

Abstract
Strong epidemiological evidence now exists that sex is an important biologic variable in immunity. Recent studies, for example, have revealed that sex differences are associated with the severity of symptoms and mortality due to coronavirus disease 2019 (COVID-19). Despite this evidence, much remains to be learned about the mechanisms underlying associations between sex differences and immune-mediated conditions. A growing body of experimental data has made significant inroads into understanding sex-influenced immune responses. As physicians seek to provide more targeted patient care, it is critical to understand how sex-defining factors (e.g., chromosomes, gonadal hormones) alter immune responses in health and disease. In this review, we highlight recent insights into sex differences in autoimmunity; virus infection, specifically severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection; and cancer immunotherapy. A deeper understanding of underlying mechanisms will allow the development of a sex-based approach to disease screening and treatment.
Keywords: immunity, chromosomal complement, sex hormones, sex differences, autoimmunity, immune-related adverse events
INTRODUCTION
Sex and gender are two critical, and often overlooked, variables that influence disease incidence and outcomes. To begin to understand this observation, it is helpful to define these terms. Sex is categorized as female, male, or intersex based on a composite of sex chromosomes (e.g., 46XX or 46XY), gonads (e.g., ovaries or testes), and gonadal hormones (e.g., estrogens or androgens) (1). Differences in sex can produce functional differences through variable expression of sex chromosome–associated genes. In addition, the presence of gonads and the levels of gonadal hormones vary by sex (e.g., testes and higher testosterone levels in males versus ovaries and higher estrogen levels in females) and shape normal physiology and disease pathogenesis (2) (Figure 1). Gender, on the other hand, is a socially determined construct best understood through a series of terms (1). Gender identity refers to an individual’s internal sense of gender, while gender expression describes how a person displays their gender outwardly, such as with clothing, mannerisms, speech, and pronouns. Gender relations describe how individuals interact with others on account of their gender expression. Each of these attributes shapes an individual’s experience with the health care system (e.g., access to care, propensity to seek care, encounters with health care providers) and influences lifestyle factors that either promote or hinder good health (e.g., stress, exercise, nutrition, smoking) (1, 3). The focus of this review is the influence of sex on immune responses, specifically immune-related pathogenic mechanisms.
Figure 1.
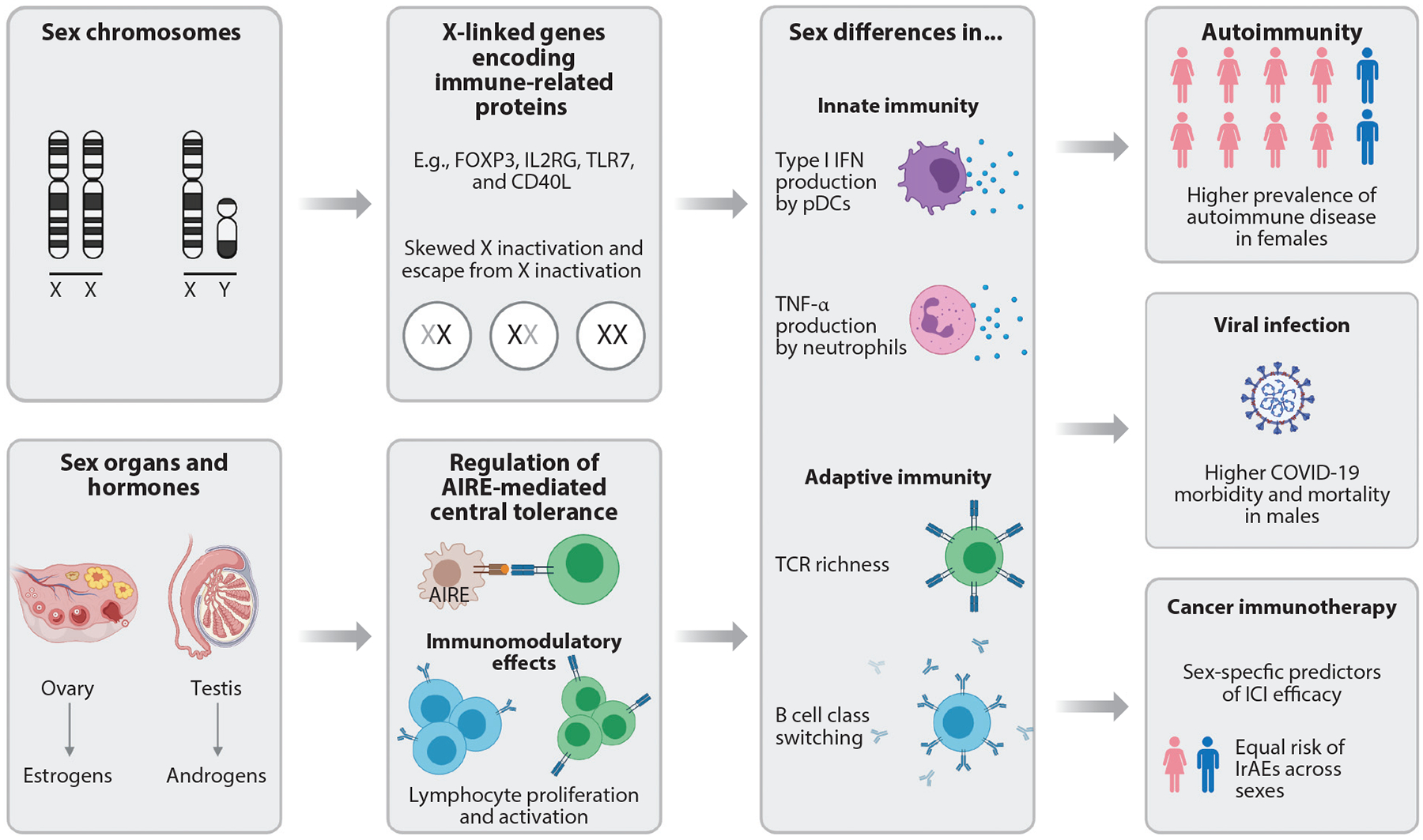
Schematic of mechanisms underlying sex-related differences in immunity. Sex chromosome complement (e.g., 46XX or 46XY), gonads (ovaries or testes), and sex steroids (e.g., estrogens or androgens) contribute to sex differences in the immune response. The X and Y chromosomes differ in gene content, with many immune-related genes located on the X chromosome. Moreover, the degree to which a subset of X chromosome genes is expressed differs between cells, tissue types, and individuals, which adds complexity to chromosomal contributions to sex differences. Sex steroids influence lymphocyte development, proliferation, and activation. Collectively, these sex-specific factors modulate innate and adaptive immunity and subsequently alter outcomes of autoimmunity, viral infection, and cancer immunotherapy. Abbreviations: AIRE, autoimmune regulator; COVID-19, coronavirus disease 2019; ICI, immune checkpoint inhibitor; IFN, interferon; IrAE, immune-related adverse event; pDC, plasmacytoid dendritic cell; TCR, T cell receptor; TLR7, Toll-like receptor 7. Figure adapted from images created with BioRender.com.
