

Abstract
Genetic divergence and the frequency of hybridization are central for defining species delimitations, especially among cryptic species where morphological differences are merely absent. Rotifers are known for their high cryptic diversity and therefore are ideal model organisms to investigate such patterns. Here, we used the recently resolved Brachionus calyciflorus species complex to investigate whether previously observed between species differences in thermotolerance and gene expression are also reflected in their genomic footprint. We identified a Heat Shock Protein gene (HSP 40 kDa) which exhibits cross species pronounced sequence variation. This gene exhibits species-specific fixed sites, alleles, and sites putatively under positive selection. These sites are located in protein binding regions involved in chaperoning and may therefore reflect adaptive diversification. By comparing three genetic markers (ITS, COI, HSP 40 kDa), we revealed hybridization events between the cryptic species. The low frequency of introgressive haplotypes/alleles suggest a tight, but not fully impermeable boundary between the cryptic species.
Subject terms: Ecological genetics, Evolutionary genetics
Introduction
According to the biological species concept1, a species is defined as a group that is reproductively isolated from other species. Reproductive isolation can be maintained either by pre-zygotic isolation, post-zygotic isolation, or a combination of both2. Pre-zygotic isolation occurs before a zygote is formed and involves physiological or systemic barriers that prevent successful mating, such as differences in mating behaviour, habitat preferences or isolation through ecological or geographical barriers3. Post-zygotic isolation mechanisms, such as increased zygote mortality or hybrid sterility, occur after zygote formation3. Reproductive isolation mechanisms are however often imperfect, so that many closely related species such as cryptic species hybridize4–7, which counteracts species divergence or may even erode species boundaries8.
Cryptic species are species that require genetic markers to inform species delimitations9–11. Such genetically identified species not only call for a revised taxonomy12–14, but also pose a major challenge to evolutionary and ecological theories. Due to the lack of morphological and physiological differences, high similarity in ecological traits and adaptations is expected15. When these species occur in sympatry16–19, the principle of competitive exclusion20 is compromised, which further challenges the understanding of co-existence and the niche concept.
Rotifers, with their approximately 2000 described species, belong to a very diverse phylum21, in which an increasing number of nominal species have been declared cryptic species complexes in the last decades22. Among them, the monogonont freshwater Brachionus calyciflorus species complex has recently been subdivided into four species using genetics and morphometrics: Brachionus calyciflorus sensu stricto (s.s.), Brachionus dorcas, Brachionus fernandoi, and Brachionus elevatus5,23. Prior to its delimitation into four species, the Brachionus calyciflorus species complex had been studied with regard to molecular phylogeny23–25, co-existence26, life history characteristics27, phylogeography28,29, and reproductive isolation28–30. More recent studies, which have taken into account the new species classification, focus on ecological processes, e.g., niche differentiation31, life history traits32, adaptation and the underlying regulatory mechanisms33–35, as well as on the robustness of species boundaries through hybridisation experiments6,7. These studies are of great importance to understand not only how these species have diverged, but also which of their adaptations are critical for the co-existence or exclusion of certain species in a specific environment.
A previous lifetable experiment36 has demonstrated diverging temperature optima between Brachionus calyciflorus s.s. (heat-tolerant) and Brachionus fernandoi (heat-sensitive). Comparison of transcriptome data from the heat-tolerant B. calyciflorus s.s. and the heat-sensitive B. fernandoi revealed differential expression of Heat Shock Protein (HSP) genes relative to temperature35. HSP gene expression levels correlated with life history traits such that the upregulation of HSP gene expression consistently occurred when the population growth rate was low. The two species were found to exhibit optimal growth at a different temperature (B. calyciflorus s.s. at 26 °C and B. fernandoi at 20/23 °C) and increased their HSP expression outside their optimal temperature range, demonstrating a stress response of the organisms when optimal growth was not feasible35. The pronounced fitness differences at different thermal conditions indicate underlying mechanisms of phenotypes’ responses to environmental conditions that allow them to occur in sympatry, but in different seasons of the year31,35.
Temperature as an environmental variable is one of the most important drivers of evolution and diversification37. Temperature adaptation is particularly interesting in aquatic invertebrates which as ectotherms are stronger influenced by temperature changes than homeotherms38. Increased temperature accelerates ontogeny, shortens the time to maturation and the generation time35,39–41. The associated faster population growth can foster competitive superiority and accelerate the underlying divergent selection that enforces niche partitioning42–44. An organism’s adaptation to a particular environment or resource availability may occur at the protein level through the regulation of gene expression45–48. This gene expression regulative mechanism can be observed in situations of rapid environmental changes (i.e., length of growing seasons) in B. plicatilis s.s.49,50, or in stress response via HSP upregulation35,51, which is a rapid and often the first response mechanism to environmental perturbation. However, a differential regulatory response among genotypes/species to the same environmental cue (genotype x environment (G x E) variation) has to be encoded somewhere in the genome, typically upstream in gene regulatory elements (e.g., transcription factors, promoters52,53). In addition, a divergent adaptation to different environmental conditions may also yield adaptive differences in protein structure, i.e., altered amino acid sequences. Such changes stem from non-synonymous substitutions in the underlying protein coding genes (as the HSP genes), such that signatures of selection can occur in specific genes or distributed over the whole genome54,55. Evolution of gene expression and protein structure do not necessarily occur independently of each other56,57, and we often find their respective contribution correlating with the progress of time, i.e., altered gene expression typically evolves more rapidly than altered protein structure53. Furthermore, discovering signs of selection in gene sequences between different species allows us to understand the genetic underpinning of the differentiation58,59. When put into perspective with the divergence time, the time frame of the adaptation/speciation can be unravelled60.
In this study, we investigated sequence/protein evolution and divergence in the HSP 40 kDa gene, which was inferred by a candidate gene selection for temperature adaptation, both by its temperature-related differential expression35 and by a transcriptome-wide SNP comparison among two species of the B. calyciflorus species complex, i.e., the heat-tolerant species B. calyciflorus s.s. and the heat-sensitive B. fernandoi. To put HSP40kDa gene evolution into perspective, this gene was further analysed in three Brachionus species outside of the species complex. We hypothesise, (I) that the adaptation of B. calyciflorus s.s. and B. fernandoi to different environments is driven by divergent selection on the structure of particular proteins (here, HSP 40 kDa) and (II) that the selection for different temperatures can promote niche partitioning, and hence constitute a pre-zygotic isolation mechanism.
Results
Candidate gene selection
To identify candidate genes within the B. calyciflorus species complex we assembled 72,165 and 94,884 super-transcripts in B. calyciflorus s.s and B. fernandoi transcriptomes, respectively. The reciprocal best blast hit identified 9655 putative orthologs which were reduced to 7976 after predicting open reading frames, locally aligning, and translating into proteins. In the selection tests, out of 7976 orthologs, 649 were significant (< 0.05) in all four pairwise Likelihood Ratio Test (LRT) comparisons, hence inferred to be putatively under positive selection. Comparing the orthologous genes under positive selection with the gene expression data35, 155 genes were differentially expressed relative to temperature (control: 20 °C, mild heat: B. calyciflorus s.s. 26 °C, B. fernandoi 23 °C, high heat: B. calyciflorus s.s. 32 °C, B. fernandoi 26 °C) in at least one of the two species (125 in B. calyciflorus s.s., 22 in B. fernandoi, and 8 in both species). Twenty-seven of these genes were assigned to the GO term GO:0006950 (response to stress), of which one gene was annotated to “response to heat” (GO:00009408). This gene (i.e., HSP 40 kDa) was differentially expressed in B. calyciflorus s.s. in cultures grown after 4 h of heat exposure to different temperature (20 °C vs. 32 °C and 20 °C vs. 26 °C), while it did not show a temperature-specific expression in B. fernandoi (Fig. 1).
Figure 1.

Annotation and differential expression of genes inferred to be under positive selection. Bar plot shows the genes which were assigned to the term GO:0006950 (response to stress). Bars represent the number genes assigned to one of the child terms of GO:0006950. More than one GO term can be assigned to the same gene. The heatmap shows the number of genes differentially expressed in each GO term category in B. calyciflorus s.s. and B. fernandoi under different temperatures regimes35.
HSP 40 kDa sequence diversity
The sequence diversity of all five Brachionus species (B. calyciflorus s.s., B. fernandoi, B. rubens, B. angularis, and B. diversicornis) was assessed by comparing HSP 40 kDa and Cytochrome Oxidase Subunit I (COI) genes. We detected an overall higher nucleotide divergence in the mitochondrial COI compared to the HSP 40 kDa gene fragment (Fig. 2). For the HSP 40 kDa sequences, the lowest nucleotide divergence was detected between B. angularis and B. diversicornis (0.103), followed by B. calyciflorus s.s. and B. fernandoi (0.121). The highest nucleotide divergence was found between B. fernandoi and B. rubens (0.198). For the COI fragment, the highest nucleotide divergence was detected between B. calyciflorus s.s. and B. diversicornis (0.243), while the lowest was between B. calyciflorus s.s. and B. fernandoi (0.154). The cross-species comparison of non-synonymous (dN) vs. synonymous (dS) substitutions in the HSP 40 kDa gene yielded an overall dN/dS: 0.049, with the the highest dN/dS ratio between B. angularis and B. diversicornis (0.085), while the lowest one was found between B. calyciflorus s.s. and B. diversicornis, followed by B. calyciflorus s.s. and B. fernandoi (0.040 and 0.042, respectively) (Supplementary Fig. s4).
