

Abstract
Given the ambiguity surrounding TBI pathophysiology and the lack of any Food and Drug Administration (FDA)-approved neurotherapeutic drugs, there is an increasing need to better understand the mechanisms of traumatic brain injury (TBI). Recently, the roles of inflammasomes have been highlighted as both potential therapeutic targets and diagnostic markers in different neurodegenerative disorders. Indeed, inflammasome activation plays a pivotal function in the central nervous system (CNS) response to many neurological conditions, as well as to several neurodegenerative disorders, specifically, TBI. This comprehensive review summarizes and critically discusses the mechanisms that govern the activation and assembly of inflammasome complexes and the major methods used to study inflammasome activation in TBI and its implication for other neurodegenerative disorders. Also, we will review how inflammasome activation is critical in CNS homeostasis and pathogenesis, and how it can impact chronic TBI sequalae and increase the risk of developing neurodegenerative diseases. Additionally, we discuss the recent updates on inflammasome-related biomarkers and the potential to utilize inflammasomes as putative therapeutic targets that hold the potential to better diagnose and treat subjects with TBI.
Keywords: Inflammasomes, traumatic brain injury, neurodegenerative diseases, caspase-1, pyroptosis, biomarkers, therapeutic targets, NLPR1
Introduction
Traumatic brain injury (TBI) is one of the leading causes of morbidity and mortality globally across all ages, where about 69 million people worldwide suffer from TBI each year, with an estimated three times more cases in low- and middle-income countries compared to high-income countries (Dewan et al., 2018). Because of the massive expenses of the direct treatment of TBI and the associated post-TBI disability, TBI imposes a substantial economic burden on society. It has been estimated that TBI costs the global economy around $400 billion annually (Maas et al., 2017). However, despite the vast impact TBI imposes on the quality of life of patients and their families and the annual cost of the healthcare system, there remains no FDA-approved therapy for the treatment of TBI (Mondello et al., 2014). In addition, according to the Centers for Disease Control and Prevention, the number of TBI patients (both adults and children) admitted to emergency departments in the USA increased by 53% from 2006 to reach 2.87 million in 2014, among whom 288,000 were hospitalized and 56,800 died (Capizzi et al., 2020).
TBI predisposes patients to several neurological complications; these post-traumatic complications may include seizures, dizziness, attention deficit, amnesia, executive dysfunction, irritability, and mood changes (Lozano et al., 2015). Depending on the head injury mode and impact severity, these complications may vary and last for a few days, months, or the rest of a patient’s life; for a detailed review on these topics please review Lozano et al (Lozano et al., 2015).
TBI is also a risk factor for post-traumatic epilepsy (Ding et al., 2016), chronic traumatic encephalopathy (CTE) (Lucke-Wold et al., 2014; McKee et al., 2009), Alzheimer’s disease (AD) (Jellinger, 2004; Uryu et al., 2007), Parkinson’s disease (PD) (Gardner et al., 2018), and amyotrophic lateral sclerosis (ALS) (Gardner and Yaffe, 2015), which constitute the leading causes of years lived with disability among TBI patients (Whiteford et al., 2015). This highlights the urge to conduct more research to gain a deeper understanding of TBI and to develop novel preventive and therapeutic rationales to diminish the substantial burden and socio-economical costs imposed by TBI.
The pathogenesis of TBI can be divided into a primary mechanical injury followed by a delayed secondary injury which is more pronounced in (DeKosky et al., 1998; Lazaridis et al., 2019; Salehi et al., 2017; Sulhan et al., 2020). The primary injury involves the direct brain damage that occurs instantly post- mechanical force, including extra parenchymal hemorrhages (epidural hematoma, subdural hematoma, subarachnoid hemorrhage, and intraventricular hemorrhage); focal contusions, and intraparenchymal hemorrhages; traumatic axonal (focal or diffuse) injury (TAI); and cerebral edema; depending on the mode of TBI injury (Jarrahi et al., 2020; Shaito et al., 2020). On the other hand, the secondary injury evolves minutes to days following the primary injury and may continue for up to several years (Galgano et al., 2017; Jarrahi et al., 2020; Kobeissy et al., 2006). It entails a series of neurochemical and metabolic events that further increase cerebral damage, such as excitotoxicity, mitochondrial dysfunction, metabolic abnormalities, oxidative stress, neuroinflammation, and blood-brain barrier (BBB) breakdown (Nasser et al., 2016; Ng and Lee, 2019; Shaito et al., 2020; Shakkour et al., 2021). Indeed, a better understanding of the underlying pathophysiological events of secondary injury may facilitate the discovery of potential therapeutic targets and diagnostic, and prognostic biomarkers of TBI (Abou-Abbass et al., 2016; Bayir et al., 2003; Kobeissy et al., 2006; Mallah et al., 2019). For this reason, there is increasing interest in understanding the role of the neuroinflammatory response as a double-edged sword following TBI.
Neuroinflammation is a pivotal innate immune response in the CNS characterized by the activation of resident immune cells of the brain (astrocytes and microglia); coupled with the release of cytokines, chemokines, and reactive oxygen species (ROS); and recruitment of peripherally derived immune cells toward the site of injury to promote neurorestorative processes (DiSabato et al., 2016; Xiong et al., 2018). When an innate inflammatory response is activated against invading pathogens and cellular damage, cytosolic multimeric protein complexes, called inflammasomes, are assembled (Broz and Dixit, 2016). Inflammasome activation in the CNS is predominant in microglia; nevertheless, expression of its components has also been demonstrated in other CNS-resident cell types, including neurons, astrocytes, perivascular CNS macrophages, oligodendrocytes, and endothelial cells (Voet et al., 2019).
Inflammasomes assemble in response to a wide range of pathogen-associated molecular (PAMPs) and damaged-associated molecular pattern molecules (DAMPs). This leads to proteolytic activation of inflammatory cysteine caspases thereby mediating the maturation of the pro-inflammatory cytokines interleukins-1β (IL-1β) and IL-18 and the initiation of a lytic programmed cell death process named pyroptosis (Broz and Dixit, 2016). Inflammasome assembly occurs in a stimulus-specific manner and can be activated via direct sensor protein-ligand binding as in the case of the “absent in melanoma 2” (AIM2) inflammasome and the “nucleotide-binding and oligomerization domain” (NOD)-like receptors (NLR) family CARD domain-containing protein 4 (NLRC4) inflammasome or by indirect sensing of distress or infection as in the case of the NLRP1, NLRP3 and pyrin inflammasomes (Christgen and Kanneganti, 2020). In TBI, several NLRP3 priming signals, such as heat shock proteins (HSPs), high mobility group box 1 (HMGB1) protein, reactive oxygen species (ROS), and Adenosine 5′-triphosphate (ATP) get recognized by Pattern Recognition Receptors (PRRs) such as toll-like receptors (TLRs). This activation triggers NF-κB-dependent upregulation of cellular NLRP3 and pro-IL-1β mRNA levels (Voet et al., 2019) and stimulates post-translational modifications (PTMs) of NLRP3 that license its activation (Swanson et al., 2019). In the following sections, we discuss the crosstalk between inflammasomes and TBI, and other neurodegenerative diseases. First, we provide an overview of inflammasome formation, maturation, and release. Then, we highlight their interaction within the CNS to modulate neurodegenerative diseases. Lastly, we discuss the novel potential therapeutics for targeting inflammasomes in TBI and their use as putative biomarkers.
Inflammasomes: Background and Cellular Expression
1. Main components of inflammasomes
Inflammasomes are cytosolic multimeric protein complexes that act as innate intracellular sensors of pathogens and foreign or host-derived danger signals aiming at maintaining tissue homeostasis (Voet et al., 2019). Inflammasomes are composed of three main components: a sensor component such as an NLR (NLRP3) (Figure 1), a procaspase-1 (the effector), and an adaptor protein. The sensor protein, which is a PRR, is responsible for recognizing pathogen-associated molecular patterns (PAMPs) or DAMPs signals (McKenzie et al., 2020). The cysteine protease procaspase-1 protein, on the other hand, mediates, upon its activation, the cleavage of pro-IL-1β and pro-IL-18 into their active forms. In addition, inflammasome activation may result in the pro-inflammatory form of cell death known as pyroptosis (McKenzie et al., 2020). The final component is the adaptor protein-ASC [apoptosis- speck-like protein containing a caspase-activating recruitment domain (CARD)] that connects the sensor protein to the protease component (McKenzie et al., 2020). In response to brain injury, glial cells produce high levels of ROS that trigger lipid peroxidation, ionic imbalance, and ATP release, thus causing cell necrosis (Irrera et al., 2020). After recognizing DAMPs and the elevated levels of ATP released from damaged cells, microglia will get activated and PRRs will stimulate transcriptional upregulation of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 (Takeuchi and Akira, 2010). TNF-α is a key player in promoting cell necrosis, which leads to membrane disruption, DAMPs release, and necrosis again; thus, exacerbating the inflammatory response and amplifying cell death mechanisms (Irrera et al., 2020). Hence, there is positive feedback between the inflammatory mediators released following PRRs activation and microglial activation leading to neuronal degeneration.
Figure 1: Structure of Inflammasome Components.
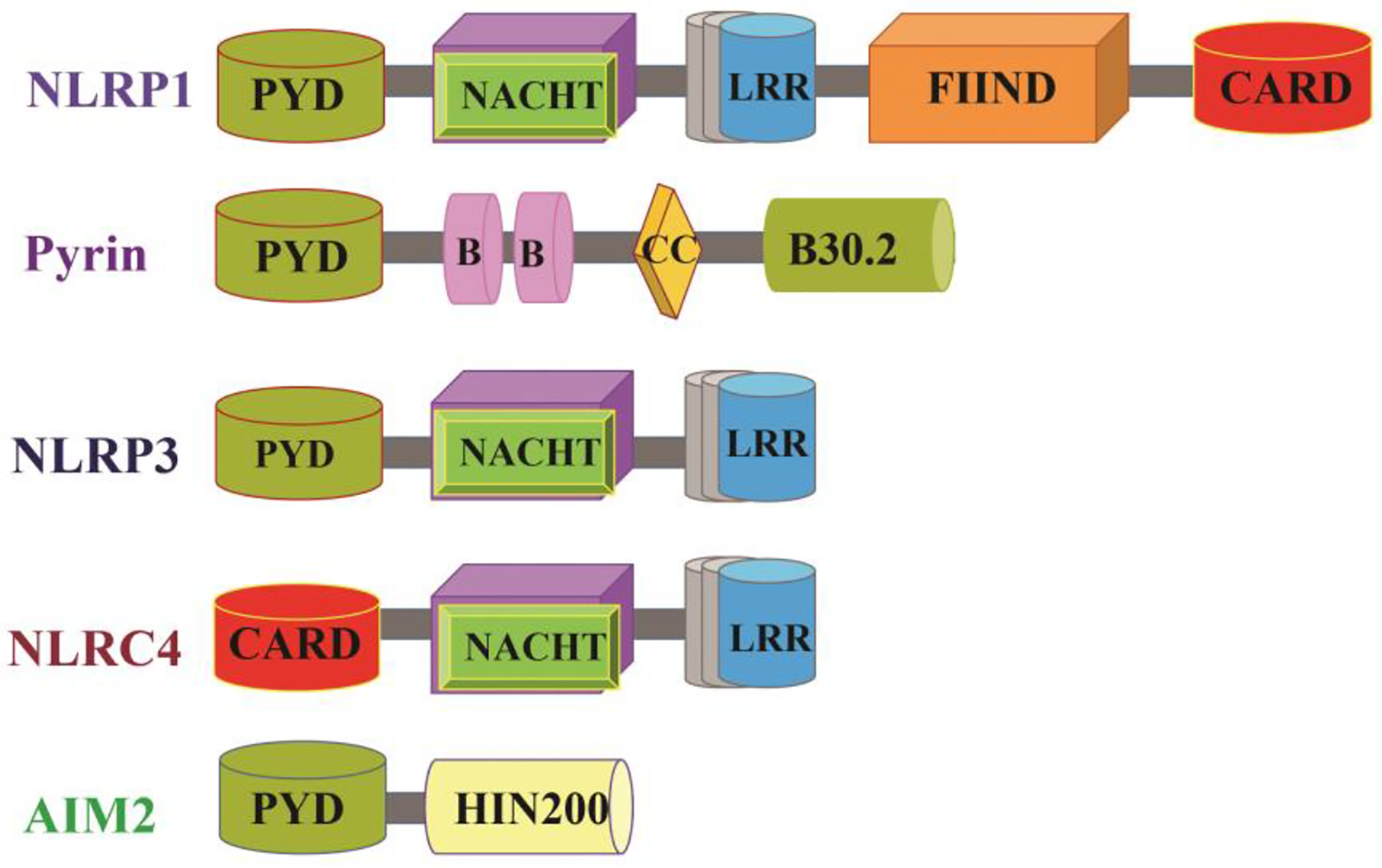
Inflammasomes comprise a sensor protein, cysteine protease caspase-1, and, an adaptor protein ASC in most cases. The inflammasome sensor proteins are classified according to their structural characteristics into NLRs, ALRs, and pyrin. NLR family members are marked by the familiar presence of nucleotide-binding and oligomerization domain (NACHT) in the center and leucine-rich repeat (LRR) motifs in the C-terminus. NLRP1 and NLRP3 have an N-terminal pyrin domain (PYD), whereas NLRC4 has N-terminal caspase activation and recruitment domain (CARD). NLRP1 has a distinct function-to-find domain (FIIND), which plays an important role in NLRP1 inflammasome activation. The adaptor protein ASC consists of a PYD and CARD, which mediates homotypic interactions with the PYD of inflammasome sensors and CARD of pro-caspase 1, respectively. Apart from the mentioned NLRs, the ALR family member AIM2 can also form an inflammasome. AIM2 is made up of an N-terminal PYD and a C-terminal DNA-binding HIN200 domain. Finally, Pyrin is composed of N-terminal PYD, two B-boxes, a coiled-coil domain, and a C-terminal B30.2 domain.
Abbreviations: NLR: Nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), ALR: absent in melanoma 2 (AIM2)-like receptor, NLRP1: NOD-, LRR- and pyrin domain-containing 1, NLRP3: NOD-, LRR- and pyrin domain-containing 3, NLRC4: NLR family CARD domain-containing protein 4, PYD: pyrin domain, FIIND: function-to-find domain, ASC: Apoptosis- speck-like protein containing a caspase-activating recruitment domain (CARD), CC: coiled-coil domain, HIN: hematopoietic expression, interferon-inducible nature, and nuclear localization
To date, five families of PRRs have been identified: Toll-like receptors (TLRs), C-type lectin receptors (CLRs), Rig-I-like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and absent in melanoma 2 (AIM2)-like receptors (ALRs) (Komada and Muruve, 2019). PRRs can be categorized based on their subcellular localization into membrane-bound PRRs (e.g., CLRs and some TLRs) and cytosolic PRRs (e.g., NLRs, ALRs, and RLRs) (Shin et al., 2019). In particular, some proteins from the NLRs and ALR families have gained considerable attention as they can form “inflammasomes”, cytosolic multiprotein complexes that are widely recognized now as essential coordinators of the initiation and prolongation of the neuroinflammatory response (Lahooti et al., 2021; Voet et al., 2019).
In addition to being present in neural cells including microglia, neurons and neurons, several PRR’s responsible for inflammasome assembly are expressed in many different types of cells such as dendritic cells, epithelial cells and neutrophils (Martinon et al., 2009). It is suggested that inflammasomes play a pathogenic role in epithelial cells (Alyaseer et al., 2020). Additionally, NLRP3 activation in M1 polarized macrophages was found to exacerbate cardiac dysfunction in relation to ischemic stroke (Lin et al., 2020). In a recently study, it was found that inflammasome activation exerted protective roles in SARS-CoV-2 infected macrophages by preventing viral amplification (Sefik et al., 2022). In the case of dendritic cells, inflammasome activation leads to either pyroptosis or hyperactivation that stimulates anti-tumor immunity (Zhivaki et al., 2020). Furthermore, Inflammasome-dependent signaling has been linked to the formation of neutrophil extracellular traps (NETs), in the progression of inflammatory diseases (Munzer et al., 2021). Based on the above-indicated findings, targeting these non-neural cells may control the pathogenesis of these inflammatory diseases which has been demonstrated by inhibiting NLRP3 which showed THP-1 macrophage foam cell formation, providing new therapeutic strategies for the management of atherosclerosis (Chen et al., 2018).
Inflammasomes: Structure and Function
As discussed previously, Inflammasomes are composed of the sensor component, the effector molecule, and the adaptor protein which together constitute the functional unit. We will discuss these components briefly to highlight their role in TBI and other disorders.
a. Sensor molecules
Several candidate inflammasome sensor proteins are classified according to their specific domain organizations (Figure 1) (Pandey et al., 2021). These sensor proteins or receptors are responsible for inflammasome assembly and include NLRs, absent in melanoma 2 (AIM2)–like receptors (ALRs), or pyrin (Malik and Kanneganti, 2017). NLR family members are all characterized by a nucleotide-binding and oligomerization domain (NACHT/NBD), a central or C-terminal leucine-rich repeat (LRR) domain, and a variable N-terminal domain (Malik and Kanneganti, 2017). The NLR family can be further subdivided into NLRP or NLRC based on whether the N terminal domain is a pyrin domain or a caspase activation and recruitment domain (CARD) (Malik and Kanneganti, 2017). Prominent NLRs, including NLRP1, NLRP3, and NLRC4, have been well-acknowledged to trigger inflammasome assembly leading to the activation of caspase-1 (Malik and Kanneganti, 2017). On the other hand, ALR sensors are characterized by an N-terminal pyrin domain (PYD) and a C-terminal DNA-binding hematopoietic interferon-inducible nuclear (HIN) domain (Latz et al., 2013). AIM2 is the only ALR family member capable of inducing inflammasome formation (Pandey et al., 2021). Lastly, pyrin, also known as “marenostrin” or TRIM20, consists of an N-terminal PYD, two B-boxes of a coiled-coil domain, and a C-terminal B30.2 domain (Malik and Kanneganti, 2017). This structure allows the recruitment of ASC at the N-terminal of the sensor molecule PYD (Fernandes-Alnemri et al., 2007).
a. Effector Pro-caspase 1
Under normal physiological conditions, caspase-1 is present in its catalytical inactivated form, pro-caspase 1. Induction of this precursor leads to its proteolytic maturation into caspase 1, which upon activation, matures and releases the pro-inflammatory cytokines IL-1β and IL-18 (Agostini et al., 2004). Furthermore, activated caspase 1 plays a crucial role in triggering pyroptosis or inflammasome-dependent cell death as shown in Figure 2 (Bergsbaken et al., 2009)
Figure 2: Assembly, Activation, and signaling of the Inflammasome Complex.