Epidemiological studies provide strong evidence for sex differences in immune responses toward self-antigens (i.e., autoimmunity), viruses, and cancer. Females generate more robust immunity against viruses (4–6). For instance, following HIV-1 infection, plasmacytoid dendritic cells (pDCs) from females produce higher levels of IFN-α, a critical cytokine in the innate antiviral response (7). Further, in a murine model of H1N1 influenza infection, more antigen-specific B cells and higher antibody titers were detected in female animals compared to males. In this model, females also generated more robust memory CD8+ T cell responses (8). As a result, many viral infections (e.g., dengue virus, hepatitis B, hepatitis C) are less prevalent in females than males (9, 10). Additionally, females who do succumb to viral infection often have lower viral loads than males (11). Furthermore, vaccine responses, which are considered a proxy for antiviral responses, are greater in females. Following influenza vaccination, females have a greater neutralizing antibody seroconversion rate than males. Interestingly, vaccine responses were positively correlated with estradiol concentrations and premenopausal status in women (12). Females are more frequently affected by autoimmunity and account for approximately 80% of these cases in the United States (13). Autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, and scleroderma have female-to-male ratios between 2:1 and 3:1, while systemic lupus erythematosus (SLE), the most sex-disparate autoimmune disease, has a 9:1 female-to-male ratio (14). Together these data suggest stronger innate and adaptive immune responses in females. The patterns of sex-associated differences in emerging viral infections and responses to newer immune-modulating therapies, such as immune checkpoint inhibitor (ICI) cancer treatment, are beginning to be defined. In this review, we highlight recent insights into sex differences in the immune response as they relate to (a) autoimmunity; (b) antiviral immunity, specifically severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2); and (c) ICI cancer immunotherapy. We also emphasize key outstanding questions in the field.
SEX DIFFERENCES IN AUTOIMMUNITY
Sex differences for multiple autoimmune conditions are well-documented and are attributed to either genetic or hormonal factors. The development of the four core genotype (FCG) mouse model has enabled researchers to isolate the effects of sex chromosomes and hormones. In the FCG mouse, the Sry gene, which is typically located on the Y chromosome and drives development of testes rather than ovaries, is translocated to an autosome (15). Thus, in this transgenic mouse, gonadal differentiation is independent of chromosome complement. Irrespective of having an XX or XY chromosomal complement, FCG mice develop testes when the Sry transgene is present (XXSry+ and XY) and ovaries when it is absent (XX and XYSry−). Thus, this model yields four genotypes: gonadal females with an XX or XY chromosome complement (both Sry−) and gonadal males with an XX or XY chromosome complement (both Sry+). The effects of sex chromosomes, therefore, can be evaluated separate from gonads and gonadal hormones (e.g., by comparison of XX and XY animals with the same gonads, either testes or ovaries).
Recent studies using the FCG mouse model have uncovered important insights about sex differences in the immune response. For instance, XX mice are more susceptible than XY mice to certain autoimmune diseases, independent of gonadal type, suggesting an important X chromosomal contribution (16, 17). Here, we discuss advances in understanding genetic and hormonal mechanisms that underlie sex differences in autoimmunity.
Chromosomal Contributions to Autoimmunity
Females and males differ in their complement of X and Y chromosomes (XX versus XY), and the X and Y chromosomes differ substantially from each other. The human X chromosome harbors more than 1,000 genes, whereas the Y chromosome contains only 45–75 genes, and there are many X-linked genes that encode immune-related proteins [e.g., FOXP3, IL2RG, Toll-like receptor 7 (TLR7), and CD40L] (18). To maintain similar levels of X-linked protein expression between sexes, females undergo random X chromosome inactivation (XCI) during early embryonic development in which the paternal or maternal X chromosome is silenced in each cell. XCI results in the formation of stable heterochromatin that is transcriptionally inert and propagated through subsequent cell divisions. This process begins with the cis coating of the future inactive X chromosome (Xi) with the long noncoding RNA (lncRNA) Xist, located on the X-inactivation center (19). Xist triggers large-scale chromosome condensation to form heterochromatic Barr bodies and recruits Polycomb proteins that induce heterochromatin modifications (20). Thus, it would seem that XX females and XY males should both express genes from a single active copy of the X chromosome and that the different number of X chromosomes should not contribute to sex differences.
The biology of X-linked gene expression, however, is much more complex, and strong evidence now exists that differences in expression of X-linked genes are important contributors to sex differences in immune responses. These differences result from multiple mechanisms that alter sex chromosome expression, including escape of X-linked genes from inactivation, dysregulation of the maintenance of X inactivation, and XCI skewing. These mechanisms and their impacts on autoimmune predisposition are discussed below.
Escape from X chromosome inactivation.
Differential gene expression in females and males can occur as a consequence of incomplete inactivation of X-linked genes in females. Incomplete XCI occurs in at least 23% of X chromosome genes (21). For those genes in which a Y chromosome homolog does not exist, incomplete XCI can result in their relative overexpression in females compared to males (22). Increased expression of escaped genes may contribute to increased autoimmune susceptibility in females (23). For instance, TLR7 does not have a Y chromosome homolog and escapes XCI, and biallelic expression is detected in primary B cells, monocytes, and pDCs in females. TLR7 encodes an endosomal sensor for single-stranded RNA that plays a key role in B cell production of type I interferons and autoantibodies. In functional tests, B cells over-expressing TLR7 were more capable of undergoing class switching and differentiating into CD27+ plasma cells, suggesting that females harboring B cells with biallelic expression of TLR7 are more prone to produce autoantibodies (24).
These findings may have relevance to female-dominant autoimmune diseases such as SLE, which is characterized by the production of B cell–derived autoantibodies targeting double-stranded DNA and autoreactive T cell–mediated inflammation. Although not directly tested using SLE patient samples, these findings support the possibility that increased TLR7 expression underlies female predisposition in B cell–mediated autoimmune conditions such as SLE. Interestingly, Klinefelter syndrome patients, who have a 47XXY chromosomal complement, also have biallelic expression of TLR7 and an increased incidence of SLE. This association between TLR7 biallelism and SLE predisposition suggests the intriguing possibility that SLE predisposition in females and Klinefelter syndrome may be attributed to TLR7 overexpression; however, this hypothesis remains to be empirically tested.