Figure 2.
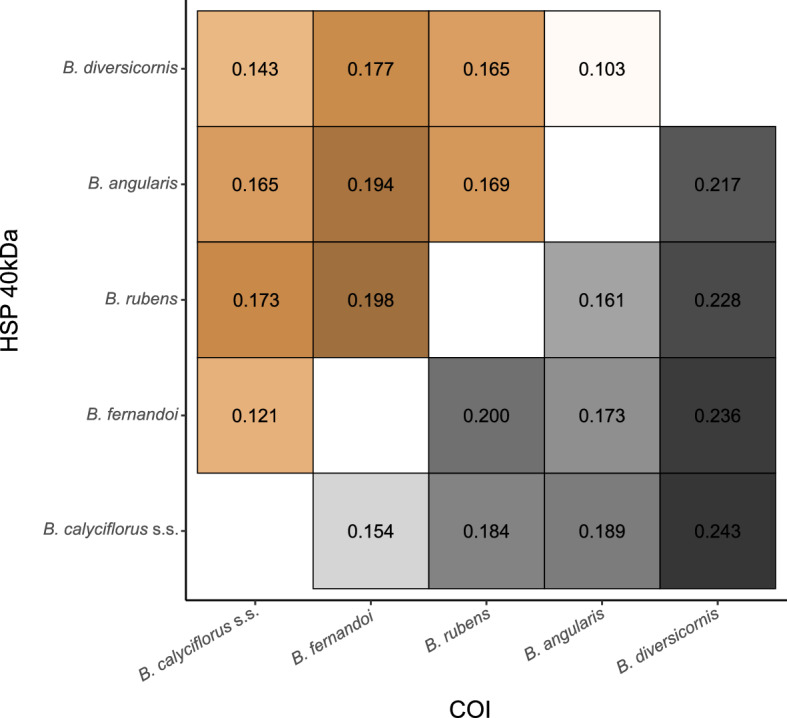
Nucleotide divergence of nuclear and mitochondrial genes. Nucleotide divergence of the nuclear HSP 40 kDa (orange) and the mitochondrial Cytochrome Oxidase Subunit I (COI) (grey) genes, based on comparisons of B. calyciflorus s.s., B. fernandoi, B. rubens, B. angularis, and B. diversicornis. The colour intensity indicates increasing nucleotide divergence between the compared sequences.
Structural variation in HSP 40 kDA between B. calyciflorus s.s. and B. fernandoi
We produced 88 genomic HSP 40kDA gene sequences for our two focus species (B. calyciflorus s.s., n = 50; B. fernandoi, n = 38) by polymerase chain reaction (PCR). The network approach based on all 88 phased sequences identified 32 unique HSP 40 kDa alleles (corresponding to 27 unique protein sequences) from which 13 alleles were exclusively found in B. fernandoi specimens and 19 alleles were solely found in B. calyciflorus s.s. specimens (Fig. 3). Only one allele, which phylogenetically clustered to the B. calyciflorus s.s. HSP 40 kDa allele type 18 (A18) was found in a B. fernandoi specimen (2484_4). The translated protein alignment (Supplementary Fig. s5) revealed 51 non-synonymous substitutions, among them the 14 found in the transcriptome data. The highest rate dN of non-synonymous substitutions per non-synonymous site among the two species was detected in position 256, followed by position 292 (Fig. 4).
Figure 3.

HSP 40 kDa allele identification. Circos plot of allele distribution among species B. calyciflorus s.s. and B. fernandoi and country of sample origin (Germany, Italy, Austria, USA). Labels A1-A32 stand for the identified alleles, red alleles are derived from B. calyciflorus s.s. and blue alleles from B. fernandoi. B. calyciflorus s.s. alleles found in more than one location are coloured orange.
Figure 4.

Number of non-synonymous substitutions per non-synonymous site among B. calyciflorus s.s. and B. fernandoi HSP 40 kDa for each amino acid alignment position of the 32 different HSP 40 kDa alleles.
Congruence among ITS1, HSP 40 kDa, and COI species affinity
The comparison of the nuclear Internal Transcribed Spacer 1 (ITS1), HSP 40 kDa, and the mitochondrial COI sequences yielded congruent species assignments for most analysed specimens. However, we identified four descendants of hybridization events between B. calyciflorus s.s. and B. fernandoi (all originating from the same pond; 2484), all of which carry the identical maternally inherited B. calyciflorus s.s. COI haplotype, but were assigned to B. fernandoi based on ITS1 sequences (Fig. 5, Supplementary Figs. s6, s7). Three of these introgressed specimens carried B. fernandoi—specific HSP 40 kDa alleles (2484_1, 2484_9, 2484_10), while one individual (2484_4) carried a B. calyciflorus s.s.—specific HSP 40 kDa allele, such that only its ITS1 sequence points towards a B. fernandoi affinity. Of the three introgressed specimens carrying B. fernandoi—specific HSP 40 kDa alleles, two were heterozygous (2484_1: A11, A1; 2484_10: A12, A2), while one was homozygous for A1 allele (2484_9). The introgressed specimen which carried a B. calyciflorus s.s. specific allele was homozygous for A18.
Figure 5.

Relationship between HSP 40 kDa and COI. Tanglegram based on the nuclear HSP 40 kDa and the mitochondrial COI of species (B. calyciflorus s.s., B. fernandoi, B. rubens, B. angularis, B. diversicornis) (n = 41). Black lines indicate connections of specimens outside of the Brachionus calyciflorus species complex, blue lines indicate connections between specimens of B. fernandoi and red lines connect specimens of B. calyciflorus s.s. Yellow coloured connections indicate specimens originating from an introgression/hybridization event between B. calyciflorus s.s. and B. fernandoi. They all occurred at the same location and all carried a B. fernandoi ITS sequence.
Inferences on selection and divergence
The conducted four LRT comparisons (M0 vs. M3, M1a vs. M2a, M7 vs. M8, M8a vs. M8) of the site substitution model in PAML significantly inferred positive selection in the HSP 40 kDa gene among the five different Brachionus species (Table 1). The Bayes Empirical Bayes (BEB) analysis detected six sites under selection (Table 1). Visual inspection of these positively selected sites in the protein alignment (Supplementary Fig. s5) showed a species-specific protein variant pattern at one of these sites (292), while variants were shared at the other sites, but differed in their relative frequencies among the species. The comparison of the positively selected sites with the functional domain description of the HSP 40kDa61 revealed that four out of the six sites are located in functional domains (J-domain and C-terminal domain; Supplementary Fig. s5). The branch site model, which was conducted to identify selection and sites under selection in specific species was able to identify selection on the CD (B. calyciflorus s.s. & B. fernandoi) and D branch (B. fernandoi), but did not detect any species-specific sites under selection (BEBs) (Table 2). The selection scenario investigation by comparing only B. calyciflorus s.s. and B. fernandoi using a McDonald-Kreitman Test (MKT), calculated a significant Neutrality Index of 12.06, p-value: ≤ 0.001 (i.e., indicative for balancing selection or slightly deleterious mutations), while Tajima’s D (1.538) was not significant (p-value: ≥ 0.10).
Table 1.
Likelihood ratio tests of seven different site models performed on 296 amino acids (888 bp) of the HSP 40 kDa, including 29 of the 32 unique alleles (A11, A12 and A18 were excluded from the analysis, as they only occurred in specimens affected by hybridization/introgression).
| Site model | LRT | 2∆l | df | p-value | l | ω | BEBs |
|---|---|---|---|---|---|---|---|
| M0 (one-ratio) | M0 vs. M3 | 262.398852 | 4 | << 0.001 | − 3598.034542 | 0.0718 | – |
| M3 (discrete) | − 3466.835116 | 0.1439 | |||||
| M1a (nearly Neutral) | M1a vs. M2a | 11.558380 | 2 | 0.003 | − 3480.993265 | 0.1328 | – |
| M2a (positive selection) | − 3475.214075 | 0.1815 | |||||
| M7 (β) | M7 vs. M8 | 21.480488 | 2 | << 0.001 | − 3477.904393 | 0.1108 | – |
| M8 (β & ω) | − 3467.164149 | 0.1450 | |||||
| M8a (β & ω = 1) | M8a vs. M8 | 31.89007 | 1 | << 0.001 | − 3483.109184 | 0.0769 | 10*; 38*; 249**; 256*; 286*; 292** |
Underlying phylogeny is based on the ITS1 (532 bp) of B. calyciflorus s.s., B. fernandoi, B. angularis, B. diversicornis and B. rubens. For each likelihood ratio test (LRT), we provide log likelihood (l) values of the compared tests (2∆l), degrees of freedom (df), p-values, estimated average ratio of non-synonymous vs. synonymous substitutions (ω), and the sites inferred to be under positive selection by Bayes Empirical Bayes (BEB) analysis, labeled with an * for a posterior probability of > 95% and ** for > 99%.
Table 2.
