCs and thus compromises their regenerative potential. Extracellular matrix (ECM) derived from MSCs has been shown to facilitate cell proliferation and multilineage differentiation. This investigation evaluated the effect of cell-deposited decellularized ECM (DECM) on oxidative stress-induced premature senescence in UC-MSCs. Sublethal dosages of H2O2, ranging from 50 μM to 200 μM, were used to induce senescence in MSCs. We found that DECM protected UC-MSCs from oxidative stress-induced premature senescence. When treated with H2O2 at the same concentration, cell proliferation of DECM-cultured UC-MSCs was two-fold higher than those on standard tissue culture polystyrene (TCPS). After exposure to 100 μM H2O2, fewer senescence-associated β-galactosidase-positive cells were observed on DECM than those on TCPS (17.6 ± 4.0% vs. 60.4 ± 6.2%). UC-MSCs cultured on DECM also showed significantly lower levels of senescence-related regulators, such as p16INK4α and p21. Most importantly, DECM preserved the osteogenic differentiation potential of UC-MSCs with premature senescence. The underlying molecular mechanisms involved the silent information regulator type 1 (SIRT1)-dependent signaling pathway, confirmed by the fact that the SIRT1 inhibitor nicotinamide counteracted the DECM-mediated anti-senescent effect. Collagen type I, rather than fibronectin, partially contributed to the protective effect of decellularized matrix. These findings provide a new strategy of using stem cell-deposited matrix to overcome the challenge of cellular senescence and to facilitate the clinical application of MSCs in regenerative medicine.
Keywords: mesenchymal stem cells, extracellular matrix, decellularization, oxidative stress, premature senescence, osteogenesis, collagen type I, SIRT1
1. Introduction
Mesenchymal stem cells (MSCs) can be harvested from a variety of adult tissues, and are of great clinical interest in cell-based therapy for regenerative medicine and tissue engineering due to their self-renewal capacity, multilineage differentiation ability, and immunological compatibility. The primary autologous MSC sources include bone marrow stroma, adipose tissue, synovial membrane, and skin tissue, all from which MSCs can be easily isolated without ethical implications related to their clinical and research uses. Recently, MSCs from extraembryonic tissues, such as human umbilical cord-derived MSCs (UC-MSCs) extracted from the Wharton’s jelly of umbilical cords, are advantageous in clinical applications because they can be collected by noninvasive procedures and their use has not raised ethical debates (Mitchell et al., 2003). Compared to MSCs from other adult tissues such as bone marrow, UC-MSCs possess superior proliferation rates and a similar gene expression profile to that of embryonic stem cells; they can also replicate for longer before reaching senescence during in vitro expansion (Hsieh et al., 2010).
Like other somatic cells, MSCs have a limited lifespan in vitro before growth arrest and the onset of senescence. Senescent cells are characterized morphologically by a flattened and enlarged appearance; cell proliferation is permanently arrested although the cells remain metabolically active (Shay and Wright, 2000). Cellular senescence in MSCs can impair their multilineage differentiation potential which can obstruct research into their therapeutic and clinical applications (Li and Pei, 2012). There is evidence that the causes of cellular senescence involve telomere shortening, DNA damage, oxidative stress, and expression of senescence related genes. For instance, over-accumulation of reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), triggers oxidative stress and induces the onset of premature senescence (Ksiazek, 2009). In particular, a deleterious level of ROS in osteoarthritic articular cartilage not only results in oxidative DNA damage, but also compromises the regenerative potential of MSCs (Zhang et al., 2014).
A number of studies have been conducted to investigate the molecular mechanisms of cellular senescence and it is thought that the dramatic change of the stem cell microenvironment from in vivo to in vitro is an essential factor in the onset of cellular senescence (Kim et al., 2016; Li et al., 2014). Once removed from the specialized in vivo environment, MSCs gradually lose their self-renewal and multilineage differentiation potentials (Vidal et al., 2012). An important component of the stem cell microenvironment is the extracellular matrix (ECM), which sustains cellular functions with complex biochemical and physical signals. A previous study has compared the effect of the three ECM proteins fibrinogen, collagen type I, and fibronectin, and suggested that fibrinogen was the strongest substrate to promote osteogenesis in MSCs (Linsley et al., 2013). However, the natural ECM not only contains an assortment of proteins, such as collagens, fibronectin, laminins, and large molecular weight proteoglycans, it also provides physical cell support and enables MSCs to respond to mechanical signals through specific cell surface integrin-ECM interactions (Watt and Huck, 2013). Therefore, the cellular behavior differs on natural cell-deposited ECM when compared to a single ECM protein-coated substrate, but the exact mechanisms by which the natural matrix modulates MSC functions needs to be fully explored.
Recently, it has been reported that decellularized ECM derived from MSCs (DECM) facilitates large-scale expansion of MSCs in vitro and promotes their lineage-specific differentiation (He et al., 2014; He et al., 2013; Pei et al., 2011). However, the effect of DECM on premature senescence of MSCs and the underlying mechanisms by which the natural ECM modulates the properties of senescent MSCs are both unknown. Silent information regulator type 1 (SIRT1), a member of the nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase protein family, was proposed to be involved in delaying oxidative stress-mediated cellular senescence (Vaziri et al., 2001; Zhou et al., 2015). In this study, the response of human UC-MSCs cultured on DECM was compared with those cultured on standard tissue culture polystyrene (TCPS). Several sublethal concentrations of H2O2 were added to create oxidative stress and to induce premature senescence in MSCs. The senescence-associated phenotypes of MSCs on DECM were evaluated and compared with those on TCPS, collagen type I-coated (COL I), and fibronectin-coated (FN) substrates. The underlying mechanisms involving the p16INK4α and SIRT1 signaling pathways were also investigated.
2. Materials and methods
2.1. Reagents and antibodies
Human UC-MSCs were obtained from Cyagen Biosciences Inc. (Guangzhou, China). Unless noted otherwise, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO). All primary antibodies were purchased from Abcam (Cambridge, MA).
2.2. Preparation and evaluation of the cell-derived matrix
2.2.1. Preparation of DECM substrates
The procedure of preparing DECM deposited by UC-MSCs was described previously (He and Pei, 2013). TCPS plates were first treated with 0.2% gelatin for 1 h at 37°C, followed by 1% glutaraldehyde and 1 M ethanolamine for 30 min at room temperature. UC-MSCs were seeded onto the pretreated surface in a standard growth medium containing alpha minimum essential medium (α-MEM), 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin (all from Thermo Fisher Scientific, Waltham, MA). When the cells reached 90% confluence, 100 μM of L-ascorbic acid was added along with the culture medium and, 8 days later, the ECM-producing cells were removed by washing with phosphate buffered saline (PBS) containing 0.5% Triton X-100 and 20 mM NH4OH (pH = 7.4) for 5 min, and subsequently incubated with 100 U/mL DNase I for 1 h at 37°C. The DECM was washed with PBS three times to remove the residual DNase I and stored under sterile conditions at 4°C.
2.2.2. Preparation of ECM protein-coated substrates
To assess the effects of ECM proteins on premature senescence in UC-MSCs, TCPS plates were pre-coated with collagen type I and fibronectin, separately. Collagen type I was dissolved in 20 mM acetic acid and coated on the TCPS surface (10 μg/cm2) at 4°C overnight. Fibronectin (Thermo Fisher Scientific) was coated on the TCPS surface (1 μg/cm2) for 1 h at 37°C. Plates pre-coated with collagen and fibronectin were rinsed with PBS before cell culturing.
2.2.3. Scanning electron microscopy (SEM) analysis
Representative samples were fixed in 2.5% glutaraldehyde for 2 h and dehydrated in increasing concentrations of ethanol (50%, 75%, 80%, 95%, and 100%). The samples were dried using an automated critical point dryer (CPD300; Leica, Vienna, Austria) and subsequently sputter-coated with gold (SC7620; Quorum Technologies, Lewes, UK). The morphology of the MSC-deposited ECM was analyzed by an SEM machine (Quanta 250, FEI, Hillsboro, OR) using magnifications of 1000× and 4000× in order to study the morphology and the microstructure of the MSC-derived matrix.
2.2.4. Immunofluorescence staining
MSC-deposited matrix (with and without decellularization) was fixed in 4% paraformaldehyde for 15 min and blocked in 1% bovine serum albumin (BSA) for 1 h. The samples were incubated in the appropriately diluted primary antibody against Col I or FN at 4°C overnight, followed by the Alexa Fluor® 594 goat anti-mouse IgG (H+L) secondary antibody (Thermo Fisher Scientific) for 30 min. The cell nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Fluorescence images were obtained with an Olympus IX51 microscope (Olympus Corporation, Tokyo, Japan). To assess F-actin, UC-MSCs cultured on TCPS and ECM substrates were incubated in CytoPainter Phalloidin-iFluor 488 Reagent (Abcam) at room temperature for 1 h. After rinsing with PBS, the cell nuclei were counterstained with DAPI and fluorescent images were captured with an Olympus IX51 microscope.