Utx (also known as KDM6a) is another example of a gene that normally escapes X inactivation and underlies female predisposition to autoimmunity. CD4+ T cells isolated from XX FCG mice had elevated expression of the X-linked gene Utx compared to those from XY FCG mice, regardless of gonadal type (25). Thus, the XX chromosome complement is responsible for increased Utx expression levels in females. Importantly, lymphocyte development differs between 46XX females (with two copies of X-linked Utx) and 45X Turner syndrome females (with one copy of Utx) (26), with alterations in the frequency of a CD4+ T follicular helper cell subset. Conditional knockout of Utx in CD4+ T cells led to the amelioration of experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis. UTX is an epigenetic regulator, and deletion of UTX from CD4+ T cells resulted in an altered transcriptional signature. Notably, Th17 cells are the predominant pathogenic T cell subset in EAE, and Rorc, the master regulator of T helper 17 (Th17) differentiation, was downregulated in the absence of UTX (25). Together, these findings suggest that increased UTX gene dosage in females due to XCI escape may be responsible for female predisposition to CNS autoimmunity. It is interesting to note that UTX does have a Y chromosome homolog, UTY. However, UTY has key differences from UTX, including attenuated histone demethylase activity (27). An open question, then, is how expression of UTY in males may contribute to male protection from autoimmunity.
Finally, increased escape of the X-linked CD40LG locus from XCI may also contribute to autoimmune predisposition in females. CD40L (encoded by CD40LG) is a tumor necrosis factor (TNF) family member that interacts with CD40 to amplify cellular immune responses. The importance of CD40L overexpression in autoimmune predisposition has recently been illustrated by the development of autoimmunity in a six-month-old male infant with CD40LG gene duplication (28). Thus, a twofold increase in CD40LG gene dosage results in early-onset autoimmunity. Relevant to more common forms of autoimmunity, CD40L RNA expression is relatively increased in B cell lines from SLE patients compared to non-SLE controls (29). The increased CD40L RNA expression was associated with a higher percentage of cells expressing CD40L from the inactive X chromosome. In line with these findings, bisulfite sequencing showed that CG pairs in the CD40LG promoter are relatively demethylated and overexpressed in women with SLE compared to non-SLE controls (30). Increased CD40/CD40L signaling has also been associated with multiple sclerosis, an autoimmune disease of the CNS with strong female bias (31). Together, these findings suggest that biallelic expression of CD40L contributes to predisposition to autoimmunity in females.
Dysregulation of maintenance of X chromosome inactivation.
Maintenance of XCI is a dynamic process, and abnormalities in this process can predispose females to autoimmunity. The epigenetic pattern of regulation of the inactive X chromosome in B cell subsets differs between healthy individuals and SLE patients, suggesting abnormal XCI maintenance in SLE (32). RNA fluorescence in situ hybridization (FISH) showed that the Xist signal in B cells isolated from SLE patients was missing or mislocalized (32). Xist RNA binds to the X chromosome through interactions with nuclear matrix proteins serving as an anchoring site. CIZ1 and hnRNPU are two Xist RNA protein partners identified from RNA-protein interaction assays, and knockout of either protein results in the failure of Xist localization (33). In SLE patients, Xist RNA–binding proteins were downregulated in SLE-activated B cells, suggesting that dysregulation of Xist location may be due to lack of binding to its partner proteins. In addition, many X-linked genes were differentially expressed, suggesting that changes in heterochromatic modifications on the inactive X lead to changes in X-linked gene expression in B cells. In addition to B cells, autoreactive T cells also likely play an important role in SLE pathogenesis. Disruption of Xist RNA localization was similarly observed in SLE T cells, leading to the upregulation of X-related genes (34). Together, these findings suggest that alterations in the maintenance of XCI in T and B lymphocytes may underlie female bias in SLE.
Skewing of X chromosome inactivation.
While it is often stated that the choice of which X chromosome is inactivated is random, it is well-documented that skewing can occur. With XCI skewing, one of the X chromosomes is preferentially inactivated so that inactivation does not occur at a 1:1 ratio. Indeed, recent work has demonstrated that the maternally derived X chromosome is preferentially expressed in CD4+ T cells (20). More DNA methylation was seen on the paternally derived X chromosome, resulting in decreased expression. Moreover, XCI skewing is tissue dependent—blood, fat, and skin tissue have different levels of skewing—whereas immune cell types (B cells, T cells, and natural killer cells) share similar XCI patterns (35). What causes XCI skewing is not completely understood, but there is now evidence that XCI skewing is associated with age and lifestyle (35). Acquired XCI skewing has been noted in blood-derived tissues of aged individuals, and increasing rates of XCI skewing are found in older smokers.
XCI skewing has been associated with the development of autoimmune disease in females. In particular, this association has been reported for scleroderma (36), rheumatoid arthritis (37), and autoimmune thyroiditis (38, 39), three autoimmune diseases with strong female predisposition. While it remains unclear how XCI skewing can lead to autoimmunity, a long-standing hypothesis is that a mismatch may occur in self-antigens expressed in target tissues and those expressed in the thymus that induce T cell tolerance (38). In the thymus, developing T cells that recognize self-antigens with high affinity undergo clonal deletion to eliminate self-reactive T cells that predispose to autoimmunity. This process is mediated by thymic expression of a large number of self-antigens, some of which may be highly polymorphic. If self-antigens expressed from the maternally versus paternally inherited allele differ, preferential silencing of one X chromosome may prevent induction of T cell tolerance toward that self-antigen variant. Thus, T cells may be incompletely tolerized to antigens that are preferentially silenced in the thymus. If this same pattern of XCI skewing does not occur in target tissues, expression of this self-antigen variant in the periphery may elicit an autoreactive T cell response. Importantly, however, this hypothesis remains to be rigorously tested. Thus, additional studies are needed to clarify the cause and consequence of XCI skewing in relation to autoimmunity.
Sex Hormone Contributions to Autoimmunity
Besides differences in sex chromosome gene expression, multiple lines of evidence suggest that sex hormones, which include estrogens and androgens, also affect autoimmune predisposition. In an SLE animal model, for example, estrogen worsens whereas removal of estrogen relieves disease progression. Meanwhile, castrated male mice have increased susceptibility to SLE (40). In addition, aged males with relatively lower serum androgens have increased incidence of rheumatoid arthritis (41). Thus, multiple instances in which increased autoimmunity is associated with lower androgen and/or higher estrogen levels have now been described.