Likelihood ratio test of branch site model performed on 296 amino acids (888 bp) of the HSP 40 kDa including the 29 of the 32 unique alleles (A11, A12 and A18 hybrid alleles are excluded from the analysis) of B. calyciflorus s.s. and B. fernandoi, B. angularis, B. diversicornis and B. rubens.
| Branch site model | Branch | 2∆l | df | p-value | l | BEBs |
|---|---|---|---|---|---|---|
| Selection | CD | 7.398576 | 1 | 0.0065 | − 3477.294043 | – |
| Neutral | − 3480.993331 | |||||
| Selection | C | 0 | 1 | 1.0000 | − 3480.993266 | – |
| Neutral | − 3480.993266 | |||||
| Selection | D | 3.859588 | 1 | 0.0495 | − 3477.544980 | – |
| Neutral | − 3479.474774 |
Phylogeny is based on 532 bp of the ITS1. For each branch selection test, we provide log likelihood (l) value of the compared tests (2∆l), degrees of freedom (df), and the p-value. No sites were inferred to be under positive selection by BEB in this test framework.
The comparison of the fixed species-specific sites revealed 11 fixed sites between B. calyciflorus s.s. and B. fernandoi (Table 3). Out of these 11 fixed sites, 9 amino acid substitutions found in B. fernandoi are considered benign in our Polyphen2 analysis, while two (site 63 and 66) may change protein function (inferred "possibly damaging", Table 3). Additionally, among the six positively selected sites, two amino acid substitutions may influence protein function (inferred "possibly damaging", Table 3). The inference of character polarity (ancestral vs. derived) showed a prevalence of B. calyciflorus s.s. to retain the ancestral variant (10/17). For position 292, the status ancestral or derived could not be identified with certainty (Table 3).
Table 3.
Prediction of impact of non-synonymous (fixed or under selection cf. Table 1) amino acid substitutions between B. calyciflorus s.s. and B. fernandoi in comparison to B. rubens, B. angularis and B. diversicornis.
| Site (aa) | Code | Bc–Bf–Br–Ba–Bd | Ancestral | Score | Sensitivity | Specificity | Prediction |
|---|---|---|---|---|---|---|---|
| 10* | H10V/I | H–V/I–H–R–-R | H | 0.002 | 0.99 | 0.30 | Benign |
| H10V/I | H–V/I–H–R–R | H | 0.004 | 0.97 | 0.59 | Benign | |
| 27 | E27G | E–G–E–E–E | E | 0.000 | 1.00 | 0.00 | Benign |
| 30 | F30- | F–F–F–F | F | – | – | – | – |
| 31 | S31- | S–S–S–S | S | – | – | – | – |
| 38* | E/K38E/K | E/K–E/K–E–E–E | E | 0.231 | 0.91 | 0.88 | Benign |
| 63 | L63M | L–M–L–M–L | L | 0.953 | 0.97 | 0.93 | Possibly damaging |
| 66 | T66S/F | T–F–T–F–T | T | 0.143 | 0.92 | 0.86 | Benign |
| T66S/F | T–F–T–F–T | T | 0.997 | 0.41 | 0.98 | Possibly damaging | |
| 127 | T127A | T–A–T–T–T | T | 0.019 | 0.95 | 0.80 | Benign |
| 175 | F175V | F–V–V–F–V | V | 0.001 | 0.99 | 0.15 | Benign |
| 177 | Y177N | Y–N–Y–Y–Y | Y | 0.000 | 1.00 | 0.00 | Benign |
| 226 | R226K | R–K–R–K–R | R | 0.000 | 1.00 | 0.00 | Benign |
| 242 | H242L | H–L–I–H–H | H | 0.000 | 1.00 | 0.00 | Benign |
| 249* | P/S249P | P/S–P–P–Q–Q | E | 0.116 | 0.93 | 0.86 | Benign |
| 256* | L/F/N256L | L/F/N–L–L–L–L | L | 0.024 | 0.95 | 0.81 | Benign |
| L/F/N256L | L/F/N–L–L–L–L | L | 0.094 | 0.93 | 0.85 | Benign | |
| 285 | D285E | D–E–D–E–D | E | 0.000 | 1.00 | 0.00 | Benign |
| 286* | I/K286I | I/K–I–I–I–I | I | 0.914 | 0.81 | 0.94 | Possibly damaging |
| 292* | T/I292R | T/I–R–A–S–V | ? | 0.001 | 0.99 | 0.15 | Possibly damaging |
| T/I292R | T/I–R–A–S–V | ? | 0.000 | 1.00 | 0.00 | Benign |
Predictions were made using Polyphen2. The code provides details on the amino acid substitution and the position. Bc–Bf–Br–Ba–Bd provides the information of the respective amino acid of B. calyciflorus s.s. (Bc), B. fernandoi (Bf), B. rubens (Br), B. angularis (Ba) and B. diversicornis (Bd). Ancestral amino acid was inferred using the most parsimonious (i.e., fewest substitutions) scenario, taking the species' phylogeny into account. In case of multiple amino acids (aa) per species, compared aa are bold in the row Code. Asteriks indicate sites under selection inferred by codeML. Note that Polyphen2 does not consider the possibility of a positive effect of an aa substitution, such that it ranks any predicted change in protein function as "damaging".
We inferred a divergence time of 25–29 Million years (Myr) between B. calyciflorus s.s. and B. fernandoi, based on mitochondrial cds, while the split between the freshwater and the marine species of Brachionus was estimated to be in a range of 41–47 Myr (Supplementary Fig. s8).
Discussion
Candidate gene selection pipeline
The utilization of available transcriptomes and different expression patterns of B. calyciflorus s.s. and B. fernandoi allowed us to identify a candidate gene which likely plays a role in the differences in temperature adaptation among the two Brachionus species. Indeed, HSPs have been shown to be involved in the response to various environmental stressors such as thermal stress, toxins, oxidative conditions62–64 and aging61. The importance of HSPs in the process of thermotolerance for invertebrates, particularly HSP 40 kDa, HSP 60 kDa and HSP 70 kDa, was previously shown51 in the monogonont rotifer B. manjavacas.
Genetic variation in the HSP 40 kDa gene among Brachionus species
The analysis of the successfully amplified HSP 40 kDa gene of the 44 specimens of the B. calyciflorus cryptic species complex (B. calyciflorus s.s. and B. fernandoi) from 14 different locations (Europe, North America) revealed 32 unique alleles, indicating divergent evolution in the HSP 40 kDa. All but one allele, which originated from descendants of a hybrid, were species-specific. The high number of alleles and expressed structural differences (27 unique protein sequences) within and between the two species might be related to their cyclical parthenogenesis65. The dominant asexual mode of reproduction via mitotic eggs from amictic females (i.e., clones) and a less frequent switch to sexual reproduction together with a short generation time66 strongly reduces genomic recombination, making purifying selection to eliminate slightly deleterious mutations less effective for those organisms, a phenomenon known as Muller’s ratchet67–69.
The comparison between all five analysed species revealed that the species from the B. calyciflorus species complex are more similar (based on nucleotide divergence of the HSP 40 kDa and COI species-wise consensus sequences) to one another than to representatives from outside that species complex (B. rubens, B. angularis and B. diversicornis), with an overall higher nucleotide diversity in the COI. The higher diversity in the mitochondrial COI compared to nuclear HSP 40 kDa gene was expected as mitochondrial DNA in general exhibits a higher mutation rate than nuclear genes70, a phenomenon observed as well in rotifers71. While HSP 40 kDa gene divergence among Brachionus species generally resembled their phylogenetic affinity, the dN/dS ratio is the smallest, when comparing the not directly related species B. diversicornis and B. calyciflorus s.s. This may have arisen from similar environmental parameters that lead to similar selection pressures72. Indeed, a recent study found the occurrence of B. diversicornis to be correlated with warmer water temperatures73, which could indicate a higher thermotolerance—as observed in B. calyciflorus s.s.35,36—also in this species.
Selection scenario of the HSP 40 kDa
Within the HSP 40KDa gene, six amino acid sites were inferred to be under positive selection. A closer inspection of those sites revealed that four of them are in functional domains of the gene. One site is located in the J-domain, a region known to be important in the chaperone activity of HSP 40 kDa and the HSP 70 kDa by interacting with the ATP domain of the HSP 70kDa74, which accelerates ATP hydrolysis75,76 and thus, regulates the ATPase activity77. Three other sites are located in the C-terminal domain of the HSP 40 kDa. Although the exact function of these sites is still undetermined, it is known that the C-terminal domain of yeast Saccharomyces cerevisiae contains a peptide binding site78,79 and is therefore enabled to interact with other proteins. Changes in amino acid composition at these sites can affect folding patterns and thus binding with other proteins (e.g., HSP 70 kDa) through suboptimal structures or polarities80,81. These suboptimal structures can affect the strength of HSP 70 kDa ATPase regulation and potentially alter an organism's downstream stress response, which is underlined by the predicted impact of amino acid substitutions on protein function at two of the six sites. However, among the inferred sites under positive selection, only one (292, cf. Supplementary Fig. s4) exhibited species-specific amino acids. One explanation could be that the large geographic range of our study correlated with different local selection pressures, such that most substitutions were not fixed among the entire distribution range of a species. Another explanation could be a low genomic recombination rate (Muller’s ratchet), whereby clonal organisms easily accumulate slightly deleterious mutations that can generate noise in the selection analysis, hindering the identification of species-specific sites under selection82. The latter is in line with the findings of the McDonald-Kreitman Test (MKT) where a high Neutrality Index of 12.05 was found between the unique alleles of B. calyciflorus s.s. and B. fernandoi. This indicates an overabundance of replacement-site polymorphisms, which can arise from slightly deleterious polymorphic variants not eliminated by purifying selection83, a phenomenon known from clonal organisms84.