2.3. Evaluation of H2O2-induced premature senescence in UC-MSCs
2.3.1. Cell culture
UC-MSCs were plated at 3,000 cells/cm2 on four substrates: TCPS, COL I, FN, and DECM in growth medium at 37°C in a humidified 5% CO2 and 21% O2 incubator. The medium was changed every 3 days. To observe the cell morphology, bright field images of UC-MSCs were collected using a Zeiss Axiovert 40 CFL phase contrast microscope (Carl Zeiss Ltd., Oberkochen, Germany).
2.3.2. Cell viability and proliferation analysis
Cell viability was assessed by staining with 5 μg/mL fluorescein diacetate (FDA) solution for 10 min at 37°C. After rinsing with PBS, fluorescent images were captured with an Olympus IX51 microscope. Cell proliferation was evaluated by a DNA assay, which represented a proxy for the number of cells, using the Quant-iT™ PicoGreen® dsDNA Assay Kit (Thermo Fisher Scientific). UC-MSCs were treated with 200 μL of papain lysis buffer (125 μg/mL in PBS), in sextuplicate wells (n = 6), and the cells were lysed for 4 h at 60°C. Equal quantities of lysates and reagent were added to a 96-well plate and the resulting samples were incubated in the dark for 5 min. Fluorescence of the samples was measured on a SynergyMx multi-mode microplate reader (BioTek, Winooski, VT) with an excitation wavelength of 485 and an emission wavelength of 520 nm alongside a standard curve.
2.3.3. Treatments with H2O2 and SIRT1 antagonist
To induce premature senescence, UC-MSCs at approximately 50% confluence were exposed to 50 μM, 100 μM, and 200 μM H2O2 (diluted in growth medium) for 2 h. The cells were washed twice with serum-free α-MEM to remove residual H2O2, re-cultured in fresh growth medium for an additional 96 h, and then subjected to subsequent tests for various durations as specified in the individual experiments. Untreated cells served as a control. For the antagonist studies, after the treatment with H2O2, 10 mM of nicotinamide (a SIRT1 inhibitor) was added into growth medium and the cells were cultured for an additional 96 h.
2.3.4. Senescence-associated β-galactosidase (SA-β-gal) staining
The staining for SA-β-gal, a typical biomarker of premature senescence, was performed using an SA-β-gal staining kit (Beyotime Institute of Biotechnology, Haimen, China) according to the manufacturer’s instructions. The cells were incubated overnight at 37°C without CO2 and the nuclei were counterstained with DAPI. To quantify the percentage of SA-β-gal-positive cells, digital images in 10 randomly chosen fields were captured by an Olympus IX51 microscope and a total of at least 200 cells from each sample were counted to calculate the percentage of senescent cells.
2.3.5. Cell cycle analysis
Cell cycle distribution was analyzed by a propidium iodide (PI) staining method. Adherent cells were dissociated, in triplicate wells (n = 3), by 0.25% trypsin-EDTA and the detached cells were fixed in 70% ethanol for 24 h at 4°C. After washing with PBS, cells were stained with 50 μg/mL PI and 50 μg/mL RNase A for 30 min at 37°C. Samples were analyzed using a Cytomics FC500 Flow Cytometer (Beckman-Coulter, Brea, CA) and at least 5,000 cells were collected per sample. Data were analyzed using the MultiCycle AV DNA analysis software (Phoenix Flow Systems, San Diego, CA, USA).
2.3.6. Apoptosis analysis
Apoptosis of UC-MSCs was detected using an Apoptosis Kit (Thermo Fisher Scientific). Cells were labeled, in triplicate (n = 3), with Alexa Fluor™ 488 dye conjugated annexin V and PI for 15 min at room temperature. Samples were measured using a Cytomics FC500 Flow Cytometer and analyzed using the WinMDI (Windows Multiple Document Interface for Flow Cytometry) 2.9 software.
2.3.7. Measurement of intracellular ROS
Intracellular accumulation of ROS in UC-MSCs was quantified by a 2′,7′-Dichlorofluorescein diacetate (DCF-DA) fluorescence method. Detached cells (2 × 105) were incubated, in quadruplicate (n = 4), in 10 μM DCF-DA for 10 min at 37°C. The fluorescence intensity was measured using a Cytomics FC500 Flow Cytometer and 10,000 events from each cell sample were analyzed using the WinMDI 2.9 software.
2.4. Osteogenic induction and evaluation of matrix mineralization
UC-MSCs cultured on TCPS and ECM were first induced to premature senescence by exposure to 100 μM H2O2 for 2 h. After washing, the cells were cultured in growth medium for an additional 96 h and then changed to osteogenic differentiation medium containing Dulbecco’s modified Eagle medium (Thermo Fisher Scientific), 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, 50 μg/mL L-ascorbic acid, 100 nM dexamethasone, and 10 mM β-glycerol phosphate for 21 days. The differentiation medium was changed every 3 days.
Mineralization of the matrix was determined by Alizarin Red S staining. Cells were fixed, in quadruplicate wells (n = 4), in 4% paraformaldehyde and incubated in 1% Alizarin Red S solution (pH=4.3) for 15 min. Images of calcium deposition were captured using an Olympus IX51 microscope. To quantify the calcified matrix, 200 μL/well of 1% hydrochloric acid was added and the absorbance was measured at 420 nm using a microplate spectrophotometer.
2.5. Total RNA extraction and real-time reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from quadruplicate samples (n = 4) using the TRIzol® reagent and 1 μg of total RNA was reverse-transcribed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). To quantify mRNA expression, an amount of cDNA equivalent to 50 ng of total RNA was amplified by real-time PCR using the iTap™ Universal SYBR® Green Supermix kit (Bio-Rad, Hercules, CA). Transcript levels of P16INK4A, SIRT1, and osteogenic marker genes, including COL1A1 (collagen type I α1), ALP (alkaline phosphatase), RUNX2 (runt-related transcription factor 2), and SPP1 (secreted phosphoprotein 1 or osteopontin) were evaluated. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) served as an internal standard. The primer sequences are listed in Table S1. Real-time PCR was performed on a CFX96™ Real-Time PCR System (Bio-Rad) following the manufacturer’s protocol. Relative transcript levels were calculated as χ = 2−ΔΔCt, in which ΔΔCt = ΔE - ΔC, ΔE = Ctexp -CtGAPDH, and ΔC = Ctct1 -CtGAPDH.
2.6. Western blot analysis
Cells were dissolved from triplicate samples (n = 3) in ice-cold cell lysis buffer (Beyotime) containing protease inhibitors; the protein concentration in cell extracts was quantified using a BCA protein assay kit (Beyotime). Equal amounts of protein from each extract were denatured and separated in a 10% polyacrylamide gel (Beyotime) and transferred by electrophoresis onto a nitrocellulose membrane (Thermo Fisher Scientific). The membrane was incubated with diluted primary antibodies against p16INK4α, SIRT1, p38, p-p38, p21, p53, Erk1/2, p-Erk1/2, or α-tubulin at 4°C overnight, followed by the secondary antibody of horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit for 1 h at room temperature. SuperSignal West Pico Substrate and CL-XPosure Film (Thermo Fisher Scientific) were used for exposure. The intensity of the bands was quantified using the ImageJ software (National Institutes of Health, Bethesda, MD).
2.7. Statistical analysis
All data were expressed as the mean ± standard error (S.E.). Statistical differences between two groups were determined by one-way analysis of variance (ANOVA) followed by the Student’s unpaired t-test using the SPSS 13.0 statistical software (SPSS Inc., Chicago, IL). Significance was indicated by a p-value of < 0.05 (*).
3. Results
3.1. Characterization of the MSC-derived matrix
From the immunofluorescence staining, we identified two ECM protein components (COL I and FN) within the structure of the native cell-deposited matrix. DAPI staining showed that the process of decellularization successfully removed the original ECM-producing cells and the cellular residues (Figure 1A). SEM was used to observe the morphology of the cell-deposited matrix at 1,000× and 4,000×, respectively. A net-like surface structure of the matrix was observed under the ECM-producing cells before decellularization. A fibrillar network of the decellularized matrix was confirmed to be composed of small bundles of nanofibrous fibrils (Figure 1B).
Figure 1.
