

Abstract
Aims:
Prostaglandin E2 (PGE2) is a lipid hormone that signals through 4 different G-protein coupled receptor subtypes which act to regulate key physiological processes. Our laboratory has previously reported that PGE2 through its EP3 receptor reduces cardiac contractility at the level of isolated cardiomyocytes and in the isolated working heart preparation. We therefore hypothesized that cardiomyocyte specific overexpression of the PGE2 EP3 receptor further decreases cardiac function in a mouse model of heart failure produced by myocardial infarction.
Main methods:
Our study tested this hypothesis using EP3 transgenic mice (EP3 TG), which overexpress the porcine analogue of human EP3 in the cardiomyocytes, and their wildtype (WT) littermates. Mice were analyzed 2 wks after myocardial infarction (MI) or sham operation by echocardiography, RT-PCR, immunohistochemistry, and histology.
Key findings:
We found that the EP3 TG sham controls had a reduced ejection fraction, reduced fractional shortening, and an increased left ventricular dimension at systole and diastole compared to the WT sham controls. Moreover, there was a further reduction in the EP3 TG mice after myocardial infarction. Additionally, single-cell analysis of cardiomyocytes isolated from EP3 TG mice showed reduced contractility under basal conditions. Overexpression of EP3 significantly increased cardiac hypertrophy, interstitial collagen fraction, macrophage, and T-cell infiltration in the sham operated group. Interestingly, after MI, there were no changes in hypertrophy but there were changes in collagen fraction, and inflammatory cell infiltration.
Significance:
Overexpression of EP3 reduces cardiac function under basal conditions and this is exacerbated after myocardial infarction.
Keywords: EP3, Myocardial Infarction, Prostaglandin E2
Introduction
Prostaglandin E2 (PGE2) is a lipid hormone that acts through 4 G-protein coupled receptor subtypes (EP1, EP2, EP3 and EP4). PGE2, through its four receptors, is widely distributed in various tissues and is involved in regulating key physiological processes which can act as a protective mechanism against injury. EP1 activates phospholipase C, resulting in an increase in intracellular calcium levels. EP2 and EP4 both activate adenylate cyclase, thereby increasing the levels of cyclic AMP (cAMP) and subsequent activation of protein kinase A. Uniquely, EP4 can also act through the PI3K-Akt signal transduction pathway (Kawahara et al., 2015). In contrast to EP2/EP4 signaling, EP3 inhibits adenylate cyclase activity, reducing cAMP levels. After PGE2 is synthesized, it is immediately released, where it acts locally, and its response is dependent upon the presence of specific EP receptors and the signaling pathways activated (Bryson and Harding, 2022).
While all four PGE2 receptors are expressed in the heart, EP3 and EP4 receptors are the most abundant. We have previously reported that PGE2 through its receptor sub-type EP3 reduces contractility in isolated cardiomyocytes and in the whole heart (Gu et al., 2016). Additionally, we demonstrated that overexpression of the EP4 receptor reduces cardiac inflammation and improves contractility after a myocardial infarction (Bryson et al., 2018).
It has been previously reported that cardiac-specific overexpression of EP3 (EP3 transgenic (TG)) in mice is protective against ischemia and reperfusion and reduces ischemic injury of the myocardium (Martin et al., 2005, Meyer-Kirchrath et al., 2009). However, the effect of EP3 overexpression in a mouse model of permanent myocardial infarction (MI) has not been studied. We examined expression levels of EP3 in the left ventricle after an MI and found that the EP3 receptor is significantly increased, suggesting the balance of the EP receptor expression is shifted towards the EP3 receptor in heart failure (Meyer-Kirchrath et al., 2009). We hypothesize that cardiomyocyte specific overexpression of the PGE2 EP3 receptor leads to decreased cardiac function in a mouse model of myocardial infarction. We test this hypothesis using EP3 transgenic mice, which overexpress the porcine analogue of human EP3 in the cardiomyocytes, and their wildtype (WT) littermates to characterize them using echocardiography, cardiomyocyte contractility, RT-PCR, immunohistochemistry, and histology.
Materials and Methods
EP3 TG Mice and WT Controls
To test our hypothesis, we used EP3 TG mice, which overexpress the full-length porcine analogue of human EP3 specifically in the cardiomyocytes as described by (Martin et al., 2005). Martin et al. have previously shown 40-fold-higher ligand binding in EP3 TG mice compared with WT littermates. Moreover, EP3 receptors in the two strains displayed similar binding affinities (Martin et al., 2005). Unpublished RNA sequencing data from our laboratory confirms EP3 TG mice have approximately a 36-fold increase in Ptger3 in left ventricle samples compared to WT littermates. EP3 TG mice and their WT controls were a generous gift from Drs Hohlfeld and Meyer-Kirchrath at the University of Dusseldorf, Germany.
Myocardial infarction Animal Model
Male EP3 TG mice or WT controls at 10–12 weeks of age were anesthetized with sodium pentobarbital (50 mg x kg−1) (Akorn, Inc., Lake Forest, IL. USA), intubated, and ventilated with a small rodent ventilator (Harvard Apparatus, Holliston, MA, USA) at a rate of 90 cycles min−1 with a tidal volume of 0.5 ml and a positive end-expiratory pressure of 2 cmH2O. A left side thoracotomy was performed, and the pericardium was incised. Myocardial infarction was then induced through permanent ligation of the left anterior descending coronary artery using 8–0 silk suture proximal to its bifurcation from the main stem. Sham surgery was performed as described above except that the suture was not tied around the artery. The chest incision was subsequently closed with a 4–0 silk suture. Mice recovered in a temperature-controlled environment. After surgery, mice were given a dose of buprenorphine (0.05 mg x kg−1 s.c.) every 12 h for 2 days. All studies involving the use of animals were approved by the animal care and use committee (IACUC) at Henry Ford Hospital and Wayne State University, in accordance with federal guidelines.
Echocardiography
Echocardiography was performed on conscious animals at 2 weeks after sham or MI surgery. Cardiac function was assessed using an Acuson 256 system (Mountain View, CA) with a 15-MHz linear transducer, as reported previously (LaPointe et al., 2004). Diastolic measurements were made at the maximum left ventricle cavity dimension, whereas systolic parameters were measured during maximum anterior motion of the posterior wall. All echocardiography was performed by the same investigator who was blinded to the experimental groups. The following parameters were obtained: Ejection fraction (LVEF), fractional shortening (FS), cardiac output (CO), left ventricular dimension at systole (LVDs), and diastole (LVDd). The left ventricle and body weights were obtained at the end of the study to calculate the left ventricle to body weight ratio (LV/BW).