Because of the potentially compromised power of selection analyses in clonal organisms82, we inspected all further amino acid substitutions as well. Indeed, among them eleven fixed positions between our species were identified, two of which (positions 63 and 66; cf. Table 3) with a potential impact on protein function. These sites are not located in known functional domains of the HSP 40 kDa, yet they may have an impact on the structure of the protein. The results of the branch site model test indicated positive (directional) selection in B. fernandoi, but not in B. calyciflorus s.s. This is corroborated by character polarity, i.e., B. fernandoi predominantly exhibiting derived substitutions, while B. calyciflorus s.s. mostly retaining the ancestral amino acids. This allows us to draw the conclusion that B. fernandoi diverged from B. calyciflorus s.s. A divergent selection scenario for both species is compatible with the positive Tajima’s D statistics (albeit not significant).
The substantial nucleotide divergence among the two formerly cryptic species B. fernandoi and B. calyciflorus s.s. suggest these lineages to be evolutionary more ancient than one might expect for morphologically hardly distinguishable sister species. We had inferred their divergence time as ~ 25–29 Myr. The same analysis dated the split between the Brachionus freshwater clade comprising B. angularis, B. fernandoi and B. calyciflorus s.s. and the marine clade comprising B. plicatilis, B. manjavacas, B. rotundiformis and B. koreanus to about 41–47 Myr. This latter estimate is considerably larger than an earlier estimate of divergence time between the freshwater species B. calyciflorus (species undefined) and the marine B. plicatilis, using COI and 18S as markers (~ 25 Myr85). This discrepancy could be due to the increased number of Brachionus species used in our analysis and the different genes and substitution rates used in the present study, relative to Tang et al. (2014)85. Tang et al. used a substitution rate of 1.76% Myr−1 for COI and 0.02% Myr−1 for 18S85, while we used the standard mitochondrial substitution rate for invertebrates (1.15% Myr−1)86. However, all these rates are derived from distant taxa and—to the best of our knowledge—no rotifer-specific substitution rates are available so far. Hence, these analyses should be repeated when substitution rate estimates for our target species (B. calyciflorus s.s. and B. fernandoi) become available. In any case, we note that estimated divergence times within the freshwater B. calyciflorus species complex are in the same order of magnitude as in the marine B. plicatilis species complex (represented by B. plicatilis and B. manjavacas in our analysis; cf. Supplementary Fig. s8).
Hybridization event
Our study revealed a natural hybridization event between B. fernandoi and B. calyciflorus s.s. individuals originating at one sampling location (a single pond): Among the specimens collected there, four exhibited B. fernandoi ITS1 alleles, but shared the same B. calyciflorus s.s COI haplotype.). This comprises—to the best of our knowledge—the first reported in-situ hybridization among these two species. The ability of the species within the B. calyciflorus species complex to hybridize has already been suggested by the widespread occurrence of mitonuclear discordance23. However, only recent studies under laboratory conditions observed hybridization between the most closely related species B. calyciflorus s.s. and B. elevatus6,7, but with a significantly lower intraspecific fertilization (i.e., pre-zygotic isolation) and higher dormant propagule mortality (i.e., post-zygotic isolation)6. Another study detected signs of hybridization between B. elevatus and B. dorcas87. The comparison of alleles/haplotypes (HSP, ITS1, COI) in our four hybrid specimens with other samples from the same location revealed the hybrids to be F2 or a subsequent generation and that genes from B. calyciflorus s.s. introgressed into the local B. fernandoi gene pool. This is because all the introgressed specimens we detected in this study were homozygous for B. fernandoi alleles at the ITS1 locus. Furthermore, three detected hybrids carried B. fernandoi specific HSP 40 kDa alleles, while in one case a B. calyciflorus s.s. specific HSP 40 kDa allele occurred. This different genetic makeup among the hybrids suggests that they either have emerged from independent hybridization events or that the hybridization was sufficiently ancient to allow for repeated sexual recombination after the hybridization event. In any case, our findings demonstrate that B. calyciflorus s.s. and B. fernandoi can naturally hybridize at sites where they occur in sympatry. It was found in the marine B. plicatilis species complex that niche differentiation of very similar species (e.g., body size, biotic niches, competition abilities) is facilitated by their response to changing physical environments in combination with life and diapause history traits16. In the B. calyciflorus species complex, a previous study reported strongest pronounced differences in life history traits of B. fernandoi compared to B. calyciflorus s.s., B. elevatus and B. dorcas, such as prolonged egg and juvenile development times and an overall lower egg production rate and mictic ratio32. The adaptation to different temperature optima34, associated with differences in HSP 40 Da expression35 and protein structure (this study), may lead to seasonal isolation of the two species. This illustrates the above example that the niche of very similar species can be determined by their response to abiotic environmental conditions. The differential niches (here preferred temperature) occupied by the respective species reflect divergent adaptation. This acts as a pre-zygotic isolation mechanism by facilitating differences in timing of reproduction and population growth based on distinct environmental cues (i.e., temperature, photoperiod) and thus maintaining the species boundaries. Such pre-zygotic isolation by season has also been observed among Daphnia species (reviewed in88). This mechanism may however be imperfect under certain environmental conditions, leading to occasional hybridization, as observed in our study.
Conclusion
We used transcriptome and expression data to infer a suitable candidate, the HSP 40 kDa gene, which may play a role in temperature adaptation in the Brachionus calyciflorus species complex. Species-specific alleles and fixed amino acid substitutions sites as well as positively selected sites located in functional domains of the protein were identified. The high number of detected alleles, the results of the branch site selection test, and the fixed sites indicate a divergent adaptation process with B. calyciflorus s.s. retaining ancestral features, from which B. fernandoi derived by positive directional selection. Furthermore, the study revealed descendants of an in-situ hybridization event between B. calyciflorus s.s. and B. fernandoi. This finding indicates that hybridization between those species is possible. We hypothesize that the temporally isolated niches they populate serve as pre-zygotic isolation mechanisms. This isolation may be imperfect under certain environmental conditions, yet hybridizations seem rare and seasonal niche separation generally prevents species boundaries from becoming blurred.
Materials and methods
Candidate gene selection
To identify candidate genes with a sequence divergence pattern related to differential temperature adaptation, previously published transcriptome data35 (GenBank accessions number SRA: SRR10426055-76) was analysed using the following customized bioinformatic pipeline (Supplementary Fig. s1): De novo transcriptome assemblies were used to generate a super transcript for each species using Trinity v. 2.11.0 gene splicer modeler89. This super transcript step was integrated to minimize redundancy in the data by inferring original genes and alternative splice forms90. A reciprocal best blast hit process was performed, using the blastn program of BLAST 2.9.0+91 to detect orthologous genes shared by B. calyciflorus s.s. and B. fernandoi. To determine the best blast hit, results were filtered using a combination of E-value, which is a size corrected measure of statistical significance, and the bit score, which is a measure of matches and mismatches between two sequences91. When E-values of the first two or more hits were the same, the best hit was chosen using the bit score. For each search, a list was created that contained one best hit per query. Orthologous gene pairs were extracted, and open reading frames were predicted using TransDecoder v 5.5.092. To calculate ratios of the rates of non-synonymous and synonymous substitutions (dN/dS) from protein-coding regions, sequences were then translated into amino acid sequences using Biopython93 and locally aligned within the Biopython integrated package pairwise2 using a Smith-Waterman algorithm. Locally aligned coding sequences and the corresponding unaligned DNA sequences were used to create codon alignments using pal2nal94. Selection tests were conducted using all available seven (M0, M1a, M2, M3, M7, M8 and M8a) codeML models (Supplementary Table s1), as implemented in PAML v. 4.995. These models where then compared in four (M0 vs. M3, M1a vs. M2, M7 vs. M8, M8 vs. M8a) predefined likelihood-ratio tests (LRT) to decide whether the different null models (i.e., models that do not allow for any codons ω > 1, corresponding to absence of positive selection) can be rejected95.
Orthologous genes that were significant (< 0.05) in all four LRT comparisons were annotated using Gene Ontology (GO) terms96. These terms are hierarchically ordered descriptions of genes or proteins molecular functions, biological processes, and cellular components. The GO term annotation was done on the online Server Argot2.5 and its batch processing function97. To use this function, the longest available amino acid sequence of each protein was used in a blast search against the Uniprot Swiss-Prot database98 and a hmmer search99,100 against the Pfam-A database100. In addition, we compared the genes exhibiting positive selection in the selection tests with those found to be differentially expressed relative to species and temperature (control: 20 °C; mild heat: B. calyciflorus s.s. 26 °C, B. fernandoi 23 °C; high heat: B. calyciflorus s.s. 32 °C, B. fernandoi 26 °C)35. Among those genes functionally related to temperature tolerance (GO:00009408; “response to heat”; GO:00009409; “response to cold”), only one orthologous gene, the HSP 40 kDa, exhibited signs of positive selection and was differentially expressed in more than one temperature category, hence chosen for in-depth analysis.