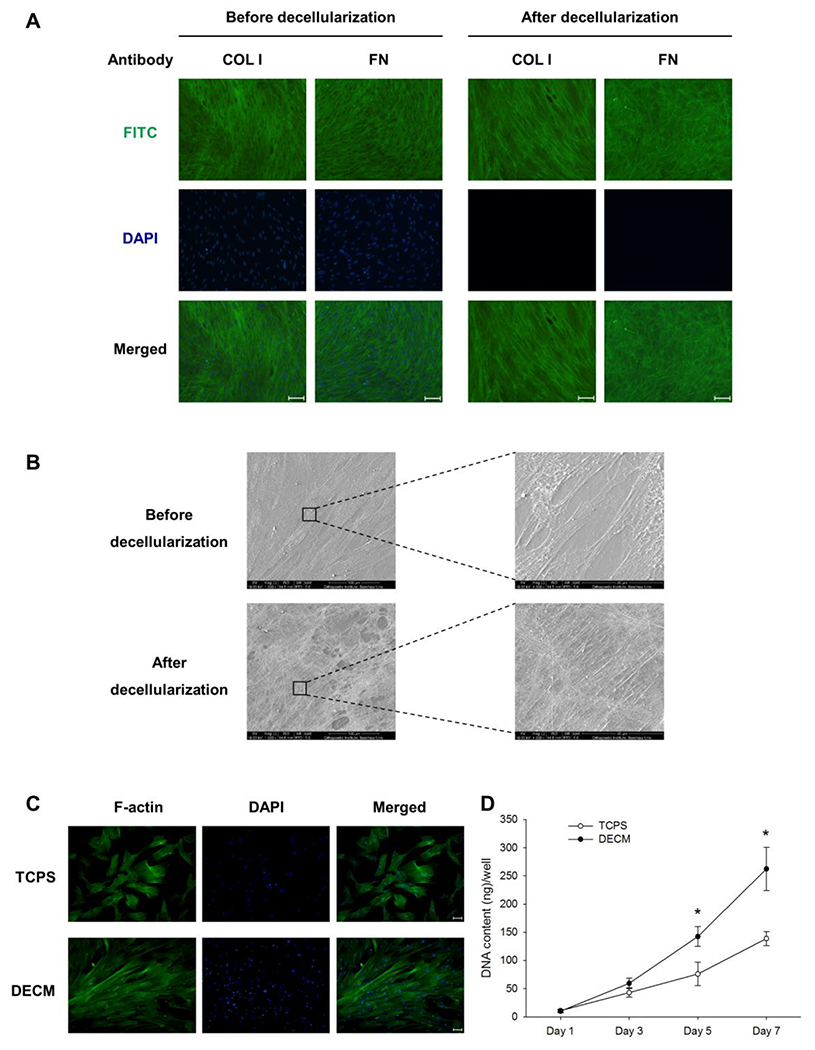
Characterization of the cell-deposited matrix and evaluation of MSC proliferation on DECM. (A) Immunofluorescence staining identified two major ECM protein components, collagen type I (COL I) and fibronectin (FN), in the cell-deposited matrix. Scale bar = 100 μm. (B) The surface topography and microstructure of cell-deposited ECM before and after decellularization was observed by SEM. Scale bar = 100 μm (left panels). Scale bar = 40 μm (right panels). (C) Immunofluorescence staining of F-actin showed that MSCs on DECM exhibited a spindle-like cell shape. Scale bar = 100 μm. (D) The DNA content assay was used to evaluate the proliferative potential of MSCs on TCPS and DECM substrates. Values are the mean ± S.E. of six independent experiments (n = 6) in the DNA content assay. Statistically significant differences are indicated by * (p < 0.05).
3.2. Evaluation of cell proliferation on DECM
To evaluate the effect of DECM on the proliferative potential, UC-MSCs were seeded on two substrates: uncoated TCPS and the DECM-coated surface. The cell morphology micrographs showed that the cell density of the DECM group was higher than that of the TCPS group (Figure S1). The immunofluorescence staining of F-actin showed that UC-MSCs on the DECM-coated surface exhibited a spindle-like cell shape and migrated along the direction of the matrix fibers; the cells cultured on TCPS substrate showed a flattened cell shape and random migration directions (Figure 1C). On days 3, 5, and 7, UC-MSCs on DECM yielded 59.4 ± 9.2 ng, 142.4 ± 17.6 ng, and 262.4 ± 38.5 ng of DNA content per well, respectively, compared to 42.9 ± 8.4 ng, 75.9 ± 20.6 ng, and 138.9 ± 12.3 ng per well from the cells on TCPS (Figure 1D).
3.3. The effect of DECM on H2O2-induced premature senescence in UC-MSCs
To evaluate the effect of DECM on oxidative stress-induced premature senescence, UC-MSCs were cultured on uncoated TCPS and DECM-coated substrates, and induced toward premature senescence by treating with H2O2. The FDA staining (Figure 2A) and the DNA assay data (Figure 2B) suggested that the cell proliferation capacity decreased in both the TCPS and DECM groups after exposure to H2O2 in a dose-dependent manner. In TCPS-cultured cells, proliferation decreased by 54.4% at 50 μM, 62.0% at 100 μM, and 69.4% at 200 μM, compared to untreated cells. In DECM-cultured cells, the proliferation decreased by 44.6% at 50 μM, 57.5% at 100 μM, and 84.3% at 200 μM, compared to untreated cells. However, the DNA content of DECM-cultured cells was at least two-fold higher than those in the TCPS group when treated with the same concentrations of H2O2. Next, we used SA-β-gal staining to label senescent UC-MSCs cultured on TCPS and DECM (Figure 2C). For the untreated cells, fewer SA-β-gal-positive cells were observed on DECM than those on TCPS (1.8 ± 0.3% vs. 10.2 ± 2.0%). After exposure to H2O2, the percentages of SA-β-gal-positive cells for the DECM groups remained at lower levels when compared to those in the TCPS groups (6.6 ± 2.7% vs. 33.6 ± 5.1% at 50 μM; 17.6 ± 4.0% vs. 60.4 ± 6.2% at 100 μM; 39.4 ± 10.0% vs. 84.7 ± 4.9% at 200 μM) (Figure 2D). The cell cycle distribution results showed that the percentage of cells in the G0/G1 phase decreased and the percentage of cells in the G2/M phase increased in both the TCPS and DECM groups after exposure to H2O2. However, culturing on DECM increased the entry of proliferating cells into S phase even after treatment with H2O2 (24.6 ± 2.4% vs. 8.7 ± 1.7%) (Figure 2E and F).
Figure 2.
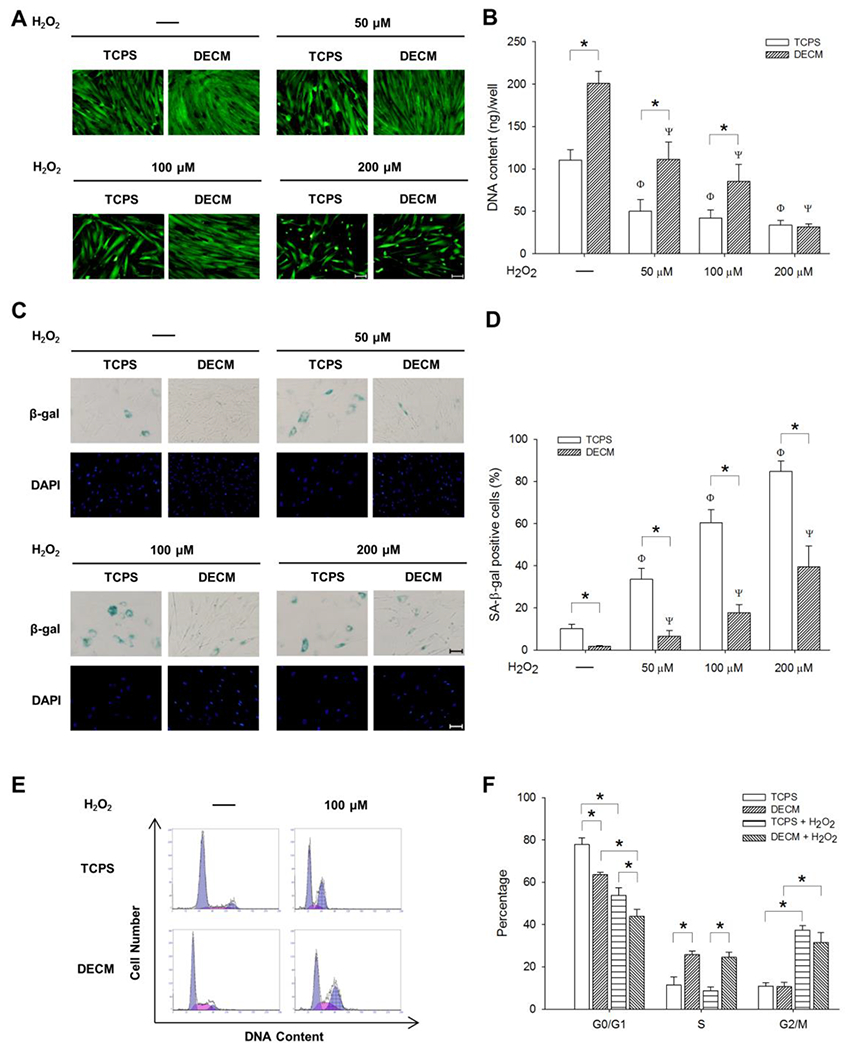
DECM improved the resistance of MSCs to H2O2-induced premature senescence. UC-MSCs were treated with different concentrations of H2O2 for 2 h and, after washing, the cells were cultured for an additional 96 h. (A) Cell morphology and density were observed in representative fluorescent images labeled by fluorescein diacetate (FDA). Scale bar = 100 μm. (B) Cell proliferation of UC-MSCs in sextuplicate wells was determined by the DNA content assay. Values are the mean ± S.E. of six independent experiments (n = 6). (C) Senescent MSCs were stained for SA-β-gal (blue) and the nuclei were counterstained with DAPI. Scale bar = 100 μm. (D) The percentage of SA-β-gal-positive cells was calculated on TCPS and DECM substrates. (E-F) The cell cycle distribution of UC-MSCs in triplicate wells was determined by flow cytometry analysis. Values are the mean ± S.E. of three independent experiments (n = 3). Statistically significant differences are indicated by * (p < 0.05); Φ (p < 0.05) versus untreated cells on TCPS; Ψ (p < 0.05) versus untreated cells on ECM.