Isolation and Pacing experiments of adult cardiomyocytes
Isolation of cardiomyocytes from 15 wk old EP3 WT and EP3 transgenic animals at baseline was performed using modifications of the method described by O’Connell et al (O’Connell et al., 2007). 10mmol/l 2,3-butanedione monoxime was omitted as it is a known inhibitor of contractility. Freshly isolated AVM prepared in Tyrode’s solution were washed and rested for 15 minutes. Cells were divided into aliquots. An aliquot of cells was added to the chamber, allowed to attach for 2 min, then superfused with Tyrode’s solution at 37°C and electrically stimulated at 3 Hz using a biphasic pulse. Contraction amplitude was recorded online using a dual excitation spectrofluorometer and video edge detection system (IonOptix) and a minimum of 30-40 transients were analyzed for each cell. As indicators of contractility, peak shortening, baseline percent peak height, and the speed of contraction and relaxation were measured.
Histological Assessment
Myocyte cross sectional area and interstitial collagen fraction
Histological assessment of myocyte cross sectional area (MCSA) and interstitial collagen fraction (ICF) was performed as previously described (Gu et al., 2015, LaPointe et al., 2004, Liu et al., 1997, Qian et al., 2008). Briefly, while under anesthesia, mouse hearts were arrested in diastole by injection of 15% potassium chloride into the apex of the heart. Hearts were harvested and sectioned transversely into three slices from apex to base (sections A-C). The sections were frozen in OCT media pre-chilled in isopentane and stored at −80°C for determination of MCSA and ICF. Hearts were sectioned at a thickness of 5 μm. All three sections of the heart were stained with fluorescein-labeled peanut agglutinin to delineate the myocytes and Rhodamine-labeled Griffonia simplicifolia lectin I to outline the glycoproteins within collagen and capillaries. Four radially oriented microscope fields were randomly selected from each section and imaged under the 20x objective. MCSA was measured by computer-based planimetry (NIH Image J) and averaged across all 4 fields of the sections. Only those myocytes in cross-sectional orientation were analyzed. Any myocytes that were in a longitudinal orientation were not analyzed. The mean cardiomyocyte area was then calculated for each animal. ICF was performed by calculating the total surface area occupied by the interstitial space (LaPointe et al., 2004). An average interstitial collagen fraction was obtained using all four images..
Picrosirius Red Staining for Infarct Size
Picrosirius red staining (PSR) was utilized to assess infarct size as we have previously described (Bryson et al., 2018). Briefly, frozen sections were postfixed in Bouin’s solution overnight at room temperature. Sections were then stained with 0.1% picrosirius red stain for 1 hour, followed by 2 washes in 0.5% acetic acid, 1 wash in 95% ethanol, 2 washes in 100% ethanol, and 2 changes of 100% Xylenes. Slides were then mounted with coverslips using a toluene-based resin mount (Permount, Fisher scientific). Bright field images of the slides were taken under 2X objective. Infarct size was analyzed using NIH image J software and averaged across two sections of the heart where we would expect to see an infarct (apex and near apex).
Triphenyltetrazolium Chloride Staining for Infarct Size
Triphenyltetrazolium chloride (TTC) staining was performed to assess infarct size of heart sections two hours post MI based on the methodology of Cohen et al. (Cohen et al., 2011). Briefly, 2 hours after MI, 15% potassium chloride was injected into the apex of the heart to cause its arrest in diastole. Hearts were removed and rinsed in ice-cold hanks buffered salt solution to remove heme-containing components. Hearts were then sectioned transversely into equal sections of about 1 mm thickness. Each section was incubated in 1% 2,3,5-Triphenyltetrazolium chloride (Sigma) in 1X phosphate buffered saline for 30 minutes at 37°C. After incubation, hearts were stored in 10% formalin overnight. Analysis was performed using NIH image J software. Infarct (white tissue) was traced, and the infarct area was calculated as a percentage of total section area. All tissue sections were averaged for each mouse and the mean data was calculated for the both groups.
All histological analyses were performed by an observer blinded to the experimental groups and any animal containing < 20% infarct subsequent to a MI surgery was excluded from further analysis.
Real Time RT-PCR
RNA expression levels of β-myosin heavy chain, Myh7 (β-MHC), brain natriuretic peptide, Nppb (BNP), calcineurin, Ppp3cb (CALN), transforming growth factor beta, Tgf-b1 and collagen 1 (Col1a1) was performed as previously described (Bryson et al., 2018). Primers and sequences are described in Table 1. RNA samples were reverse transcribed using random primers and omniscript reverse transcriptase (Qiagen, Valencia, CA). 2 μL of the reverse transcription reaction was amplified in a Roche version 2.0 lightcycler PCR instrument (Roche, Indianapolis, IN) using SYBR green dye (SA Biosciences, Frederick, MD) and specific primers. At the end of PCR cycling, melting curve analysis was performed. A relative quantitation method (ΔΔCt) was used to evaluate expression of each gene, corrected to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and presented as fold of WT + Sham group (Bryson et al., 2018).
Table 1.
Real Time RT-PCR Primer Sequences
| Gene | Source | Sense | Antisense | Cat. No. |
|---|---|---|---|---|
| Collagen 1 (Col1a1) | TIB | gACCgATggATTCCMgTTCg | gTAggCTACgTTCTTgCA | |
| β-MHC (Myh7) | TIB | ACAggAAgAACCTACTgCgRC | AgCTTgTTgACCTgggACT | |
| TGF-B1 (Tgf-b1) | TIB | AAgggCTACCATgCCAACT | TCAgCTgCACTTgCAggAg | |
| BNP (Nppb) | TIB | CCTACAACAACTTCAgTgCgTT | CCCAAAAAgAgTCCTTCggT | |
| Calcineurin (Ppp3cb) | QIAGEN | QT00161280 |
Primer sequences used from TIB MolBiol and Qiagen. Primer sequences from Qiagen (Ppp3cb) are proprietary, thus only the catalog number is available.
Immunohistochemistry for macrophages, and T cells (CD3+, CD4+, CD8+)
The total number of macrophages was analyzed using CD68+ staining (rat anti-CD68+ antibody, BioRad) on frozen heart sections as we previously described (Gu et al., 2015). The total T cell population was visualized using CD3+ staining (rabbit-anti CD3+ antibody, Abcam). CD4+ T helper cells and cytotoxic CD8+ T cells were visualized using antibodies against CD4 and CD8, respectively (rabbit anti-CD4, Abcam and rabbit anti-CD8 alpha, Abcam). Frozen sections were thawed and fixed in acetone for 10 min, followed by incubation with fresh 0.3% hydrogen peroxide for 30 min. Sections were then blocked in 5% centrifuged milk in TBS (Tris- buffered saline) blocking buffer and incubated with primary antibody in blocking buffer (anti-CD68+;1:200. anti-CD3+; 1:100. Anti-CD4+; 1:100, anti-CD8+; 1:100) overnight at 4 °C in a humidified chamber. Biotinylated secondary antibodies (1:200 dilution of anti-mouse IgG for CD68+, 1:200 dilution of goat anti-rabbit IgG for CD3+, CD8+, and CD4+) were placed onto sections for 1 h at room temperature. Sections were then incubated with horseradish peroxidase reagent for 40 min at room temperature and then visualized by AEC single solution (Vector laboratories; Burlingame, CA). After a rinse in tap water, slides were counterstained with Harris’ hematoxylin solution for 1 min, then rinsed again in tap water and mounted onto slides using Aquamount (Lerner laboratories). Negative controls consisted of sections lacking the primary antibody. Photographs were obtained by choosing four random fields per stained section. Images were taken under the 20X objective, and the number of positively stained cells were counted by a blinded observer using Image J software. For analysis, the infarct, peri-infarct, and remote regions were averaged.