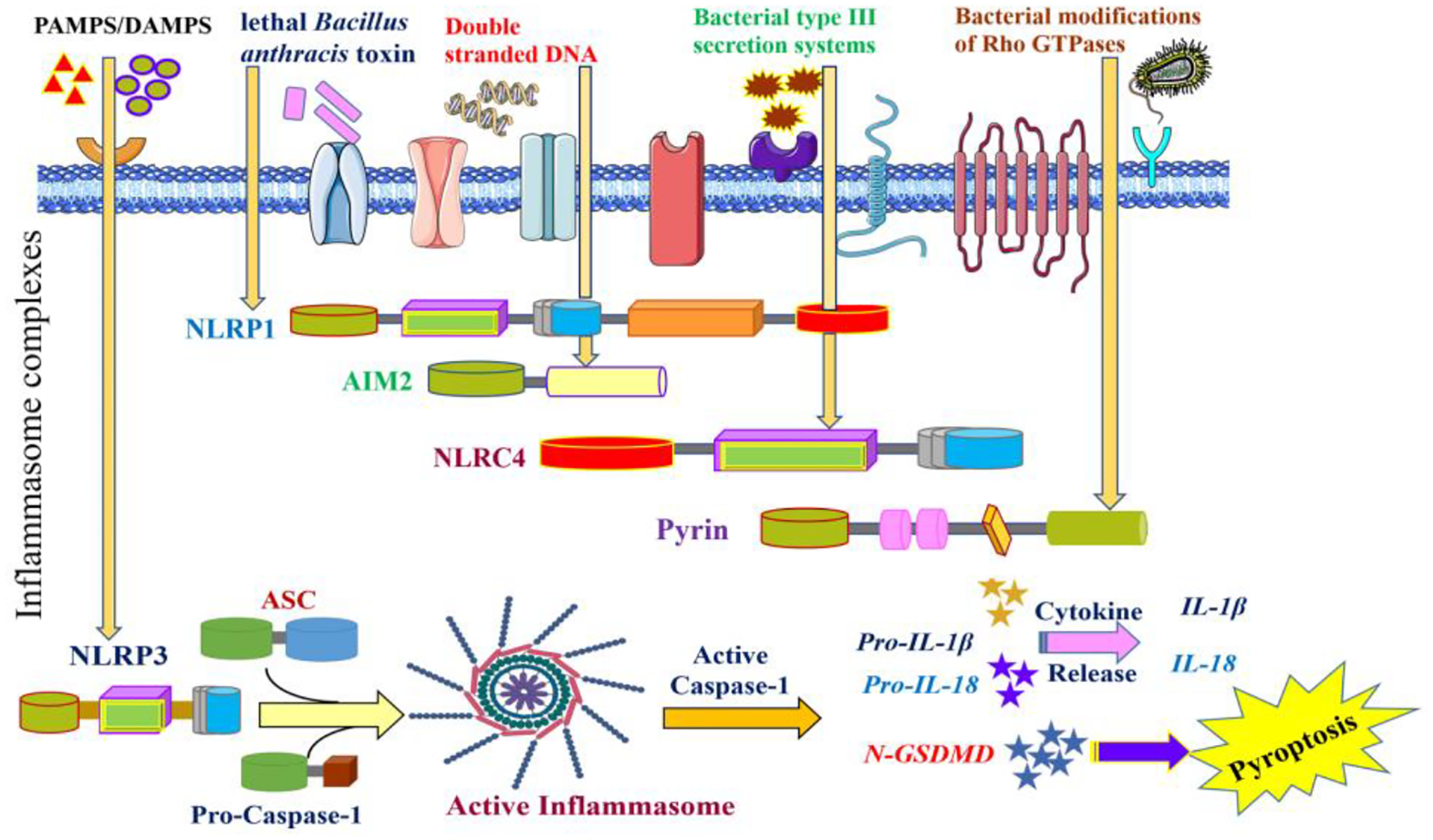
Inflammasomes assemble in response to different stimuli. NLRC4 responds to bacterial flagellin or type III secretion system of bacterial pathogens, NLRP1 recognizes Bacillus anthracis lethal toxin, AIM2 senses dsDNA, and pyrin detects the inactivation of RhoA by bacterial toxins and effector proteins. NLRP3 may be activated by different signals, including but not limited to potassium efflux, lysosomal disruption, and mitochondrial dysfunction; however, NLRP3 is unique as it requires a priming step before its activation. The priming step involves the binding of DAMPs/PAMPs to TLRs, which induces NF-kB-dependent transcriptional upregulation of NLRP3 and pro-IL1b. Following activation, the inflammasome sensors oligomerize to form a platform that recruits caspase-1 and the adaptor protein ASC. As a result, inflammasome assembly facilitates proximity-induced autocatalysis of pro-caspase 1 into active caspase-1. Caspase-1 cleaves pro-IL-1b and pro-IL-18 into the mature pro-inflammatory cytokines IL-1b and IL-18, respectively. Also, caspase-1 releases the GSDMD N-terminal domain, which translocates to and forms transmembrane pores in the plasma membrane through which IL-1b and IL-18 get released. The permeability of the GSDMD pores to ions and water causes cell swelling, leading to a pro-inflammatory form of lytic cell death named pyroptosis.
Abbreviations: NLRC4: NLR family CARD domain-containing protein 4, NLRP1: NOD-, LRR- and pyrin domain-containing, AIM2: absent in melanoma 2, dsDNA: double-stranded deoxyribonucleic acid, NLRP3: NOD-, LRR- and pyrin domain-containing 3, DAMPs: Damage-associated molecular patterns, PAMPs: Pathogen-associated molecular patterns, TLR: Toll-like receptor, NF-kB: nuclear factor kappa B, ASC: Apoptosis- speck-like protein containing a caspase-activating recruitment domain (CARD), GSDMD: Gasdermin D, LDH: lactate dehydrogenase, HSP: heat shock protein, HMGB1: high mobility group box 1, ATP: adenosine triphosphate
b. Adaptor ASC
Structural analysis of ASC reveals the presence of two domains: PYD and CARD (Broz and Dixit, 2016). PYD aids in homotypic interactions with PYD-containing inflammasome sensors that induce the oligomerization of ASC into a single large molecular complex called ASC speck (Broz and Dixit, 2016). On the other hand, the CARD domain of the adaptor allows the recruitment of monomers of procaspase-1 to the inflammasome complex through CARD-CARD interactions (Broz and Dixit, 2016). As a result, procaspase-1 undergoes proximity-induced autocleavage into caspase 1, which is the active form (Latz et al., 2013). NLRP1 and NLRC4 can recruit pro-caspase 1 independent of ASC via their CARD domain (Yang et al., 2019b). Nevertheless, ASC greatly improves the efficiency of cytokine processing, supporting its crucial role in the activity of inflammasomes (Latz et al., 2013).
2. Mechanisms of Inflammasome Activation
Inflammasome assembly occurs in a stimulus-specific manner, (Voet et al., 2019) stimulated by PAMPS or by DAMPs (Zheng et al., 2020) as shown in Figure 2. Inflammasome activation can be induced indirectly, by sensing any distress signal or infection (NLRP1, NLRP3, pyrin), or directly by sensor protein-ligand binding (AIM2, NLRC4) resulting in the assembly of their different components (Christgen and Kanneganti, 2020).
The NLRP3 inflammasome is stimulated by a wide range of exogenous stimuli such as foreign infectious antigens, crystalline substances, and particulate matter crystals such as silica (Broz and Dixit, 2016). In addition, NLRP3 can sense endogenous signals downstream of these exogenous stimuli including ATP, mitochondrial dysfunction, nucleic acids, lysosomal rupture, potassium efflux, as well as ROS elevation (Fusco et al., 2020). NLRP3 activation is unique in that it requires a priming signal recognized by the PRRs, including the TLRs and C-type lectin receptors (CLRs) (Voet et al., 2019). The priming signal will then trigger NF-κB-dependent upregulation of cellular NLRP3 and pro-IL-1β transcription (Voet et al., 2019), as well as several PTMs, including ubiquitylation, phosphorylation, and SUMOylation of NLRP3 (Swanson et al., 2019). This is followed by a second activation signal by various PAMPs and DAMPs that induces full NLRP3 inflammasome activation and assembly (Heneka et al., 2018).
Unlike NLRP3, other inflammasome sensors do not require the initial priming step for inflammasome activation (Voet et al., 2019). For instance, flagellin and the gram-negative type III secretion systems of Salmonella induce the NLRC4 inflammasome by binding and activating the NLR family apoptosis inhibitory proteins (NAIPs) (Yang et al., 2019a). Similarly, NLRP1 can induce inflammasome assembly and pyroptosis unassisted by the adaptor ASC when the Bacillus-produced anthracis lethal toxin is detected (Lamkanfi and Dixit, 2014).
Following the activation of inflammasome sensors by endogenous or exogenous stimuli, they undergo oligomerization to allow the recruitment of ASC and pro-caspase 1, resulting in a full inflammasome complex (Walsh et al., 2014). The latter facilitates a proximity-induced auto-cleavage of pro-caspase 1 into active caspase-1 (Latz et al., 2013) that will cleave pro-IL-1β and pro-IL-18 into the mature pro-inflammatory cytokines IL-1β and IL-18, respectively (Xue et al., 2019). In addition, caspase-1 cleaves Gasdermin-D (GSMD) protein to liberate the N-terminal domain (N-GSDMD) that oligomerizes and perforates the plasma membrane resulting in osmotic swelling and cell rupture (Xue et al., 2019). GSDM-mediated cell lysis is a pro-inflammatory form of cell death named pyroptosis (McKenzie et al., 2020). It involves the extracellular release of the pro-inflammatory cytokines IL-1β and IL-18, and other intracellular components such as the HSPs, HMGB1, and ATP, as well as other molecules (McKenzie et al., 2020).
Inflammasome Activation and Cell Death
Inflammasome activation can be induced using a vast array of stimulants that play a significant role in infectious or inflammatory diseases, as well as molecules that occur during tissue damage or metabolic imbalances. Since inflammasomes play a critical role in the inflammatory response, their activation requires a finely regulated process consisting of two steps: priming and activation (Latz et al., 2013). For instance, different priming stimuli can be used to stimulate NLRP3 and IL-1β upregulation (normally existing in low concentrations at basal levels) such as ligands for TLRs (Bauernfeind et al., 2009). The second activation step can be induced using a wide array of activators such as ATP, glucose, monosodium urate crystals (MSU), phospholipid platelet-activating factor (PAF), and nigericin (Amores-Iniesta et al., 2017; Deng et al., 2019; Mouasni et al., 2019; Piancone et al., 2018a).
Furthermore, as previously mentioned, ASC oligomerization serves as a distinguishing feature for inflammasome activation and has been commonly used as a reliable downstream readout for inflammasome activity (Stutz et al., 2013). Regulated cell death mechanisms, such as pyroptosis, have been studied considering their close association with inflammation. Generally, pyroptosis can be identified through staining for annexin V and propidium iodide (Fink and Cookson, 2006; Koopman et al., 1994). Moreover, enzymatic assays that can be used to detect pyroptotic cells include Lactate Dehydrogenase (LDH) assay and Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay (Jia et al., 2019; Tajima et al., 2019), although they are not specific to pyroptosis. Above all approaches, the discovery of GSDMD is what redefined the known paradigm of pyroptosis (Shi et al., 2015).
Inflammasome Activation Following TBI
The degree of the neuroinflammatory process initiated by neural traumatic events is dependent on the mechanism of injury (focal, diffuse, blast), degree of injury (mild, moderate, severe), secondary insults (hypoxemia, hypotension, uncontrolled immunological response); in addition to external factors such as the patient’s age, sex, lifestyle and genetic variability (Schimmel et al., 2017; Xiong et al., 2018). Inflammasomes have gained considerable attention as essential coordinators and regulators of the neuroinflammatory response. Consequently, extensive research has been conducted to understand their roles in TBI (Figure 3).
Figure 3: NLRP3 Inflammasome Activation and Assembly in response to traumatic brain injury.
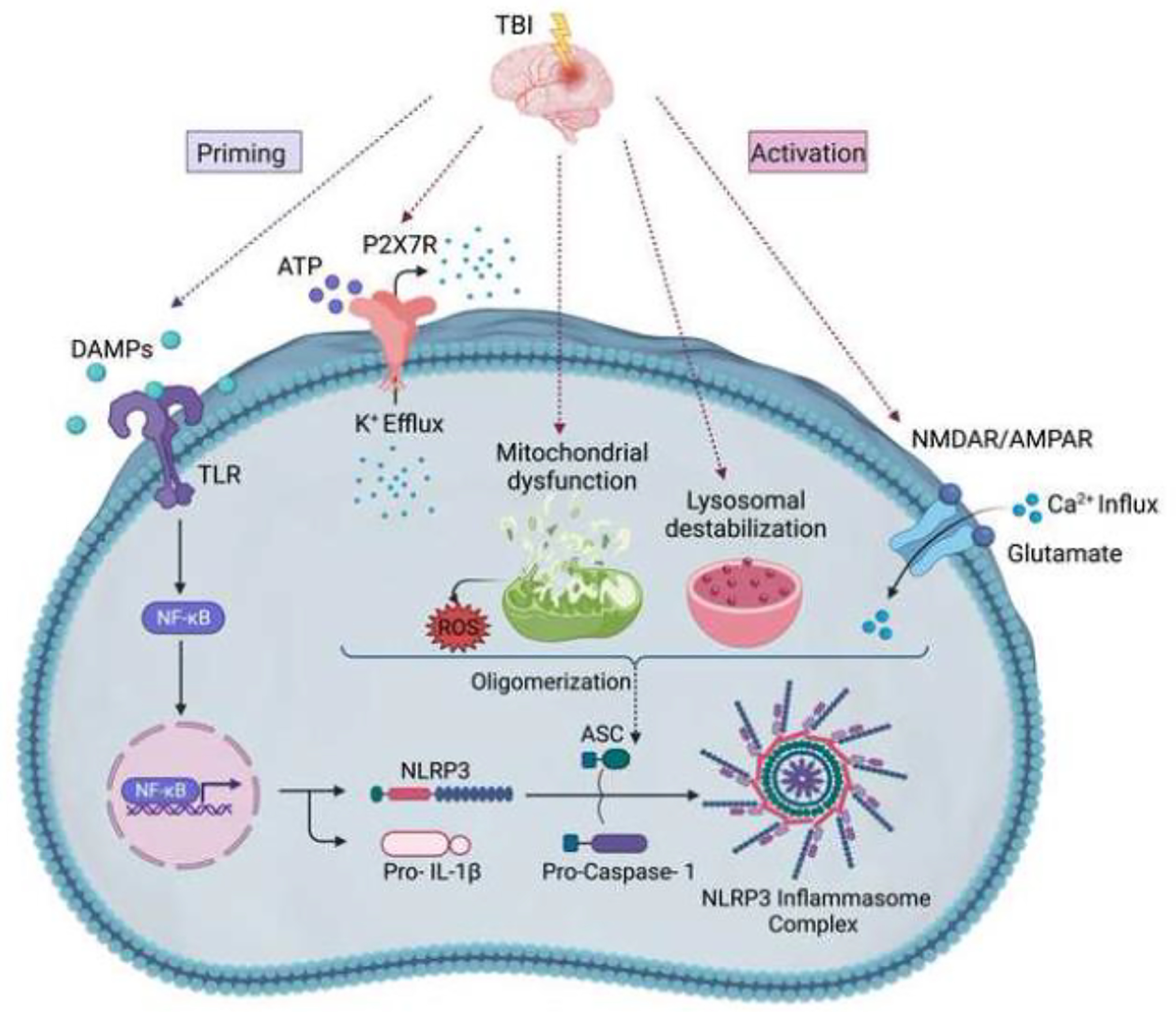
Traumatic brain injury-induced brain damage involves the release of several damage-associated molecular patterns (DAMPs) that act as priming signals and trigger transcriptional upregulation of NLRP3 and pro-IL-1β through the TLR/NF-κB pathway. Following priming, several activating signals induce the oligomerization of the inflammasome sensors (NLRP3), followed by the recruitment of ASC and pro-caspase-1, resulting in a full NLRP3 inflammasome complex. These activating signals include the potassium efflux after the activation of purinergic P2X7 receptor channels by ATP; massive calcium influx upon glutamate binding to its ionic receptors (NMDAR/AMPAR); mitochondrial injury-induced ROS release; and lysosomal destabilization.
Abbreviations: DAMPs: Damage-associated molecular patterns, ATP: Adenosine triphosphate, TLR: Toll-like receptor, NF-κB: Nuclear factor kappa B, NLRP3: NOD-, LRR- and pyrin domain-containing 3, ASC: Apoptosis- speck-like protein containing a caspase-activating recruitment domain (CARD), P2X7R: purinergic P2X7 receptor channels, NMDAR: N-methyl-D-aspartate receptor, AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor, ROS: reactive oxygen species, IL-1β: Interleukin-1β, IL-18: Interleukin-18.
Several experimental studies have reported inflammasome activation in TBI-induced inflammatory response (Table 1). The first inflammasome to be described post-TBI was the NLRP1 inflammasome in neurons in a rat model of fluid-percussion traumatic brain injury (FPI) (de Rivero Vaccari et al., 2009). The NLRP1 inflammasome, post-TBI, was shown to be comprised of NLRP1, caspase-1, ASC, and XIAP (de Rivero Vaccari et al., 2009) that form protein-protein interactions with pannexin-1 (Silverman et al., 2009) in neurons and astrocytes. Similarly, Liu et al. were the first to investigate NLRP3 inflammasome expression following the closed head weight drop TBI model in rats (Liu et al., 2013). They found out that TBI induced NLRP3 inflammasome activation and upregulation of ASC and cleaved caspase-1, which increased the maturation and release of IL-1β and IL-18. They detected NLRP3 inflammasome in neurons, astrocytes, and microglia in the pericontusional cortex 3 days after TBI. Moreover, their data demonstrated that IL-1β exacerbates the inflammatory response in the early phase of TBI; however, IL-18 is involved in neuronal damage in the late phase post-TBI. This is consistent with the results of another study that detected increased IL-18 levels at 7 days following a mouse model of closed head injury, suggesting its involvement in the perpetuation of the inflammatory response following TBI (Yatsiv et al., 2002).
Table 1:
Experimental studies reporting inflammasome activation following traumatic brain injury.
| Species | TBI model | Inflammasome identified | Components of inflammasomes identified | Method of detection | Timepoints | Site of detection | Major findings | Ref |
|---|---|---|---|---|---|---|---|---|
| Adult male Sprague–Dawley rats | Closed head weight drop using 40 g steel weight. | NLRP3 | NLRP3, ASC, Caspase-1 |
|
6 h, 24 h, 3d, 7d post-injury |
|
|
(Liu et al., 2013) |
| Adult male Sprague–Dawley rats | Closed moderate (1.7–2.2 atmospheres) Parasagittal FPI | NLRP1 | NLRP1, ASC, Caspase-1, Caspase-11 |
|
15 min, 30 min, 1 h, 3 h, 6 h, and 24 h post-injury | Cortical neurons at 4 h post-trauma |
|
(de de de Rivero Vaccari et al., 2009) |
| Male C57/BL6 mice |
|
NLRP3 | NLRP3, ASC, Caspase-1 |
|
1d, 3d, 7d post-injury | Microglia in the pericontusional cortex at 3-day post-trauma. |
|
(Xu et al., 2018) |
| Adult 3-month-old C57BL/6 J male mice |
|
NLRP3 | NLRP3, ASC, Caspase-1 |
|
1d, 2d, 4d, 7d, 14d post-injury | Cerebral cortex |
|
(Ma et al., 2017a) |
| Male C57/BL6 mice |
|
NLRP3, NLRC4, NLRP1, AIM2 | NLRP3, NLRC4, NLRP1, AIM2, Caspase-1 |
|
1d, 2d, 3d, 7d post-injury | Microglia in the peri-injury cortex at 3-day post-trauma. |
|
(Du et al., 2022) |
| C57/BL6 mice |
|
NLRP3 | NLRP3, ASC, Caspase-1 |
|
1d and 3d post-injury | Pericontusional cortex |
|
(Ismael et al., 2018a) |
TBI:Traumatic Brain Injury, CCI:Controlled Cortical Impact, FPI:Fluid Percussion Injury, NLRP3:NOD-, LRR- and pyrin domain-containing 3, NLRC4:NLR family CARD domain-containing protein 4, NLRP1:NOD-, LRR- and pyrin domain-containing, AIM2:absent in melanoma 2, ASC:Apoptosis- speck-like protein containing a caspase-activating recruitment domain (CARD), IL-1β:Interleukin-1β, IL-18:Interleukin-18, TXNIP:thioredoxin-interacting protein, XIAP:X-linked inhibitor of apoptosis protein
Later studies conducted later consistently reported upregulation of NLRP3, ASC, and caspase-1 following moderate penetrating in a mouse model of controlled cortical impact (CCI) experimental TBI (Ismael et al., 2018a; Ma et al., 2017a; Xu et al., 2018). While Liu et al. reported that the components of NLRP3 inflammasome were upregulated in a time-dependent manner from 6 hours to 7 days post-TBI in a rat model (Liu et al., 2013), Xu et al. indicated that CCI significantly increased the expression of NLRP3, ASC, and caspase-1 from one to seven days, peaking at three days post-TBI in mice (Xu et al., 2018). Indeed, this difference could be explained by the fact that the two studies used different TBI models, animals, and injury severity. Interestingly, Xu et al. also confirmed using flow cytometry and double immunofluorescence staining that NLRP3 was found primarily in microglia and not in astrocytes or neurons (Xu et al., 2018), which is different from what Liu et al. reported (Liu et al., 2013).