Sex hormones and Aire-mediated thymic tolerance.
Recent studies have delineated mechanisms by which sex hormones may control autoimmune predisposition. An important sex hormone–regulated self-tolerance mechanism occurs within the thymus, where T cells are educated after developing from bone marrow–derived T cell precursors. As discussed above, negative selection removes developing T cells in the thymus that recognize self-antigens with high affinity. Expression of these self-antigens is normally limited to a specific tissue, but they are also ectopically expressed by rare medullary thymic epithelial cells (mTECs) within the thymus. Insulin, for example, is a tissue-specific antigen expressed specifically by pancreatic beta cells that is also ectopically expressed by mTECs. Expression of large proportion of these tissue-specific antigens is under the control of the autoimmune regulator (Aire) gene (42). The importance of AIRE in preventing autoimmunity is illustrated by the development of multiorgan autoimmunity in Aire-deficient humans, rats, and mice.
Both androgens and estrogens play a crucial role in controlling Aire-mediated thymic tolerance (43, 44). Comparison of mTEC transcriptomes in males and females revealed higher Aire expression in male mTECs compared to female mTECs. As a consequence, Aire-regulated tissue-specific antigen expression was higher in males, and self-reactive T cells underwent more efficient negative selection in the thymus. This increase in Aire expression is due to androgen-mediated upregulation of Aire expression. Multiple androgen-response elements are found in the Aire promoter, and the androgen promoter is localized to the Aire locus in an androgen-dependent manner (44). At the same time, AIRE expression is downregulated by estrogen treatment through regulation of CpG methylation at the AIRE promoter (43). This push-and-pull effect of sex hormones on Aire expression enforces sex differences in self-tolerance and may underlie the relative protection seen in males against CNS autoimmunity (44) and thyroid autoimmunity (43).
Sex hormones and peripheral tolerance.
In addition to thymic tolerance, sex hormones also play a major role in peripheral T cell tolerance mechanisms. Estrogen’s effect on cellular function is mediated by estrogen receptors alpha and beta (ERα and ERβ), and ERα is highly expressed on T cells. In a mouse model of colitis, T cell–specific deletion of ERα was associated with reduced weight loss, suggesting that ERα normally promotes the pathogenic potential of CD4+ T cells (45). Moreover, deletion of ERα in T cells reduced T cell activation and proliferation, decreased inflammatory Th1 and Th17 subsets, and increased numbers of T regulatory cells (Tregs), a T helper cell type important in preventing autoimmunity. Thus, estrogen promotes autoimmune T helper cell subsets while limiting suppressive T helper cell subsets.
Conversely, recent evidence suggests that androgens promote Treg development. Androgens can do so either indirectly through their effects on stromal cells that modulate Treg differentiation (46) or directly through the control of transcription factors important in Treg differentiation (47). In visceral adipose tissue, androgens stimulate increases in IL-33-producing CD31−Gp38+ stromal cells, and IL-33 in turn leads to expansion of Tregs. Additionally, androgens alter the transcriptional profile of Tregs through upregulation of Foxp3, a transcription factor important for CD4+ Treg differentiation. Androgens increase Foxp3 expression via direct binding of androgen–androgen receptor (AR) to the Foxp3 locus. AR binding was associated with increased acetylation of histone H4 at the Foxp3 locus of human primary Tregs, suggesting that androgens alter epigenetic regulation at the locus. An important consideration with these findings, however, is that the effect of androgen on the Foxp3 locus was seen only in Tregs of females. The low circulating levels of gonad-derived androgens in females bring into question the physiologic relevance of this finding. Finally, androgen also ameliorates EAE severity by shifting T helper subsets away from a pathogenic response (Th17 to Th2). Androgens stimulate mast cell production of IL-33, which acts through group 2 innate lymphoid cells to elicit this shift (48).
Sex hormones also play a major role in modulating B cell numbers, including pathogenic B cells in autoimmune disease. Over 40 years ago, testosterone levels were associated with protection from SLE (49). Castration of male mice resulted in a decline in serum testosterone, increased IgM–to–pathogenic IgG antibody switching and premature death. In male subjects with low androgen levels due to hypogonadotropic hypogonadism, B cell numbers and immunoglobulin levels were increased compared to age-matched healthy controls (50). Furthermore, treatment with exogenous androgens normalized these numbers, suggesting that androgens normally downregulate B cells and B cell–derived antibodies. Similarly, in subjects with Klinefelter syndrome, a condition associated with low androgen levels, B cell numbers and immunoglobulin levels were higher than those in control males, and testosterone replacement therapy decreased these numbers (51). Studies in androgen receptor knockout mice suggest that testosterone limits B cell numbers through downregulation of the cytokine BAFF, an essential survival factor for B cells (52). Taken together, these results suggest that androgens act to attenuate immune responses by increasing immune regulation and countering immune activation. In a groundbreaking report, Markle et al. (53) demonstrated that sex differences in the gut microbiome may also underlie autoimmune susceptibility. Male nonobese diabetic (NOD) mice housed in specific-pathogen free conditions were relatively protected from type 1 diabetes compared to female NOD mice. Conversely, mice housed in germ-free conditions exhibited less difference in type 1 diabetes incidence. Transfer of cecal contents from males to females resulted in reduced islet inflammation, autoantibody production, and diabetes development. Interestingly, this effect was mediated through androgen, since female recipients had increased testosterone levels in the serum and the protective effect was abrogated with androgen receptor blockade. These findings raise a number of questions, including the role of the gut microbiome in other sex-biased autoimmune conditions and how the microbiome may be manipulated for therapeutic benefit.
SEX DIFFERENCES IN SARS-CoV-2 INFECTION
The coronavirus disease 2019 (COVID-19) pandemic has highlighted sex disparities in infectious disease outcomes. In a meta-analysis of 3,111,714 patients from 46 countries, males were more likely than females to require intensive care unit admission [odds ratio (OR) = 2.84, 95% confidence interval (CI) = 2.06–3.92, p = 1.86 × 10−10] and die (OR = 1.39, 95% CI = 1.31–1.47, p = 5 × 10−30) owing to severe acute respiratory coronavirus 2 (SARS-CoV-2) infection (54). Analysis of plasma and circulating immune cells from male and female patients with COVID-19 identified distinct factors associated with disease progression in each sex, suggesting that sex-specific variability in the immune response may contribute to differences in COVID-19 outcomes (55). In this section, we discuss key steps in the defense against SARS-CoV-2 infection, highlighting evidence of sex differences that may occur at each stage. Additionally, we discuss new data regarding the roles of sex chromosomes and gonadal hormones in mediating these differences (Figure 2).