DNA extraction
To extract DNA, single rotifer specimens were collected from laboratory cultures, washed in 96% EtOH and stored in a 1.5 mL reaction tube in10µL HPLC-H20. In total, 56 rotifer individuals (B. calyciflorus s.s., n = 25; B. fernandoi, n = 19; B. rubens, n = 4; B. diversicornis, n = 3; B. angularis, n = 5), comprising 56 clonal cultures (WC medium at 20 °C under 16:8 light:dark photoperiod) which were established from one individual isolated from the field, were collected from different locations in Germany. For B. calyciflorus s.s., additional samples from USA, Italy and Austria were available (Supplementary Fig. s2, Supplementary Table s2). To prevent that the DNA from single individuals is lost during the extraction process, 3 µL carrier RNA [1 µg/µL] (QIAGEN, Germany) was added to the sample during the lysis. DNA was extracted using the NucleoSpin®Tissue extraction kit (Macherey–Nagel, Germany) following the manufacturer’s protocol for animal tissue (page 12–14).
Primer design HSP 40 kDa
To amplify the HSP 40 kDa from all five different Brachionus species, primers were designed using the candidate gene sequences (B. calyciflorus s.s. and B. fernandoi) as well as the published HSP 40 kDa sequence of B. calyciflorus (unknown species assignment regarding current taxonomy; Genbank: KC176712.161). Sequences were aligned in Geneious v. 8.1.9101 using ClustalW102 and the implemented primer3 algorithm103 was used to design primer pairs.
Amplification and sequencing of the ITS1, COI and HSP 40 kDa
To determine the species and clonal diversity, both the nuclear internal transcribed spacer (ITS1) and the mitochondrial Cytochrome Oxidase Subunit I (COI) were amplified, using universal invertebrate primers104,105. PCR conditions, primer sequences, and used PCR chemicals mixes can be found in the Supplementary Tables s3, s4 and s5. The HSP 40 kDa gene of all five Brachionus species was amplified using newly designed primer pairs (Supplementary Tables s3, s6 and s7). Successfully amplified HSP 40 kDa, COI and ITS1 products were purified using an enzymatic (ExoAP) procedure and sequenced on a Sanger sequencing platform (Applied Biosystems™ 3500 Genetic Analyzer). Sequences from all specimens used in this study (Supplementary Table s2) were visually inspected using Geneious v. 8.1.9 and heterozygous positions (for ITS1 and HSP 40 kDa) were encoded with the standard IUPAC code for ambiguity106. To reconstruct both alleles of the diploid nuclear DNA, the PHASE algorithm version 2.1107,108 was used, as implemented in DNAsp v. 6109.
HSP 40 kDa sequence diversity
To compare sequence divergence in the HSP 40 kDa gene among the five different species, consensus sequences per species were generated. In order to avoid any bias which may arise from uneven geographic sampling across species, we only used specimens originating from the same region “Uckermark” for this analysis: B. calyciflorus s.s. (n = 11), B. fernandoi (n = 8), B. rubens (n = 4), B. diversicornis (n = 3) and B. angularis (n = 5). Sequence ambiguities in the consensus sequence were coded as N’s (see Supplementary Table s8). Pairwise nucleotide diversities of the 1,025 bp long HSP 40 kDa gene sequence and the 435 bp long COI sequences between the five species were calculated. For an 888 bp fragment of the HSP 40 kDa gene (see below), non-synonymous resp. synonymous substitutions were identified using DNAsp v. 6.
Expressed variation in the HSP 40 kDa gene between B. calyciflorus s.s. and B. fernandoi
Sequences originating from multiple individuals (B. calyciflorus s.s., n = 50; B. fernandoi, n = 38) were translated using the published translation reading frame of the HSP 40 kDa gene of B. calyciflorus (Genbank: KC176712.1). As we did not yield the complete sequence for all specimens, the alignment was truncated to 888 bp present in all sequences. Number of non-synonymous substitutions per non-synonymous site between the two species were compared using SLAC analysis implemented in Datamonkey110,111.
HSP 40 kDa allele identification
To identify different allele types within the B. calyciflorus species complex, allele networks were generated from the resulting 88 phased sequences (B. calyciflorus s.s. n = 50 and B. fernandoi n = 38) using the program popART version 1.7112. Subsequently, a Circos plot113 was generated using the web interface based on allele types identified by popART, species, and specimens’ origin.
Sequence divergence in the HSP 40 kDa gene relative to established barcoding markers
Species recognition within the B. calyciflorus complex relies on species-specific ITS1 sequences and clonal lineages are further characterized by their mitochondrial COI haplotype. For 41 specimens (B. fernandoi n = 18, B. calyciflorus s.s. n = 20, and consensus sequences of B. rubens, B. angularis, and B. diversicornis), sequences were available for both these marker genes and the HSP 40 kDa gene. For these specimens, congruence in genetic affinity across genetic markers was assessed using a tanglegram approach. The respective phylogenies were calculated using RaxML v.8114, performing 1000 bootstrap iterations. Dendrograms and the tanglegrams were generated in R v.4.0.5 with the package dendextend v.1.15.1115.
Selection tests and divergence time
To test whether the expressed genetic variation among the the five different Brachionus species is under selection, a selection test was performed based on the 29 HSP 40 kDa species-specific alleles using the Program PAML v.4.995. A specific focus was on the divergence of the two species B. calyciflorus s.s. and B. fernandoi. This analysis needs a phylogeny as input which was based on the 532 bp long ITS1 fragment and calculated using RaxML v.8, performing 1000 bootstrap iterations. The phylogeny was viewed in FigTree version 1.4.4.116 and adapted using Inkscape version 1.0.1.117 (Supplementary Fig. s3). This ITS1 phylogeny as well as the HSP 40 kDa unique alleles were used to infer sites under selection according to the codeML site model implemented in PAML, testing all seven (M0, M1a, M2, M3, M7, M8 and M8a) different substitution models. The different models vary regarding substitution rates of synonymous and non-synonymous sites (ω values; see Supplementary Table s1) with a subsequent site under selection inference via Bayes Empirical Bayes (BEBs) (for parameter setting see Supplementary Table s9). The models were compared in four (M0 vs. M3, M1a vs. M2, M7 vs. M8, M8a vs. M8) predefined Likelihood Ratio Tests (LRTs) to decide whether the respective null models can be rejected. Significance of the M0 vs. M3 comparison indicates variation in ω, a prerequisite for further tests on positive selection. The null model M1a allows only negatively selected and neutral sites (2 ω values), to which M2a adds a third ω for positively selected sites. M7 and M8 resemble M1a and M2a, but allow for rate variation among negatively selected sites (10 ω values < 1 taken from a β distribution), without consideration of neutral sites (ω = 1). The last comparison of M8a vs. M8 tests for positive selection against a null model (M8a) resembling M7, but with an additional fixed ω = 1 (allowing for sites under selective neutrality); this comparison has been found to increase robustness by yielding fewer false positive results118. An additional branch site test was performed to identify patterns of selection on three different branches: (I) the shared branch of B. calyciflorus s.s. and B. fernandoi, (II) B. calyciflorus s.s. only and (III) B. fernandoi only (Supplementary Fig. s3). Subsequently, to further compare the two formerly cryptic species of B. calyciflorus s.s. and B. fernandoi, a codon-based McDonald–Kreitman test (MKT) and Tajima’s D statistics (allele frequency based) were calculated based on the unique inferred alleles using DNAsp v. 6.
Species-specific fixed amino acid substitutions within the B. calyciflorus species complex were identified (Supplementary Fig. s5) and the ancestral amino acid was inferred under parsimony, using sequence information from B. rubens, B. angularis and B. diversicornis and the ITS1 phylogeny (Supplementary Fig. s3). The functional effects of the specific amino acid (aa) substitutions were predicted with Polyphen2119 using B. calyciflorus s.s. as a reference. Note here that Polyphen2 does not consider the possibility of a positive effect of an aa substitution, such that it ranks any substitution without a predicted change in protein function as "benign" (i.e., no effect), while any predicted change is ranked as "damaging" (i.e., predicted to impact protein function).
To estimate the within B. calyciflorus species complex divergence time between B. calyciflorus s.s. and B. fernandoi the program BEAST v.1.10.4120 was used. Whole mtGenome coding sequences (cds) of different rotifer species were downloaded from NCBI: B. calyciflorus s.s.121, B. fernandoi121, B. angularis122, B. manjavacas123, B. plicatilis124, B. koreanus125, B. rotundiformis126, B. rubens127 and P. similis128 (for Genebank accession numbers cf. Supplementary figure s8). In absence of a published substitution rate for rotifers, the standard divergence rate for invertebrates/insects (2.3% Myr−1) with a constant substitution rate of 0.0115 per million years per lineages86 was used to calibrate the phylogeny. Cds were aligned in Geneious v. 8.1.9 using ClustalW and the IQ-Tree webserver129 was used to determine the best fitting substitution model. The BEAST run was conducted with 10 million MCMC iterations, with GTR + G4 + I chosen as the substitution model. Convergence was checked with Tracer v.1.7.2130, using the first million MCMC iterations as the burn-in value and tree was visualized using FigTree version 1.4.4. and adapted in Inkscape version 1.0.1.