Flow cytometry was used to measure the percentage of early (Annexin V-positive) and late (PI-positive) apoptotic cells (Figure S2A). The data showed that the treatments with H2O2 (100 μM or 200 μM) induced a slight increase of Annexin V-positive cells in the DECM group (Figure S2B) and that DECM-cultured cells showed a higher percentage of PI-positive cells than those on TCPS after treatment with H2O2 (Figure S2C). Furthermore, the levels of intracellular ROS in the cells cultured on DECM were only 40.1% in the control group and 24.4% upon treatment with 100 μM H2O2, respectively, compared with those on TCPS (Figure S3).
3.4. Molecular mechanisms of DECM suppressing MSC senescence
We next examined the mRNA levels of the cell cycle regulator p16INK4α. The expression in the TCPS group was significantly higher than that in the DECM group (2.8-fold in untreated cells, 4.9-fold at 50 μM, 2.6-fold at 100 μM, and 2.5-fold at 200 μM) (Figure 3A). The mRNA levels of SIRT1 decreased after exposing the cells to H2O2, but UC-MSCs cultured on DECM showed significantly higher mRNA levels than those cultured on TCPS (by 44.9% in untreated cells, 31.5% at 50 μM, 116.0% at 100 μM, and 38.5% at 200 μM) (Figure 3B).
Figure 3.
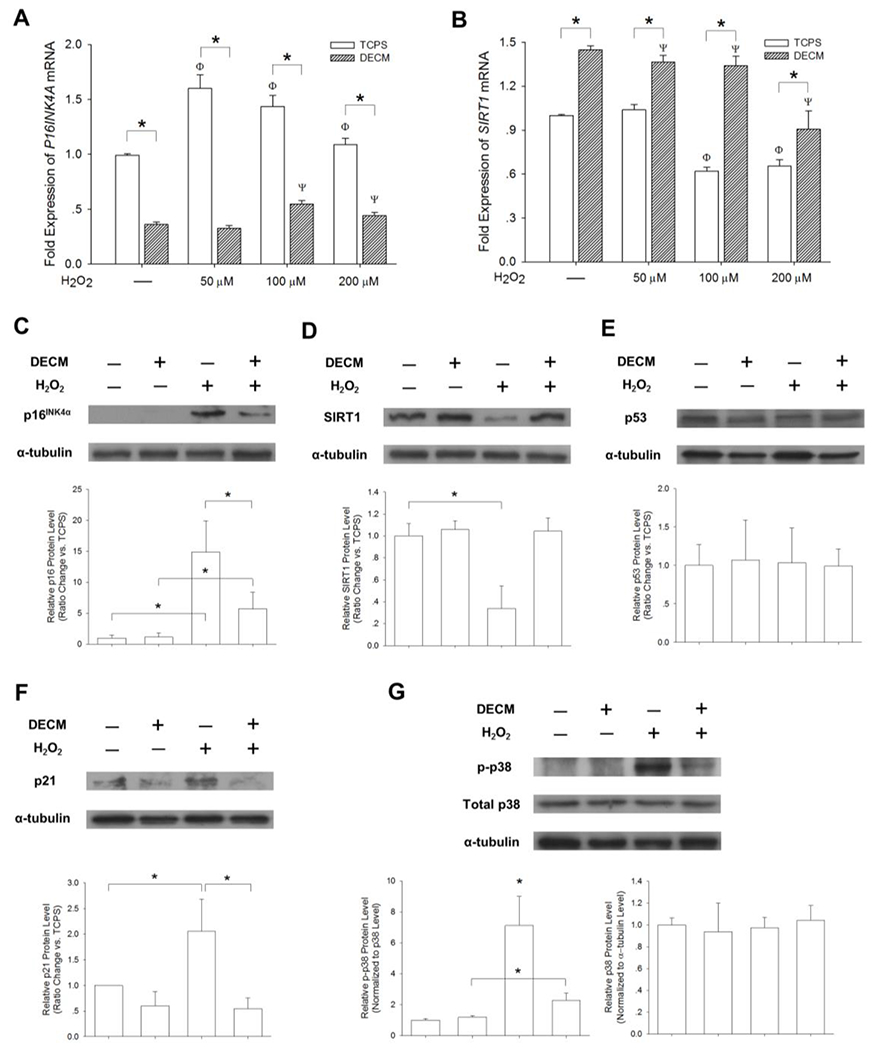
The involvement of the SIRT1/p38/p16INK4α pathway in DECM-mediated anti-senescence. UC-MSCs were pretreated with 100 μM H2O2 for 2 h to induce premature senescence. The mRNA levels of P16INK4A (A) and SIRT1 (B) were measured in quadruplicate samples by real-time RT-PCR. Values are the mean ± S.E. of four independent experiments (n = 4). Western blotting was used to measure the protein levels of p16INK4α (C), SIRT1 (D), p53 (E), and p21 (F) in triplicate samples. (G) The activation of the p38 signaling pathway was evaluated. The levels of p-p38 were normalized to total p38 protein. The levels of p38 were normalized to α-tubulin protein. Values are the mean ± S.E. of three independent experiments (n = 3). Statistically significant differences are indicated by * (p < 0.05).
To determine whether SIRT1-related signals were involved in DECM-mediated anti-senescence, Western blotting was used to measure the levels of p16INK4α, SIRT1, p53, p21, p-p38, p38, p-Erk1/2, and Erk1/2 in UC-MSCs after inducing senescence. In agreement with the real-time reverse transcription-polymerase chain reaction (RT-PCR) data, Western blot analysis confirmed that, after exposure to H2O2, UC-MSCs on TCPS showed the highest expression of p16INK4α which was 2.7-fold of that on DECM (Figure 3C). The levels of SIRT1 decreased in H2O2-treated cells, but DECM preserved the expression of SIRT1 (Figure 3D). The levels of p53 showed no significant difference (Figure 3E) but, as its transcriptional target, p21 was significantly upregulated in TCPS-cultured cells (Figure 3F). To determine the roles of p38 and Erk1/2 in H2O2-induced premature senescence, we measured the phosphorylated levels of p38 and Erk1/2. We found that treatment with H2O2 significantly enhanced phosphorylation of p38 by 6.1-fold compared with the untreated cells on TCPS, whereas DECM attenuated p-p38 by 67.8% compared with H2O2-treated cells on TCPS. However, we found that the total p38 protein expression did not vary in all the groups (Figure 3G). The differences of phosphorylated and total levels of Erk1/2 were insignificant. Before treating with H2O2, the levels of p-Erk1/2 were higher in DECM-cultured cells than in the TCPS-cultured cells (p = 0.28) and, after exposure to H2O2, the levels of p-Erk1/2 showed an opposite tendency (p = 0.21) (Figure S4).