Figure 2.
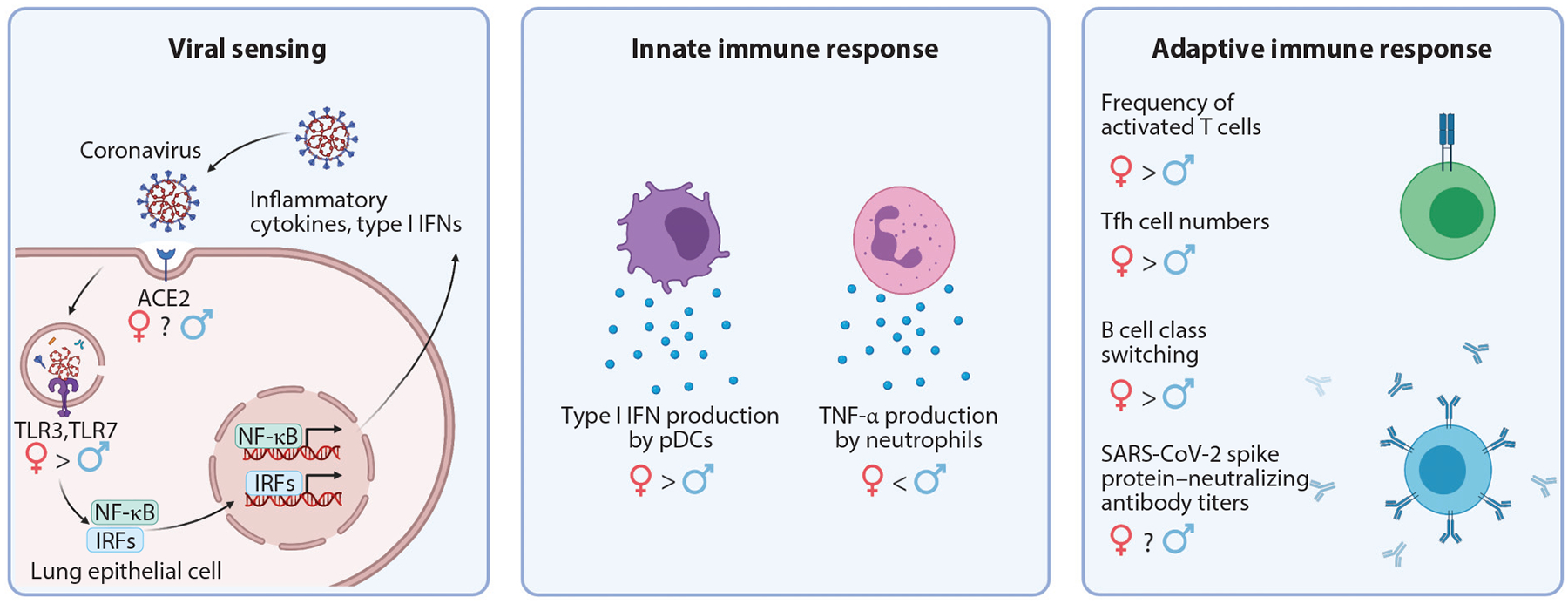
Schematic of sex differences identified during COVID-19 infection. SARS-CoV-2 is internalized through interaction with ACE2. Following host cell entry, viral nucleic acids are detected by pattern recognition receptors, such as TLRs, and antiviral transcriptional programs are initiated. The innate and adaptive immune systems are successively activated to control viral infection. Sex differences have been identified at each stage of the immune response to SARS-CoV-2 infection. TLR7 escapes X inactivation and is more highly expressed in females. Type I interferon production by pDCs, T cell activation, and B cell class switching are also enhanced in females. TNF-α production, however, is enhanced in males. Abbreviations: ACE2, angiotensin-converting enzyme 2; COVID-19, coronavirus disease 2019; IFN, interferon; IRF, interferon regulatory factor; pDC, plasmacytoid dendritic cell; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; Tfh, T follicular helper; TLR, Toll-like receptor. Figure adapted from images created with BioRender.com.
Viral Entry
SARS-CoV-2 infects host cells through interaction with angiotensin-converting enzyme 2 (ACE2) and transmembrane protease serine 2 (TMPRSS2) (56). Both ACE2 and TMPRSS2 are expressed by various cell types of the respiratory, cardiovascular, and gastrointestinal systems (57). Emerging evidence suggests that ACE2 and TMPRSS2 are differentially regulated between males and females. ACE2 is encoded on the X chromosome and has been shown to escape X inactivation so that it is expressed from both sex chromosomes in females (21). This alone suggests that ACE2 expression is elevated in women. However, estrogens also modulate ACE2 expression. Following treatment with 17β-estradiol, cultured human bronchial epithelial cells downregulate ACE2 (58). How the opposing effects of sex chromosomes and hormones interact is unclear and may differ across the lifespan as sex hormone levels fluctuate. TMPRSS2 is also modulated by sex hormones. In various cell lines, androgen treatment induced TMPRSS2 expression (59, 60). Although ACE2 and TMPRSS2 are modulated by sex hormones, direct comparison of protein levels in male and female lung tissue surprisingly revealed no difference in expression in healthy individuals (61). One possible explanation for this discrepancy is that additional variables that alter receptor expression may override these sex-linked factors during different stages of infection. In support of this possibility, Ziegler and colleagues (62) recently reported that IFN-α induced ACE2 expression in cultured primary human nasal epithelial cells. This suggests ACE2 is upregulated in the early antiviral response, when type I interferons are induced to control viral replication. Thus, the presence of virus, and potentially the extent and duration of infection, likely works in concert with sex factors to drive differences in ACE2 and TMPRSS2 expression in COVID-19.
Early Type I Interferon Response
Differences in type I interferon production, an important component of the early antiviral response, may also underlie sex differences in COVID-19 outcomes. Following host cell entry, viral nucleic acids are detected by various pattern recognition receptors (PRRs). Engagement of intracellular PRRs induces antiviral transcriptional programs, which include production of type I interferons. In COVID-19 infection, type I interferon production also plays an important role in effective antiviral responses, since patients with severe COVID-19 have significantly lower plasma IFN-α2 than individuals with mild COVID-19 (63).