Statistical analysis
All statistics were performed by a statistician in the Department of Public Health Sciences of Henry Ford Hospital using the statistical package SAS Version 9.4. All data are shown as mean ± standard error of the mean (SEM). The EP3 overexpression on cardiac function and cardiomyocyte hypertrophy and isolated cardiomyocyte contractility was analyzed using Student’s t-test. If the standard deviations were different, a Satterthwaite correction was used. All other variables were examined using a two-sample Wilcoxon test as the data did not appear to be normally distributed. If the standard deviations were different a Fligner-Policello version was used. We used two-sample tests as we were not interested in the overall test nor all pairwise comparisons. We note these tests are independent of the results of the overall test.
Results
Effect of EP3 overexpression on cardiac function
Echocardiography data are presented in Figure 1. Under sham-operated conditions, the EP3 WT mice had a significantly higher ejection fraction than the EP3 TG mice (Fig. 1A, 72.5 ± 1.25 for WT Sham vs 62.4 ± 3.95 for TG Sham, p=0.06). As expected, when the WT mice were subject to MI, their ejection fraction decreased significantly compared to their sham operated littermates (72.5 ± 1.25 in WT sham vs. 51.17 ± 4.88 in WT MI, p< 0.01). Similarly, when EP3 TG mice were subject to MI, there was also a reduction in EF albeit started at a lower baseline (35.26 ± 8.10 in TG MI vs. 62.4 ± 3.95 in TG sham, p < 0.001). Importantly, EF in EP3 TG mice subject to MI was significantly lower than the WT MI group (p<0.01). In addition to reduced ejection fraction in the TG mice at baseline, they have a significantly lower fractional shortening (SF) and a further reduction after MI (p<0.001), to a level that is significantly different from that of the WT MI group (Fig. 1B). The significant reduction in fractional shortening was coupled with significant increases in dilation of the ventricle in TG mice at both systole and diastole (Fig. 1C, Fig. 1D). EP3 TG mice had a further exaggerated increase in left ventricle dimensions at systole compared to their WT littermates (3.03 ± 1.15 for TG MI vs. 2.01 ± 0.50 in WT MI; p<0.01). Altogether, the data suggests that overexpression of EP3 reduces cardiac function at baseline and causes ventricular remodeling in both the absence and presence of a myocardial infarction.
Figure 1.
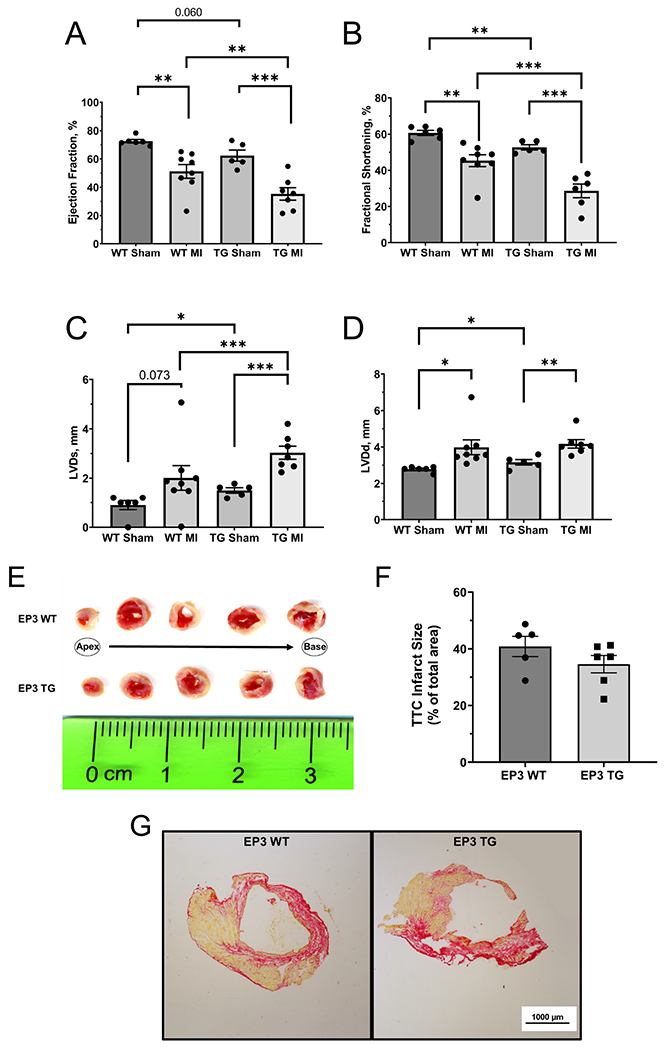
Quantitative analysis of echocardiography data. (A) Ejection fraction. (B) fractional shortening. (C) left ventricle dimension at systole, LVDs. (D) left ventricle dimension at diastole, LVDd. * p<0.05, ** p<0.01, *** p<0.001. N = 6 for WT sham, N = 5 for TG sham, N = 8 for WT MI, N = 7 for TG MI. (E) Representative TTC staining images 2 hours post LAD ligation. Top row is EP3 WT, bottom row is EP3 TG. (F) Mean TTC staining – infarct area quantified as a percentage of total area 2 hours post LAD ligation. N=5 for WT + 2 hrs MI, N=6 for EP3 TG + 2 hrs MI(G) Representative picrosirius red staining (PSR) images taken under 2x objective lens 2 weeks post MI. Red staining represents collagen deposition, yellow is non-collagen tissue. All data presented as mean ± SEM
Effect of EP3 Overexpression on Infarct Size
To verify that the EP3 TG mice and their WT littermates have equivalent infarct sizes after MI, we performed TTC staining two hours post LAD ligation. As shown in the representative heart sections in figure 1E and mean quantitative data in figure 1F, there was no difference in infarct size between groups after 2 hours of MI (34.6% ± 3.06 in EP3 TG vs. 40.8% ± 3.57 in EP3 WT). Moreover, after two weeks of MI, we performed PSR staining to assess infarct size and collagen deposition. Representative images of the PSR staining are shown in figure 1G. There was no significant difference between EP3 TG mice and their WT littermates (37.8 ± 4.45 in EP3 TG vs. 27.1% ± 3.71 in EP3 WT). Altogether, these data suggest that the exaggerated reduction in cardiac function is not due to an increased infarct size in the EP3 TG mice.