3.5. The effect of DECM on osteogenic differentiation of senescent MSCs
The differentiation capacity of MSCs is crucial to their therapeutic application, thus we analyzed whether DECM could retain the osteogenic differentiation potential for senescent MSCs. UC-MSCs were first treated with 100 μM H2O2 and then induced toward an osteoblast-lineage differentiation. Alizarin Red S staining results revealed mineralized matrix deposition 21-days after induction, and no positive results were detected either on the TCPS or DECM-coated substrates when incubating in standard growth medium (Figure 4A). The level of matrix mineralization in the TCPS group decreased by 63.7% after induction of premature senescence by H2O2, but the DECM group yielded an eight-fold increase of calcified matrix in the senescent UC-MSCs (Figure 4B). Real-time RT-PCR data showed that DECM increased COL1A1 transcription by 5.4-fold and 12.8-fold, with and without H2O2 treatment, respectively, compared to the cells cultured on TCPS (Figure 4C). The mRNA levels of ALP (Figure 4D), RUNX2 (Figure 4E), and SPP1 (Figure 4F) showed a similar tendency.
Figure 4.
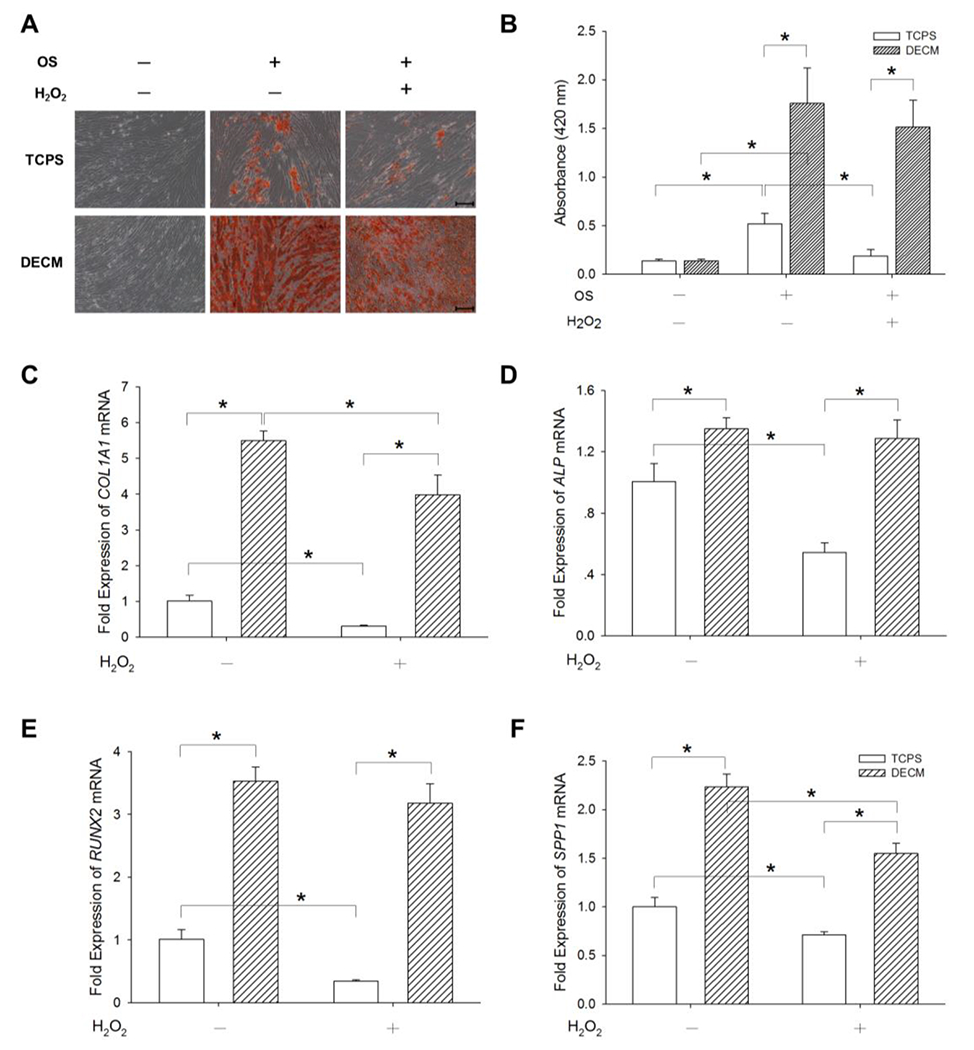
The effect of DECM on osteogenic (OS) differentiation of senescent MSCs. UC-MSCs were first pretreated with 100 μM H2O2 for 2 h to cause premature senescence and then induced toward osteogenic differentiation. (A) Calcium deposition during osteogenesis was assessed by Alizarin Red S staining. Scale bar = 200 μm. (B) The stained mineral layers in quadruplicate wells were quantified via a spectrophotometer. Values are the mean ± S.E. of four independent experiments (n = 4). The mRNA levels of osteoblast-specific marker genes, including COL1A1 (C), ALP (D), RUNX2 (E), and SPP1 (F) were measured in quadruplicate samples using real-time RT-PCR. Values are the mean ± S.E. of four independent experiments (n = 4). Statistically significant differences are indicated by * (p < 0.05).
3.6. Evaluation of different ECM proteins in modulating premature senescence
To evaluate the role of ECM protein components in modulating the anti-senescent effect of DECM, UC-MSCs were seeded on four substrates: uncoated TCPS, COL I, FN, or DECM. The cells were then subjected to 100 μM H2O2. The SA-β-gal staining results showed that, after exposure to H2O2, the percentage of positive cells in the DECM groups was significantly lower than that of the other three groups and that COL I significantly reduced the number of positive cells compared with those cultured on TCPS or FN (58.4 ± 4.4% on TCPS, 28.2 ± 3.9% on COL I, 54.6 ± 7.8% on FN, and 15.7 ± 4.3% on ECM) (Figure 5A and B). The DNA content data confirmed that cells cultured on COL I or DECM preserved their proliferation capacity (Figure 5C). The cell cycle distribution data (Figure 5D) showed that the percentage of cells in the S phase was significantly higher in the DECM group than that of the other groups (Figure 5E). Upon stimulation with H2O2, the substrates of COL I and DECM promoted entry of proliferating cells into the S phase compared with TCPS and FN-coated surfaces (11.8 ± 1.4% on TCPS, 15.8 ± 1.8% on COL I, 9.5 ± 2.1% on FN, and 25.8 ± 1.0% on ECM) (Figure 5F).
Figure 5.
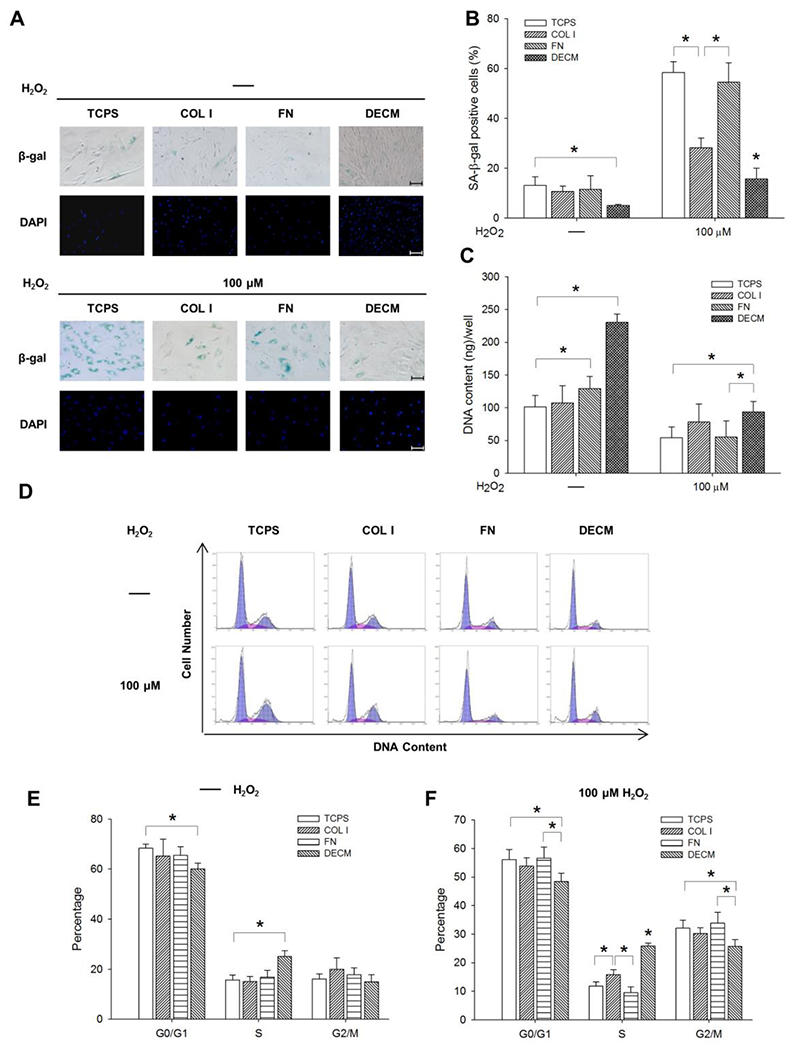
The effect of different ECM proteins on MSC premature senescence. UC-MSCs were seeded on TCPS, COL I, FN, and DECM and exposed to 100 μM H2O2 to induce premature senescence. (A) Representative SA-β-gal (blue) staining images indicated senescent cells. Scale bar = 100 μm. (B) The percentage of SA-β-gal-positive cells was suppressed by the substrates of COL I and DECM. (C) Cell proliferation in sextuplicate wells was determined by the DNA content assay. Values are the mean ± S.E. of six independent experiments (n = 6). (D) The cell cycle distribution was determined by flow cytometry analysis. (E) Analysis of the cell cycle distribution in UC-MSCs without H2O2 treatment. (F) Analysis of the cell cycle distribution in UC-MSCs treated with 100 μM H2O2 in triplicate wells. Values are the mean ± S.E. of three independent experiments (n = 3). Statistically significant differences are indicated by * (p < 0.05).