Supplementary Information
Acknowledgements
The authors would like to acknowledge Magdalena Litwin for providing rotifer specimens, Dr. Stefanie Hartmann for her assistance in analysing the transcriptome data, Anja Ernst for her valuable help in the laboratory, and Christina Schirmer, Sabine Donath and Claudia Wahl for their support in cultivating the rotifers.
Author contributions
K.K., R.T. and G.W. designed the research. K.K. and S.P. collected the samples. K.K. performed laboratory processing of samples with support from K.H. K.K., M.G. and S.P. analysed the data with input from R.T. and G.W. K.K. produced the graphics and wrote the manuscript with the support of R.T. S.P., M.G. and G.W. All authors approved the final version of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by Deutsche Forschungsgemeinschaft (DFG) in the framework of the BioMove Research Training Group (DFG-GRK 2118).
Data availability
The genome sequence data (ITS1, COI and HSP 40 kDa) that supports the findings of this study are openly available on Genbank of NCBI (http://www.ncbi.nlm.nih.gov/) under the Accession numbers: ITS1: OP868747-OP868802, COI: OP861581-OP861626 and HSP 40 kDa: OP888060-OP888091. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA544636, SRR10426055—SRR10426076 and SAMN11845726—SAMN11845747.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-27137-3.
References
- 1.Mayr E. Systematics and the Origin of Species, from the Viewpoint of a Zoologist. Harvard University Press; 1942. [Google Scholar]
- 2.Ostevik KL, Andrew RL, Otto SP, Rieseberg LH. Multiple reproductive barriers separate recently diverged sunflower ecotypes. Evolution. 2016;70:2322–2335. doi: 10.1111/evo.13027. [DOI] [PubMed] [Google Scholar]
- 3.Seehausen O, et al. Genomics and the origin of species. Nat. Rev. Genet. 2014;15:176–192. doi: 10.1038/nrg3644. [DOI] [PubMed] [Google Scholar]
- 4.Cheng J, Sha Z-L. Cryptic diversity in the Japanese mantis shrimp (Crustacea: Squillidae): Allopatric diversification, secondary contact and hybridization. Sci. Rep. 2017;7:1972. doi: 10.1038/s41598-017-02059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michaloudi E, et al. Reverse taxonomy applied to the Brachionus calyciflorus cryptic species complex: Morphometric analysis confirms species delimitations revealed by molecular phylogenetic analysis and allows the (re)description of four species. PLoS ONE. 2018;13:e0203168. doi: 10.1371/journal.pone.0203168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang W, Declerck SAJ. Intrinsic postzygotic barriers constrain cross-fertilisation between two hybridising sibling rotifer species of the Brachionus calyciflorus species complex. Freshw. Biol. 2022;67:240–249. doi: 10.1111/fwb.13727. [DOI] [Google Scholar]
- 7.Zhang W, Declerck SAJ. Reduced fertilization constitutes an important prezygotic reproductive barrier between two sibling species of the hybridizing Brachionus calyciflorus species complex. Hydrobiologia. 2022;849:1701–1711. doi: 10.1007/s10750-022-04814-y. [DOI] [Google Scholar]
- 8.Seehausen O, van Alphen JJM, Witte F. Cichlid fish diversity threatened by eutrophication that curbs sexual selection. Science. 1997;277:1808–1811. doi: 10.1126/science.277.5333.1808. [DOI] [Google Scholar]
- 9.Bickford D, et al. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 2007;22:148–155. doi: 10.1016/j.tree.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Gill BA, et al. Cryptic species diversity reveals biogeographic support for the ’mountain passes are higher in the tropics’ hypothesis. Proc. R. Soc. B. 2016;283:20160553. doi: 10.1098/rspb.2016.0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sáez AG, Lozano E. Body doubles. Nature. 2005;433:111. doi: 10.1038/433111a. [DOI] [PubMed] [Google Scholar]
- 12.Fišer C, Robinson CT, Malard F. Cryptic species as a window into the paradigm shift of the species concept. Mol. Ecol. 2018;27:613–635. doi: 10.1111/mec.14486. [DOI] [PubMed] [Google Scholar]
- 13.Mills S, et al. Fifteen species in one: deciphering the Brachionus plicatilis species complex (Rotifera, Monogononta) through DNA taxonomy. Hydrobiologia. 2017;796:39–58. doi: 10.1007/s10750-016-2725-7. [DOI] [Google Scholar]
- 14.Struck TH, et al. Finding evolutionary processes hidden in cryptic species. Trends Ecol. Evol. 2018;33:153–163. doi: 10.1016/j.tree.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Leibold MA, McPeek MA. Coexistence of the niche and neutral perspectives in community ecology. Ecology. 2006;87:1399–1410. doi: 10.1890/0012-9658(2006)87[1399:COTNAN]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 16.Gabaldón C, Fontaneto D, Carmona MJ, Montero-Pau J, Serra M. Ecological differentiation in cryptic rotifer species: What we can learn from the Brachionus plicatilis complex. Hydrobiologia. 2017;796:7–18. doi: 10.1007/s10750-016-2723-9. [DOI] [Google Scholar]
- 17.Nicholls B, Racey PA. Contrasting home-range size and spatial partitioning in cryptic and sympatric pipistrelle bats. Behav. Ecol. Sociobiol. 2006;61:131–142. doi: 10.1007/s00265-006-0244-7. [DOI] [Google Scholar]
- 18.Ortells R, Gómez A, Serra M. Coexistence of cryptic rotifer species: Ecological and genetic characterisation of Brachionus plicatilis. Freshw. Biol. 2003;48:2194–2202. doi: 10.1046/j.1365-2427.2003.01159.x. [DOI] [Google Scholar]
- 19.Wellborn GA, Cothran RD. Niche diversity in crustacean cryptic species: Complementarity in spatial distribution and predation risk. Oecologia. 2007;154:175–183. doi: 10.1007/s00442-007-0816-x. [DOI] [PubMed] [Google Scholar]
- 20.Gause GF. The struggle for existence. Williams and Wilkins; 1934. [DOI] [PubMed] [Google Scholar]
- 21.Segers H. Global diversity of rotifers (Rotifera) in freshwater. Hydrobiologia. 2008;595:49–59. doi: 10.1007/s10750-007-9003-7. [DOI] [Google Scholar]
- 22.Fontaneto D. Molecular phylogenies as a tool to understand diversity in rotifers. Int. Rev. Hydrobiol. 2014;99:178–187. doi: 10.1002/iroh.201301719. [DOI] [Google Scholar]
- 23.Papakostas S, et al. Integrative taxonomy recognizes evolutionary units despite widespread mitonuclear discordance: Evidence from a rotifer cryptic species complex. Syst. Biol. 2016;65:508–524. doi: 10.1093/sysbio/syw016. [DOI] [PubMed] [Google Scholar]
- 24.García-Morales AE, Elías-Gutiérrez M. DNA barcoding of freshwater rotifera in Mexico: Evidence of cryptic speciation in common rotifers. Mol. Ecol. Resour. 2013;13:1097–1107. doi: 10.1111/1755-0998.12080. [DOI] [PubMed] [Google Scholar]
- 25.Wang XL, et al. Differences in life history characteristics between two sibling species in Brachionus calyciflorus complex from tropical shallow lakes. Ann. Limnol. Int. J. Lim. 2014;50:289–298. doi: 10.1051/limn/2014024. [DOI] [Google Scholar]
- 26.Wen X, Xi Y, Zhang G, Xue Y, Xiang X. Coexistence of cryptic Brachionus calyciflorus (Rotifera) species: Roles of environmental variables. J. Plankton Res. 2016;38:478–489. doi: 10.1093/plankt/fbw006. [DOI] [Google Scholar]
- 27.Xiang X-L, Chen Y-Y, Han Y, Wang X-L, Xi Y-L. Comparative studies on the life history characteristics of two Brachionus calyciflorus strains belonging to the same cryptic species. Biochem. Syst. Ecol. 2016;69:138–144. doi: 10.1016/j.bse.2016.09.003. [DOI] [Google Scholar]
- 28.Xiang X-L, et al. Patterns and processes in the genetic differentiation of the Brachionus calyciflorus complex, a passively dispersing freshwater zooplankton. Mol. Phylogenet. Evol. 2011;59:386–398. doi: 10.1016/j.ympev.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Xiang X-L, et al. Genetic differentiation and phylogeographical structure of the Brachionus calyciflorus complex in eastern China. Mol. Ecol. 2011;20:3027–3044. doi: 10.1111/j.1365-294X.2011.05147.x. [DOI] [PubMed] [Google Scholar]
- 30.Gilbert JJ, Walsh EJ. Brachionus calyciflorus is a species complex: Mating behavior and genetic differentiation among four geographically isolated strains. Hydrobiologia. 2005;546:257–265. doi: 10.1007/s10750-005-4205-3. [DOI] [Google Scholar]
- 31.Zhang Y, et al. Temporal patterns and processes of genetic differentiation of the Brachionus calyciflorus (Rotifera) complex in a subtropical shallow lake. Hydrobiologia. 2018;807:313–331. doi: 10.1007/s10750-017-3407-9. [DOI] [Google Scholar]
- 32.Zhang W, Lemmen KD, Zhou L, Papakostas S, Declerck SAJ. Patterns of differentiation in the life history and demography of four recently described species of the Brachionus calyciflorus cryptic species complex. Freshw. Biol. 2019;64:1994–2005. doi: 10.1111/fwb.13388. [DOI] [Google Scholar]
- 33.Lemmen KD, Verhoeven KJF, Declerck SAJ. Experimental evidence of rapid heritable adaptation in the absence of initial standing genetic variation. Funct. Ecol. 2022;36:226–238. doi: 10.1111/1365-2435.13943. [DOI] [Google Scholar]