Isolated Cardiomyocyte Contractility in EP3 TG and WT Mice
Figure 2A shows a representative transient tracing from EP3 WT and EP3 TG cardiomyocytes under basal conditions, paced at 3 Hz. The parameters of peak height (4.56 ± 0.25 mm for WT vs. 2.10 ± 0.24 mm for TG, p<0.001), departure velocity, (−235.2 ± 13.1 mm/sec for WT vs. −107 ± 12.15 mm/sec for TG, p<0.001), Bl% peak height (3.47 ± 0.20 % for WT vs. 1.37 ± 0.25 mm for TG, p<0.001), and return velocity, (106.3 ± 8.21 mm/sec for WT vs. 47.21 ± 8.15 mm/sec for TG, p<0.001) were all reduced in cardiomyocytes obtained from EP3 TG mice. This mean data is presented in figure 2B–E. Overall, these results indicate that the EP3 TG cardiomyocytes have impaired contractility and relaxation.
Figure 2.
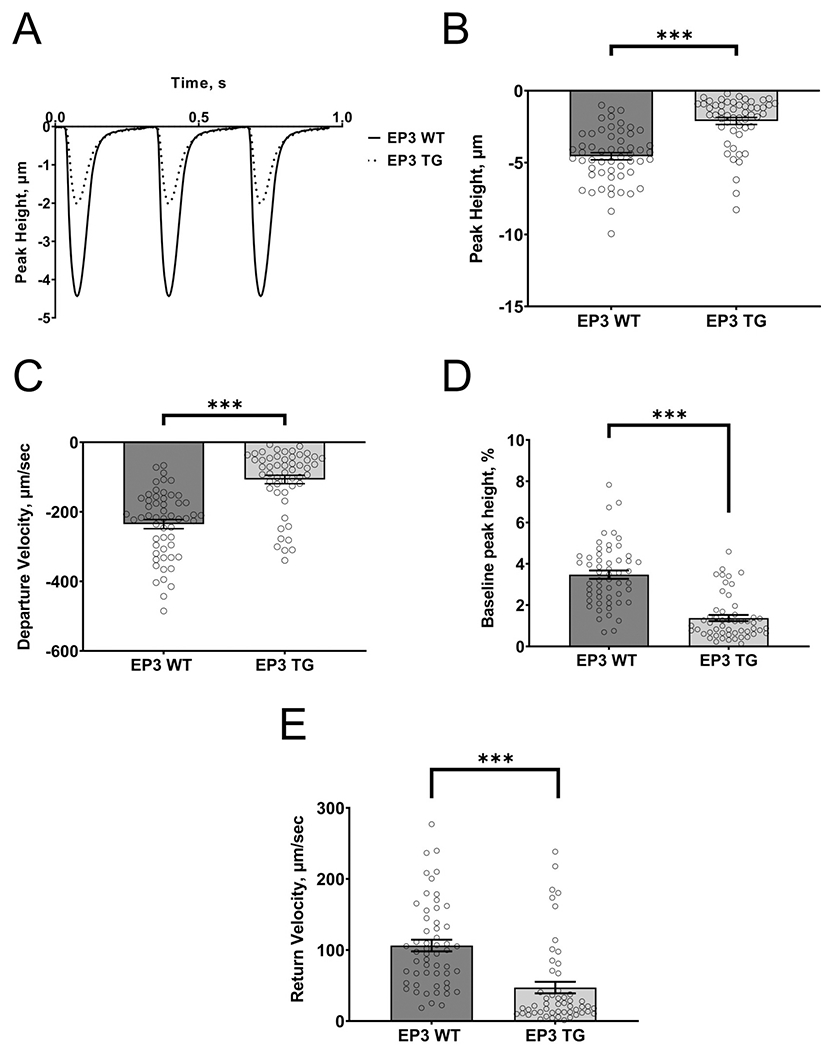
Effect of EP3 overexpression in cardiac myocytes on cell shortening. (A) Representative tracings of transients from EP3 WT (dotted line) and EP3 TG (solid line) cells paced at 3 Hz. (B) Mean quantitative data of peak height. (C) Mean quantitative data of the speed of contraction (departure velocity). (D) The baseline as a percentage of peak height. (E) Mean quantitative data of the speed of relaxation (return velocity). ***p<0.001. N=56 cells from 7 mice for EP3 WT and N=53 cells from 5 mice for EP3 TG.
Effect of EP3 overexpression on cardiomyocyte hypertrophy
Myocyte cross sectional area (MCSA) is a method used to examine cardiac hypertrophy at the cellular level (Bryson et al., 2018, Liu et al., 1997). Representative images of the peanut agglutinin stain used to calculate MCSA are shown in figure 3A under the 20X objective from the different experimental groups. MCSA in EP3 TG mice was significantly higher than WT mice under sham operated conditions (fig. 3B: 142.9 ± 2.65 mm2 for WT Sham vs. 167.7 ± 4.01 mm2 for TG Sham, p<0.001). As expected, MI increased MCSA in the WT mice (142.9 ± 2.65 mm2 for WT Sham vs. 167.7 ± 2.00 mm2 for WT MI, p<0.001). Interestingly however, MI did not increase MCSA in EP3 TG mice (168.4 ± 4.02mm2 for TG Sham vs. 164.2 ± 1.44 mm2 for TG MI). In agreement with the MCSA data, there was a tendency towards an increase in the left ventricle to body weight ratio after MI in WT animals, although this failed to reach statistical significance (3.88 ± 0.37 mg/10gBW in WT MI vs. 3.04 ± 0.14 mg/10gBW in WT Sham, p=0.062). The left ventricle to body weight ratio was significantly increased under sham operated conditions in EP3 TG versus WT controls (4.87 ± 0.25 mg/g BW in TG sham vs. 3.04 ± 0.14 mg/g BW in WT sham, p<0.001). These results suggest that EP3 TG cardiac myocytes are already hypertrophied at baseline and have an impaired hypertrophic response to MI.
Figure 3.