3.7. Reversal of the ECM-mediated anti-senescent effect by nicotinamide
To further determine the involvement of SIRT1 in modulating DECM-mediated anti-senescence, UC-MSCs were cultured on DECM and nicotinamide was used to specifically inhibit SIRT1 activity. When treating with nicotinamide alone, the percentage of SA-β-gal-positive cells increased to 12.3 ± 2.5%; after treating with H2O2, nicotinamide significantly counteracted DECM-mediated anti-senescent effects by raising the percentage of SA-β-gal-positive cells to 57.2 ± 5.5% (Figure 6A and B). The treatment with nicotinamide significantly increased p16INK4α in the H2O2-treated cells by 52.4% at the mRNA level (Figure 6C) and by 4.2-fold at the protein level (Figure 6D) compared with the untreated cells. The nicotinamide supplement also decreased SIRT1 in the H2O2-treated cells by 22.8% at the mRNA level (Figure 6E) and by 68.9% at the protein level (Figure 6F). The western blot assay showed that exposure to nicotinamide and H2O2 increased phosphorylation of p38 by 4.2-fold compared with the control cells, but we found that the levels of total p38 protein were not significantly different in all groups (Figure 6G).
Figure 6.
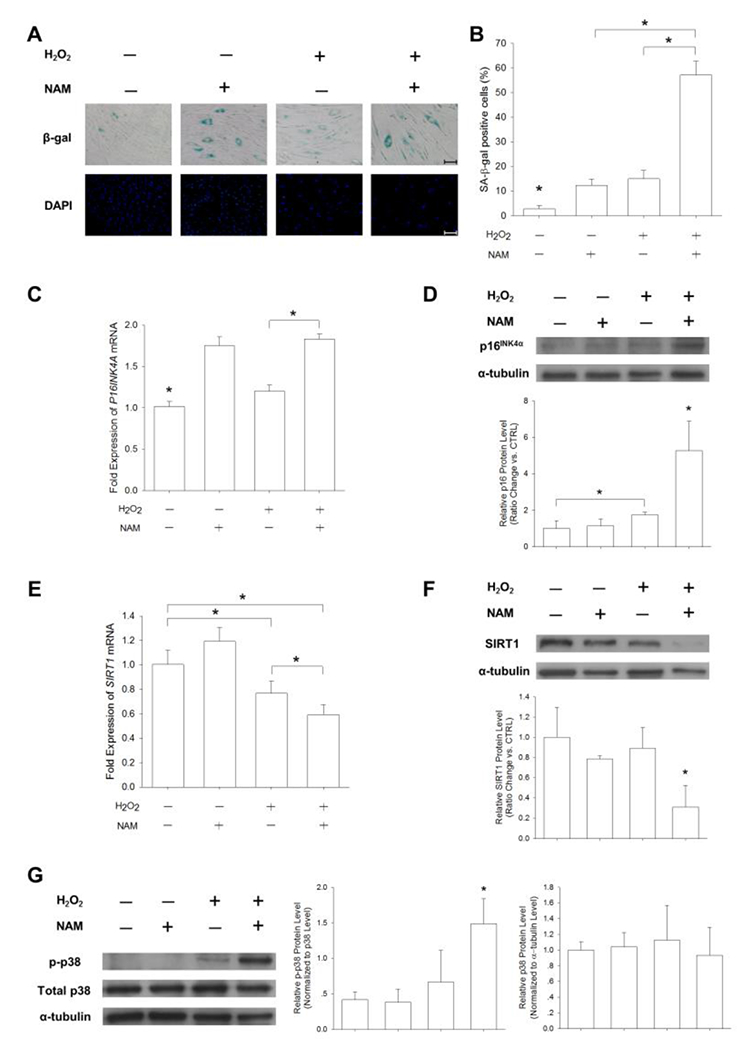
Nicotinamide (NAM) reversed the DECM-mediated anti-senescent effect. UC-MSCs were seeded onto the DECM-coated surface and treated with 100 μM H2O2 or 10 mM NAM (a SIRT1 inhibitor). Cells left untreated in growth medium served as a control. (A) Representative SA-β-gal (blue) staining images indicated senescent cells. Scale bar = 100 μm. (B) The percentage of SA-β-gal-positive cells increased in the presence of NAM. (C-D) The mRNA levels and the protein levels of p16INK4α were measured in DECM-cultured cells in the presence of NAM and H2O2. (E-F) The mRNA levels and the protein levels of SIRT1 were measured. (G) The phosphorylation and total expression of p38 were measured by the Western blot assay. The level of p-p38 was normalized to total p38 protein and the level of p38 was normalized to α-tubulin protein. Values are the mean ± S.E. of four independent experiments (n = 4) in real-time RT-PCR and of three independent experiments (n = 3) in the Western blot assay. Statistically significant differences are indicated by * (p < 0.05).
4. Discussion
Previous studies have demonstrated that the intracellular accumulation of ROS in MSCs decreased significantly when culturing on DECM (He et al., 2014; He and Pei, 2013); however, it was unknown whether DECM could resist oxidative stress-induced premature senescence in MSCs. In this study, we have shown that DECM, compared with the uncoated plastic surface, is sufficient in suppressing H2O2-induced premature senescence in UC-MSCs, as evidenced by improved cell proliferation, decreased SA β-gal activity, and higher levels of proliferating cells in the S phase. As a major component of MSC-derived ECM, collagen type I, but not fibronectin, partially contributed to the resistance of premature senescence. Our data also suggest that even MSCs with senescent phenotypes acquired a superior osteogenic differentiation potential on DECM. Additionally, we discovered that the SIRT1-dependent signaling pathway was actively involved in DECM-mediated protection against oxidative stress-induced premature senescence.
Increased levels of ROS, observed in damaged tissue environments, such as in an arthritic joint (Henrotin et al., 2003), are believed to cause DNA damage in progenitor cells or transplanted MSCs. Damaged DNA that has been improperly repaired is a major cause of induction of senescence-associated secretory phenotypes of MSCs. Thus, to maintain their regenerative potentials, MSCs have to cope with cellular senescence by resisting oxidative damage. Based on our previous studies, exposing MSCs to a high dosage of H2O2 (400 μM), a major component of ROS, resulted in apoptosis and eventually cell death (Zhou et al., 2015). In this study, we used intermediate concentrations of H2O2 ranging from 50 μM to 200 μM to create the oxidative environment and to induce premature senescence in UC-MSCs. As compared to those cells cultured on the uncoated plastic surface, cells cultured on DECM showed a delay of premature senescence, especially after exposure to 50 μM or 100 μM H2O2. However, when exposed to 200 μM H2O2, DECM failed to prevent premature senescence in MSCs, suggesting that DECM was able to protect MSCs from premature senescence when oxidative stress was at a relatively moderate level. However, abnormally high levels of oxidative stress beyond cell tolerance eventually resulted in irreversible cellular senescence.