Berghöfer et al. (64) reported enhanced type I interferon responses to viral infection and autoimmunity in females compared to males, and these may explain worse outcomes in males with SARS-CoV-2 infection. Indeed, direct comparison of plasma from males and females with COVID-19 revealed elevated IFN-α2 among females (55). Studies in animal models and human samples have identified both chromosomal and hormonal underpinnings for these differences. As discussed above, TLR7, an endosomal immune sensor that activates type I interferon signaling, is encoded on the X chromosome and escapes X inactivation in pDCs, monocytes, and B cells (24). pDCs with biallelic TLR7 expression have higher levels of IFN-α and IFN-β transcripts at rest and upon TLR7 stimulation (65). Estrogen has also been shown to enhance TLR7-mediated IFN-α production by pDCs (66). Collectively, these data highlight key mechanisms by which the immune system in females is poised to generate stronger type I interferon responses.
There is emerging evidence that type I interferon responses may have been impaired in men with severe COVID-19. Type I interferon–neutralizing autoantibodies were detected in 10% of patients with severe, but not mild, COVID-19 (67). Interestingly, among severely affected individuals, men have 5.22-higher odds of harboring type I interferon–specific autoantibodies compared to women. It is unknown whether men are more likely to have type I interferon–neutralizing autoantibodies at baseline or whether both sexes have autoantibodies at baseline but the presence of type I interferon–neutralizing autoantibodies is more deleterious in men with COVID-19 infection. Clues may come from patients with APS1 (autoimmune polyendocrinopathy syndrome type 1). This disease results from mutations in the AIRE gene involved in thymic education of T cells and is defined by multi-organ autoimmunity and production of type I interferon antibodies in both male and female affected individuals. In a recent study of 22 APS1 patients by Bastard et al. (68), not only was autoantibody production similar between males and females, but patients of both sexes with type I interferon–neutralizing antibodies were at elevated risk of developing severe COVID-19. This suggests that the presence of type 1interferon antibodies is deleterious, regardless of sex. Thus, additional investigation is needed to understand the basis for the preponderance of males among patients with severe COVID-19 who have type I interferon autoantibodies but not APS1.
T Cell Response
T lymphocytes are critical players in the antiviral response. However, when dysregulated, they may also contribute to pathogenic inflammation. T cell responses differ between males and females (69). Following stimulation with phorbol myristate acetate and ionomycin, peripheral T cells from females upregulate more antiviral and proinflammatory genes than T cells from men (70). Polarization of T helper subsets also differs between males and females (71, 72). Given these differences in T cell biology between sexes, there is interest in comparing male and female T cell responses during SARS-CoV-2 infection.
In patients with COVID-19, total circulating T cell numbers and the relative frequencies of naive T cells, memory T cells, and Tregs were comparable among females and males (73). Females, however, had a higher frequency of activated T cells, as defined by expression of CD38 and HLA-DR. It is not clear how this observation might relate to sex differences in COVID-19 outcomes, however, since the association between T cell activation and COVID-19 severity is not established. For instance, Mathew and colleagues (74) found that while a subset of patients with severe COVID-19 had a significantly higher frequency of activated T cells, 20% of severely affected patients had no increase in activated T cells compared to healthy controls.
COVID-19 is a systemic inflammatory disease, but much of the morbidity and mortality is associated with pulmonary inflammation. Thus, analysis of bronchoalveolar lavage fluid (BALF) may provide insight into important immune processes contributing to poor patient outcomes. BALF analysis showed reduced clonal expansion of T cells in patients with severe disease (75). Based on this, one might speculate that greater T cell activation may protect from progression to severe COVID-19. Indeed in males, poor T cell activation was associated with disease progression, perhaps due to ineffective viral clearance. Paradoxically, females with severe COVID-19 and clinical decompensation generated robust peripheral T cell responses. Further investigation is required to reconcile these overtly conflicting findings.
Humoral Response
In addition to T cells, B cells help to clear viral infections through the production of immunoglobulins. Within two weeks of symptom onset, SARS-CoV-2-specific antibodies are detectable in sera from COVID-19 patients (76). In the early stages of disease, higher levels of IgG and IgM specific for the SARS-CoV-2 spike and nucleocapsid proteins are associated with greater viral control (77). Results from a randomized controlled trial (RCT) showed that convalescent plasma with high IgG titers prevents progression to severe disease when given to patients with mild COVID-19 within 72 h of symptom onset. Collectively, these data highlight the importance of the antibody response in the control of SARS-CoV-2 infection.
COVID-19 survival has been associated with greater antibody affinity maturation. Tang and colleagues (78) found that antibodies from sera of patients who survived COVID-19 formed more stable interactions with recombinant SARS-CoV-2 spike protein than antibodies from sera of expired COVID-19 patients. This finding suggests that failure to develop high-affinity antibodies against the SARS-CoV-2 spike protein may contribute to mortality. Further, an absence of BCL6+ germinal center B cells and Tfh cells was noted in postmortem spleen and thoracic lymph node samples from COVID-19 patients (79). One might speculate based on this finding that defective germinal center B cell responses contribute to progression of SARS-CoV-2 infection.
Humoral immunity may be an important sex-dichotomous feature of the response to SARS-CoV-2 infection. Females generate enhanced germinal center B cell responses in various contexts. In a mouse model of rheumatoid arthritis, females have more numerous Tfh and germinal center B cells, higher levels of serum IgG, and more severe joint inflammation than male mice (80). Sex differences in both B cell and Tfh cell biology likely contribute to these phenotypic differences. Mechanistic studies reveal a critical role for UTX, a histone demethylase encoded on the X chromosome, in Tfh cell development. T cell–specific UTX deletion results in reduced Tfh cell numbers and effector function (26). Notably, UTX escapes X inactivation and shows dose-dependent function, as evidenced by studies in individuals with Turner syndrome, who lack all or part of an X chromosome (26, 81). TLR7, which has important roles in B cell maturation, is also expressed on the X chromosome and has been shown to escape X inactivation. B cells with biallelic TLR7 expression have 2.4-higher odds of class switching in the presence of T cell help and TLR7 agonism compared to B cells with monoallelic TLR7 expression (24). Collectively, these data demonstrate mechanisms by which the female immune system is poised to generate stronger antibody responses. Whether sex differences in the humoral immune response occur in COVID-19 is an important unanswered question.