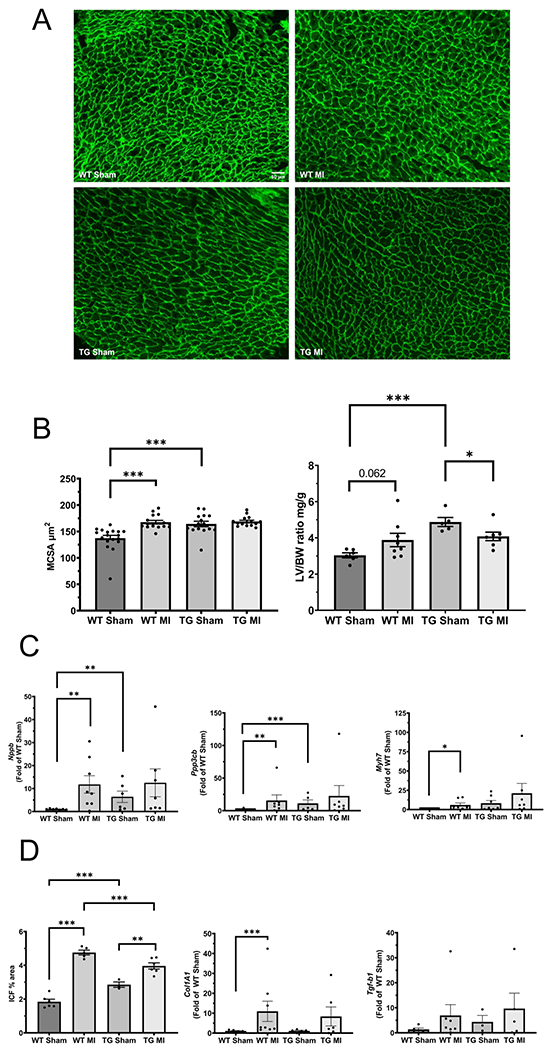
Effect of EP3 overexpression on cardiac hypertrophy and fibrosis. (A) Representative images of peanut agglutinin staining for myocyte cross sectional area analysis taken under the 20x objective (scale bar indicates 50 μm). (B) Left panel-quantitative analysis of MCSA, and Right panel – an additional measure of hypertrophy, left ventricle to body weight ratio. (C) mRNA expression of hypertrophic gene markers; left panel –brain natriuretic peptide (Nppb), middle panel – calcineurin (Ppp3cb), and right panel – myosin heavy chain (Myh7). (D) Measurements of fibrosis; left panel – quantitative analysis of interstitial collagen fraction (ICF), middle panel – mRNA expression of collagen 1A (Col1A1), right panel – mRNA expression of transforming growth factor β (Tgf-b1). MCSA and ICF quantitative analysis was performed using NIH Image J software. * p<0.05, *** p<0.001 N=5-6/group. All mRNA expression data was corrected to GAPDH and presented as a fold of wildtype sham control group. *p<0.05, **p<0.01, ***p< 0.001. N = 5-7 for wildtype sham, N = 3-7 for transgenic sham, N = 5-8 for wildtype MI, N = 4-6 for transgenic MI.
Effect of EP3 overexpression on gene markers of cardiac hypertrophy and fibrosis
In addition to MCSA, cellular hypertrophy can be assessed by analyzing the mRNA expression of specific genes involved in the cardiac hypertrophic response. To analyze changes in their expression, we measured mRNA expression of brain natriuretic peptide (Nppb), calcineurin (Ppp3cb), and β-myosin heavy chain (Myh7). All expression levels were corrected to GAPDH as a loading control and are presented as fold of EP3 WT sham group. As shown in figure 3C, Nppb and Ppp3cb were significantly higher in the TG sham group when compared to the WT sham group (1.02 ± 0.08 for WT sham vs. 6.41 ± 2.47 for TG sham, p<0.01) and (1.37 ± 0.53 for WT sham vs. 11.22 ± 5.24 for TG Sham, p<0.001) respectively. β-MHC, while not significant, also increased in the TG sham mice. As expected, after MI, there were significant increases in the hypertrophic markers Nppb (1.02 ± 0.083 for WT sham vs. 11.77 ± 3.81 for WT MI, p< 0.01) Ppp3cb (1.37 ± 0.53 for WT sham vs. 15.62 ± 8.53 for WT MI, p<0.01) and Myh7 (1.04 ± 0.13 for WT sham vs. 6.02 ± 2.51 for WT MI, p< 0.05) in wildtype mice. However, these increases were not seen in the TG mice after MI and the data are consistent with the speculation that EP3 TG mice may have an impaired hypertrophic response. Importantly, these results are also consistent with the MCSA data.
Effect of EP3 overexpression on cardiac fibrosis
Cardiac fibrosis was assessed by analyzing the total interstitial collagen fraction (ICF) area in the hearts and by performing real time RT-PCR to analyze mRNA expression of collagen 1 (Col1A1) and transforming growth factor β (Tgf-b1). The ICF in the EP3 TG group was significantly higher than WT controls in sham operated mice, (fig 3D. 1.84 ± 0.15 % for WT Sham vs. 2.98 ± 0.15 % for TG Sham, p<0.001). As anticipated, MI increased ICF significantly in both WT (1.84 ± 0.15% for WT Sham vs. 4.76± 0.11 % for WT MI, p<0.001) and EP3 TG groups (2.98 ± 0.15% for TG Sham vs. 4.09 ± 0.14% for TG MI, p<0.01) when compared to their sham operated littermates. ICF was significantly different in EP3 TG MI when compared with WT MI (p<0.001), although this reduction may reflect the fact that EP3 TG sham have higher ICF at baseline and there is less of an increase after MI. In addition, we analyzed mRNA expression of collagen 1 (Col1a1) and TGF-β (Tgf-b1) to further explore the role of EP3 overexpression on fibrosis (fig 3D). The mRNA expression of Col1a1 was significantly increased in the WT group after MI (1.10 ± 0.2 for WT Sham vs. 10.94 ± 5.1 for WT MI, p<0.05). Similarly, expression was increased after MI in the EP3 Tg group 8-fold, however this change did not reach statistical significance. Interestingly, Col1a1 mRNA expression was unchanged between WT and EP3 Tg groups under sham operated conditions (1.10 ± 0.2 for WT sham vs. 1.13 ± 0.3 for EP3 Tg sham). The mRNA expression of Tgf-b1 followed a similar trend as Col1a1.
Effect of EP3 overexpression on macrophage and T cell infiltration
Myocardial infarction is characterized by an influx of macrophages and T cells into the myocardium and simultaneous activation of resident macrophages (Chen and Frangogiannis, 2016). CD68+ macrophage cells were observed in the infarct/peri-infarct region 2 weeks after sham or MI operation. As expected, after MI there were increases for both the WT (126.3 ± 2.32 for WT Sham vs. 159.2 ± 9.42 for WT MI, p< 0.01) and EP3 TG mice (144.3 ± 5.18 for TG Sham vs. 214.5 ± 8.95 for TG MI, p< 0.001) when compared to their respective sham operated controls (fig 4A).
Figure 4.