Cellular senescence results in irreversible cell cycle arrest and the underlying mechanisms involve two major molecular signaling pathways: the p53/p21 pathway and the p38 mitogen-activated protein kinase/p16INK4α pathway. The activation of p53 and its transcriptional target p21 play an important role in responding to DNA damage as well as cellular senescence (Cheng et al., 2011). Another essential regulator is p16INK4α, which regulates the progression from the G1 phase to the S phase in the cell cycle by controlling cell cycle kinases. The protein p16INK4α, a cyclin-dependent kinase inhibitor that deactivates the cell cycle kinases CDK4 and CDK6, showed increased levels in H2O2-induced senescent cells but those levels decreased in the presence of DECM (Krishnamurthy et al., 2004). Our cell cycle distribution data showed decreased percentage levels of cells in the G0/G1 phase in contrast to increased levels in the G2 phase, indicating that oxidative stress possibly induced UC-MSCs into G2 cell cycle arrest, which was in agreement with a previous study (Li et al., 2009). Most importantly, the fact that the SIRT1 inhibitor nicotinamide nullifies DECM-mediated protection indicates that SIRT1 catalytic activity accounts for the protection of MSCs from premature senescence. MSCs expanded on DECM showed increased expression of SIRT1 at both the mRNA and protein levels, which may prevent oxidative stress-induced cellular damage by upregulating antioxidant enzymes via the Forkhead box O3 (FoxO3a) signaling pathway (Hasegawa et al., 2008).
In addition, the stress-induced protein kinases p38 and Erk1/2 have been reported to be activated in the presence of oxidative stress and inflammatory cytokines (Kefaloyianni et al., 2006). We observed p38 activation in senescent cells cultured on TCPS, but DECM attenuated the phosphorylated levels of p38 even after H2O2 treatment, in agreement with previous studies that a sustained activation of p38 contributed to H2O2-induced senescence in MSCs (Borodkina et al., 2014) and that the pharmacological inhibition of p38 by sb203580 enhanced MSC proliferation (Zhang et al., 2015). However, the phosphorylated levels of Erk1/2 between the TCPS group and the DECM group were not significantly different. The results differed from a previous study, in which 50 μM of H2O2 stimulated a sustained activation of Erk1/2 in DECM-expanded MSCs (Pei et al., 2013). This difference could be because the cells were treated with continuous exposure to H2O2 and phosphorylation of Erk1/2 was measured at the time point of 24 h; in this study, we treated MSCs with H2O2 for only 2 h and cultured the cells for an additional 96 h. The oxidative stress-induced activation of Erk1/2 may have increased for a short time, but returned to a baseline level after the removal of H2O2. Further work is necessary to elucidate the role of stress-induced Erk1/2 in the onset of premature senescence and in the DECM-mediated anti-senescent effect. Therefore, our results strongly suggested that DECM could protect MSCs from oxidative stress-induced premature senescence and decreased p16INK4α expression through the p38/SIRT1-mediated signaling pathway.
It has been demonstrated that the p53/p21 pathway is involved in initiating replicative senescence. Unlike premature senescence, replicative senescence in MSCs is accompanied by telomere shortening and activation of p53 and its downstream target p21. Yew et al. demonstrated that the increased expression of p21 in late-passage MSCs was responsible for their replicative senescence and knockdown of p21 by lentivirus-mediated shRNAs restored their proliferation capacity and expression of stemness markers (Yew et al., 2011). In this study, we found that the levels of p53 did not change and that the expression of p21 increased after induced premature senescence, which was attributed to cell cycle arrest rather than activation of apoptosis. However, we observed that the percentages of late apoptotic cells, which were labeled by PI, were significantly higher in the DECM group than those in the TCPS group, although the absolute values of PI-positive cells were maintained at low levels. We speculated that DECM-cultured UC-MSCs might be more sensitive to apoptosis triggered by moderate oxidative stress and this high apoptotic response of cells cultured on DECM may involve a p53-mediated mechanism. Tavana and colleagues demonstrated that abrogation of p53-dependent apoptosis resulted in an enhanced cellular senescence in mouse fibroblasts by the treatment with ultraviolet (Tavana et al., 2010). A recent study also confirmed that inhibition of apoptosis potentiated senescence in K562 leukemia cells, as evidenced by decreased caspase-3 cleavage and increased SA-β-gal-positive cell population (Drullion et al., 2012). In the absence of H2O2, we also found that the protein level of p21 was higher in TCPS-cultured cells than in DECM-cultured cells. Consequently, further experiments are required to investigate the role of the p53/p21 pathway in ECM-mediated anti-senescence or pro-apoptosis. In addition, DNA damage repair usually involves several proteins such as Ku 70 and Ku 80, both of which bind to telomeres and maintain telomere length. The ECM has been shown to rescue senescent human diploid fibroblasts by activating SIRT1 and subsequently deacetylating Ku70 (Choi et al., 2011). However, the evidence for telomerase activity in DECM-cultured cells is controversial. Lai et al. found that telomerase activity remained highly stable in DECM-expanded MSCs (Lai et al., 2010), but another report demonstrated that telomerase expression of DECM-restored cells was undetectable (Choi et al., 2011). Therefore, our future work will focus on the telomere-dependent mechanism that might contribute to DECM-mediated prevention of MSC senescence.
The multilineage differentiation potential of MSCs is essential for their clinical application and we found that H2O2-induced senescent UC-MSCs lost their osteogenic differentiation capacity and over-accumulation of intracellular H2O2 was responsible for the decline of osteogenic capacity. We further showed that DECM maintained osteogenic potential of MSCs at a stable high level. However, a previous study suggested that the propensity for adipogenic rather than osteogenic differentiation increased in MSCs when the cells were cultured on DECM, possibly due to MSC source and the high expression of peroxisome proliferator-activated receptor γ (PPARγ) that promoted the adipogenic lineage commitment (Li et al., 2014). We speculate that, in senescent MSCs, SIRT1 interacts with several transcriptional factors such as PPARγ via a deacetylation pathway and subsequently shifts the balance of osteogenesis and adipogenesis (Han et al., 2010).
The mechanisms by which DECM protects MSCs from H2O2-induced premature senescence are not yet fully understood, especially with the kind of ECM protein components that might account for its prevention. In this study, we identified and compared two major ECM proteins, collagen type I and fibronectin, in the process of MSC senescence. Surprisingly, MSCs cultured on the collagen substrate, rather than the fibronectin substrate, showed attenuated levels of senescence-associated secretory phenotypes, although this suppression effect was weaker than that of DECM. Although not studied here, one possible explanation could be that the attachment of MSCs to collagen type I induces integrin-collagen interactions, such as the activation of α2β1 integrins. Overexpression of microRNA 183, which increased in senescent human diploid fibroblasts, has been proven to result in a decrease of integrin β1 and to reduce attachment to collagen substrates (Li et al., 2010). Although collagen type I has the potential to attenuate premature senescence of MSCs, the results suggest that the signaling from collagen type I alone is insufficient for the complete prevention of senescence and that other signals from DECM might also be necessary. Soluble non-collagenous proteins extracted from the native matrix have been shown to produce greater effects on MSC attachment, proliferation, migration, and differentiation than insoluble collagenous proteins (Lin et al., 2012). Both fibrillin-2 and tenascin C have been identified in the native matrix derived from fetal synovium MSCs (Li et al., 2014) and the enhanced expression of these two ECM proteins during the wound healing process was observed (Brinckmann et al., 2010), suggesting that these proteins might be involved in ECM-mediated anti-senescence. Furthermore, the degree of hydrophobicity of biomaterials was also found to be associated with senescence-related gene expression (Lou et al., 2010) and the matrix stiffness was a potential regulator of stem cell differentiation (Ye et al., 2015). Our results revealed that, after decellularization, the cell-derived ECM consisted of multiple matrix proteins and retained the fibrous microstructure of nanoscale dimensions, suggesting that the mechanical properties of DECM might also contribute to the protection of MSCs from premature senescence. Therefore, the roles of protein components and matrix biophysical properties of DECM in suppressing premature senescence of MSCs will be investigated in our future studies.
5. Conclusion
Our study demonstrated that DECM from human UC-MSCs could enhance resistance to oxidative stress-induced premature senescence in MSCs. Senescence-associated phenotypes including SA-β-gal-positive staining, cell cycle arrest, intracellular ROS levels, and expression of senescence-related genes were significantly suppressed when cells were cultured on DECM. The underlying molecular mechanisms involved the SIRT1 signaling pathway, evidenced by the fact that the SIRT1 inhibitor nicotinamide counteracted the DECM-mediated anti-senescent effect. Collagen type I, rather than fibronectin, both of which were identified as major protein components of cell-deposited ECM, partially contributed to the protective effect of DECM against premature senescence. Future work should focus on illustrating the influence of bioactive ECM components and matrix biophysical properties on DECM-mediated anti-senescence. DECM derived from MSCs provides a new strategy to solve the challenge of cellular senescence and to facilitate the clinical application of MSCs in cell-based tissue regeneration.