Because of the accumulating evidence of inflammasome activation following TBI, several studies of genetically manipulated inflammasome-associated proteins were evaluated to understand their contribution to brain damage (Table 2). For instance, the genetic ablation of NLRP3 was investigated to attenuate the inflammatory response and neuronal death in the mouse impact-acceleration model of diffuse TBI. It was shown that following TBI, the NLRP3 knock-out mice had more preserved brain tissue, diminished brain edema, better cognitive function, and reduced release of pro-inflammatory cytokines and apoptosis. Their results validate the critical role that NLRP3 plays in the pathogenesis of TBI and implicate the possibility of using it as a potential target to mitigate the course of injury (Irrera et al., 2017). As for NLRP1 inflammasome, de Rivero Vaccari et al. showed that moderate FPI triggers NLRP1 inflammasome activation leading to caspase-1, IL-1β, and XIAP processing (de Rivero Vaccari et al., 2009). They detected NLRP1 inflammasome in the cerebral cortical neurons of rats suggesting that microglia may not be the major source of IL-1β in the brain (de Rivero Vaccari et al., 2009).
Table 2:
Experimental studies assessing the effects of genetically manipulating inflammasome-associated proteins on traumatic brain injury-induced inflammatory response and brain damage.
| Subject | Subject average | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | H-scan US | B-scan US | |
| Kernel size = 160 × 160 μm | |||||||
| p-value | < 0.001 | < 0.001 | < 0.001 | < 0.001 | < 0.001 | < 0.001 | < 0.001 |
| R 2 | 0.48 | 0.29 | 0.33 | 0.52 | 0.37 | 0.31 | 0.09 |
| 95% CI | 0.45 to 0.51 | 0.26 to 0.32 | 0.29 to 0.36 | 0.49 to 0.56 | 0.34 to 0.41 | 0.30 to 0.33 | 0.08 to 0.10 |
| Slope | 2.9 | 3.9 | 3.7 | 3.4 | 3.5 | 3.2 | 2.1 |
| 95% CI | 3.1 | 3.6 to 4.1 | 3.5 to 3.9 | 3.2 to 3.5 | 3.3 to 3.7 | 3.1 to 3.3 | 2.0 to 2.2 |
| Kernel size = 320 × 320 μm | |||||||
| p-value | < 0.001 | < 0.001 | < 0.001 | < 0.001 | < 0.001 | < 0.001 | < 0.001 |
| R 2 | 0.50 | 0.30 | 0.32 | 0.60 | 0.28 | 0.40 | 0.12 |
| 95% CI | 0.43 to 0.55 | 0.24 to 0.37 | 0.26 to 0.38 | 0.54 to 0.65 | 0.21 to 0.35 | 0.37 to 0.43 | 0.10 to 0.15 |
| Slope | 3.1 | 4.9 | 4.2 | 3.5 | 3.3 | 2.2 | 2.6 |
| 95% CI | 2.9 to 3.4 | 4.3 to 5.5 | 3.7 to 4.7 | 3.2 to 3.8 | 2.8 to 3.8 | 2.1 to 2.3 | 2.3 to 2.8 |
| Kernel size = 800 × 800 μm | |||||||
| p-value | < 0.001 | < 0.001 | < 0.001 | < 0.001 | < 0.001 | < 0.001 | < 0.001 |
| R 2 | 0.44 | 0.43 | 0.19 | 0.73 | 0.21 | 0.27 | 0.18 |
| 95% CI | 0.25 to 0.60 | 0.26 to 0.59 | 0.05 to 0.36 | 0.58 to 0.83 | 0.05 to 0.40 | 0.19 to 0.35 | 0.11 to 0.26 |
| Slope | 3.3 | 6.8 | 3.1 | 3.7 | 2.7 | 3.0 | 3.3 |
| 95% CI | 2.4 to 4.2 | 5.0 to 8.5 | 1.6 to 4.6 | 3.1 to 4.3 | 1.4 to 4.1 | 2.5 to 3.5 | 2.6 to 4.1 |
TBI:Traumatic Brain Injury, CCI:Controlled Cortical Impact, NLRP3:NOD-, LRR- and pyrin domain-containing 3, NLRP1:NOD-, LRR- and pyrin domain-containing, ASC:Apoptosis- speck-like protein containing a caspase-activating recruitment domain (CARD), IL-1β:Interleukin-1β, IL-18:Interleukin-18, IFNγ: Interferon γ, TNF-α:Tumor Necrosis Factor α, TGF-β1:Tumor Growth Factor β1, NSS:Neurological Severity Score, mNSS:modified Neurological Severity Score, LDH:Lactate Dehydrogenase, CD68:Cluster of Differentiation 68, GFAP:Glial Fibrillary Acidic Protein, MAP2:Microtubule-associated protein 2, NeuN:Neuronal Nuclear, PSD95:Postsynaptic density protein 9, VAMP:vesicle-associated membrane protein, SNAP25:Synaptosomal-Associated Protein, 25kDa, TUNEL:Terminal deoxynucleotidyl transferase dUTP nick end labeling, KO:Knock Out, WT:wild-type, GSDMD:Gasdermin D, BAX:Bcl-2-associated X
Furthermore, in the same study, ASC neutralization with a neutralizing antibody against ASC reduced caspase-1 cleavage and enhanced tissue sparing, demonstrating how inflammasomes play a pivotal role in acute neural injury and suggesting a potential therapeutic target for decreasing the inflammatory responses induced by TBI (de Rivero Vaccari et al., 2009). Surprisingly, Brickler et al. were the first to show a non-essential role for the NLRP1 inflammasome in mice moderate CCI model with the genetic deletion of NLRP1 or ACS (Brickler et al., 2016). No significant changes in contusion volume, motor recovery, histopathology, or hippocampal cell death were detected (Table 2) (Brickler et al., 2016). However, in that study, protein levels of caspase-1 were not addressed. Thus, it remains unknown whether the main effector of canonical inflammasome activation (caspase-1) was decreased by the genetic ablation of NLRP1 or ASC. Nonetheless, This discrepancy between the effects of ASC neutralization (de Rivero Vaccari et al., 2009) and NLRP1 or ASC knockout (Brickler et al., 2016) could be due to using different species and TBI models, highlighting the need to further explore the role of NLRP1 inflammasome in other TBI models to develop an effective therapeutic strategy.
As mentioned earlier, many studies have described the participation of several inflammasomes, including NLRP3, NLRP1, NLRC4, and AIM2, in the progression of the neuroinflammatory response and the development of neuropathological changes following TBI (de Rivero Vaccari et al., 2009; Ge et al., 2018; Ismael et al., 2018a; Liu et al., 2013; Ma et al., 2017a; Sun et al., 2020; Xu et al., 2018); however, none of these studies attempted to identify the most prominent inflammasome involved in inducing pyroptosis. Interestingly, a very recent study performed by Du et al. tried to fill this gap of knowledge by exploring the role of GSDMD, a pyroptosis executor, in modulating the pathogenesis of TBI (Figure 4) (Du et al., 2022). First, they demonstrated that GSDMD knockout showed striking differences in improving the behavioral outcomes, reducing synaptic protein loss, decreasing the release of pro-inflammatory cytokines (IL-1β and TNF-α), and increasing the release of anti-inflammatory cytokines (IL-10 and TGF-β1) 3 days following CCI in mice. These findings support the potential of exploiting GSDMD as a therapeutic target for TBI. Second, they explored the temporal expression patterns of NLRP3, NLRP1, NLRC4, and AIM2 inflammasome and found that NLRP3 was significantly high from 1 to 7 days after TBI, whereas AIM2, NLRP1, and NLRC4 increased significantly only one day following TBI. This showed that the NLRP3 inflammasome is the dominant contributor among all the other inflammasomes to the progression of the neuroinflammatory response following TBI. Finally, they showed that NLRP3 knockout suppressed the expression and cleavage of GSDMD and exhibited the exact effects that were obtained upon knocking out GSDMD. Building upon the similarity in the temporal expression patterns of NLRP3 and N-GSDMD and the mentioned findings, this study confirmed that the inhibition of GSDMD is a promising therapeutic target that is primarily mediated by the NLRP3 inflammasome. However, it should be noted that further studies are needed to explore the long-term effects of GSDMD inhibition on the damage induced by TBI in the chronic stage.
Figure 4: GSDMD-mediated Pyroptosis Following NLRP3 Inflammasome Activation in Traumatic Brain Injury.
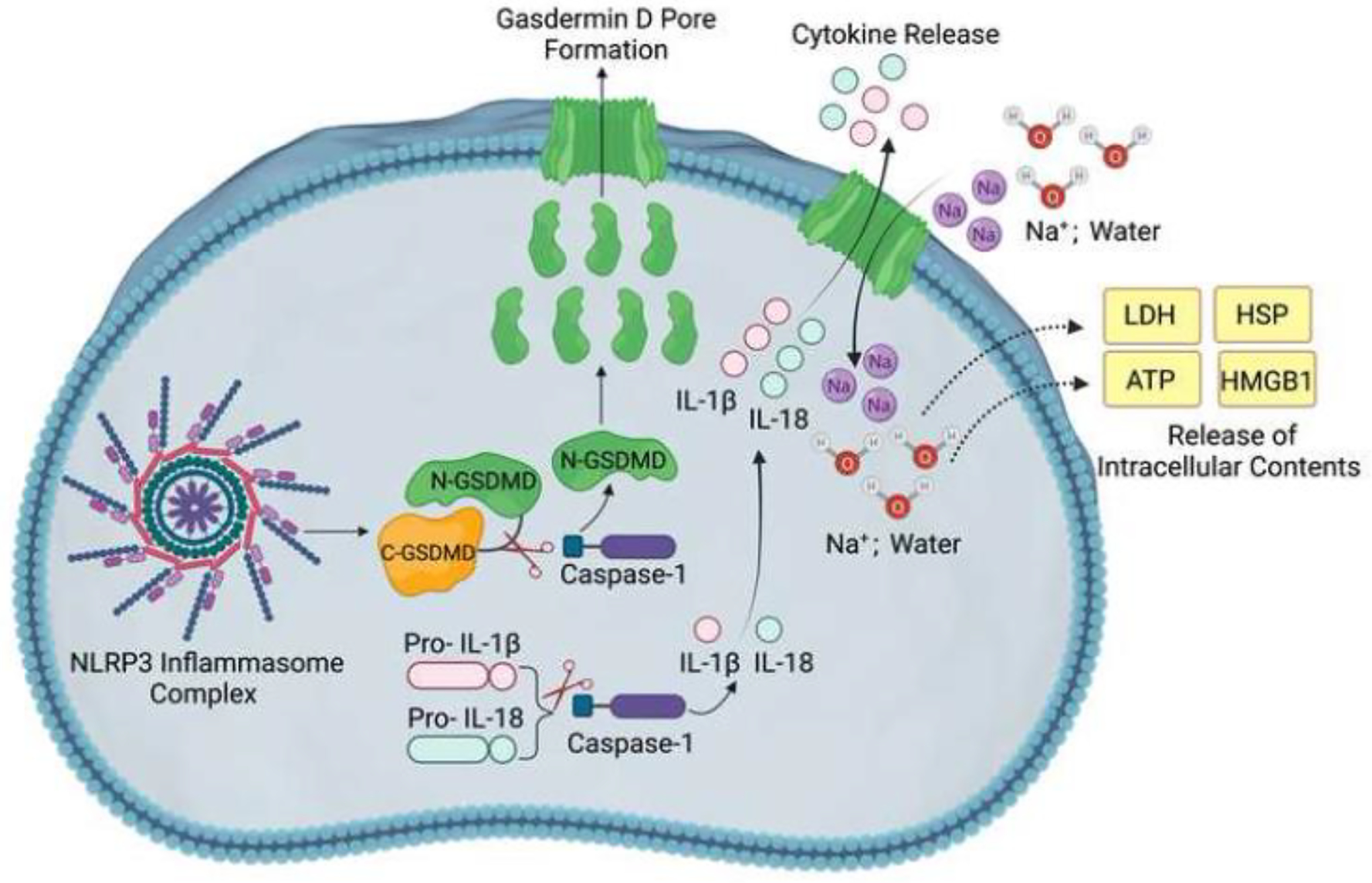
Inflammasome assembly facilitates autocatalysis of pro-caspase 1 into active caspase-1. Caspase-1 cleaves pro-IL-1β and pro-IL-18 into the mature pro-inflammatory cytokines IL-1β and IL-18, respectively. Also, caspase-1 releases GSDMD N-terminal domain, which translocates to and forms transmembrane pores in the plasma membrane through which IL-1β and IL-18 get released. The permeability of the GSDMD pores to ions and water causes cell swelling, leading to membrane rupture and the release of intracellular contents, including LDH, HSP, HMGB1, and ATP.
Abbreviations: NLRP3: NOD-, LRR- and pyrin domain-containing 3, GSDMD: Gasdermin D, IL-1β: Interleukin-1β, IL-18: Interleukin-18, LDH: lactate dehydrogenase, HSP: heat shock protein, HMGB1: high mobility group box 1, ATP: adenosine triphosphate.
Besides ASC, GSDMD, and NLRP3 as being potential therapeutic targets for the treatment of TBI, Liu et al. were interested in investigating whether the genetic ablation or the expression reduction of caspase-1 through siRNA knockdown also exerts neuroprotective effects on TBI in a mouse model of moderate CCI (Liu et al., 2018a). it was found that caspase-1 deficiency mitigated neurological and behavioral deficits, attenuated the release of pro-inflammatory cytokines (IL-1β and TNF-α) and LDH, amplified the release of anti-inflammatory cytokines (IL-10 and TGF-β1), and reduced the expression of pyroptosis-related proteins (caspase-1 p45, caspase-11, and GSDMD). This implies that caspase-1 is a potent therapeutic target as its inhibition may reduce TBI-induced neuroinflammation and pyroptosis.
Moreover, the inflammasome not only plays a role in the innate immune response taking place in the brain after TBI, but it also contributes to systemic inflammation post-brain injury. TBI results in systemic complications such as sepsis, pneumonia, and multiple organ dysfunction, which are associated with worse outcomes after TBI (Kinoshita, 2016). Extracranial organ failure after TBI has generally been associated with a catecholamine surge that originates in the hypothalamic-pituitary axis that releases epinephrine and norepinephrine (Nguyen and Zaroff, 2009); hence, activating the adrenal glands and increasing the arterial pressure due to peripheral vessel vasoconstriction (Kinoshita, 2016); thus, affecting the cardiovascular system. In addition, following TBI, increased intracranial pressure results in sympathetic activation, cardiovascular complications, and systemic inflammation (Wijayatilake et al., 2015). Furthermore, a systemic consequence of TBI is acute respiratory distress syndrome (ARDS). To this regard, a brain-lung-axis has been described through extracellular vesicles (EVs) containing inflammasome proteins are carried from the brain to the lungs after TBI, resulting in acute lung injury (ALI) in an animal model of severe TBI (Adamczak et al., 2012; Kerr et al., 2020; Kerr et al., 2018c; Kerr et al., 2019; Kerr et al., 2021). Accordingly, the adoptive transfer of EVs containing inflammasome proteins from TBI mice into naïve mice resulted in ALI. An effect that was in part decreased by inhibition of the inflammasome with a monoclonal antibody against ASC and Enoxaparin, the latter of which interferes with EV trafficking (Kerr et al., 2021). Thus, the inflammasome contributes to the systemic inflammatory response after TBI. Whether the inflammasome is involved in the inflammatory response present in other organs after TBI is under investigation.
In addition to the direct effects of TBI on the brain and on the effects on peripheral organs such as the lung and the heart secondary to TBI, pre-existing conditions and peripheral trauma such as in cases of polytrauma can also prime the inflammasome, which has the potential to worsen outcomes after TBI. These pre-existing conditions capable of priming the inflammasome are characterized by a heightened chronic inflammatory response such as in patients with obesity, hypertension, atherosclerosis, or diabetes as well as acute inflammatory responses due to active infections (Kwan et al., 2013; Murray et al., 2015). Similarly, a heightened inflammatory response occurs as a result of aging, which also activates the inflammasome and primes the CNS for a more exacerbated inflammatory response in the event that a TBI is to take place (Cyr et al., 2022; Mejias et al., 2018). Hence, a primed immune system by either aging, infections, polytrauma or other comorbidities, make the CNS a more vulnerable target after TBI, which could potentially result in worse outcome measures in these patients.”
Role of Inflammasomes in Neurodegenerative Disorders
While the links and associations between TBI and neurodegenerative diseases have been somewhat controversial and inconsistent, hypotheses remain to be validated or rejected relating processes of neuroinflammation, chronic microglial activation, aggregation, decreased degradation, and/or reduced cognitive reserve following TBI in predisposing or leading to certain neurodegenerative diseases (Brett et al., 2022; DeKosky and Asken, 2017; Wilson et al., 2017). Neuroinflammation is characterized by the activation of resident innate immune and astrocytic cells as well as the recruitment of peripheral immune cells to nervous system tissues where it plays a major role in the progression and the pathogenesis of neurodegenerative diseases (DiSabato et al., 2016). Glial cells (mainly microglia and astrocytes), endothelial cells, and recruited immune cells release pro and anti-inflammatory signals (DiSabato et al., 2016). These can also alter the BBB permeability and recruit peripheral immune cells post-neural damage (Piancone et al., 2021; Ravichandran and Heneka, 2021). Because of the substantial contribution of neuroinflammation and the inflammasome to the pathogenesis of brain diseases, it follows that targeting inflammasome activity following TBI is a clinically relevant strategy to attenuate neuronal damage and possibly delay or reduce the risk of developing neurodegenerative diseases (Brett et al., 2022; DeKosky and Asken, 2017). There are also calls to consider neurodegenerative diseases post-TBI, such as traumatic encephalopathy or trauma-induced neurodegeneration (Washington et al., 2016). In fact, several studies on human samples have found that the NLRP3, IL-1β, IL-18, and caspase-1 levels are elevated in Alzheimer’s Disease (AD), Parkinson’s Disease (PD), Multiple Sclerosis (MS), and Amyotrophic Lateral Sclerosis (ALS), while other studies have found no such significant differences (Piancone et al., 2018a; Ravichandran and Heneka, 2021). Despite some studies finding little to no evidence of increased inflammasomal activation in studies with humans, many have found a link between inflammasomal activation in clinical and post-mortem studies, as well as studies using rodent models or in vitro cultures (Gordon et al., 2018; Ising et al., 2019; Johann et al., 2015; Kadhim et al., 2016; Keane et al., 2018; Moreno-Garcia et al., 2021; Peelen et al., 2015; Piancone et al., 2018b; Pontillo et al., 2012; Saresella et al., 2014; Sarkar et al., 2017; Trudler et al., 2021). NLRP3, for example, has been implicated in AD, PD, MS, and ALS. Findings of elevated levels of NLRP3 and/or its downstream pro-inflammatory response have been somewhat consistent. Additionally, studies have found improved outcomes (e.g., decreased pro-inflammatory interleukins or ASC) through the administration of inflammasome inhibitors or in NLRP3 or ASC-deficient mice. Adding these findings to evidence pointing towards a role for inflammasomal activation in driving certain pathologies of neurodegenerative diseases (Ising et al., 2019), it becomes necessary to investigate the possible mechanisms (inflammasomal or otherwise) that can link those two sets of findings. While the involvement of specific inflammasomal pathways differs by disease and even within disease subcategories (e.g., sporadic ALS), it remains to be seen whether targeting these inflammasomal pathways can slow down the chronic sequelae following TBI or even be used to predict predisposition to certain neurodegenerative diseases over others.