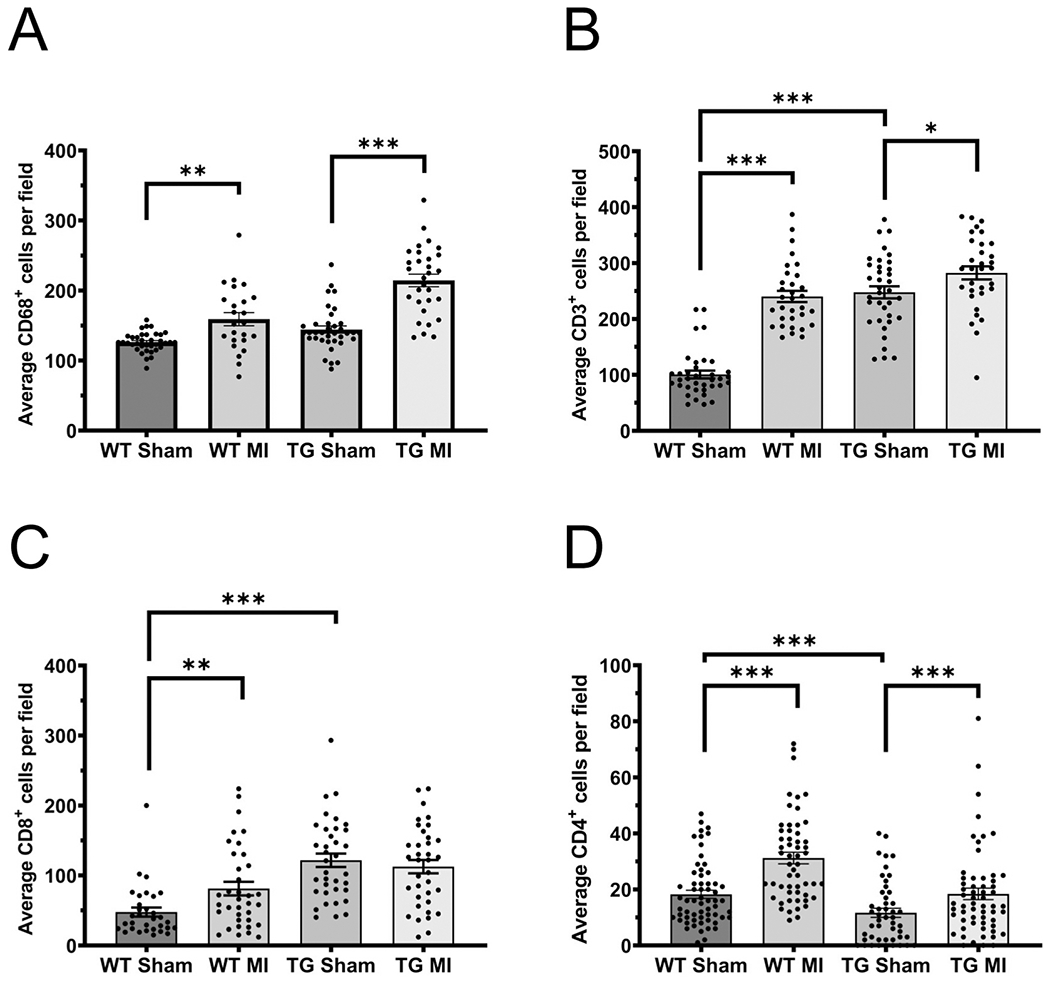
Effect of EP3 overexpression on macrophage and T cell infiltration into the heart. (A) Mean number of total macrophages (CD68+). (B) Mean number of total T cells (CD3+) cells. (C) Mean number of cytotoxic (CD8+) cells. (D) Mean number of CD4+ cells. All immunohistochemistry was performed on sections from the infarct, border zone, and remote zone of the left ventricles. Quantitative analysis was performed using NIH image J software by a blinded observer and all three regions of the heart were averaged. Data presented as mean ± SEM. * p<0.05, ** p<0.01, *** p<0.001 N = 5-7 for wildtype sham, N = 3-7 for transgenic sham, N = 5-8 for wildtype MI, N = 4-6 for transgenic MI.
CD3+ T cells were observed in left ventricles 2 weeks after sham or MI operation. In the sham operated animals, there were significant increases in the number of CD3+ cells in TG mice compared to WT mice (fig 4B. 100.6 ± 6.98 for WT Sham vs. 247.7 ± 10.86 for TG Sham, p<0.001). As anticipated, after MI there were significant increases in the number of CD3+ cells in WT mice (100.6 ± 6.98 for WT Sham vs. 240.3 ± 10.03 for WT MI, p<0.001) and in the EP3 TG mice (282.4 ± 11.77 in TG MI vs. 247.7 ± 10.86 in TG sham, p<0.05). To further evaluate which subset of T-cells were present, we stained for cytotoxic CD8+ cells and CD4+ cells (fig. 4C). Under sham operated conditions, there was a 2.5-fold increase in CD8+ T cells in EP3 TG mice compared to WT mice (47.56 ± 6.49 for WT Sham vs. 121.6 ± 9.65 for TG Sham, p<0.001). After MI, the number of CD8+ cells increased (47.56 ± 6.49 for WT Sham vs. 81.14 ± 9.74 for WT MI, p<0.05). There was no change in CD8+ cells in the EP3 TG mice after MI (121.6 ± 9.65 for TG Sham vs. 112.5 ± 9.75 for TG MI). Figure 4D shows that CD4+ cells were significantly decreased in the EP3 TG mice under sham conditions (18.3 ± 1.44 for WT Sham vs. 11.7 ± 1.61 for TG Sham, p<0.001). Myocardial infarction resulted in a significant increase in CD4+ cells in both the WT (31.2 ± 2.06 for WT MI vs. 18.3 ± 1.44 for WT Sham, p<0.001) and in the EP3 TG mice (18.4 ± 2.04 for TG MI vs. 11.7 ± 1.61 for TG Sham, p<0.001).
Discussion
The results of this study show that cardiac-specific overexpression of prostaglandin E2 EP3 receptor negatively affects cardiac function. Furthermore, it shows that contractility in the EP3 TG mouse model is reduced in the whole heart, and in single cardiomyocytes. Currently, the mechanism responsible for the impaired contractility of isolated cardiomyocytes is under investigation. We have previously reported that the prostaglandin E2 EP3 receptor reduced contractility in male mice and that expression of EP3 was greatly increased after a myocardial infarction (Gu et al., 2016). Recently, Bosma et al. reported that EP3 blockade and/or EP4 activation improves contractility and calcium handling in mouse cardiomyocytes in a diabetic model (Bosma et al., 2022), consistent with previous findings from our laboratory showing that overexpression of EP4 using AAV9 improves cardiac function in a mouse model of MI (Bryson et al., 2018). In the present study, we hypothesized that overexpression of EP3 in the heart would reduce cardiac function at baseline and would further reduce cardiac function after MI. Our experiments described here are novel in that they are the first to subject EP3 TG mice to permanent myocardial infarction for 2 weeks. Previous studies with this mouse model investigated hypertension via Angiotensin II infusion and myocardial ischemia reperfusion injury. It was previously reported that the left ventricles of these mice are hypertrophied and that a decline in heart function was observed as early as 5-6 weeks of age (Martin et al., 2005, Meyer-Kirchrath et al., 2009). Our results support those previous findings but also add new and important additional information regarding the influence of EP3 overexpression in a model of myocardial infarction, with significant reductions in cardiac function after MI and increased hypertrophy at baseline. Importantly, our novel findings show that these changes cannot be ascribed to differences in infarct size between the EP3 TG mice and their WT littermates either at the early time point of 2 hours after MI or 2 weeks post MI. Our results also add substantial new mechanistic information to those previously reported in that we find significant defects in single cardiomyocyte contractility, suggesting that alterations in the cellular contractile apparatus may be partly responsible for the altered cardiac function observed at baseline in the EP3 TG mice.