Supplementary Material
Acknowledgments
The authors are grateful to Suzanne Danley (West Virginia University, USA) and Paula Sahyoun (University of Waterloo, Canada) for carefully reviewing and editing the manuscript. This work was supported by the National Natural Science Foundation of China (No.31570978, No.51203194, No.21574091, No.81320108018, No.51303120); the Natural Science Foundation of Jiangsu Province (No.BK20140323, No.BK20130335); the National Institutes of Health (NIH) (AR062763-01A1 and AR067747-01A1) and Established Investigator Grant from Musculoskeletal Transplant Foundation (MTF) to M.P.; and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Grant information
National Natural Science Foundation of China: 31570978, 51203194, 21574091, 51303120;
Natural Science Foundation of Jiangsu Province: BK20140323, BK20160056;
National Institutes of Health: AR062763-01A1;
Musculoskeletal Transplant Foundation;
Priority Academic Program Development of Jiangsu Higher Education Institutions.
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
References
- Borodkina A, Shatrova A, Abushik P et al. 2014; Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging 6: 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinckmann J, Hunzelmann N, Kahle B et al. 2010; Enhanced fibrillin-2 expression is a general feature of wound healing and sclerosis: potential alteration of cell attachment and storage of TGF-beta. Lab Invest 90: 739–752. [DOI] [PubMed] [Google Scholar]
- Cheng H, Qiu L, Ma J et al. 2011; Replicative senescence of human bone marrow and umbilical cord derived mesenchymal stem cells and their differentiation to adipocytes and osteoblasts. Mol Biol Rep 38: 5161–5168. [DOI] [PubMed] [Google Scholar]
- Choi HR, Cho KA, Kang HT et al. 2011; Restoration of senescent human diploid fibroblasts by modulation of the extracellular matrix. Aging Cell 10: 148–157. [DOI] [PubMed] [Google Scholar]
- Drullion C, Tregoat C, Lagarde V et al. 2012; Apoptosis and autophagy have opposite roles on imatinib-induced K562 leukemia cell senescence. Cell Death Dis 3:e373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Zhou R, Niu J et al. 2010; SIRT1 is regulated by a PPAR{γ}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res 38: 7458–7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa K, Wakino S, Yoshioka K et al. 2008; Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun 372: 51–56. [DOI] [PubMed] [Google Scholar]
- He F, Liu X, Xiong K et al. 2014; Extracellular matrix modulates the biological effects of melatonin in mesenchymal stem cells. J Endocrinol 223: 167–180. [DOI] [PubMed] [Google Scholar]
- He F, Pei M. 2013; Extracellular matrix enhances differentiation of adipose stem cells from infrapatellar fat pad toward chondrogenesis. J Tissue Eng Regen Med 7: 73–84. [DOI] [PubMed] [Google Scholar]
- He H, Liu X, Peng L et al. 2013; Promotion of hepatic differentiation of bone marrow mesenchymal stem cells on decellularized cell-deposited extracellular matrix. Biomed Res Int 2013: 406871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrotin YE, Bruckner P, Pujol JP. 2003; The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage 11: 747–755. [DOI] [PubMed] [Google Scholar]
- Hsieh JY, Fu YS, Chang SJ et al. 2010; Functional module analysis reveals differential osteogenic and stemness potentials in human mesenchymal stem cells from bone marrow and Wharton’s jelly of umbilical cord. Stem Cells Dev 19: 1895–1910. [DOI] [PubMed] [Google Scholar]
- Kefaloyianni E, Gaitanaki C, Beis I. 2006; ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal 18: 2238–2251. [DOI] [PubMed] [Google Scholar]
- Kim JH, Shin SH, Li TZ et al. 2016; Influence of in vitro biomimicked stem cell ‘niche’ for regulation of proliferation and differentiation of human bone marrow-derived mesenchymal stem cells to myocardial phenotypes: serum starvation without aid of chemical agents and prevention of spontaneous stem cell transformation enhanced by the matrix environment. J Tissue Eng Regen Med 10: E1–13. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey MR et al. 2004; Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114: 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek K. 2009; A comprehensive review on mesenchymal stem cell growth and senescence. Rejuvenation Res 12: 105–116. [DOI] [PubMed] [Google Scholar]
- Lai Y, Sun Y, Skinner CM et al. 2010; Reconstitution of marrow-derived extracellular matrix ex vivo: a robust culture system for expanding large-scale highly functional human mesenchymal stem cells. Stem Cells Dev 19: 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Luna C, Qiu J et al. 2010; Targeting of integrin beta1 and kinesin 2alpha by microRNA 183. J Biol Chem 285: 5461–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Hansen KC, Zhang Y et al. 2014; Rejuvenation of chondrogenic potential in a young stem cell microenvironment. Biomaterials 35: 642–653. [DOI] [PubMed] [Google Scholar]
- Li J, Pei M. 2012; Cell senescence: a challenge in cartilage engineering and regeneration. Tissue Eng Part B Rev 18: 270–287. [DOI] [PubMed] [Google Scholar]
- Li M, Zhao L, Liu J et al. 2009; Hydrogen peroxide induces G2 cell cycle arrest and inhibits cell proliferation in osteoblasts. Anat Rec 292: 1107–1113. [DOI] [PubMed] [Google Scholar]
- Lin H, Yang G, Tan J et al. 2012; Influence of decellularized matrix derived from human mesenchymal stem cells on their proliferation, migration and multi-lineage differentiation potential. Biomaterials 33: 4480–4489. [DOI] [PubMed] [Google Scholar]
- Linsley C, Wu B, Tawil B. 2013; The effect of fibrinogen, collagen type I, and fibronectin on mesenchymal stem cell growth and differentiation into osteoblasts. Tissue Eng Part A 19: 1416–1423. [DOI] [PubMed] [Google Scholar]
- Lou PJ, Chiu MY, Chou CC et al. 2010; The effect of poly (ethylene-co-vinyl alcohol) on senescence-associated alterations of human dermal fibroblasts. Biomaterials 31: 1568–1577. [DOI] [PubMed] [Google Scholar]
- Mitchell KE, Weiss ML, Mitchell BM et al. 2003; Matrix cells from Wharton’s jelly form neurons and glia. Stem Cells 21: 50–60. [DOI] [PubMed] [Google Scholar]
- Pei M, Li JT, Shoukry M et al. 2011; A review of decellularized stem cell matrix: a novel cell expansion system for cartilage tissue engineering. Eur Cell Mater 22: 333–343. [DOI] [PubMed] [Google Scholar]
- Pei M, Zhang Y, Li J et al. 2013; Antioxidation of decellularized stem cell matrix promotes human synovium-derived stem cell-based chondrogenesis. Stem Cells Dev 22: 889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, Wright WE. 2000; Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol 1: 72–76. [DOI] [PubMed] [Google Scholar]
- Tavana O, Benjamin CL, Puebla-Osorio N et al. 2010; Absence of p53-dependent apoptosis leads to UV radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle 9:3328–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E et al. 2001; hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107: 149–159. [DOI] [PubMed] [Google Scholar]
- Vidal MA, Walker NJ, Napoli E et al. 2012; Evaluation of senescence in mesenchymal stem cells isolated from equine bone marrow, adipose tissue, and umbilical cord tissue. Stem Cells Dev 21: 273–283. [DOI] [PubMed] [Google Scholar]
- Watt FM, Huck WT. 2013; Role of the extracellular matrix in regulating stem cell fate. Nat Rev Mol Cell Biol 14: 467–473. [DOI] [PubMed] [Google Scholar]
- Ye K, Wang X, Cao L et al. 2015; Matrix Stiffness and Nanoscale Spatial Organization of Cell-Adhesive Ligands Direct Stem Cell Fate. Nano Lett 15: 4720–4729. [DOI] [PubMed] [Google Scholar]
- Yew TL, Chiu FY, Tsai CC et al. 2011; Knockdown of p21(Cip1/Waf1) enhances proliferation, the expression of stemness markers, and osteogenic potential in human mesenchymal stem cells. Aging Cell 10: 349–361. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Pizzute T, Li J et al. 2015; sb203580 preconditioning recharges matrix-expanded human adult stem cells for chondrogenesis in an inflammatory environment - A feasible approach for autologous stem cell based osteoarthritic cartilage repair. Biomaterials 64: 88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pizzute T, Pei M. 2014; Anti-inflammatory strategies in cartilage repair. Tissue Eng Part B Rev 20: 655–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Chen X, Liu T et al. 2015; Melatonin reverses H2O2-induced premature senescence in mesenchymal stem cells via the SIRT1-dependent pathway. J Pineal Res 59: 190–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.