- 34.Paraskevopoulou S, Dennis AB, Weithoff G, Hartmann S, Tiedemann R. Within species expressed genetic variability and gene expression response to different temperatures in the rotifer Brachionus calyciflorus sensu stricto. PLoS ONE. 2019;14:e0223134. doi: 10.1371/journal.pone.0223134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paraskevopoulou S, Dennis AB, Weithoff G, Tiedemann R. Temperature-dependent life history and transcriptomic responses in heat-tolerant versus heat-sensitive Brachionus rotifers. Sci. Rep. 2020;10:13281. doi: 10.1038/s41598-020-70173-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paraskevopoulou S, Tiedemann R, Weithoff G. Differential response to heat stress among evolutionary lineages of an aquatic invertebrate species complex. Biol. Lett. 2018;14:20180498. doi: 10.1098/rsbl.2018.0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takemoto K, Akutsu T. Origin of structural difference in metabolic networks with respect to temperature. BMC Syst. Biol. 2008;2:82. doi: 10.1186/1752-0509-2-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Angilletta MJ. Thermal Adaptation: A Theoretical and Empirical Synthesis. Oxford University Press; 2009. [Google Scholar]
- 39.Atkinson D. Temperature and organism size: A biological law for ectotherms? Adv. Ecol. Res. 1994;25:1–58. doi: 10.1016/S0065-2504(08)60212-3. [DOI] [Google Scholar]
- 40.Gillooly JF, Brown JH, West GB, Savage VM, Charnov EL. Effects of size and temperature on metabolic rate. Science. 2001;293:2248–2251. doi: 10.1126/science.1061967. [DOI] [PubMed] [Google Scholar]
- 41.Walczyńska A, Franch-Gras L, Serra M. Empirical evidence for fast temperature-dependent body size evolution in rotifers. Hydrobiologia. 2017;796:191–200. doi: 10.1007/s10750-017-3206-3. [DOI] [Google Scholar]
- 42.Brown WL, Wilson EO. Character displacement. Syst. Zool. 1956;5:49–64. doi: 10.2307/2411924. [DOI] [Google Scholar]
- 43.Marrone F, Fontaneto D, Naselli-Flores L. Cryptic diversity, niche displacement and our poor understanding of taxonomy and ecology of aquatic microorganisms. Hydrobiologia. 2022 doi: 10.1007/s10750-022-04904-x. [DOI] [Google Scholar]
- 44.Pekkonen M, Ketola T, Laakso JT. Resource availability and competition shape the evolution of survival and growth ability in a bacterial community. PLoS ONE. 2013;8:e76471. doi: 10.1371/journal.pone.0076471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brawand D, et al. The evolution of gene expression levels in mammalian organs. Nature. 2011;478:343–348. doi: 10.1038/nature10532. [DOI] [PubMed] [Google Scholar]
- 46.Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat. Rev. Genet. 2009;10:715–724. doi: 10.1038/nrg2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fraser HB. Genome-wide approaches to the study of adaptive gene expression evolution: Systematic studies of evolutionary adaptations involving gene expression will allow many fundamental questions in evolutionary biology to be addressed. BioEssays. 2011;33:469–477. doi: 10.1002/bies.201000094. [DOI] [PubMed] [Google Scholar]
- 48.Fraser HB. Gene expression drives local adaptation in humans. Genome Res. 2013;23:1089–1096. doi: 10.1101/gr.152710.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Franch-Gras L, et al. Rotifer adaptation to the unpredictability of the growing season. Hydrobiologia. 2019;844:257–273. doi: 10.1007/s10750-019-3886-y. [DOI] [Google Scholar]
- 50.Tarazona E, Lucas-Lledó JI, Carmona MJ, García-Roger EM. Gene expression in diapausing rotifer eggs in response to divergent environmental predictability regimes. Sci. Rep. 2020;10:21366. doi: 10.1038/s41598-020-77727-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith HA, Burns AR, Shearer TL, Snell TW. Three heat shock proteins are essential for rotifer thermotolerance. J. Exp. Mar. Biol. Ecol. 2012;413:1–6. doi: 10.1016/j.jembe.2011.11.027. [DOI] [Google Scholar]
- 52.Alonso CR, Wilkins AS. The molecular elements that underlie developmental evolution. Nat. Rev. Genet. 2005;6:709–715. doi: 10.1038/nrg1676. [DOI] [PubMed] [Google Scholar]
- 53.Romero IG, Ruvinsky I, Gilad Y. Comparative studies of gene expression and the evolution of gene regulation. Nat. Rev. Genet. 2012;13:505–516. doi: 10.1038/nrg3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franch-Gras L, et al. Genomic signatures of local adaptation to the degree of environmental predictability in rotifers. Sci. Rep. 2018;8:16051. doi: 10.1038/s41598-018-34188-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nowell RW, et al. Comparative genomics of bdelloid rotifers: Insights from desiccating and nondesiccating species. PLoS Biol. 2018;16:e2004830. doi: 10.1371/journal.pbio.2004830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feugeas J-P, et al. Links between transcription, environmental adaptation and gene variability in Escherichia coli: Correlations between gene expression and gene variability reflect growth efficiencies. Mol. Biol. Evol. 2016;33:2515–2529. doi: 10.1093/molbev/msw105. [DOI] [PubMed] [Google Scholar]
- 57.Pai AA, Pritchard JK, Gilad Y. The genetic and mechanistic basis for variation in gene regulation. PLoS Genet. 2015;11:e1004857. doi: 10.1371/journal.pgen.1004857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gribble KE, Mark Welch DB. The mate recognition protein gene mediates reproductive isolation and speciation in the Brachionus plicatilis cryptic species complex. BMC Evol. Biol. 2012;12:134. doi: 10.1186/1471-2148-12-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Via S. Natural selection in action during speciation. Proc. Natl. Acad. Sci. USA. 2009;106:9939–9946. doi: 10.1073/pnas.0901397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ho SYW, Duchêne S. Molecular-clock methods for estimating evolutionary rates and timescales. Mol. Ecol. 2014;23:5947–5965. doi: 10.1111/mec.12953. [DOI] [PubMed] [Google Scholar]
- 61.Yang J, Mu Y, Dong S, Jiang Q, Yang J. Changes in the expression of four heat shock proteins during the aging process in Brachionus calyciflorus (Rotifera) Cell Stress Chaperones. 2014;19:33–52. doi: 10.1007/s12192-013-0432-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahmood K, Jadoon S, Mahmood Q, Irshad M, Hussain J. Synergistic effects of toxic elements on heat shock proteins. Biomed. Res. Int. 2014;2014:564136. doi: 10.1155/2014/564136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park JC, et al. Genome-wide identification and structural analysis of heat shock protein gene families in the marine rotifer Brachionus spp.: Potential application in molecular ecotoxicology. Comp. Biochem. Physiol. D. 2020;36:100749. doi: 10.1016/j.cbd.2020.100749. [DOI] [PubMed] [Google Scholar]
- 64.Santoro M. Heat shock factors and the control of the stress response. Biochem. Pharmacol. 2000;59:55–63. doi: 10.1016/S0006-2952(99)00299-3. [DOI] [PubMed] [Google Scholar]
- 65.Birky CW, Gilbert JJ. Parthenogenesis in rotifers: The control of sexual and asexual reproduction. Am. Zool. 1971;11:245–266. doi: 10.1093/icb/11.2.245. [DOI] [Google Scholar]
- 66.Snell TW. Rotifers as models for the biology of aging. Int. Rev. Hydrobiol. 2014;99:84–95. doi: 10.1002/iroh.201301707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78:737–756. doi: 10.1093/genetics/78.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muller HJ. Some genetic aspects of sex. Am. Nat. 1932;66:118–138. doi: 10.1086/280418. [DOI] [Google Scholar]
- 69.Muller HJ. The relation of recombination to mutational advance. Mut. Res. 1964;1:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- 70.Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Mol. Ecol. 2004;13:729–744. doi: 10.1046/j.1365-294X.2003.02063.x. [DOI] [PubMed] [Google Scholar]
- 71.Zhang Y, Xu S, Sun C, Dumont H, Han B-P. A new set of highly efficient primers for COI amplification in rotifers. Mitochondrial DNA B. 2021;6:636–640. doi: 10.1080/23802359.2021.1878951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Turner CB, Marshall CW, Cooper VS. Parallel genetic adaptation across environments differing in mode of growth or resource availability. Evol. Lett. 2018;2:355–367. doi: 10.1002/evl3.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lan B, et al. Tempo-spatial variations of zooplankton communities in relation to environmental factors and the ecological implications: A case study in the hinterland of the Three Gorges Reservoir area. China. PLoS ONE. 2021;16:e0256313. doi: 10.1371/journal.pone.0256313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pellecchia M, Szyperski T, Wall D, Georgopoulos C, Wüthrich K. NMR structure of the J-domain and the Gly/Phe-rich region of the Escherichia coli DnaJ chaperone. Mol. Biol. 1996;260:236–250. doi: 10.1006/jmbi.1996.0395. [DOI] [PubMed] [Google Scholar]