Overexpression of EP3 significantly increased cardiac hypertrophy even under sham-operated conditions. Interestingly, there was no increase in hypertrophy after MI suggesting to us that the hearts of EP3 TG mice have an impaired hypertrophic response to MI. This postulation is evidenced by changes in the MCSA and confirmed by changes in hypertrophic gene markers BNP, Calcineurin, and β-MHC. Fibrosis was increased post MI in both groups as shown by increases in ICF staining and the EP3 TG mice had an increase in ICF under sham operated conditions. Contrary to these results, collagen 1 mRNA was not increased in the EP3 TG mice under sham operated conditions. We speculate that the increased ICF may be due to other collagen types (e.g., collage type 3), rather than collagen type 1 alone. Additionally, there was no exacerbated increase in collagen deposition in the EP3 TG mice according to our picrosirius red staining 2 weeks post MI.
Previously other studies have shown a protective role for the EP3 receptor in ischemia-reperfusion injury. Martin, M., et al. (Martin et al., 2005) reported that overexpression of EP3 receptors in cardiomyocytes is protective against ischemia/reperfusion-induced myocardial injury via a reduction in left ventricle contractility. A study by Meyer-Kirchrath et al. (Meyer-Kirchrath et al., 2009), provided similar findings and direct evidence that EP3 receptors expressed by cardiomyocytes mediate pro-hypertrophic signaling, demonstrated by increased cardiomyocyte size and left ventricle dimension. Our data using a mouse model of left anterior descending coronary artery ligation, coupled with overexpression of EP3 in cardiomyocytes, agrees with the previously mentioned pro-hypertrophic effects. However, our results do not suggest a cardioprotective effect of EP3 overexpression in our MI model. The controversial role of the EP3 receptor may be due to the different mouse models employed. Martin et al. (Martin et al., 2005) used an ex-vivo isolated heart perfusion method compared with our current in-vivo permanent MI study.
There are several previous studies demonstrating the critical role that T cells have in heart failure. Our data shows increased CD3+ T-cells in both the EP3 transgenic and wildtype mice following a myocardial infarction. Furthermore, the number of cytotoxic T-cells (CD8+) present in the EP3 TG sham mice was significantly increased whereas, the number of helper T-cells (CD4+) was significantly decreased. Cytotoxic CD8+ cells have been shown to play an important role in cardiac remodeling and additionally, impair cardiac function, alluding to a dual role in cardiac pathology (Laroumanie et al., 2014, Rai et al., 2020, Varda-Bloom et al., 2000). Under sham-operated conditions CD8+ cells were significantly increased in the EP3 TG mice, suggesting a potential mechanism of the impaired cardiac function observed in these animals. Helper CD4+ T cells have been shown to play an important role in wound healing, scar limitation and repair of infarcted cardiac muscle in the early stages following a MI (van der Heide et al., 2016) (van der Heide et al., 2016). As expected, CD4+ cells were increased in both strains of mice after MI. Importantly, there were no increases in CD4+ cells in EP3 TG mice under sham-operated conditions compared to WT controls, suggesting that hearts of TG mice may be subject to pro-inflammatory environment, contributing to reduced cardiac function.
Previous studies examining the role of T-cells in various pathologies have also examined cytokine levels such as IL-6, IL-17, IL-10, TNFα and interferon γ (Rai et al., 2020, van der Heide et al., 2016, van Hamburg and Tas, 2018). These particular chemokines/cytokines promote cardiac fibrosis and hypertrophy and cross talk among the adaptive immune cells and innate immune cells which play a role in heart failure(Norlander et al., 2018, Rai et al., 2020, Xin et al., 2019). Chemokines such as monocyte chemoattractant protein (MCP)-1/CCL2 are activated in ischemic environments. Furthermore, when MCP-1 is activated, it results in fibrosis (Dobaczewski and Frangogiannis, 2009). Our lab previously reported that EP3 TG mice subjected to Angiotensin II-induced hypertension have reduced cardiac function, in association with increased cardiac cytokines and chemokines under basal conditions (Bryson et al., 2020)
Although there were significant changes after MI in both the WT and TG mice in the number of macrophages, the phenotypes of the macrophages have yet to be identified (M1 pro inflammatory vs. M2 anti-inflammatory). In recent years, the role of PGE2 in macrophage polarization has surfaced (Sanin et al., 2018), however, in this study we did not distinguish between the polarity states of the macrophages.
A potential limitation of this study is the timepoint at which we ended the study. We concluded the study 2-weeks post sham or MI operations. Alternative time points might help identify the timeline of immune cell infiltration and at what time points the hypertrophic markers reach their peak. Future experiments to examine the chemokine and cytokine profile in these mice will help to gain a better understanding of the role the adaptive immune system and EP3 in heart failure. Additionally, we did not assess the area at risk immediately after MI between the two groups. However, we measured infarct size at 2 hours post MI and in another group of animals, we measured it 2 weeks post MI. Importantly, infarct sizes were equivalent between the two groups at both time points. Thus, we consider that the area at risk would be equal between the two groups. Although it is well known that the mouse coronary artery anatomy is highly variable, providing, arguably, unreliable reproducibility in one’s measurement of area at risk (Degabriele et al., 2004, Kanno et al., 2002, Kumar et al., 2005, Patten et al., 1998, Salto-Tellez et al., 2004), our laboratory has performed these studies for nearly 25 years (Liu et al., 2002, Nakagawa et al., 2018, Qian et al., 2008, Wang et al., 1999, Xu et al., 2007, Xu et al., 2002, Yang et al., 2002) and the variation in measures of cardiac function are remarkably small, suggesting a high degree of reproducibility in placement of the ligature. Thus, the results of our current studies are not likely due to alterations in the infarct size and area at risk between WT and TG mice.
Conclusion
Overall, the results of our experiments confirm our in vitro studies and demonstrate the deleterious role of the EP3 receptor in the heart. Thus, potential therapeutics that target and inhibit EP3 receptors in cardiomyocytes after MI may be a new strategy for the treatment of heart failure.
Acknowledgements
We thank Drs. Thomas Hohlfield and Jutta Meyer-Kirchrath at the University of Dusseldorf for providing us with the EP3 transgenic mouse strain.