The Inflammasome after TBI and Alzheimer’s Disease and the missing link: Implications for Chronic Traumatic Encephalopathy (CTE)
AD is characterized by the accumulation of extracellular aggregated proteins known as amyloid β (Aβ) plaques, the aggregation of neurofibrillary tau tangles (NFTs), and gliosis (Knopman et al., 2021). These toxic self-aggregation products are accompanied by neuroinflammation and neuronal cell death (Piancone et al., 2021; Ravichandran and Heneka, 2021). Halle et al. investigated treating immortalized microglial cells isolated from C57BL/6 mice with fibrillary Aβ (fAβ) and studied its effect on the NLRP3 inflammasome (Halle et al., 2008). In that study, fAβ-treated cells exhibited increased caspase-1 activation and higher IL-1β levels compared to the untreated group. The authors attributed caspase-1 activation and IL-1β release to NLRP3 activation because bone-marrow-derived macrophages (BMDM) from NLRP3-deficient (Nlrp3−/−) or ASC-deficient (Pycard−/−) mice did not secrete IL-1β in response to fAβ (Halle et al., 2008). Moreover, they also observed increased lysosomal damage accompanied by a greater release of cathepsin-B, a lysosomal enzyme required for NLRP3 activation (Chevriaux et al., 2020), and higher levels of IL-1β and caspase-1. Likewise, Heneka et al. found increased cleaved caspase-1 in human cortical and hippocampal lysates collected from patients with mild cognitive impairment, early-onset AD, or AD (Heneka et al., 2013). Similar findings were obtained in a mouse model of AD expressing human APP and presenilin-1 (Heneka et al., 2013). NLRP3 deficiency caused a decrease in neuroinflammation and Aβ aggregation, with APP/PS1 mice exhibiting improved locomotion, habituation, hippocampal-related memory, and synaptic plasticity. Additionally, microglia activation in NLRP3-deficient and caspase-1-deficient APP/PS1 mice caused an increase in Aβ phagocytosis and promoted an M2 phenotype, which is an anti-inflammatory functional phenotype. The latter induces a decrease in cerebral nitric oxide synthase 2 (NOS2) expression and nitrated Aβ formation (Heneka et al., 2013). Hence, targeting the inflammasome is a viable strategy to inhibit AD pathogenesis.
A study by Dempsey et al. investigated the promising use of the MCC950, a small molecule inhibitor of NLRP3, for alleviating AD pathogenesis and symptomatology (Dempsey et al., 2017a). Incubation of primary mouse glia cells with MCC950 following LPS and ATP co-treatment or LPS and Aβ co-treatment significantly decreased IL-1β gene expression, caspase-1 immunoreactivity, and LDH release. Also, MCC950 injections on APP/PS1 mice significantly lowered IL-1β and TNFα levels, as well as Aβ1–40 and Aβ1–42 levels in brain tissue when compared to controls. This was accompanied by a reduction in the percentage of microglia immunoreactive for caspase-1, a decrease in CD11b and CD68 mRNA expression, and an improvement in hippocampal-related memory (Dempsey et al., 2017a), further supporting the potential use of MCC950 to target inflammasome activation in AD. Similar results have been observed with other mouse models of AD (APP/PS1 and TgCRND8 AD) using different NLRP3 inhibitors such as B-hydroxybutyrate (BHB) and an NF-kB inhibitor in 5XFAD and APP23 mice; respectively (Lonnemann et al., 2020; Ruan et al., 2019; Shippy et al., 2020; Yin et al., 2018).
The NLRP3 inflammasome is not the only complex mounting an inflammasome response in AD, with many studies showing that NLRP1 genetic variations are associated with AD (Pontillo et al., 2012). Additionally, Tan et al. showed Aβ-induced caspase-1-dependent pyroptosis in vitro and in vivo (Tan et al., 2014). Moreover, Kaushal et al. found increased NLRP1 mRNA levels in human primary CNS neurons, which colocalized with caspase-6, revealing that the NLRP1 inflammasome is implicated in AD pathogenesis (Kaushal et al., 2015). To confirm these results, in vitro experiments also revealed that the activation of an NLRP1/Casp1/Casp6 pathway that results from LPS stimulation plays a role in Aβ aggregation in human serum-deprived neurons (Kaushal et al., 2015). Taken together, both NLRP3 (ASC-dependent) and NLRP1 (ASC-independent) are activated in AD, with amyloid plaques being the primary triggers for neuroinflammation and NFTs subsequently exacerbating the pathogenesis of AD.
Moreover, ASC is elevated in the blood of patients with mild cognitive impairment and AD (Scott et al., 2020). Interestingly, the levels of ASC were higher in patients with MCI than in AD, suggesting that the inflammasome plays an early role in the pathology of AD and that ASC is a promising biomarker of the early stages of AD (Scott et al., 2020).
Importantly, TBI presents as a risk factor for AD and CTE (Mohamed et al., 2022; Van Den Heuvel et al., 2007). Taken together, it is important to highlight the overlap between inflammasome signaling activation in TBI and AD (Johnson et al., 2022a). For example, several inflammasomes such as the NLPR1, NLRP3, and AIM2 inflammasomes are activated after TBI (de Rivero Vaccari et al., 2009; Kerr et al., 2018a; Lee et al., 2019; Lee et al., 2018) and in AD (Ennerfelt and Lukens, 2020; Heneka et al., 2013; Venegas and Heneka, 2019; Venegas et al., 2017), which is consistent with studies indicating that approximately 30% of TBI patients develop Aβ after injury (Kokiko-Cochran and Godbout, 2018; Tran et al., 2011), through a mechanism that could be in part mediated by the inflammasome due to the overlap of signaling cascades between these conditions.
Inflammasome proteins as biomarkers of TBI
As previously mentioned, neuroinflammation is a critical cellular and molecular feature that enables the CNS to respond to insults such as trauma (O’Brien et al., 2020). To date, several inflammasome-related entities such as caspase-1, NLRP1, NLRP3, or ASC have been assessed as potential neuro-biomarkers in a variety of diseases and tissue injuries (Perez-Barcena et al., 2020). The first inflammasomes described in the nervous system were the NLRP1 inflammasome in neurons and the NLRP2 inflammasome in astrocytes. Nowadays, the most studied is the NLRP3 inflammasome that is present in microglia and astrocytes. However, due to the early availability of NLRP3 knockout mice, the most studied inflammasome is the NLRP3 inflammasome that is present in microglia and astrocytes. Additionally, other studies describe the importance of the NLRC4 inflammasome present in astrocytes and the AIM-2 inflammasome present in neurons (Kerr et al., 2018b). Inflammasome proteins can be found in CSF and serum of patients with TBI (Johnson et al., 2022b; Kerr et al., 2018b). Despite the limited number of studies, inflammasome proteins are potential biomarkers to assess TBI severity, outcome, and secondary injury mechanisms (Adamczak et al., 2012) (Table 3).
Table 3:
Human studies highlighting the use of inflammasome-related proteins as promising biomarkers of traumatic brain injury.
| Biomarker | Study Design | Patient Population | Levels in Control | Levels in TBI patients | Outcomes | Clinical Significance | Limitations | Ref |
|---|---|---|---|---|---|---|---|---|
| NLRP3, Caspase-1, IL-1β, IL-18 |
|
Control brain tissues: n = 3 TBI brain tissues: n = 5 |
– | – |
|
– | – | (Lin et al., 2017) |
| ASC, Caspase-1, IL-18 |
|
Control CSF samples: n = 120 TBI CSF samples: n = 21 Control serum samples: n = 30 TBI serum samples: n = 18 |
Mean ACS serum level: 236.6 pg/ml Mean Caspase-1 serum level: 1.436 pg/ml Mean IL-18 serum level: 213.5 pg/ml |
Mean ACS serum level: 629 pg/ml Mean Caspase-1 serum level: 4.540 pg/ml Mean IL-18 serum level: 218.625 pg/ml |
|
|
|
(Kerr et al., 2018b) |
| ASC, Caspase-1, NLRP1 |
|
Control CSF samples: n = 9 TBI CSF samples: n = 23 |
– | – |
|
|
|
(Adamczak et al., 2012) |
TBI:Traumatic Brain Injury, CSF:Cerebrospinal Fluid, GCS:Glasgow Coma Scale, AUC:Area Under Curve, ROC:Receiver operating characteristic. NLRP3:NOD-, LRR- and pyrin domain-containing 3, NLRP1:NOD-, LRR- and pyrin domain-containing, ASC:Apoptosis- speck-like protein containing a caspase-activating recruitment domain (CARD), IL-1β:Interleukin-1β, IL-18:Interleukin-18
a. Cerebrospinal fluid biomarkers
Adamczak et al. reported a quantitative analysis of inflammasome components in the CSF from TBI patients (n=23) and followed them for five months post-the following injury to investigate any correlations between inflammasome activity and neurocognitive outcomes (Adamczak et al., 2012). CSF samples were collected within 12 hours of injury and up to 72 hours after injury. They found that higher expression of ASC, caspase-1 (p20), and NLRP-1 correlates with unfavorable outcomes at five months, including death and disability with complete dependence (P<0.0001) compared to patients with moderate to no disability. The data suggest that inflammasome components play a role in the acute phase of TBI and produce chronic neuroinflammation contributing to secondary injury and unfavorable outcomes 5 months after the primary lesion (Adamczak et al., 2012). In addition, Perez-Barcena et al. reported an early elevation of caspase-1 in patients with TBI and its correlation with high increased intracranial pressure and association with unfavorable outcomes (Perez-Barcena et al., 2020). Lastly, Wallisch et al. studied CSF biofluid from children with severe TBI (n = 34) who had externalized ventricular drains placed for routine care (Wallisch et al., 2017). The study evaluated the levels of NLRP1 and NLRP3 at 0–24, 25–48, 49–72, and >72 h post-TBI comparing it to infection-free controls that underwent lumbar puncture to rule out CNS infection (n = 8) (Wallisch et al., 2017). Patient age, sex, initial Glasgow Coma Scale (GCS), injury mechanism, therapeutic hypothermia treatment, and six-month GCS were collected (Wallisch et al., 2017). The study revealed that CSF NLRP3 is elevated in children with severe TBI and is independently associated with younger age and poor outcomes (Wallisch et al., 2017). These studies demonstrate evidence of the presence and role of the inflammasome complex after TBI. They also pave the way for more extensive, and well-designed clinical trials.
b. Serum biomarkers
Serum biomarkers play an essential role in the diagnosis and prognosis of TBI patients that are both brain-derived (Mondello et al., 2022; Ottens et al., 2006; Wang et al., 2021) or within the inflammasome context (Keane et al., 2018; Kerr et al., 2020; Kerr et al., 2018b). Existing data comparing biomarkers in the CSF (n=18) versus serum (n=21) showed that protein levels of ASC (area under curve “AUC” =0.90) and caspase-1 (AUC=0.93) are elevated after TBI (Kerr et al., 2018b). Furthermore, the specificity and sensitivity of these proteins as serum biomarkers for TBI were calculated and found to have 94% sensitivity and 89% specificity for caspase-1 and 85% sensitivity and 99% specificity for ASC (Kerr et al., 2018b). Improving their performance by raising the cut-off point indicates that these proteins are reliable serum biomarkers for TBI. ASC was also higher in the serum of TBI patients with unfavorable outcomes compared to the samples obtained from patients with favorable outcomes whereas caspase-1 and IL-18 levels were not statistically different between the two groups. The performance as a serum predictive TBI biomarker with a cut-off point of 547.6 pg/ml was promising (86% sensitivity and 100% specificity) and suggested that it could be used as a predictive biomarker of unfavorable outcomes (Kerr et al., 2018b). Thus, in regards to blood biomarkers, caspase-1 and ASC are reliable biomarkers of the inflammatory response after TBI.
c. MicroRNAs
Recently, the field of microRNA (miRNA), which are short non-coding strands of RNA are receiving interest as putative regulators of inflammasomes formation and activation (Cetin, 2021; Houshmandfar et al., 2021; Li et al., 2022; Zamani et al., 2020) as well as putative biomarkers of inflammation which intersect with their roles in TBI (Gerber et al., 2022). To the best of our knowledge, there are no studies that assessed miRNA to target the inflammasome in TBI, there are other studies that have demonstrated the effectiveness of certain miRNAs targeting inflammasomal components in related diseases. For example, (Chen et al., 2020) found that miR-374a-5p is down-regulated in neonatal hypoxic-ischemic encephalopathy, and overexpressing it regulated the NLRP3 inflammasome and Smad6 leading to a decrease in pro-inflammatory cytokines production (IL-1β, IL-6, and TNFα). Similarly, miR-9a-5p overexpression decreased protein levels of NLRP1, ASC, pro and cleaved-caspase-1, IL-1β and IL-18 in rats in rats subjected to middle cerebral artery occlusion and SY-5Y cells exposed to oxygen-glucose deprivation (Cao et al., 2020). miRNAs have also been successful in ameliorating certain markers of symptomatology in PD rodent models by targeting NLRP3 (miR-7) or other components (miR-135b: FoxO1) (Zhou et al., 2016), as well as AD models (miR-22) (Han et al., 2020) while in another study miR-7 increased neurogenesis (Fan et al., 2016). These findings denote the importance of developing such methods and further testing them before they are translated to the clinic.
In the TBI arena, Redell et al. analyzed MicroRNAs (miRNAs) plasma levels from patients with severe TBI and found that 27 samples were significantly elevated (Redell et al., 2010). From these altered hits, they chose five candidates for further analysis (miR-16, miR-26a, miR-92a, miR-638, and miR-765) reporting significant elevation for miR-16, miR-92a and miR-765 (AUC=1.00, indicating 100% specificity and 100% sensitivity for the biomarker combination of miR-92a, miR-765, and miR-16, to correctly distinguish severe TBI patients from polytrauma (Redell et al., 2010). Likewise, Qin et al., analyzed (n=120) a microarray containing probes of 2549 human miRNAs (Qin et al., 2018). Among these, they found 13 miRNAs present in all TBI groups and selected seven candidates for further analysis (miR-6867–5p, miR-3665, miR-328–5p, miR-762, miR-3195, miR-4669, and miR-2861). Plasma levels of these miRNAs were upregulated in all TBI groups with AUC values: miR-6867–5p (0.854, P<0.001), miR-3665 (0.877, P<0.001), miR-328–5p (0.888, P<0.001), miR-762 (0.916, P<0.001), miR-3195 (0.899, P<0.001), miR-4669 (0.907, P<0.001), and miR-2861 (0.913, P<0.001) (Qin et al., 2018).
Similarly, Di Pietro et al., analyzed (n=120) the expression of 754 miRNA to find candidate biomarkers to diagnose and discriminate between mild and severe TBI (Di Pietro et al., 2017). They found miR-21 and miR-33, which were significantly upregulated and could act as valid biomarkers for TBI diagnosis and discrimination between mild and severe TBI. In addition, they reported that miR-425–5p and miR-21 were strong predictors of six-month outcomes, suggesting that these miRNAs may support early clinical decision-making and patient stratification for treatment or research (Di Pietro et al., 2017). Later, Tas et al., analyzed (n=59) serum levels of miR93 and miR191 in patients with mild TBI (Tas et al., 2020). They found that miR191 levels were markedly elevated in patients with TBI compared to controls suggesting that circulating miRNA levels increased after minor trauma while differentiating between patients with intracranial versus extracranial lesions (Tas et al., 2020). These findings were consistent with one study that analyzed miR-28–3p and miR-339–3p in patients with cumulative concussions that found a significant correlation (p < 0.05) in the number of prior concussions (Hicks et al., 2020). Together, these data indicate that the combination of miR-16, miR-92a, and miR-765 have a strong diagnostic role in the inflammatory response post-TBI. As with many diseases, relevant miRNAs can be used as therapeutic agents as well.
d. Extracellular vesicles (EVs)
EVs are lipid-bound vesicles secreted from various cell types that can be found in all bodily fluids and the extracellular matrix (Zaborowski et al., 2015). EVs play an important role in intracellular communication and can induce a wide range of effects on the target cells (Kerr et al., 2020). Inflammasome proteins have been found to be elevated in EVs acutely in patients after TBI (de Rivero Vaccari et al., 2016a). Furthermore, it has been reported that inflammasome activation can also be triggered by the release of EV-containing pro-inflammatory cytokines taken up by the lungs after TBI (Kerr et al., 2018c). It is hypothesized that this signaling contributes to the physiopathology of TBI-induced acute lung injury (Kerr et al., 2018c). In addition, Atai et al. demonstrated that EV uptake is inhibited in vitro using heparin and enoxaparin (Atai et al., 2013). Kerr et al. showed that treatment with enoxaparin and ASC monoclonal antibody reduces acute lung injury after the adoptive transfer of serum-derived EV from TBI mice (Kerr et al., 2018c). This area of research is exciting and currently evolving. Therefore, findings indicate that EVs are promising biomarkers of TBI that can be associated not only with CNS damage but also with systemic inflammation after TBI.
Benefits and Limitations of Blood-Based Biomarkers
Whether we refer to imaging or fluid biomarkers, biomarkers are primordial tools in personalized medicine. However, it is imperative to identify a battery of biomarkers that can be used reliably in the care of TBI patients. To date, ubiquitin carboxyl-terminal esterase L1 (UCH-L1) and glial fibrillary acidic protein (GFAP) are the only two biomarkers that are FDA-approved for the care of this patient population (Korley et al., 2022). These biomarkers provide information regarding damage to neurons and astrocytes in the CNS. However, it is imperative to identify reliable biomarkers of the inflammatory status acutely after TBI. To this extent, the inflammasome proteins are reliable biomarkers of inflammation after TBI. CSF biomarkers, although more specific to CNS status, the mode of collection is too invasive. On the other hand, these biomarkers, although they are more prone to be affected by systemic effects taking place after TBI or other conditions, they are also less invasive. Thus, the sensitivity of these biomarkers may be high; however, the specificity may be affected due to contributions from peripheral levels of these biomarkers. Thus, linear predictor models need to be established where these biomarkers are combined with other clinical TBI data to increase the specificity of these markers in TBI and use this linear predictor as a diagnostic, prognostic, and theranostic tool for the care of TBI patients.
e. Inflammasomes and Sex Differences
Sex differences in response to TBI have been relatively well-documented, with hormones 17β-estradiol (E2) and progesterone (P) providing a neuroprotective and anti-inflammatory effect at least in the acute phase of TBI (Engler-Chiurazzi et al., 2017; Villapol et al., 2017). These hormones have a neuroprotective effect due to their presence in glial cells and neurons where they downregulate pro-inflammatory cascades by changing microglial phenotype, affecting inflammasome assembly, and the network of associated miRNAs (Slowik and Beyer, 2015). However, there have been no studies-to the best of our knowledge-that have specifically looked at sex differences in inflammasomal activation due to TBI. In a related study on spinal cord injury in male rats, E2 mitigated the gliosis and inflammatory effects with reduced expression of ASC, NLRP1b, caspase-1, and NLRP3 as well as cytokines IL-1β and IL-18 (Zendedel et al., 2018). Similar findings have been reached of a protective effect of estrogens following ischemic stroke in ovariectomized rats and mice by reducing the expression of inflammasomal components such NLRC4, AIM2, NLRP3, and caspase-1 as well as pro-inflammatory cytokines among others (Habib et al., 2020; Suzuki et al., 2009; Thakkar et al., 2016).
Furthermore, a study looking at the serum of 65 male and 40 female Australian footballers during pre-season and during the season to monitor the occurrence of concussion and correlate it with the levels of IL-1β and IL-18 found that IL-1β levels were higher in males who presented a prior history of concussion when compared to individuals with no history of prior concussions, an effect that was not found in the female cohort (O’Brien et al., 2021). Similarly, the study found a positive correlation between years of participating in this contact sport and protein levels of IL-18 in males (O’Brien et al., 2021). Taken together, these data indicate that inflammasome protein expression differs between males and females and that the reliable role of inflammasome proteins as biomarkers differs in males and females, consistent with previous studies showing sex differences in regards to inflammasome regulation (de Rivero Vaccari, 2020; de Rivero Vaccari et al., 2019; Raval et al., 2019).
Inflammasomes as potential therapeutic targets for TBI
The innate immune system functions as the first line of host defense against pathogens and tissue damage (Marshall et al., 2018). Inflammasome complexes play a critical role in innate immune inflammatory responses and contribute to various pathologies and metabolic dysfunctions, including the innate responses to TBI (de Rivero Vaccari et al., 2016b). Therefore, a clear understanding of the pathophysiology leading to secondary injury in TBI is imperative for developing novel therapeutic targets. Unfortunately, despite preclinical trials demonstrating therapeutic success with various treatments, most clinical trials have failed to show positive outcomes in TBI patients (Kabadi and Faden, 2014). The advent of new information and understanding regarding inflammasome activation and regulation has opened a window for exploring potential therapeutic targets to modulate inflammation after TBI (de Rivero Vaccari et al., 2016b; Ismael et al., 2021). Evidence shows inflammasome activation in various TBI models (de Rivero Vaccari et al., 2009; Liu et al., 2013). Moreover, inflammasome proteins have also been identified in adult serum and CSF as well as in pediatric CSF samples, correlating with neurological outcomes and injury severity (Adamczak et al., 2012; Kerr et al., 2018b; Wallisch et al., 2017). Inflammasome regulation occurs in several places where the inactivation of specific proteins could interfere with signaling platforms. This supports their role as specific therapeutic targets.
a. Inhibition of the NLRP3 inflammasome: Non-specific inhibitors vs. Specific Inhibitors
Currently, several studies targeting the NLRP3 inflammasome were able to decrease NLRP3 inflammasome activity providing neuroprotective effects. This protection can be against naturally occurring molecules/targets and repurposed medications such as inhibitors of NLRP-associated molecules and specific NLRP3 inflammasome inhibitors (Kuwar et al., 2019; O’Brien et al., 2020). Based on these observations, several studies evaluated the effects of several therapeutic agents that directly or indirectly target inflammasomes on the neurological outcome following TBI.