Direct studies of sex differences in the humoral immune response to SARS-CoV-2 are limited and provide mixed results. Analysis of sera from patients with ongoing COVID-19 symptoms revealed no significant difference in spike protein–specific IgG or IgM titers between males and females (73). Analysis of plasma from convalescent donors, however, showed male sex was associated with higher antibody titers (82). This observation, which contradicts the general idea that females generate more robust antibody responses, may be explained by the fact that men are more likely to have severe disease. Lending support to this idea is the observation that hospitalization is an even stronger predictor of increased antibody titers than male sex (82). Also possible is that increased antibody titers in convalescent plasma from males reflect a general increase in antibody production in the setting of COVID-19 infection, which could account for both increased antiviral and anti-IFN antibodies seen in males. Further investigation is needed to understand this observation and how the quality and quantity of the antibody response differ between men and women across the course of SARS-CoV-2 infection.
SEX DIFFERENCES IN CANCER
Females and males display distinct immune responses to microbial pathogens and immune-modulating interventions, such as vaccines, and have distinct propensities to autoimmunity. It is not surprising, then, that sex differences might also be observed in cancer-related immune responses. In response to a variety of foreign pathogens, vaccines, and self-antigens, females have increased innate and adaptive immunity, including increased interferon production and antigen presentation. This is hypothesized to contribute to greater tumor immune surveillance and immune editing during cancer development and progression. Analyses from The Cancer Genome Atlas (TCGA) showed greater intratumoral accumulation of activated T cells, as well as counterbalancing immune suppressor cells, in females compared to males (83). This topic is further reviewed by Klein & Morgan (84). Here we turn our attention to sex differences observed in a new area of cancer therapy: ICIs.
Cancer Immunotherapy Efficacy
Cancer immunotherapy uses the body’s own immune system to eliminate tumor cells. This approach leverages the immune system’s inherent ability to find and destroy abnormal cells in the body. Specifically, cancer cells expressing new antigens or causing local tissue damage through rapid growth or invasion will elicit an immune response, termed an antitumor immune response, that comprises innate and adaptive cells destroying cancer cells. However, many tumors develop mechanisms to escape or suppress antitumor immunity (85). Tumor cells may downregulate antigen expression, such as MHC gene expression, to escape immune detection. Tumors may also induce tolerance by the immune system through recruitment of immune suppressor cells (e.g., Tregs, myeloid-derived suppressor cells) and expression of ligands that attenuate or turn off effector responses. Checkpoint proteins [e.g., programmed death protein 1 (PD-1) and cytotoxic T lymphocyte antigen 4 (CTLA-4)] are natural regulatory proteins expressed on T cells to terminate immune responses (86). Aberrant expression of checkpoint proteins on tumor-infiltrating T effector cells contributes to tumor immune tolerance. Immune responses generated to tumor antigens are impaired due to early termination by the regulatory signals from checkpoint proteins, such as PD-1 and CTLA-4, on T cells that shut off T cell effector function. ICIs reverse this tumor immune tolerance by blocking checkpoint protein regulatory signals and increasing activation of T effector cells (86). Compared to conventional chemotherapy, ICIs are associated with significantly improved outcomes in many advanced cancers, including melanoma; renal cancer; bladder cancer; lung cancer; head-and-neck cancers; and some breast, colon, and prostate cancers (86, 87). Biomarkers are needed to predict which patients with cancer will respond to ICIs and to guide treatment selection. Given the well-recognized differences in immune responses between females and males, sex has been evaluated as one such biomarker in recent meta-analyses. Conforti et al. (88) evaluated 10 RCTs including 11,351 patients (67% male, period ending November 2017) treated with ICIs [ipilimumab and tremelimumab (anti-CTLA-4) and nivolumab and pembrolizumab (anti-PD-1)] and reported overall survival stratified by sex. The pooled hazard ratio (HR) for overall survival was lower for both sexes compared to controls, suggesting a survival benefit from ICI therapy over conventional therapy. This analysis also found that the reduction in HR with ICI treatment was greater for males than for females [HR = 0.72 (95% CI = 0.65–0.79) versus HR = 0.86 (95% CI = 0.79–0.93), p = 0.0019]. This difference in HR reduction would suggest that male patients benefitted more from ICI therapy. A subsequent meta-analysis by Wallis et al. (89) found no difference in efficacy between males and females (83). This study included 23 RCTs with 13,721 individuals (68% male, period ending October 2018) treated with ICIs [ipilimumab or tremelimumab (anti-CTLA-4); pembrolizumab or nivolumab (anti-PD-1); atezolizumab, avelumab, or durvalumab (anti-PD-L1); or a combination of ipilimumab and nivolumab] and found an overall survival benefit of immunotherapy for both men and women [HR = 0.75 (95% CI = 0.69–0.81), p < 0.01, and 0.77 (95% CI 0.67–0.88), p = 0.002; respectively], with no difference by sex (I2 = 38%, p = 0.6). One important difference between these studies was the inclusion by Wallis et al. (89), but exclusion by Conforti et al. (85), of four studies evaluating ICIs combined with chemotherapy, which showed greater ICI efficacy in females. Studies of ICI alone may be expected to show less benefit in females. The greater adaptive immune activation in females is thought to lead to greater immune editing during tumor development such that highly antigenic tumors are less likely to persist in females (83). Previous studies have shown that ICI treatment is more effective in tumors with high antigen expression (90). On the other hand, studies of ICI in combination with chemotherapy may have fewer sex-associated differences in efficacy. This is because the addition of chemotherapy, which increases tumor mutational burden and antigenicity, likely influences ICI efficacy and perhaps more so in females. In summary, the specific context of ICI therapy likely influences the outcomes of studies comparing efficacy in males versus females.
Indeed, this nuanced relationship of sex-modulated immune molecular predictors of ICI response was confirmed by a more recent analysis. Ye et al. (91) revisited the combined 27 RCTs and found a nonsignificant HR for sex (female versus male) with respect to ICI efficacy (HR = 1.07, 95% CI = 0.95–1.19, p = 0.28). However, the authors noted a consistent sex benefit in some cancers (e.g., male in melanoma and colon cancer and female in non–small cell lung cancer and esophageal cancer) and variance of molecular biomarkers for ICI efficacy by sex [e.g., TMB, T cell inflamed gene expression profile (GEP), cytolytic activity, and expression of checkpoint proteins] (91). Using the TCGA database, as well as validation in several independent data sets, these authors then delineated sex- and cancer-specific patterns in molecular markers for ICI efficacy. Tumor mutational burden positively associated with ICI efficacy in male patients for melanoma and bladder, liver, head-and-neck, and renal cell cancers. This male-specific association of tumor mutational burden with positive ICI response was also seen in patients with lung cancer (92). In contrast, the frequency of activated CD4+ and CD8+ T cells, GEP and cytolytic activity, T cell receptor richness, and immune checkpoint protein expression (including both inhibitory PD-1, CTLA-4, and Lag3 and stimulatory OX40, ICOS, and CD27) were associated with ICI efficacy in female patients with lung cancer. These results suggest that pooled meta-analyses may not fully capture the nuanced interactions of sex and ICI response.