Grants
This study was funded by the National Institute of Health R01 HL148060 to P. Harding. D. L. Maxwell was supported by the Interdisciplinary Biomedical Sciences fellowship at Wayne State University School of Medicine Physiology department.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- Bosma KJ, Ghosh M, Andrei SR, Zhong L, Dunn JC, Ricciardi VF, et al. Pharmacological modulation of prostaglandin E(2) (PGE(2)) EP receptors improves cardiomyocyte function under hyperglycemic conditions. Physiol Rep. 2022;10:e15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson TD, Gu X, Khalil RM, Khan S, Zhu L, Xu J, et al. Overexpression of prostaglandin E2 EP4 receptor improves cardiac function after myocardial infarction. J Mol Cell Cardiol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson TD, Harding P. Prostaglandin E2 EP receptors in cardiovascular disease: An update. Biochem Pharmacol. 2022;195:114858. [DOI] [PubMed] [Google Scholar]
- Bryson TD, Pandrangi TS, Khan SZ, Xu J, Pavlov TS, Ortiz PA, et al. The Deleterious Role of the Prostaglandin E2 EP3 Receptor in Angiotensin II Hypertension. Am J Physiol Heart Circ Physiol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Frangogiannis NG. Macrophages in the Remodeling Failing Heart. Circ Res. 2016;119:776–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen R, Shainberg A, Hochhauser E, Cheporko Y, Tobar A, Birk E, et al. UTP reduces infarct size and improves mice heart function after myocardial infarct via P2Y2 receptor. Biochem Pharmacol. 2011;82:1126–33. [DOI] [PubMed] [Google Scholar]
- Degabriele NM, Griesenbach U, Sato K, Post MJ, Zhu J, Williams J, et al. Critical appraisal of the mouse model of myocardial infarction. Experimental physiology. 2004;89:497–505. [DOI] [PubMed] [Google Scholar]
- Dobaczewski M, Frangogiannis NG. Chemokines and cardiac fibrosis. Front Biosci (Schol Ed). 2009;1:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Xu J, Yang XP, Peterson E, Harding P. Fractalkine neutralization improves cardiac function after myocardial infarction. Experimental physiology. 2015;100:805–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Xu J, Zhu L, Bryson T, Yang XP, Peterson E, et al. Prostaglandin E2 Reduces Cardiac Contractility via EP3 Receptor. Circ Heart Fail. 2016;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno S, Lerner DL, Schuessler RB, Betsuyaku T, Yamada KA, Saffitz JE, et al. Echocardiographic evaluation of ventricular remodeling in a mouse model of myocardial infarction. J Am Soc Echocardiogr. 2002;15:601–9. [DOI] [PubMed] [Google Scholar]
- Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochimica et biophysica acta. 2015;1851:414–21. [DOI] [PubMed] [Google Scholar]
- Kumar D, Hacker TA, Buck J, Whitesell LF, Kaji EH, Douglas PS, et al. Distinct mouse coronary anatomy and myocardial infarction consequent to ligation. Coronary artery disease. 2005;16:41–4. [DOI] [PubMed] [Google Scholar]
- LaPointe MC, Mendez M, Leung A, Tao Z, Yang XP. Inhibition of cyclooxygenase-2 improves cardiac function after myocardial infarction in the mouse. Am J Physiol Heart Circ Physiol. 2004;286:H1416–24. [DOI] [PubMed] [Google Scholar]
- Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111–24. [DOI] [PubMed] [Google Scholar]
- Liu YH, Xu J, Yang XP, Yang F, Shesely E, Carretero OA. Effect of ACE inhibitors and angiotensin II type 1 receptor antagonists on endothelial NO synthase knockout mice with heart failure. Hypertension. 2002;39:375–81. [DOI] [PubMed] [Google Scholar]
- Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E, et al. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Invest. 1997;99:1926–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Meyer-Kirchrath J, Kaber G, Jacoby C, Flogel U, Schrader J, et al. Cardiospecific overexpression of the prostaglandin EP3 receptor attenuates ischemia-induced myocardial injury. Circulation. 2005;112:400–6. [DOI] [PubMed] [Google Scholar]
- Meyer-Kirchrath J, Martin M, Schooss C, Jacoby C, Flogel U, Marzoll A, et al. Overexpression of prostaglandin EP3 receptors activates calcineurin and promotes hypertrophy in the murine heart. Cardiovasc Res. 2009;81:310–8. [DOI] [PubMed] [Google Scholar]
- Nakagawa P, Romero CA, Jiang X, D’Ambrosio M, Bordcoch G, Peterson EL, et al. Ac-SDKP decreases mortality and cardiac rupture after acute myocardial infarction. PLoS One. 2018;13:e0190300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. The Journal of experimental medicine. 2018;215:21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271–96. [DOI] [PubMed] [Google Scholar]
- Patten RD, Aronovitz MJ, Deras-Mejia L, Pandian NG, Hanak GG, Smith JJ, et al. Ventricular remodeling in a mouse model of myocardial infarction. Am J Physiol. 1998;274:H1812–20. [DOI] [PubMed] [Google Scholar]
- Qian JY, Harding P, Liu Y, Shesely E, Yang XP, LaPointe MC. Reduced cardiac remodeling and function in cardiac-specific EP4 receptor knockout mice with myocardial infarction. Hypertension. 2008;51:560–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A, Narisawa M, Li P, Piao L, Li Y, Yang G, et al. Adaptive immune disorders in hypertension and heart failure: focusing on T-cell subset activation and clinical implications. J Hypertens. 2020;38:1878–89. [DOI] [PubMed] [Google Scholar]
- Salto-Tellez M, Yung Lim S, El-Oakley RM, Tang TP, ZA AL, Lim SK. Myocardial infarction in the C57BL/6J mouse: a quantifiable and highly reproducible experimental model. Cardiovasc Pathol. 2004;13:91–7. [DOI] [PubMed] [Google Scholar]
- Sanin DE, Matsushita M, Klein Geltink RI, Grzes KM, van Teijlingen Bakker N, Corrado M, et al. Mitochondrial Membrane Potential Regulates Nuclear Gene Expression in Macrophages Exposed to Prostaglandin E2. Immunity. 2018;49:1021–33.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heide V, Möhnle P, Rink J, Briegel J, Kreth S. Down-regulation of MicroRNA-31 in CD4+ T Cells Contributes to Immunosuppression in Human Sepsis by Promoting TH2 Skewing. Anesthesiology. 2016;124:908–22. [DOI] [PubMed] [Google Scholar]
- van Hamburg JP, Tas SW. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J Autoimmun. 2018;87:69–81. [DOI] [PubMed] [Google Scholar]
- Varda-Bloom N, Leor J, Ohad DG, Hasin Y, Amar M, Fixler R, et al. Cytotoxic T lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro. J Mol Cell Cardiol. 2000;32:2141–9. [DOI] [PubMed] [Google Scholar]
- Wang D, Yang XP, Liu YH, Carretero OA, LaPointe MC. Reduction of myocardial infarct size by inhibition of inducible nitric oxide synthase. Am J Hypertens. 1999;12:174–82. [DOI] [PubMed] [Google Scholar]
- Xin M, Jin X, Cui X, Jin C, Piao L, Wan Y, et al. Dipeptidyl peptidase-4 inhibition prevents vascular aging in mice under chronic stress: Modulation of oxidative stress and inflammation. Chem Biol Interact. 2019;314:108842. [DOI] [PubMed] [Google Scholar]
- Xu J, Carretero OA, Lin CX, Cavasin MA, Shesely EG, Yang JJ, et al. Role of cardiac overexpression of ANG II in the regulation of cardiac function and remodeling postmyocardial infarction. Am J Physiol Heart Circ Physiol. 2007;293:H1900–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Carretero OA, Liu YH, Shesely EG, Yang F, Kapke A, et al. Role of AT2 receptors in the cardioprotective effect of AT1 antagonists in mice. Hypertension. 2002;40:244–50. [DOI] [PubMed] [Google Scholar]
- Yang F, Liu YH, Yang XP, Xu J, Kapke A, Carretero OA. Myocardial infarction and cardiac remodelling in mice. Experimental physiology. 2002;87:547–55. [DOI] [PubMed] [Google Scholar]