Inflammasome Non-specific Inhibitors
As described previously, several candidate inhibitors are shown to exhibit neuroprotection post-TBI that indirectly target inflammasomes ameliorating neurological outcomes. Propofol (2,6-diisopropyl phenol) is a lipid-soluble intravenous anesthetic that has been proven to exert neuroprotective effects in various brain injury models without fully establishing the molecular mechanisms underlying these effects (Cai et al., 2011; Ding et al., 2013; Ma et al., 2009; Xi et al., 2011). In particular, one study postulated that the neuroprotective effects of propofol were associated with reduced expression of IL-1β and TNF-α in a controlled cortical injury rat model (Ding et al., 2013). Ma et al. were the first to demonstrate the role of NLRP3 inflammasome activation in a rat model of blast-induced TBI (Ma et al., 2016), and because IL-1β maturation and release are primarily regulated by the NLRP3 inflammasome, Ma et al. were interested in studying the effect of propofol on NLRP3 inflammasome activation and expression (Ma et al., 2016). Indeed, propofol treatment significantly reduced the expression of NLRP3, active caspase-1, and thioredoxin-interacting protein (TXNIP) (Ma et al., 2016), an important ROS-sensitive protein that regulates NLRP3 activation by detaching from thioredoxin and binding to NLRP3 (Zhou et al., 2010). Furthermore, propofol treatment attenuated pro-inflammatory cytokine (IL-1β, and TNF-α) release in the cerebral cortex, reduced oxidative stress and alleviated cerebral cortex damage (Ma et al., 2016). This allows us to speculate that the mechanism through which propofol suppresses NLRP3 inflammasome activation could be through the reduction of ROS, one of NLRP3’s major activating signals; thus, preventing TXNIP upregulation to induce NLRP3 activation. Interestingly, another study performed by Fan et al. (Fan et al., 2017) treated rats suffering from blast-induced injury with Mangiferin, a traditional Chinese medicine, and obtained the same results reported by Ma et al. (Ma et al., 2016). Similarly, they postulated that Mangiferin (1,3,6,7-tetrahydroxyxanthone-C2-β-D-glucoside) inhibited NLRP3 inflammasome activation by reducing oxidative stress and ultimately attenuating the ROS-TXNIP pathway (Fan et al., 2017). However, further studies are needed to elucidate the detailed molecular mechanism underlying the beneficial effect of propofol and Mangiferin in animal models of TBI.
In line with these findings, a recent study confirmed the crosstalk between oxidative stress and NLRP3 inflammasome activation through TXINP in the context of TBI (Ma et al., 2017a). NADPH oxidase 2 (NOX2) has been reported to be the major producer of ROS and contributor to oxidative stress, which plays a very important role in the pathogenesis of TBI (Cooney et al., 2013; Dohi et al., 2010; Ma et al., 2017b; Zhang et al., 2012). This study showed that NOX2 deletion or inhibition using apocynin (NOX2 inhibitor) caused a significant attenuation of NLRP3 and ASC expression, cleavage of caspase-1, and IL-1β levels in the mouse CCI model (Ma et al., 2017a). In addition, NOX2 deletion significantly decreased lesion volume, neuronal death, and TXNIP-NLRP3 interaction, suggesting that NOX2 absence exerts neuroprotective effects because of the decrease in the oxidative stress sensed by TXINP (Ma et al., 2017a). Further studies are needed to determine whether NOX plays a role in the regulation of other inflammasomes. Also, it is noteworthy that apocynin has been demonstrated as an effective and safe drug for asthma (Stefanska et al., 2012b) and chronic obstructive pulmonary disease patients (Stefanska et al., 2012a), which strengthens its potential translational value for therapeutically improving neurological outcomes and attenuating TBI-induced inflammatory responses.
Several studies have reported evidence of anti-inflammatory and neuroprotective effects conferred by angiotensin receptor blockers (ARBs) (Panahpour et al., 2014a, b; Villapol et al., 2015). Telmisartan, a highly lipophilic ARB that is capable of penetrating the brain, has been shown to exert anti-edemic effects and reduce the IL-1β-induced inflammatory response in rat models of ischemic stroke (Kono et al., 2015) and intracerebral hemorrhage (Jung et al., 2007). For this reason, Wei et al. wanted to examine the effects of telmisartan on TBI-induced cerebral edema and investigate whether these effects are mediated by inhibiting IL-1β release through the NLRP3 inflammasome in a cold injury-induced TBI model (Wei et al., 2016). Accordingly, they found out that oral administration of telmisartan 1h prior to TBI improved neurological outcome, lowered lesion volume, decreased BBB permeability, and reduced cerebral edema, which was associated with a reduction in NLRP3, ASC, caspase-1, IL-1β, and IL-18 levels in the pericontusional cortex 24h post-TBI (Wei et al., 2016). It should be noted that the mechanism by which telmisartan inhibited the NLRP3 inflammasome is still elusive; thus, more studies are needed to identify the molecular mechanism by which telmisartan regulates NLRP3 inflammasome expression.
Pioglitazone, a peroxisome proliferator-activated receptor agonist, has been shown to promote neuroprotective effects in different animal models of TBI, including moderate midline FPI in rats (Liu et al., 2017) and CCI in mice (Thal et al., 2011). A recent study evaluated the effects of pioglitazone on modulating NLRP3 in a mouse weight drop model of TBI and found that Pioglitazone administration reduced peri-lesional edema, which was associated with attenuation of activation of microglial cells, astrocytes, as well as the NLRP3 inflammasome activation 14 days post-TBI (Yi et al., 2020). Therefore, Pioglitazone seems to be a potential therapeutic drug for the treatment of TBI through modulating NLRP3, microglial, and astrocyte activity.
Gastrodin is the main bioactive constituent of rhizome, the main medicinal part of Gastrodia elata that has demonstrated significant therapeutic effects in treating dizziness, headache, epilepsy, amnesia, and stroke (Liu et al., 2018b). In specific, Gastrodin has shown neuroprotective activity via anti-inflammatory, anti-oxidant, and anti-apoptosis effects in brain injury and neurodegenerative diseases (Hu et al., 2014; Kumar et al., 2013; Li et al., 2019; Liu et al., 2020; Wang et al., 2019). However, the mechanism through which Gastrodin improves neurological outcomes and alleviates brain injury is still unknown. That being the case, a very recent study performed by Yang et al. investigated the role of Gastrodin in a rat model of closed head weight drop (Yang et al., 2022). Gastrodin administration alleviated brain injury; enhanced functional recovery; and suppressed NLRP3 inflammasome activation via reducing the expression of NLRP3, ASC, and caspase-1 in a dose-dependent manner. As a result, Gastrodin treatment diminished the expression of GSDMD and the production of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-18) in a dose-dependent manner (Yang et al., 2022). In conclusion, Gastrodin is a promising therapeutic drug for TBI as it exerts neuroprotective effects by inhibiting NLRP3 activation and suppressing pyroptosis.
Artesunate is a more stable derivative of artemisinin that is obtained from the Chinese plant Artemisia annua that has been used in traditional Chinese medicine to treat severe malaria (Rosenthal, 2008; Woodrow et al., 2005). Artesunate has been shown to exhibit anti-inflammatory properties in cerebral malaria (Clemmer et al., 2011; Miranda et al., 2013), stroke, and neurodegenerative diseases with the ability to reach and sustain a high concentration in the brain without any side effects (Wang et al., 2015; Zuo et al., 2016). Artesunate administration in a mouse CCI model alleviated the TBI-induced inflammatory response by significantly reducing NLRP3 inflammasome, astrocyte, and microglial activation which was associated with a decrease in TNF-α, NF-kB, IL-1β, and iNOS expression 24h post-TBI (Gugliandolo et al., 2018). Furthermore, Artesunate treatment inhibited neuronal apoptosis and improved post-TBI reparative processes by increasing the expression of Brain-Derived Neurotrophic Factor (BDNF), Glial cell-Derived Neurotrophic Factor (GDNF), and Vascular Endothelial Growth Factor (VEGF) (Gugliandolo et al., 2018). Thus, Artesunate is a promising multitarget drug for the treatment of TBI that modulates neuroinflammation, oxidative stress, and neuronal death.
As illustrated in Figure 2, NF-kB-dependent transcriptional upregulation of NLRP3 and pro- IL-1β is an essential priming step before other activating signals trigger full inflammasome activation and assembly, which makes NF-kB a target for NLRP3 inflammasome inhibition. Building up on this, Irrera et al. treated mice with BAY 11–7082 (NF-kB inhibitor), which significantly improved behavioral deficits; reduced inflammatory infiltrates and edema in the perilesional area, and decreased IL-1β and caspase-1 expression levels (Irrera et al., 2017). In another study, the administration of Dexmedetomidine (a highly selective α−2 adrenergic receptor agonist) in rats enhanced cognitive deficits following CCI, which was associated with reduced microglial and NLRP3 inflammasome activation in the hippocampus (Zheng et al., 2018). Interestingly, this study has shown that the suppressing effects of Dexmedetomidine and BAY-11–7082 on the NLRP3 inflammasome were comparable. Moreover, BAY-11–7082 and Dexmedetomidine demonstrated a synergistic blocking effect on NLRP3 inflammasome activation upon co-administering them together (Zheng et al., 2018). This means that Dexmedetomidine may be a powerful candidate for the treatment of TBI through the modulation of inflammasome activation.
Inflammasomes Specific Inhibitors
Up till now, all the mentioned inhibitors have shown neuroprotective effects in the context of experimental models of TBI. However, it must be taken into consideration that these agents inhibited NLRP3 activation either in an indirect or non-specific way, which makes it challenging to determine whether the enhanced outcomes are resulting from inhibiting the inflammasome itself or inhibiting an upstream activating stimulus of inflammasomes. Furthermore, the use of non-specific inhibitors in a clinical setting poses a high risk of side effects. Indeed, MCC950, JC-124, and Oridonin are the only specific NLRP3 inhibitors whose effects have been evaluated following TBI which are illustrated in Figure 5.
Figure 5: Pharmacological Modulation of Inflammasome Following Traumatic Brain Injury and its Potential Use as a Therapeutic Target.
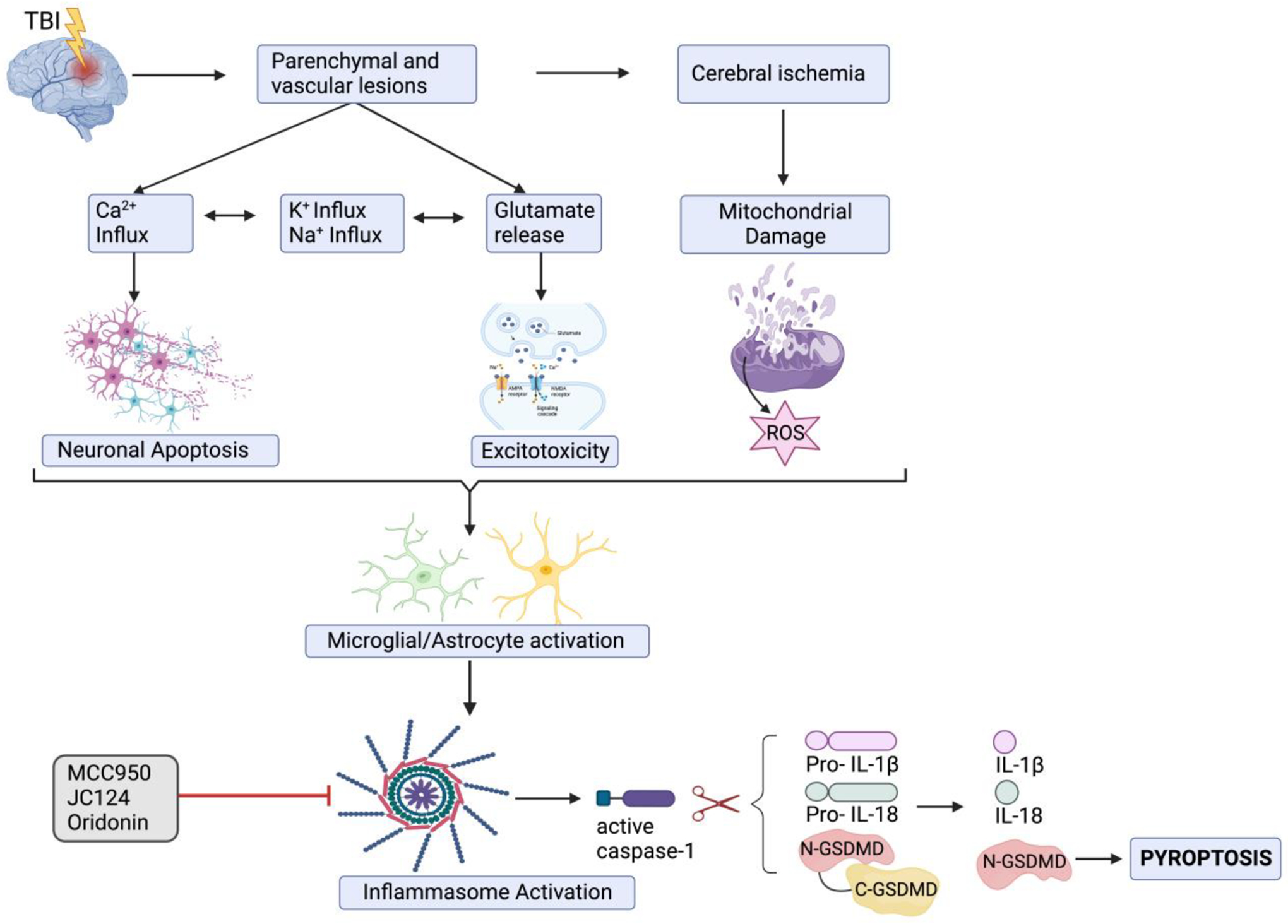
Following a primary brain injury, parenchymal and vascular lesions cause a massive release of excitatory amino acids such as glutamate, and prolonged activation of glutamate receptors, followed by an excessive influx of Ca2+ ions, ultimately leading to cellular excitotoxicity. Aberrant calcium overload stimulates the activation of calpains and caspases, causing neuronal apoptosis. Additionally, cerebral ischemia or calcium influx into the mitochondria induces the production of ROS. Consequently, the ionic changes (calcium/sodium influx and potassium efflux), ROS released following mitochondrial damage and DAMPs released following neuronal apoptosis or necrosis act as activating signals for the inflammasome sensors expressed on microglia or astrocytes. This induces full inflammasome complex assembly, leading to a proximity-induced auto-cleavage of pro-caspase 1 into active caspase-1. Active caspase-1 proteolytically cleaves pro-IL-1β and pro-IL-18 into the mature pro-inflammatory cytokines IL-1β and IL-18. It also liberates the N-terminal fragment of GSDMD, leading to a pro-inflammatory form of cell death named pyroptosis. MCC950, JC124, and oridonin are specific NLRP3 inflammasome inhibitors that have been shown to enhance neurological outcomes and improved motor and cognitive deficits following TBI.
Abbreviations: TBI: Traumatic brain injury, ROS: Reactive oxygen species, DAMPs: Damage-associated molecular patterns, GSDMD: Gasdermin D, IL: Interleukin
MCC950 is a diarylsulphonylurea-containing compound that is a highly selective and potent NLRP3 inflammasome inhibitor (Coll et al., 2015). It acts by binding directly to the NACHT domain of the NLRP3 protein, blocking the homotypic interactions between NACHT domains of NLRP3 proteins, thereby preventing NLRP3 oligomerization and assembly (Coll et al., 2019). Experimental studies provided evidence of neuroprotective effects exerted by MCC950 in multiple sclerosis, Alzheimer’s disease, stroke, and intracerebral hemorrhage (Coll et al., 2015; Dempsey et al., 2017b; Ismael et al., 2018b; Ren et al., 2018). Ismael et al. were the first to evaluate the effects of MCC950 treatment on the acute phase of injury (24h-72h) in a CCI mouse model (Ismael et al., 2018a). Intraperitoneal administration of 50mg/kg MCC950 at 1h and 3h post-TBI significantly inhibited NLRP3 inflammasome activation as evident by the decrease in NLRP3, ASC, cleaved caspase-1, and IL-1β levels in the peri-lesional area at 24h and 72h post-TBI. Of note, MCC950 treatment reduced NF-kB/p65 upregulation in TBI mice, which is very important for the priming signal of NLRP3 inflammasome activation. Also, MCC950 improved neurological function, reduced TBI-induced cerebral edema, and decreased apoptosis (Ismael et al., 2018a). These findings provide clear evidence of the beneficial effects of MCC950 in the acute phase of TBI in a mouse model.
Following this study, Xu et al. were interested in evaluating the long-term efficacy of MCC950 on the inflammatory response and neurological outcome in a CCI mouse model (Xu et al., 2018). Intraperitoneal administration of MCC950 (10mg/kg) daily for the first 3d and then every other day up to 21d enhanced long-term motor and cognitive function. This was evident by the decrease in modified neurological severity score and improvement in the performance in the rotarod test (at 3d, 7d, and 14d post-TBI) and Morris water maze (at 17d, 18d, and 19d post-TBI). Furthermore, MCC950 increased the levels of anti-inflammatory cytokines (IL-10) and diminished BBB disruption, lesion volume, cerebral edema, apoptosis, microglial activation, leukocyte infiltration, and the number of NLRP3-expressing microglia. Interestingly, all these neuroprotective effects were lost in microglia-depleted mice, supporting that microglial cells are the major source of pro-inflammatory cytokines (IL-1β and IL-18) in the brain. Finally, they demonstrated that MCC950 had a wide therapeutic time window of up to 6h in protecting against TBI, which can be very helpful when translated into clinical practice. Altogether, these findings suggest that MCC950 is a promising and potent therapeutic drug for TBI treatment; however, it should be noted that the Xu et al. selected a dose that was beneficial in another setting without conducting a dose-response study, highlighting the urge for more research to find the optimal dose and frequency of administration.
Glyburide, an FDA-approved sulfonylurea derivative used for the treatment of Type 2 diabetes, has been shown to inhibit NLRP3 inflammasome activation (Lamkanfi et al., 2009). Because of the risk of inducing hypoglycemia upon using high doses of Glyburide in animal models, Fulp et al. structurally optimized Glyburide and developed a more selective NLRP3 inflammasome inhibitor named JC124 (Fulp et al., 2018). In fact, JC124 demonstrated multiple neuroprotective effects as a selective NLRP3 inflammasome inhibitor in an AD mouse model (Yin et al., 2018). For this reason, Kuwar et al. wanted to evaluate the effects of JC124 treatment in rats following CCI (Kuwar et al., 2019). Intraperitoneal administration of 100mg/kg JC124 at 30min, 6h, 24h, and 30h post-TBI significantly decreased the expression of NLRP3, ASC, and caspase-1; reduced cortical lesion volume; and lowered the number of injury-induced degenerating neurons in the ipsilateral cortex and hippocampal dentate gyrus. Additionally, JC124 decreased the levels of IL-1β, TNF-α, and iNOS in the ipsilateral cortex. These findings suggest that JC124 may be a potential therapeutic drug; however, further studies are needed to evaluate the long-term JC124 efficacy on the neurological outcome and understand the mechanism underlying NLRP3 inflammasome inhibition to enhance its translational value.