- 75.Greene MK, Maskos K, Landry SJ. Role of the J-domain in the cooperation of Hsp40 with Hsp70. Proc. Natl. Acad. Sci. USA. 1998;95:6108–6113. doi: 10.1073/pnas.95.11.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wittung-Stafshede P, Guidry J, Horne BE, Landry SJ. The J-domain of Hsp40 couples ATP hydrolysis to substrate capture in Hsp70. Biochemistry. 2003;42:4937–4944. doi: 10.1021/bi027333o. [DOI] [PubMed] [Google Scholar]
- 77.Cintron NS, Toft D. Defining the requirements for Hsp40 and Hsp70 in the Hsp90 chaperone pathway. J. Biol. Chem. 2006;281:26235–26244. doi: 10.1074/jbc.M605417200. [DOI] [PubMed] [Google Scholar]
- 78.Li J, Qian X, Sha B. The crystal structure of the yeast Hsp40 Ydj1 complexed with its peptide substrate. Structure. 2003;11:1475–1483. doi: 10.1016/j.str.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 79.Sha B, Lee S, Cyr DM. The crystal structure of the peptide-binding fragment from the yeast Hsp40 protein Sis1. Structure. 2000;8:799–807. doi: 10.1016/S0969-2126(00)00170-2. [DOI] [PubMed] [Google Scholar]
- 80.Brender JR, Zhang Y. Predicting the effect of mutations on protein-protein binding interactions through structure-based interface profiles. PLoS Comput. Biol. 2015;11:e1004494. doi: 10.1371/journal.pcbi.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shortle D. One sequence plus one mutation equals two folds. Proc. Natl. Acad. Sci. USA. 2009;106:21011–21012. doi: 10.1073/pnas.0912370107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Charlesworth B. The effects of deleterious mutations on evolution at linked sites. Genetics. 2012;190:5–22. doi: 10.1534/genetics.111.134288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cutter AD. A Primer of Molecular Population Genetics. Oxford University Press; 2019. [Google Scholar]
- 84.Barraclough TG, Fontaneto D, Ricci C, Herniou EA. Evidence for inefficient selection against deleterious mutations in cytochrome oxidase I of asexual bdelloid rotifers. Mol. Biol. Evol. 2007;24:1952–1962. doi: 10.1093/molbev/msm123. [DOI] [PubMed] [Google Scholar]
- 85.Tang CQ, Obertegger U, Fontaneto D, Barraclough TG. Sexual species are separated by larger genetic gaps than asexual species in rotifers. Evol. Int. J. Org. Evol. 2014;68:2901–2916. doi: 10.1111/evo.12483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brower AV. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc. Natl. Acad. Sci. U.S.A. 1994;91:6491–6495. doi: 10.1073/pnas.91.14.6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang W, Deng Z, Blair D, Hu W, Yin M. Phylogeography of the freshwater rotifer Brachionus calyciflorus species complex in China. Hydrobiologia. 2022;849:2813–2829. doi: 10.1007/s10750-022-04897-7. [DOI] [Google Scholar]
- 88.Chin TA, Cristescu ME. Speciation in Daphnia. Mol. Ecol. 2021;30:1398–1418. doi: 10.1111/mec.15824. [DOI] [PubMed] [Google Scholar]
- 89.Grabherr MG, et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Davidson NM, Hawkins ADK, Oshlack A. SuperTranscripts: A data driven reference for analysis and visualisation of transcriptomes. Genome Biol. 2017;18:148. doi: 10.1186/s13059-017-1284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Altschul SF, Gish WP, Miller W, Myers EW, Lipman DL. Basic local alignment search tool. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 92.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cock PJA, et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009;25:1422–1423. doi: 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Suyama M, Torrents D, Bork P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 96.Ashburner M, et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lavezzo E, Falda M, Fontana P, Bianco L, Toppo S. Enhancing protein function prediction with taxonomic constraints: The Argot2.5 web server. Methods. 2016;93:15–23. doi: 10.1016/j.ymeth.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 98.The UniProt Consortium UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–D489. doi: 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Finn RD, Clements J, Eddy SR. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kearse M, et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Untergasser A, et al. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Palumbi SR. The polymerase chain reaction. Mol. Syst. 1996;2:205–247. [Google Scholar]
- 105.Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994;3:294–299. [PubMed] [Google Scholar]
- 106.Cornish-Bowden A. Nomenclature for incompletely specified bases in nucleic acid sequences: Recommendations. Nucleic Acids Res. 1985;39:3021–3030. doi: 10.1093/nar/13.9.3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stephens M, Donnelly P. A Comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rozas J, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017;34:3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- 110.Kosakovsky Pond SL, Frost SDW. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 111.Weaver S, et al. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018;35:773–777. doi: 10.1093/molbev/msx335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Leigh JW, Bryant D. popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015;6:1110–1116. doi: 10.1111/2041-210X.12410. [DOI] [Google Scholar]
- 113.Krzywinski M, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Galili T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics. 2015;31:3718–3720. doi: 10.1093/bioinformatics/btv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Andrew Rambaut Group. FigTree. (2022). http://tree.bio.ed.ac.uk/software/.
- 117.Inkscape Project. Inkscape. (2020). https://inkscape.org.
- 118.Wong WSW, Yang Z, Goldman N, Nielsen R. Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004;168:1041–1051. doi: 10.1534/genetics.104.031153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Suchard MA, et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4:016. doi: 10.1093/ve/vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kiemel K, de Cahsan B, Paraskevopoulou S, Weithoff G, Tiedemann R. Mitochondrial genomes of the freshwater monogonont rotifer Brachionus fernandoi and of two additional B. calyciflorus sensu stricto lineages from Germany and the USA (Rotifera, Brachionidae) Mitochondrial DNA B. 2022;7:646–648. doi: 10.1080/23802359.2022.2060765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim M-S, et al. Complete mitochondrial genome of the freshwater monogonont rotifer Brachionus angularis (Rotifera, Brachionidae) Mitochondrial DNA B. 2020;5:3754–3755. doi: 10.1080/23802359.2020.1835578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kim M-S, et al. Complete mitochondrial genomes of two marine monogonont rotifer Brachionus manjavacas strains. Mitochondrial DNA B. 2021;6:1921–1923. doi: 10.1080/23802359.2021.1935349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Suga K, Mark Welch DB, Tanaka Y, Sakakura Y, Hagiwara A. Two circular chromosomes of unequal copy number make up the mitochondrial genome of the rotifer Brachionus plicatilis. Mol. Biol. Evol. 2008;25:1129–1137. doi: 10.1093/molbev/msn058. [DOI] [PubMed] [Google Scholar]
- 125.Hwang D-S, et al. Complete mitochondrial genome of the monogonont rotifer, Brachionus koreanus (Rotifera, Brachionidae) Mitochondrial DNA B. 2014;25:29–30. doi: 10.3109/19401736.2013.775274. [DOI] [PubMed] [Google Scholar]
- 126.Kim H-S, et al. Complete mitochondrial genome of the monogonont rotifer Brachionus rotundiformis (Rotifera, Brachionidae) Mitochondrial DNA B. 2017;2:39–40. doi: 10.1080/23802359.2016.1202743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Choi B-S, et al. Complete mitochondrial genome of the freshwater monogonont rotifer Brachionus rubens (Rotifera, Brachionidae) Mitochondrial DNA B. 2019;5:5–6. doi: 10.1080/23802359.2019.1694853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Choi B-S, et al. Complete mitochondrial genome of the marine monogonont rotifer Proales similis (Rotifera, Proalidae) Mitochondrial DNA B. 2020;5:1151–1152. doi: 10.1080/23802359.2020.1730265. [DOI] [Google Scholar]
- 129.Trifinopoulos J, Nguyen L-T, von Haeseler A, Minh BQ. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016;44:W232–W235. doi: 10.1093/nar/gkw256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genome sequence data (ITS1, COI and HSP 40 kDa) that supports the findings of this study are openly available on Genbank of NCBI (http://www.ncbi.nlm.nih.gov/) under the Accession numbers: ITS1: OP868747-OP868802, COI: OP861581-OP861626 and HSP 40 kDa: OP888060-OP888091. The associated BioProject, SRA, and Bio-Sample numbers are PRJNA544636, SRR10426055—SRR10426076 and SAMN11845726—SAMN11845747.