The last specific NLRP3 inflammasome inhibitor to be discussed in this article is Oridonin, a bioactive ingredient extracted from the Chinese herbal medicine Rabdosia rubescens (He et al., 2018). Oridonin inhibits NLRP3 by binding covalently with cysteine 279 in the NACHT domain of NLRP3, thereby preventing NLRP3 inflammasome activation and assembly (He et al., 2018). A very recent study showed that the intraperitoneal administration of 10mg/kg Oridonin within 30 min after TBI in mice and once daily up to 14 days significantly inhibited NLRP3 inflammasome activation and the maturation of pro-inflammatory cytokines (IL-1β, and IL-18) in the pericontusional cerebral cortex (Yan et al., 2020). Furthermore, Oridonin treatment enhanced neurobehavioral performance and functional recovery; diminished lesion volume and TBI-induced cerebral edema; inhibited apoptosis, and maintained BBB integrity. These results imply that Oridonin is a promising therapeutic drug; however, further studies are required to determine its therapeutic window.
Neutralization of ASC
de Rivero et al. demonstrated that antibody neutralization of ASC interferes with inflammasome signaling after TBI (de Rivero Vaccari et al., 2009). Currently, this antibody approach is being developed for testing in clinical trials under the name of IC 100 (De Rivero Vaccari et al., 2022). IC100 binds to ASC specks that are released into the extracellular space after inflammasome activation preventing the extracellular activation of caspase-1 and IL-1b (de Rivero Vaccari et al., 2016b; Franklin et al., 2014). IC 100 readily penetrates several organs including the brain and spinal cord, and it binds to intracellular ASC while avoiding degradation by the atypical antibody receptor Tripartite motif-containing protein-21 (TRIM21) antibody-dependent intracellular neutralization pathway (De Rivero Vaccari et al., 2022). Moreover, since ASC is present in several inflammasomes such as the NLRP1, NLRP2, and NLRP3 inflammasomes (de Rivero Vaccari et al., 2016b), targeting ASC offers the potential of inhibiting multiple inflammasomes.
Caspase-1 Inhibition
The adapter protein ASC binds directly to caspase-1 and is critical for caspase-1 activation in response to a broad range of stimuli (Fernandes-Alnemri et al., 2007). Since pyroptosis is caspase-1 dependent, it could potentially be inhibited with a caspase −1 blocker. Indeed, numerous caspase inhibitors have been identified and developed for therapeutic use. Current endogenous inhibitors include cytokine response modifier A (CrmA), p35 (viral caspase inhibitor), and family members of the Inhibitor of apoptosis (IAP) proteins (Dhani et al., 2021) such as X-linked IAP (XIAP) (de Rivero Vaccari et al., 2008). In contrast, synthetic caspase inhibitors are divided between peptide-based inhibitors, peptidomimetic inhibitors, non-peptidic compounds, and allosteric caspase inhibitors (Dhani et al., 2021). However, due to poor efficacy or toxic effects, only a limited number of synthetic caspase inhibitors have advanced into clinical trials, with none of them being successful for clinical use yet (Dhani et al., 2021).
Conclusion and Future directions
Our progress in advancing methods and developing our knowledge base concerning TBI and inflammasomes has brought up many promising questions to answer. Despite these advances, avenues of investigation related to the relationship between TBI and neurodegenerative diseases, the roles of different cell types in the inflammasomal response, the feasibility of miRNA inflammasomal regulation, as well as the need to delve much deeper into sex differences would represent new areas of investigation in the area of brain injury. This highlights the necessity to systematically develop effective therapeutic interventions and neuroprotective strategies to reduce short- and long-term complications following TBI and diminish its substantial direct and indirect global burden. In recent years, inflammasomes have gained considerable attention as essential coordinators of the initiation and prolongation of the neuroinflammatory response; however, their role in the pathogenesis of TBI has only been recently identified as several animal and human studies have reported inflammasome activation and inflammasome-related proteins upregulation following TBI (Table 2 & 3). As discussed, and presented in this review, we aimed to highlight the current inflammasome inhibitors, both specific and non-specific ones, that would hold promise as second-generation therapeutic-pharmacological targets for mitigating the course of TBI. Moreover, clinical evidence has evolved to consider the use of inflammasome protein components as potential diagnostic and predictive biomarkers of TBI severity and clinical outcomes; an area that carries a lot of promise in research for the general public as well as for the military and Veteran population sustaining chronic brain injury incidents. Thus, this updated review calls for conducting future research in the areas of inflammasome biochemistry and their integral role in orchestrating the pathomechanisms of TBI and its related neurodegenerative disorders.
Research Search Strategy.
For this review, studies on the topic were identified by searches of PubMed for articles published between 1950 and 2022 using a mixture of keyword-based search and controlled vocabulary search strategies. Key concepts were identified as Medical Subject Heading (MeSH) and combined with key terms as follows:
(“brain injuries, traumatic”[MeSH Terms] OR (“brain”[All Fields] AND “injuries”[All Fields] AND “traumatic”[All Fields]) OR “traumatic brain injuries”[All Fields] OR (“traumatic”[All Fields] AND “brain”[All Fields] AND “injury”[All Fields]) OR “traumatic brain injury”[All Fields]) AND (“Chronic microglial activation”[All Fields] OR “Microglia”[MeSH Terms] OR (“inflammation”[MeSH Terms] OR “inflammation”[All Fields] OR “inflammations”[All Fields] OR “inflammation s”[All Fields]) OR (“neuroinflammatory diseases”[MeSH Terms] OR (“neuroinflammatory”[All Fields] AND “diseases”[All Fields]) OR “neuroinflammatory diseases”[All Fields] OR “neuroinflammation”[All Fields])) OR (“brain injuries, traumatic”[MeSH Terms] OR (“brain”[All Fields] AND “injuries”[All Fields] AND “traumatic”[All Fields]) OR “traumatic brain injuries”[All Fields] OR (“traumatic”[All Fields] AND “brain”[All Fields] AND “injury”[All Fields]) OR “traumatic brain injury”[All Fields] OR (“neurodegenerative diseases”[MeSH Terms] OR (“neurodegenerative”[All Fields] AND “diseases”[All Fields]) OR “neurodegenerative diseases”[All Fields]) OR (“central nervous system diseases”[MeSH Terms] OR (“central”[All Fields] AND “nervous”[All Fields] AND “system”[All Fields] AND “diseases”[All Fields]) OR “central nervous system diseases”[All Fields] OR (“cns”[All Fields] AND “diseases”[All Fields]) OR “cns diseases”[All Fields])) AND (“inflammasomes”[MeSH Terms] OR “inflammasomes”[All Fields] OR “inflammasome”[All Fields] OR “NLRP3”[All Fields] OR “nlr family, pyrin domain containing 3 protein”[MeSH Terms] OR “NLRP1”[All Fields] OR “AIM2”[All Fields] OR “gsdmd”[All Fields] OR “Gasdermin D”[All Fields] OR (“pyroptosis”[MeSH Terms] OR “pyroptosis”[All Fields]))
Highlights.
Traumatic brain injury (TBI) is a leading cause for morbidity and mortality worldwide with huge socioeconomic burden and no FDA-approved drug
The pathophysiology underlying TBI is complicated and multifactorial, which makes it hard to diagnose and to treat.
Current approaches for treating TBI focus on limiting the secondary damages followed by the injury, such as oxidative stress, edema, excitotoxicity, mitochondrial dysfunction, and neuroinflammation.
Neuroinflammation is a hallmark of both acute and chronic TBI. In early stages, neuroinflammation is beneficial and has a significant protective role; however, a dysregulated or chronic response can result in neurological impairments and neurodegeneration
Inflammasomes are crucial players in the innate immune response to tissue damages and abnormality in the inflammasome activity has been associated with several neurological conditions. In TBI, it has been shown that inflammasome activation and inflammasome-related proteins upregulation occur after injury
Accumulated evidence has suggested the use of inflammasome proteins as potential diagnostic and predictive biomarkers of TBI severity and clinical outcomes
Acknowledgment
This work is partially supported by the grant “UL1TR002736, Miami Clinical, and Translational Science Institute, from the National Center for Advancing Translational Sciences and the National Institute on Minority Health and Health Disparities” (PI: J C. Munoz Pareja). Additionally, this work was also supported by grants from NIH for (PI: Yehia Mechref) (1R01GM112490-09, 1U01CA225753-05, and 1R01GM130091-04) and JPdRV (1R01NS113969-01, 1RF1NS125578-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest
JPdRV is a co-founder and managing member of InflamaCORE, LLC, and has licensed patents on inflammasome proteins as biomarkers of injury and disease as well as on targeting inflammasome proteins for therapeutic purposes. JPdRV is a Scientific Advisory Board Member of ZyVersa Therapeutics Inc.
References
- Abou-Abbass H, Bahmad H, Abou-El-Hassan H, Zhu R, Zhou S, Dong X, Hamade E, Mallah K, Zebian A, Ramadan N, Mondello S, Fares J, Comair Y, Atweh S, Darwish H, Zibara K, Mechref Y, Kobeissy F, 2016. Deciphering glycomics and neuroproteomic alterations in experimental traumatic brain injury: Comparative analysis of aspirin and clopidogrel treatment. Electrophoresis 37, 1562–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamczak S, Dale G, de Rivero Vaccari JP, Bullock MR, Dietrich WD, Keane RW, 2012. Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: clinical article. J Neurosurg 117, 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J, 2004. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325. [DOI] [PubMed] [Google Scholar]
- Alyaseer AAA, de Lima MHS, Braga TT, 2020. The Role of NLRP3 Inflammasome Activation in the Epithelial to Mesenchymal Transition Process During the Fibrosis. Front Immunol 11, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amores-Iniesta J, Barbera-Cremades M, Martinez CM, Pons JA, Revilla-Nuin B, Martinez-Alarcon L, Di Virgilio F, Parrilla P, Baroja-Mazo A, Pelegrin P, 2017. Extracellular ATP Activates the NLRP3 Inflammasome and Is an Early Danger Signal of Skin Allograft Rejection. Cell Rep 21, 3414–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atai NA, Balaj L, van Veen H, Breakefield XO, Jarzyna PA, Van Noorden CJ, Skog J, Maguire CA, 2013. Heparin blocks transfer of extracellular vesicles between donor and recipient cells. J Neurooncol 115, 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E, 2009. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183, 787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayir H, Clark RS, Kochanek PM, 2003. Promising strategies to minimize secondary brain injury after head trauma. Crit Care Med 31, S112–117. [DOI] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, Cookson BT, 2009. Pyroptosis: host cell death and inflammation. Nature reviews. Microbiology 7, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett BL, Gardner RC, Godbout J, Dams-O’Connor K, Keene CD, 2022. Traumatic Brain Injury and Risk of Neurodegenerative Disorder. Biol Psychiatry 91, 498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickler T, Gresham K, Meza A, Coutermarsh-Ott S, Williams TM, Rothschild DE, Allen IC, Theus MH, 2016. Nonessential Role for the NLRP1 Inflammasome Complex in a Murine Model of Traumatic Brain Injury. Mediators Inflamm 2016, 6373506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Dixit VM, 2016. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16, 407–420. [DOI] [PubMed] [Google Scholar]
- Cai J, Hu Y, Li W, Li L, Li S, Zhang M, Li Q, 2011. The neuroprotective effect of propofol against brain ischemia mediated by the glutamatergic signaling pathway in rats. Neurochem Res 36, 1724–1731. [DOI] [PubMed] [Google Scholar]
- Cao Y, Zhang H, Lu X, Wang J, Zhang X, Sun S, Bao Z, Tian W, Ning S, Wang L, Cui L, 2020. Overexpression of MicroRNA-9a-5p Ameliorates NLRP1 Inflammasome-mediated Ischemic Injury in Rats Following Ischemic Stroke. Neuroscience 444, 106–117. [DOI] [PubMed] [Google Scholar]
- Capizzi A, Woo J, Verduzco-Gutierrez M, 2020. Traumatic Brain Injury: An Overview of Epidemiology, Pathophysiology, and Medical Management. Med Clin North Am 104, 213–238. [DOI] [PubMed] [Google Scholar]
- Cetin Z, 2021. Targeting the PANoptosome with miRNA Loaded Mesenchymal Stem Cell Derived Extracellular Vesicles; a New Path to Fight Against the Covid-19? Stem Cell Rev Rep 17, 1074–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Yao Q, Xu S, Wang H, Qu P, 2018. Inhibition of the NLRP3 inflammasome attenuates foam cell formation of THP-1 macrophages by suppressing ox-LDL uptake and promoting cholesterol efflux. Biochem Biophys Res Commun 495, 382–387. [DOI] [PubMed] [Google Scholar]
- Chen Z, Hu Y, Lu R, Ge M, Zhang L, 2020. MicroRNA-374a-5p inhibits neuroinflammation in neonatal hypoxic-ischemic encephalopathy via regulating NLRP3 inflammasome targeted Smad6. Life Sci 252, 117664. [DOI] [PubMed] [Google Scholar]
- Chevriaux A, Pilot T, Derangere V, Simonin H, Martine P, Chalmin F, Ghiringhelli F, Rebe C, 2020. Cathepsin B Is Required for NLRP3 Inflammasome Activation in Macrophages, Through NLRP3 Interaction. Front Cell Dev Biol 8, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christgen S, Kanneganti TD, 2020. Inflammasomes and the fine line between defense and disease. Curr Opin Immunol 62, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemmer L, Martins YC, Zanini GM, Frangos JA, Carvalho LJ, 2011. Artemether and artesunate show the highest efficacies in rescuing mice with late-stage cerebral malaria and rapidly decrease leukocyte accumulation in the brain. Antimicrob Agents Chemother 55, 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, Chitty JL, Fraser JA, Jennings MP, Robertson AAB, Schroder K, 2019. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol 15, 556–559. [DOI] [PubMed] [Google Scholar]
- Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Núñez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O’Neill LA, 2015. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21, 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney SJ, Bermudez-Sabogal SL, Byrnes KR, 2013. Cellular and temporal expression of NADPH oxidase (NOX) isotypes after brain injury. J Neuroinflammation 10, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr B, Hadad R, Keane RW, de Rivero Vaccari JP, 2022. The Role of Non-canonical and Canonical Inflammasomes in Inflammaging. Front Mol Neurosci 15, 774014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, 2020. The Inflammasome in Reproductive Biology: A Promising Target for Novel Therapies. Front Endocrinol (Lausanne) 11, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Bramlett HM, Perez-Pinzon MA, Raval AP, 2019. Estrogen preconditioning: A promising strategy to reduce inflammation in the ischemic brain. Cond Med 2, 106–113. [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Brand F 3rd, Adamczak S, Lee SW, Perez-Barcena J, Wang MY, Bullock MR, Dietrich WD, Keane RW, 2016a. Exosome-mediated inflammasome signaling after central nervous system injury. J Neurochem 136 Suppl 1, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Dietrich WD, Keane RW, 2016b. Therapeutics targeting the inflammasome after central nervous system injury. Transl Res 167, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW, 2009. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab 29, 1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW, 2008. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci 28, 3404–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rivero Vaccari JP, Mim C, Hadad R, Cyr B, Stefansdottir TA, Keane RW, 2022. Mechanism of action of IC 100, a humanized IgG4 monoclonal antibody targeting apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). Transl Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Asken BM, 2017. Injury cascades in TBI-related neurodegeneration. Brain Inj 31, 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Kochanek PM, Clark RS, Ciallella JR, Dixon CE, 1998. Secondary Injury After Head Trauma: Subacute and Long-term Mechanisms. Semin Clin Neuropsychiatry 3, 176–185. [PubMed] [Google Scholar]
- Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, Robertson AAB, Cooper MA, O’Neill LAJ, Lynch MA, 2017a. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav Immun 61, 306–316. [DOI] [PubMed] [Google Scholar]
- Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, Robertson AAB, Cooper MA, O’Neill LAJ, Lynch MA, 2017b. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav Immun 61, 306–316. [DOI] [PubMed] [Google Scholar]
- Deng M, Guo H, Tam JW, Johnson BM, Brickey WJ, New JS, Lenox A, Shi H, Golenbock DT, Koller BH, McKinnon KP, Beutler B, Ting JP, 2019. Platelet-activating factor (PAF) mediates NLRP3-NEK7 inflammasome induction independently of PAFR. J Exp Med 216, 2838–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, Agrawal A, Adeleye AO, Shrime MG, Rubiano AM, Rosenfeld JV, Park KB, 2018. Estimating the global incidence of traumatic brain injury. J Neurosurg, 1–18. [DOI] [PubMed] [Google Scholar]
- Dhani S, Zhao Y, Zhivotovsky B, 2021. A long way to go: caspase inhibitors in clinical use. Cell Death Dis 12, 949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pietro V, Ragusa M, Davies D, Su Z, Hazeldine J, Lazzarino G, Hill LJ, Crombie N, Foster M, Purrello M, Logan A, Belli A, 2017. MicroRNAs as Novel Biomarkers for the Diagnosis and Prognosis of Mild and Severe Traumatic Brain Injury. J Neurotrauma 34, 1948–1956. [DOI] [PubMed] [Google Scholar]
- Ding K, Gupta PK, Diaz-Arrastia R, 2016. Frontiers in Neuroscience Epilepsy after Traumatic Brain Injury, in: Laskowitz D, Grant G (Eds.), Translational Research in Traumatic Brain Injury. CRC Press/Taylor and Francis Group; © 2016 by Taylor & Francis Group, LLC., Boca Raton (FL). [Google Scholar]
- Ding Z, Zhang J, Xu J, Sheng G, Huang G, 2013. Propofol administration modulates AQP-4 expression and brain edema after traumatic brain injury. Cell Biochem Biophys 67, 615–622. [DOI] [PubMed] [Google Scholar]
- DiSabato DJ, Quan N, Godbout JP, 2016. Neuroinflammation: the devil is in the details. J Neurochem 139 Suppl 2, 136–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohi K, Ohtaki H, Nakamachi T, Yofu S, Satoh K, Miyamoto K, Song D, Tsunawaki S, Shioda S, Aruga T, 2010. Gp91phox (NOX2) in classically activated microglia exacerbates traumatic brain injury. J Neuroinflammation 7, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Li CH, Gao RB, Cen XQ, Li P, 2022. Ablation of GSDMD Attenuates Neurological Deficits and Neuropathological Alterations After Traumatic Brain Injury. Front Cell Neurosci 16, 915969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler-Chiurazzi EB, Brown CM, Povroznik JM, Simpkins JW, 2017. Estrogens as neuroprotectants: Estrogenic actions in the context of cognitive aging and brain injury. Prog Neurobiol 157, 188–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennerfelt HE, Lukens JR, 2020. The role of innate immunity in Alzheimer’s disease. Immunol Rev 297, 225–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan K, Ma J, Xiao W, Chen J, Wu J, Ren J, Hou J, Hu Y, Gu J, Yu B, 2017. Mangiferin attenuates blast-induced traumatic brain injury via inhibiting NLRP3 inflammasome. Chem Biol Interact 271, 15–23. [DOI] [PubMed] [Google Scholar]
- Fan Z, Lu M, Qiao C, Zhou Y, Ding JH, Hu G, 2016. MicroRNA-7 Enhances Subventricular Zone Neurogenesis by Inhibiting NLRP3/Caspase-1 Axis in Adult Neural Stem Cells. Mol Neurobiol 53, 7057–7069. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES, 2007. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 14, 1590–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, Cookson BT, 2006. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cellular Microbiology 8, 1812–1825. [DOI] [PubMed] [Google Scholar]
- Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al-Amoudi A, Mangan MS, Zimmer S, Monks BG, Fricke M, Schmidt RE, Espevik T, Jones B, Jarnicki AG, Hansbro PM, Busto P, Marshak-Rothstein A, Hornemann S, Aguzzi A, Kastenmuller W, Latz E, 2014. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 15, 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulp J, He L, Toldo S, Jiang Y, Boice A, Guo C, Li X, Rolfe A, Sun D, Abbate A, Wang XY, Zhang S, 2018. Structural Insights of Benzenesulfonamide Analogues as NLRP3 Inflammasome Inhibitors: Design, Synthesis, and Biological Characterization. J Med Chem 61, 5412–5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco R, Siracusa R, Genovese T, Cuzzocrea S, Di Paola R, 2020. Focus on the Role of NLRP3 Inflammasome in Diseases. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgano M, Toshkezi G, Qiu X, Russell T, Chin L, Zhao LR, 2017. Traumatic Brain Injury: Current Treatment Strategies and Future Endeavors. Cell Transplant 26, 1118–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Byers AL, Barnes DE, Li Y, Boscardin J, Yaffe K, 2018. Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study. Neurology 90, e1771–e1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Yaffe K, 2015. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol Cell Neurosci 66, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Li W, Huang S, Yin Z, Xu X, Chen F, Kong X, Wang H, Zhang J, Lei P, 2018. The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Res 1697, 10–20. [DOI] [PubMed] [Google Scholar]
- Gerber KS, Alvarez G, Alamian A, Behar-Zusman V, Downs CA, 2022. Biomarkers of Neuroinflammation in Traumatic Brain Injury. Clin Nurs Res 31, 1203–1218. [DOI] [PubMed] [Google Scholar]
- Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, Robertson AAB, Butler MS, Rowe DB, O’Neill LA, Kanthasamy AG, Schroder K, Cooper MA, Woodruff TM, 2018. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gugliandolo E, D’Amico R, Cordaro M, Fusco R, Siracusa R, Crupi R, Impellizzeri D, Cuzzocrea S, Di Paola R, 2018. Neuroprotective Effect of Artesunate in Experimental Model of Traumatic Brain Injury. Frontiers in Neurology 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib P, Harms J, Zendedel A, Beyer C, Slowik A, 2020. Gonadal Hormones E2 and P Mitigate Cerebral Ischemia-Induced Upregulation of the AIM2 and NLRC4 Inflammasomes in Rats. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT, 2008. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Guo L, Yang Y, Guan Q, Shen H, Sheng Y, Jiao Q, 2020. Mechanism of microRNA-22 in regulating neuroinflammation in Alzheimer’s disease. Brain Behav 10, e01627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, Liu Q, Liang G, Deng X, Jiang W, Zhou R, 2018. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun 9, 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT, 2013. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, McManus RM, Latz E, 2018. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 19, 610–621. [DOI] [PubMed] [Google Scholar]
- Hicks SD, Olympia RP, Onks C, Kim RY, Zhen KJ, Fedorchak G, DeVita S, Rangnekar A, Heller M, Zwibel H, Monteith C, Gagnon Z, McLoughlin CD, Randall J, Madeira M, Campbell TR, Fengler E, Dretsch MN, Neville C, Middleton FA, 2020. Saliva microRNA Biomarkers of Cumulative Concussion. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houshmandfar S, Saeedi-Boroujeni A, Rashno M, Khodadadi A, Mahmoudian-Sani MR, 2021. miRNA-223 as a regulator of inflammation and NLRP3 inflammasome, the main fragments in the puzzle of immunopathogenesis of different inflammatory diseases and COVID-19. Naunyn Schmiedebergs Arch Pharmacol 394, 2187–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Li C, Shen W, 2014. Gastrodin alleviates memory deficits and reduces neuropathology in a mouse model of Alzheimer’s disease. Neuropathology 34, 370–377. [DOI] [PubMed] [Google Scholar]
- Irrera N, Pizzino G, Calò M, Pallio G, Mannino F, Famà F, Arcoraci V, Fodale V, David A, Francesca C, Minutoli L, Mazzon E, Bramanti P, Squadrito F, Altavilla D, Bitto A, 2017. Lack of the Nlrp3 Inflammasome Improves Mice Recovery Following Traumatic Brain Injury. Front Pharmacol 8, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irrera N, Russo M, Pallio G, Bitto A, Mannino F, Minutoli L, Altavilla D, Squadrito F, 2020. The Role of NLRP3 Inflammasome in the Pathogenesis of Traumatic Brain Injury. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, Griep A, Santarelli F, Brosseron F, Opitz S, Stunden J, Merten M, Kayed R, Golenbock DT, Blum D, Latz E, Buee L, Heneka MT, 2019. NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismael S, Ahmed HA, Adris T, Parveen K, Thakor P, Ishrat T, 2021. The NLRP3 inflammasome: a potential therapeutic target for traumatic brain injury. Neural regeneration research 16, 49–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismael S, Nasoohi S, Ishrat T, 2018a. MCC950, the Selective Inhibitor of Nucleotide Oligomerization Domain-Like Receptor Protein-3 Inflammasome, Protects Mice against Traumatic Brain Injury. J Neurotrauma 35, 1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismael S, Zhao L, Nasoohi S, Ishrat T, 2018b. Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci Rep 8, 5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrahi A, Braun M, Ahluwalia M, Gupta RV, Wilson M, Munie S, Ahluwalia P, Vender JR, Vale FL, Dhandapani KM, Vaibhav K, 2020. Revisiting Traumatic Brain Injury: From Molecular Mechanisms to Therapeutic Interventions. Biomedicines 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, 2004. Traumatic brain injury as a risk factor for Alzheimer’s disease. J Neurol Neurosurg Psychiatry 75, 511–512. [PMC free article] [PubMed] [Google Scholar]
- Jia C, Zhang J, Chen H, Zhuge Y, Chen H, Qian F, Zhou K, Niu C, Wang F, Qiu H, Wang Z, Xiao J, Rong X, Chu M, 2019. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death & Disease 10, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, Troost D, Beyer C, 2015. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63, 2260–2273. [DOI] [PubMed] [Google Scholar]
- Johnson NH, de Rivero Vaccari JP, Bramlett HM, Keane RW, Dietrich WD, 2022a. Inflammasome activation in traumatic brain injury and Alzheimer’s disease. Transl Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NH, Hadad R, Taylor RR, Rodríguez Pilar J, Salazar O, Llompart-Pou JA, 2022b. Inflammatory Biomarkers of Traumatic Brain Injury. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KH, Chu K, Lee ST, Kim SJ, Song EC, Kim EH, Park DK, Sinn DI, Kim JM, Kim M, Roh JK, 2007. Blockade of AT1 receptor reduces apoptosis, inflammation, and oxidative stress in normotensive rats with intracerebral hemorrhage. J Pharmacol Exp Ther 322, 1051–1058. [DOI] [PubMed] [Google Scholar]
- Kabadi SV, Faden AI, 2014. Neuroprotective strategies for traumatic brain injury: improving clinical translation. Int J Mol Sci 15, 1216–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadhim H, Deltenre P, Martin JJ, Sebire G, 2016. In-situ expression of Interleukin-18 and associated mediators in the human brain of sALS patients: Hypothesis for a role for immune-inflammatory mechanisms. Med Hypotheses 86, 14–17. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Dye R, Pakavathkumar P, Foveau B, Flores J, Hyman B, Ghetti B, Koller BH, LeBlanc AC, 2015. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ 22, 1676–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane RW, Dietrich WD, de Rivero Vaccari JP, 2018. Inflammasome Proteins As Biomarkers of Multiple Sclerosis. Front Neurol 9, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr N, de Rivero Vaccari JP, Dietrich WD, Keane RW, 2020. Neural-respiratory inflammasome axis in traumatic brain injury. Exp Neurol 323, 113080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr N, Lee SW, Perez-Barcena J, Crespi C, Ibanez J, Bullock MR, Dietrich WD, Keane RW, de Rivero Vaccari JP, 2018a. Inflammasome proteins as biomarkers of traumatic brain injury. PLoS One 13, e0210128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr N, Lee SW, Perez-Barcena J, Crespi C, Ibañez J, Bullock MR, Dietrich WD, Keane RW, de Rivero Vaccari JP, 2018b. Inflammasome proteins as biomarkers of traumatic brain injury. PLoS One 13, e0210128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr NA, de Rivero Vaccari JP, Abbassi S, Kaur H, Zambrano R, Wu S, Dietrich WD, Keane RW, 2018c. Traumatic Brain Injury-Induced Acute Lung Injury: Evidence for Activation and Inhibition of a Neural-Respiratory-Inflammasome Axis. J Neurotrauma 35, 2067–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr NA, de Rivero Vaccari JP, Umland O, Bullock MR, Conner GE, Dietrich WD, Keane RW, 2019. Human Lung Cell Pyroptosis Following Traumatic Brain Injury. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr NA, de Rivero Vaccari JP, Weaver C, Dietrich WD, Ahmed T, Keane RW, 2021. Enoxaparin Attenuates Acute Lung Injury and Inflammasome Activation after Traumatic Brain Injury. J Neurotrauma 38, 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita K, 2016. Traumatic brain injury: pathophysiology for neurocritical care. J Intensive Care 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Amieva H, Petersen RC, Chetelat G, Holtzman DM, Hyman BT, Nixon RA, Jones DT, 2021. Alzheimer disease. Nat Rev Dis Primers 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobeissy FH, Ottens AK, Zhang Z, Liu MC, Denslow ND, Dave JR, Tortella FC, Hayes RL, Wang KK, 2006. Novel differential neuroproteomics analysis of traumatic brain injury in rats. Mol Cell Proteomics 5, 1887–1898. [DOI] [PubMed] [Google Scholar]
- Kokiko-Cochran ON, Godbout JP, 2018. The Inflammatory Continuum of Traumatic Brain Injury and Alzheimer’s Disease. Front Immunol 9, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada T, Muruve DA, 2019. The role of inflammasomes in kidney disease. Nat Rev Nephrol 15, 501–520. [DOI] [PubMed] [Google Scholar]
- Kono S, Kurata T, Sato K, Omote Y, Hishikawa N, Yamashita T, Deguchi K, Abe K, 2015. Neurovascular protection by telmisartan via reducing neuroinflammation in stroke-resistant spontaneously hypertensive rat brain after ischemic stroke. J Stroke Cerebrovasc Dis 24, 537–547. [DOI] [PubMed] [Google Scholar]
- Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH, 1994. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84, 1415–1420. [PubMed] [Google Scholar]
- Korley FK, Jain S, Sun X, Puccio AM, Yue JK, Gardner RC, Wang KKW, Okonkwo DO, Yuh EL, Mukherjee P, Nelson LD, Taylor SR, Markowitz AJ, Diaz-Arrastia R, Manley GT, Investigators T-TS, 2022. Prognostic value of day-of-injury plasma GFAP and UCH-L1 concentrations for predicting functional recovery after traumatic brain injury in patients from the US TRACK-TBI cohort: an observational cohort study. Lancet Neurol 21, 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Kim IS, More SV, Kim BW, Bahk YY, Choi DK, 2013. Gastrodin protects apoptotic dopaminergic neurons in a toxin-induced Parkinson’s disease model. Evid Based Complement Alternat Med 2013, 514095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwar R, Rolfe A, Di L, Xu H, He L, Jiang Y, Zhang S, Sun D, 2019. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J Neuroinflammation 16, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan J, Horsfield G, Bryant T, Gawne-Cain M, Durward G, Byrne CD, Englyst NA, 2013. IL-6 is a predictive biomarker for stroke associated infection and future mortality in the elderly after an ischemic stroke. Exp Gerontol 48, 960–965. [DOI] [PubMed] [Google Scholar]
- Lahooti B, Chhibber T, Bagchi S, Varahachalam SP, Jayant RD, 2021. Therapeutic role of inflammasome inhibitors in neurodegenerative disorders. Brain Behav Immun 91, 771–783. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM, 2014. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM, 2009. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 187, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A, 2013. Activation and regulation of the inflammasomes. Nat Rev Immunol 13, 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis C, Rusin CG, Robertson CS, 2019. Secondary brain injury: Predicting and preventing insults. Neuropharmacology 145, 145–152. [DOI] [PubMed] [Google Scholar]
- Lee SW, de Rivero Vaccari JP, Truettner JS, Dietrich WD, Keane RW, 2019. The role of microglial inflammasome activation in pyroptotic cell death following penetrating traumatic brain injury. J Neuroinflammation 16, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Gajavelli S, Spurlock MS, Andreoni C, de Rivero Vaccari JP, Bullock MR, Keane RW, Dietrich WD, 2018. Microglial Inflammasome Activation in Penetrating Ballistic-Like Brain Injury. J Neurotrauma 35, 1681–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Zong X, Cheng Y, Xu J, Deng J, Huang Y, Ma C, Fu Q, 2022. miR-223–3p contributes to suppressing NLRP3 inflammasome activation in Streptococcus equi ssp. zooepidemicus infection. Vet Microbiol 269, 109430. [DOI] [PubMed] [Google Scholar]
- Li S, Bian L, Fu X, Ai Q, Sui Y, Zhang A, Gao H, Zhong L, Lu D, 2019. Gastrodin pretreatment alleviates rat brain injury caused by cerebral ischemic-reperfusion. Brain Res 1712, 207–216. [DOI] [PubMed] [Google Scholar]
- Lin C, Chao H, Li Z, Xu X, Liu Y, Bao Z, Hou L, Liu Y, Wang X, You Y, Liu N, Ji J, 2017. Omega-3 fatty acids regulate NLRP3 inflammasome activation and prevent behavior deficits after traumatic brain injury. Exp Neurol 290, 115–122. [DOI] [PubMed] [Google Scholar]
- Lin HB, Wei GS, Li FX, Guo WJ, Hong P, Weng YQ, Zhang QQ, Xu SY, Liang WB, You ZJ, Zhang HF, 2020. Macrophage-NLRP3 Inflammasome Activation Exacerbates Cardiac Dysfunction after Ischemic Stroke in a Mouse Model of Diabetes. Neurosci Bull 36, 1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CM, Tian ZK, Zhang YJ, Ming QL, Ma JQ, Ji LP, 2020. Effects of Gastrodin against Lead-Induced Brain Injury in Mice Associated with the Wnt/Nrf2 Pathway. Nutrients 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HD, Li W, Chen ZR, Hu YC, Zhang DD, Shen W, Zhou ML, Zhu L, Hang CH, 2013. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res 38, 2072–2083. [DOI] [PubMed] [Google Scholar]
- Liu M, Bachstetter AD, Cass WA, Lifshitz J, Bing G, 2017. Pioglitazone Attenuates Neuroinflammation and Promotes Dopaminergic Neuronal Survival in the Nigrostriatal System of Rats after Diffuse Brain Injury. J Neurotrauma 34, 414–422. [DOI] [PubMed] [Google Scholar]
- Liu W, Chen Y, Meng J, Wu M, Bi F, Chang C, Li H, Zhang L, 2018a. Ablation of caspase-1 protects against TBI-induced pyroptosis in vitro and in vivo. J Neuroinflammation 15, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gao J, Peng M, Meng H, Ma H, Cai P, Xu Y, Zhao Q, Si G, 2018b. A Review on Central Nervous System Effects of Gastrodin. Front Pharmacol 9, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonnemann N, Hosseini S, Marchetti C, Skouras DB, Stefanoni D, D’Alessandro A, Dinarello CA, Korte M, 2020. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 117, 32145–32154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV, 2015. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat 11, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucke-Wold BP, Turner RC, Logsdon AF, Bailes JE, Huber JD, Rosen CL, 2014. Linking traumatic brain injury to chronic traumatic encephalopathy: identification of potential mechanisms leading to neurofibrillary tangle development. J Neurotrauma 31, 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Dong Z, Li QG, Wang JR, 2009. Protective effect of propofol against intracerebral hemorrhage injury in rats. Yao Xue Xue Bao 44, 344–349. [PubMed] [Google Scholar]
- Ma J, Xiao W, Wang J, Wu J, Ren J, Hou J, Gu J, Fan K, Yu B, 2016. Propofol Inhibits NLRP3 Inflammasome and Attenuates Blast-Induced Traumatic Brain Injury in Rats. Inflammation 39, 2094–2103. [DOI] [PubMed] [Google Scholar]
- Ma MW, Wang J, Dhandapani KM, Brann DW, 2017a. NADPH Oxidase 2 Regulates NLRP3 Inflammasome Activation in the Brain after Traumatic Brain Injury. Oxid Med Cell Longev 2017, 6057609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MW, Wang J, Zhang Q, Wang R, Dhandapani KM, Vadlamudi RK, Brann DW, 2017b. NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener 12, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AIR, Menon DK, Adelson PD, Andelic N, Bell MJ, Belli A, Bragge P, Brazinova A, Büki A, Chesnut RM, Citerio G, Coburn M, Cooper DJ, Crowder AT, Czeiter E, Czosnyka M, Diaz-Arrastia R, Dreier JP, Duhaime AC, Ercole A, van Essen TA, Feigin VL, Gao G, Giacino J, Gonzalez-Lara LE, Gruen RL, Gupta D, Hartings JA, Hill S, Jiang JY, Ketharanathan N, Kompanje EJO, Lanyon L, Laureys S, Lecky F, Levin H, Lingsma HF, Maegele M, Majdan M, Manley G, Marsteller J, Mascia L, McFadyen C, Mondello S, Newcombe V, Palotie A, Parizel PM, Peul W, Piercy J, Polinder S, Puybasset L, Rasmussen TE, Rossaint R, Smielewski P, Söderberg J, Stanworth SJ, Stein MB, von Steinbüchel N, Stewart W, Steyerberg EW, Stocchetti N, Synnot A, Te Ao B, Tenovuo O, Theadom A, Tibboel D, Videtta W, Wang KKW, Williams WH, Wilson L, Yaffe K, 2017. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol 16, 987–1048. [DOI] [PubMed] [Google Scholar]
- Malik A, Kanneganti TD, 2017. Inflammasome activation and assembly at a glance. J Cell Sci 130, 3955–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallah K, Quanico J, Raffo-Romero A, Cardon T, Aboulouard S, Devos D, Kobeissy F, Zibara K, Salzet M, Fournier I, 2019. Mapping Spatiotemporal Microproteomics Landscape in Experimental Model of Traumatic Brain Injury Unveils a link to Parkinson’s Disease. Mol Cell Proteomics 18, 1669–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JS, Warrington R, Watson W, Kim HL, 2018. An introduction to immunology and immunopathology. Allergy, Asthma & Clinical Immunology 14, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J, 2009. The inflammasomes: guardians of the body. Annu Rev Immunol 27, 229–265. [DOI] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA, 2009. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68, 709–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie BA, Dixit VM, Power C, 2020. Fiery Cell Death: Pyroptosis in the Central Nervous System. Trends Neurosci 43, 55–73. [DOI] [PubMed] [Google Scholar]
- Mejias NH, Martinez CC, Stephens ME, de Rivero Vaccari JP, 2018. Contribution of the inflammasome to inflammaging. J Inflamm (Lond) 15, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda AS, Brant F, Rocha NP, Cisalpino D, Rodrigues DH, Souza DG, Machado FS, Rachid MA, Teixeira AL Jr., Campos AC, 2013. Further evidence for an anti-inflammatory role of artesunate in experimental cerebral malaria. Malar J 12, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed AZ, Nestor PJ, Cumming P, Nasrallah FA, Alzheimer’s Disease Neuroimaging I, 2022. Traumatic brain injury fast-forwards Alzheimer’s pathology: evidence from amyloid positron emission tomorgraphy imaging. J Neurol 269, 873–884. [DOI] [PubMed] [Google Scholar]
- Mondello S, Sandner V, Goli M, Czeiter E, Amrein K, Kochanek PM, Gautam S, Cho BG, Morgan R, Nehme A, Fiumara G, Eid AH, Barsa C, Haidar MA, Buki A, Kobeissy FH, Mechref Y, 2022. Exploring serum glycome patterns after moderate to severe traumatic brain injury: A prospective pilot study. EClinicalMedicine 50, 101494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondello S, Schmid K, Berger RP, Kobeissy F, Italiano D, Jeromin A, Hayes RL, Tortella FC, Buki A, 2014. The challenge of mild traumatic brain injury: role of biochemical markers in diagnosis of brain damage. Med Res Rev 34, 503–531. [DOI] [PubMed] [Google Scholar]
- Moreno-Garcia L, Miana-Mena FJ, Moreno-Martinez L, de la Torre M, Lunetta C, Tarlarini C, Zaragoza P, Calvo AC, Osta R, 2021. Inflammasome in ALS Skeletal Muscle: NLRP3 as a Potential Biomarker. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouasni S, Gonzalez V, Schmitt A, Bennana E, Guillonneau F, Mistou S, Avouac J, Ea HK, Devauchelle V, Gottenberg J-E, Chiocchia G, Tourneur L, 2019. The classical NLRP3 inflammasome controls FADD unconventional secretion through microvesicle shedding. Cell Death & Disease 10, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzer P, Negro R, Fukui S, di Meglio L, Aymonnier K, Chu L, Cherpokova D, Gutch S, Sorvillo N, Shi L, Magupalli VG, Weber ANR, Scharf RE, Waterman CM, Wu H, Wagner DD, 2021. NLRP3 Inflammasome Assembly in Neutrophils Is Supported by PAD4 and Promotes NETosis Under Sterile Conditions. Front Immunol 12, 683803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KN, Parry-Jones AR, Allan SM, 2015. Interleukin-1 and acute brain injury. Front Cell Neurosci 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasser M, Bejjani F, Raad M, Abou-El-Hassan H, Mantash S, Nokkari A, Ramadan N, Kassem N, Mondello S, Hamade E, Darwish H, Zibara K, Kobeissy F, 2016. Traumatic Brain Injury and Blood-Brain Barrier Cross-Talk. CNS Neurol Disord Drug Targets 15, 1030–1044. [DOI] [PubMed] [Google Scholar]
- Ng SY, Lee AYW, 2019. Traumatic Brain Injuries: Pathophysiology and Potential Therapeutic Targets. Front Cell Neurosci 13, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Zaroff JG, 2009. Neurogenic stunned myocardium. Curr Neurol Neurosci Rep 9, 486–491. [DOI] [PubMed] [Google Scholar]
- O’Brien WT, Pham L, Symons GF, Monif M, Shultz SR, McDonald SJ, 2020. The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J Neuroinflammation 17, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien WT, Symons GF, Bain J, Major BP, Costello DM, Sun M, Kimpton JS, Chen Z, Brady RD, Mychasiuk R, O’Brien TJ, Monif M, Shultz SR, McDonald SJ, 2021. Elevated Serum Interleukin-1beta Levels in Male, but not Female, Collision Sport Athletes with a Concussion History. J Neurotrauma 38, 1350–1357. [DOI] [PubMed] [Google Scholar]
- Ottens AK, Kobeissy FH, Golden EC, Zhang Z, Haskins WE, Chen SS, Hayes RL, Wang KK, Denslow ND, 2006. Neuroproteomics in neurotrauma. Mass Spectrom Rev 25, 380–408. [DOI] [PubMed] [Google Scholar]
- Panahpour H, Nekooeian AA, Dehghani GA, 2014a. Blockade of Central Angiotensin II AT1 Receptor Protects the Brain from Ischemia/Reperfusion Injury in Normotensive Rats. Iran J Med Sci 39, 536–542. [PMC free article] [PubMed] [Google Scholar]
- Panahpour H, Nekooeian AA, Dehghani GA, 2014b. Candesartan attenuates ischemic brain edema and protects the blood-brain barrier integrity from ischemia/reperfusion injury in rats. Iran Biomed J 18, 232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A, Shen C, Feng S, Man SM, 2021. Cell biology of inflammasome activation. Trends Cell Biol 31, 924–939. [DOI] [PubMed] [Google Scholar]
- Peelen E, Damoiseaux J, Muris AH, Knippenberg S, Smolders J, Hupperts R, Thewissen M, 2015. Increased inflammasome related gene expression profile in PBMC may facilitate T helper 17 cell induction in multiple sclerosis. Mol Immunol 63, 521–529. [DOI] [PubMed] [Google Scholar]
- Perez-Barcena J, Crespi C, Frontera G, Llompart-Pou JA, Salazar O, Goliney V, Ibanez J, Bullock MR, de Rivero Vaccari JP, 2020. Levels of caspase-1 in cerebrospinal fluid of patients with traumatic brain injury: correlation with intracranial pressure and outcome. J Neurosurg 134, 1644–1649. [DOI] [PubMed] [Google Scholar]
- Piancone F, La Rosa F, Marventano I, Saresella M, Clerici M, 2021. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piancone F, Saresella M, Marventano I, La Rosa F, Santangelo MA, Caputo D, Mendozzi L, Rovaris M, Clerici M, 2018a. Monosodium Urate Crystals Activate the Inflammasome in Primary Progressive Multiple Sclerosis. Front Immunol 9, 983–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piancone F, Saresella M, Marventano I, La Rosa F, Santangelo MA, Caputo D, Mendozzi L, Rovaris M, Clerici M, 2018b. Monosodium Urate Crystals Activate the Inflammasome in Primary Progressive Multiple Sclerosis. Front Immunol 9, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontillo A, Catamo E, Arosio B, Mari D, Crovella S, 2012. NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis Assoc Disord 26, 277–281. [DOI] [PubMed] [Google Scholar]
- Qin X, Li L, Lv Q, Shu Q, Zhang Y, Wang Y, 2018. Expression profile of plasma microRNAs and their roles in diagnosis of mild to severe traumatic brain injury. PLoS One 13, e0204051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raval AP, Martinez CC, Mejias NH, de Rivero Vaccari JP, 2019. Sexual dimorphism in inflammasome-containing extracellular vesicles and the regulation of innate immunity in the brain of reproductive senescent females. Neurochem Int 127, 29–37. [DOI] [PubMed] [Google Scholar]
- Ravichandran KA, Heneka MT, 2021. Inflammasome activation in neurodegenerative diseases. Essays Biochem 65, 885–904. [DOI] [PubMed] [Google Scholar]
- Redell JB, Moore AN, Ward NH 3rd, Hergenroeder GW, Dash PK, 2010. Human traumatic brain injury alters plasma microRNA levels. J Neurotrauma 27, 2147–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H, Kong Y, Liu Z, Zang D, Yang X, Wood K, Li M, Liu Q, 2018. Selective NLRP3 (Pyrin Domain-Containing Protein 3) Inflammasome Inhibitor Reduces Brain Injury After Intracerebral Hemorrhage. Stroke 49, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PJ, 2008. Artesunate for the treatment of severe falciparum malaria. N Engl J Med 358, 1829–1836. [DOI] [PubMed] [Google Scholar]
- Ruan Y, Qiu X, Lv YD, Dong D, Wu XJ, Zhu J, Zheng XY, 2019. Kainic acid Induces production and aggregation of amyloid beta-protein and memory deficits by activating inflammasomes in NLRP3- and NF-kappaB-stimulated pathways. Aging (Albany NY) 11, 3795–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Zhang JH, Obenaus A, 2017. Response of the cerebral vasculature following traumatic brain injury. J Cereb Blood Flow Metab 37, 2320–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saresella M, Marventano I, Calabrese E, Piancone F, Rainone V, Gatti A, Alberoni M, Nemni R, Clerici M, 2014. A complex proinflammatory role for peripheral monocytes in Alzheimer’s disease. J Alzheimers Dis 38, 403–413. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Malovic E, Harishchandra DS, Ghaisas S, Panicker N, Charli A, Palanisamy BN, Rokad D, Jin H, Anantharam V, Kanthasamy A, Kanthasamy AG, 2017. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis 3, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmel SJ, Acosta S, Lozano D, 2017. Neuroinflammation in traumatic brain injury: A chronic response to an acute injury. Brain Circ 3, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott XO, Stephens ME, Desir MC, Dietrich WD, Keane RW, de Rivero Vaccari JP, 2020. The Inflammasome Adaptor Protein ASC in Mild Cognitive Impairment and Alzheimer’s Disease. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefik E, Qu R, Junqueira C, Kaffe E, Mirza H, Zhao J, Brewer JR, Han A, Steach HR, Israelow B, Blackburn HN, Velazquez SE, Chen YG, Halene S, Iwasaki A, Meffre E, Nussenzweig M, Lieberman J, Wilen CB, Kluger Y, Flavell RA, 2022. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 606, 585–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaito A, Hasan H, Habashy KJ, Fakih W, Abdelhady S, Ahmad F, Zibara K, Eid AH, El-Yazbi AF, Kobeissy FH, 2020. Western diet aggravates neuronal insult in post-traumatic brain injury: Proposed pathways for interplay. EBioMedicine 57, 102829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkour Z, Habashy KJ, Berro M, Takkoush S, Abdelhady S, Koleilat N, Eid AH, Zibara K, Obeid M, Shear D, Mondello S, Wang KK, Kobeissy F, 2021. Drug Repurposing in Neurological Disorders: Implications for Neurotherapy in Traumatic Brain Injury. Neuroscientist 27, 620–649. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F, 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Shin JI, Lee KH, Joo YH, Lee JM, Jeon J, Jung HJ, Shin M, Cho S, Kim TH, Park S, Jeon BY, Jeong H, Lee K, Kang K, Oh M, Lee H, Lee S, Kwon Y, Oh GH, Kronbichler A, 2019. Inflammasomes and autoimmune and rheumatic diseases: A comprehensive review. J Autoimmun 103, 102299. [DOI] [PubMed] [Google Scholar]
- Shippy DC, Wilhelm C, Viharkumar PA, Raife TJ, Ulland TK, 2020. beta-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J Neuroinflammation 17, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, Keane RW, Dahl G, 2009. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J Biol Chem 284, 18143–18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slowik A, Beyer C, 2015. Inflammasomes are neuroprotective targets for sex steroids. J Steroid Biochem Mol Biol 153, 135–143. [DOI] [PubMed] [Google Scholar]
- Stefanska J, Sarniak A, Wlodarczyk A, Sokolowska M, Doniec Z, Bialasiewicz P, Nowak D, Pawliczak R, 2012a. Hydrogen peroxide and nitrite reduction in exhaled breath condensate of COPD patients. Pulm Pharmacol Ther 25, 343–348. [DOI] [PubMed] [Google Scholar]
- Stefanska J, Sarniak A, Wlodarczyk A, Sokolowska M, Pniewska E, Doniec Z, Nowak D, Pawliczak R, 2012b. Apocynin reduces reactive oxygen species concentrations in exhaled breath condensate in asthmatics. Exp Lung Res 38, 90–99. [DOI] [PubMed] [Google Scholar]
- Stutz A, Horvath GL, Monks BG, Latz E, 2013. ASC Speck Formation as a Readout for Inflammasome Activation, in: De Nardo CM, Latz E (Eds.), The Inflammasome: Methods and Protocols. Humana Press, Totowa, NJ, pp. 91–101. [DOI] [PubMed] [Google Scholar]
- Sulhan S, Lyon KA, Shapiro LA, Huang JH, 2020. Neuroinflammation and blood-brain barrier disruption following traumatic brain injury: Pathophysiology and potential therapeutic targets. J Neurosci Res 98, 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Nyanzu M, Yang S, Zhu X, Wang K, Ru J, Yu E, Zhang H, Wang Z, Shen J, Zhuge Q, Huang L, 2020. VX765 Attenuates Pyroptosis and HMGB1/TLR4/NF-κB Pathways to Improve Functional Outcomes in TBI Mice. Oxid Med Cell Longev 2020, 7879629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Brown CM, Wise PM, 2009. Neuroprotective effects of estrogens following ischemic stroke. Front Neuroendocrinol 30, 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson KV, Deng M, Ting JP, 2019. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima T, Yoshifuji A, Matsui A, Itoh T, Uchiyama K, Kanda T, Tokuyama H, Wakino S, Itoh H, 2019. β-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney International 95, 1120–1137. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S, 2010. Pattern recognition receptors and inflammation. Cell 140, 805–820. [DOI] [PubMed] [Google Scholar]
- Tan MS, Tan L, Jiang T, Zhu XC, Wang HF, Jia CD, Yu JT, 2014. Amyloid-beta induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis 5, e1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tas D, Kaplan O, Sogut O, 2020. Validity of Serum miRNA 93 and miRNA 191 to Reduce Unnecessary Computed Tomography in Patients With Mild Head Trauma. J Clin Med Res 12, 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakkar R, Wang R, Sareddy G, Wang J, Thiruvaiyaru D, Vadlamudi R, Zhang Q, Brann D, 2016. NLRP3 Inflammasome Activation in the Brain after Global Cerebral Ischemia and Regulation by 17beta-Estradiol. Oxid Med Cell Longev 2016, 8309031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal SC, Heinemann M, Luh C, Pieter D, Werner C, Engelhard K, 2011. Pioglitazone reduces secondary brain damage after experimental brain trauma by PPAR-γ-independent mechanisms. J Neurotrauma 28, 983–993. [DOI] [PubMed] [Google Scholar]
- Tran HT, LaFerla FM, Holtzman DM, Brody DL, 2011. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci 31, 9513–9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudler D, Nazor KL, Eisele YS, Grabauskas T, Dolatabadi N, Parker J, Sultan A, Zhong Z, Goodwin MS, Levites Y, Golde TE, Kelly JW, Sierks MR, Schork NJ, Karin M, Ambasudhan R, Lipton SA, 2021. Soluble alpha-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc Natl Acad Sci U S A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH, 2007. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol 208, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Heuvel C, Thornton E, Vink R, 2007. Traumatic brain injury and Alzheimer’s disease: a review. Prog Brain Res 161, 303–316. [DOI] [PubMed] [Google Scholar]
- Venegas C, Heneka MT, 2019. Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J 33, 13075–13084. [DOI] [PubMed] [Google Scholar]
- Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz M, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, Heneka MT, 2017. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 552, 355–361. [DOI] [PubMed] [Google Scholar]
- Villapol S, Balarezo MG, Affram K, Saavedra JM, Symes AJ, 2015. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 138, 3299–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Loane DJ, Burns MP, 2017. Sexual dimorphism in the inflammatory response to traumatic brain injury. Glia 65, 1423–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet S, Srinivasan S, Lamkanfi M, van Loo G, 2019. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallisch JS, Simon DW, Bayir H, Bell MJ, Kochanek PM, Clark RSB, 2017. Cerebrospinal Fluid NLRP3 is Increased After Severe Traumatic Brain Injury in Infants and Children. Neurocrit Care 27, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JG, Muruve DA, Power C, 2014. Inflammasomes in the CNS. Nat Rev Neurosci 15, 84–97. [DOI] [PubMed] [Google Scholar]
- Wang D, Shi J, Lv S, Xu W, Li J, Ge W, Xiao C, Geng D, Liu Y, 2015. Artesunate Attenuates Lipopolysaccharide-Stimulated Proinflammatory Responses by Suppressing TLR4, MyD88 Expression, and NF-κB Activation in Microglial Cells. Inflammation 38, 1925–1932. [DOI] [PubMed] [Google Scholar]
- Wang KK, Munoz Pareja JC, Mondello S, Diaz-Arrastia R, Wellington C, Kenney K, Puccio AM, Hutchison J, McKinnon N, Okonkwo DO, Yang Z, Kobeissy F, Tyndall JA, Buki A, Czeiter E, Pareja Zabala MC, Gandham N, Berman R, 2021. Blood-based traumatic brain injury biomarkers - Clinical utilities and regulatory pathways in the United States, Europe and Canada. Expert Rev Mol Diagn 21, 1303–1321. [DOI] [PubMed] [Google Scholar]
- Wang X, Li S, Ma J, Wang C, Chen A, Xin Z, Zhang J, 2019. Effect of Gastrodin on Early Brain Injury and Neurological Outcome After Subarachnoid Hemorrhage in Rats. Neurosci Bull 35, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington PM, Villapol S, Burns MP, 2016. Polypathology and dementia after brain trauma: Does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp Neurol 275 Pt 3, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Hu CC, Zhang YL, Yao SL, Mao WK, 2016. Telmisartan reduced cerebral edema by inhibiting NLRP3 inflammasome in mice with cold brain injury. J Huazhong Univ Sci Technolog Med Sci 36, 576–583. [DOI] [PubMed] [Google Scholar]
- Whiteford HA, Ferrari AJ, Degenhardt L, Feigin V, Vos T, 2015. The global burden of mental, neurological and substance use disorders: an analysis from the Global Burden of Disease Study 2010. PLoS One 10, e0116820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijayatilake DS, Sherren PB, Jigajinni SV, 2015. Systemic complications of traumatic brain injury. Curr Opin Anaesthesiol 28, 525–531. [DOI] [PubMed] [Google Scholar]
- Wilson L, Stewart W, Dams-O’Connor K, Diaz-Arrastia R, Horton L, Menon DK, Polinder S, 2017. The chronic and evolving neurological consequences of traumatic brain injury. The Lancet Neurology 16, 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodrow CJ, Haynes RK, Krishna S, 2005. Artemisinins. Postgrad Med J 81, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi HJ, Zhang TH, Tao T, Song CY, Lu SJ, Cui XG, Yue ZY, 2011. Propofol improved neurobehavioral outcome of cerebral ischemia-reperfusion rats by regulating Bcl-2 and Bax expression. Brain Res 1410, 24–32. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M, 2018. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chin J Traumatol 21, 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Yin D, Ren H, Gao W, Li F, Sun D, Wu Y, Zhou S, Lyu L, Yang M, Xiong J, Han L, Jiang R, Zhang J, 2018. Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis 117, 15–27. [DOI] [PubMed] [Google Scholar]
- Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM, 2019. Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol 40, 1035–1052. [DOI] [PubMed] [Google Scholar]
- Yan C, Yan H, Mao J, Liu Y, Xu L, Zhao H, Shen J, Cao Y, Gao Y, Li K, Jin W, 2020. Neuroprotective Effect of Oridonin on Traumatic Brain Injury via Inhibiting NLRP3 Inflammasome in Experimental Mice. Front Neurosci 14, 557170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Li G, Lin B, Zhang K, 2022. Gastrodin suppresses pyroptosis and exerts neuroprotective effect in traumatic brain injury model by inhibiting NLRP3 inflammasome signaling pathway. J Integr Neurosci 21, 72. [DOI] [PubMed] [Google Scholar]
- Yang Q, Liu R, Yu Q, Bi Y, Liu G, 2019a. Metabolic regulation of inflammasomes in inflammation. Immunology 157, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang H, Kouadir M, Song H, Shi F, 2019b. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis 10, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsiv I, Morganti-Kossmann MC, Perez D, Dinarello CA, Novick D, Rubinstein M, Otto VI, Rancan M, Kossmann T, Redaelli CA, Trentz O, Shohami E, Stahel PF, 2002. Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J Cereb Blood Flow Metab 22, 971–978. [DOI] [PubMed] [Google Scholar]
- Yi HJ, Lee JE, Lee DH, Kim YI, Cho CB, Kim I, Sung JH, Yang SH, 2020. The role of NLRP3 in traumatic brain injury and its regulation by pioglitazone. Journal of Neurosurgery 133, 1083–1091. [DOI] [PubMed] [Google Scholar]
- Yin J, Zhao F, Chojnacki JE, Fulp J, Klein WL, Zhang S, Zhu X, 2018. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol Neurobiol 55, 1977–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborowski MP, Balaj L, Breakefield XO, Lai CP, 2015. Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience 65, 783–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamani P, Oskuee RK, Atkin SL, Navashenaq JG, Sahebkar A, 2020. MicroRNAs as important regulators of the NLRP3 inflammasome. Prog Biophys Mol Biol 150, 50–61. [DOI] [PubMed] [Google Scholar]
- Zendedel A, Monnink F, Hassanzadeh G, Zaminy A, Ansar MM, Habib P, Slowik A, Kipp M, Beyer C, 2018. Estrogen Attenuates Local Inflammasome Expression and Activation after Spinal Cord Injury. Mol Neurobiol 55, 1364–1375. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, Dhandapani KM, Brann DW, 2012. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS One 7, e34504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Zhang S, Ying Y, Guo X, Li H, Xu L, Ruan X, 2018. Administration of Dexmedetomidine inhibited NLRP3 inflammasome and microglial cell activities in hippocampus of traumatic brain injury rats. Biosci Rep 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Liwinski T, Elinav E, 2020. Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov 6, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivaki D, Borriello F, Chow OA, Doran B, Fleming I, Theisen DJ, Pallis P, Shalek AK, Sokol CL, Zanoni I, Kagan JC, 2020. Inflammasomes within Hyperactive Murine Dendritic Cells Stimulate Long-Lived T Cell-Mediated Anti-tumor Immunity. Cell Rep 33, 108381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J, 2010. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11, 136–140. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Lu M, Du RH, Qiao C, Jiang CY, Zhang KZ, Ding JH, Hu G, 2016. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegener 11, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo S, Li Q, Liu X, Feng H, Chen Y, 2016. The Potential Therapeutic Effects of Artesunate on Stroke and Other Central Nervous System Diseases. Biomed Res Int 2016, 1489050. [DOI] [PMC free article] [PubMed] [Google Scholar]