Immune-Related Adverse Events
Autoimmune responses against healthy tissues can occur as a side effect of immune activation by ICIs. Such immune-related adverse events (IrAEs) occur in approximately 30% of patients treated with a single agent (e.g., anti-PD-1/PD-L1) and nearly 60% of patients treated with a combination of ICIs (e.g., anti-PD-1 plus anti-CTLA-4) (93). Organs most frequently affected by IrAEs are skin, intestines, liver, lung, and endocrine glands (94), but ICI-associated autoimmunity has been observed in nearly all tissues. While spontaneous autoimmunity is generally more common in females, as discussed above (69), IrAEs appear to have a more equal sex distribution (95).
No significant sex differences were seen in the rate of ICI-associated IrAEs affecting skin (96), gastrointestinal (97–99), salivary gland, or lacrimal gland (100), or rheumatologic tissues (e.g., joints, muscles) (101). Endocrine IrAEs, similarly, occur in both male and female patients, but the target tissues affected show clear sex dimorphism. Thyroid dysfunction is more frequent in females (95, 102), mirroring spontaneous Hashimoto thyroiditis and Graves disease. One possible explanation for this association is the high background prevalence of spontaneous thyroid autoimmunity in females (e.g., due to antithyroid peroxidase antibodies) and activation of preexisting thyroid-reactive T cells by ICI treatment (103–105). ICI-associated hypophysitis, on the other hand, is more common in males (1:4 female-to-male ratio) and does not mirror the female predominance seen in primary lymphocytic hypophysitis (2:1 female-to-male ratio) (106, 107).
While the cause of IrAEs is unknown, recent data suggest mechanisms that are similar to those that mediate spontaneous autoimmune disease as well as those with distinct etiologies. In particular, we and others have shown a role for T cell–mediated autoimmunity and proinflammatory cytokines (105, 108, 109). Production of TNF-α in particular is modulated by sex hormones and is increased in males (69). Peripheral blood immune cells from human males produced more TNF-α following TLR4 stimuli (e.g., lipopolysaccharide) than those from females. In animal models, TNF-α levels and subsequent joint inflammation were increased following ovariectomy (producing experimental estrogen deficiency) and reduced by exogenous administration of estrogen (e.g., estradiol) (110). Females classically have increased humoral and interferon-mediated immune responses (84). In many IrAEs, in contrast to spontaneous autoimmunity, production of self-reactive immunoglobulins (i.e., autoantibodies) is markedly absent (111, 112). The fact that males are more likely to be affected by IrAEs and the more equal sex distribution of IrAEs compared to many spontaneous autoimmune diseases may be due to activation of these proinflammatory cytokine pathways during ICI treatment and less dependence upon humoral immunity.
Current data suggest that antitumor and self-reactive immune responses during ICI treatment vary between sexes. However, several contextualizing points are worth mention. The routine exclusion of individuals with preexisting autoimmune disease, which is more prevalent in females, and the historical underrepresentation of women in clinical trials (84) must be considered as they may lead to bias in the evaluation of sex differences in ICI anticancer efficacy and IrAE risk. In addition, certain behaviors associated with cancer incidence, prognosis, and treatment response may be more common in males or females. For example, smoking is more prevalent in males and correlates with improved response to ICIs for non–small cell lung cancer (113). On the other hand, oncogenic driver mutations, such as EGFR and ALK mutations, are more frequently found in women with lung cancer and are known to be independently associated with reduced benefit from ICI treatment (113). Finally, health-seeking behavior, access to care, treatment adherence, and reporting of adverse events are different for men and women and, like racial, ethnic, and socioeconomic factors, likely impact the reporting of ICI efficacy and side effects (84). Future studies evaluating molecular immune biomarkers and ICI efficacy and toxicity should delineate results by sex and other confounding variables. However, in the present absence of compelling data showing lack of efficacy in one sex, patients should not be excluded from ICI treatment based upon sex or sex hormone status. After accounting for tumor-specific differences, the data reviewed here generally suggest that ICI works equally well for both sexes.
CONCLUSIONS
The topic of sex differences in health and disease has historically been overlooked, and even intentionally sidelined. In 1977, the United States Food and Drug Administration recommended that women of childbearing age be excluded from phase 1 and early phase 2 clinical trials. This mandate was ostensibly made to protect women and children; however, it did the opposite, creating a gap in knowledge surrounding women’s health. Major steps have since been undertaken to rectify this situation. In 1993, a National Institutes of Health (NIH) policy that mandated inclusion of women and minorities in all clinical research became federal law (114), and in 2015, the NIH called on scientists to account for sex as a biological variable (SABV) in studies involving animals and cells (115, 116). These policies have been associated with measurable changes in how NIH-funded science is conducted (117). Continued progress in defining sex-based mechanisms will pave the path to refining vaccine development and the treatment and management of immune-mediated diseases. For example, a deeper understanding of how sex influences protective immunity could be key to optimizing vaccination strategies. Different dosing regimens for males and females may maximize efficacy while minimizing side effects and/or unnecessary doses. Along these lines, targeting overexpressed X-linked genes may be an effective therapeutic strategy in individuals with two copies of the X chromosome but not in those with a single X chromosome. Thus, a better understanding of sex differences in immunity will likely have myriad benefits to the population as a whole.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Mauvais-Jarvis F, Bairey Merz N, Barnes PJ, Brinton RD, Carrero J-J, et al. 2020. Sex and gender: modifiers of health, disease, and medicine. Lancet 396(10250):565–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dill-Garlow R, Chen KHE, Walker AM. 2019. Sex differences in mouse popliteal lymph nodes. Sci. Rep 9(1):965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clayton JA, Tannenbaum C. 2016. Reporting sex, gender, or both in clinical research? JAMA 316(18):1863–64 [DOI] [PubMed] [Google Scholar]
- 4.Klein SL. 2012. Sex differences in prophylaxis and therapeutic treatments for viral diseases. Sex Gend. Differ. Pharmacol 214:499–522 [DOI] [PubMed] [Google Scholar]
- 5.Ghosh S, Klein RS. 2017. Sex drives dimorphic immune responses to viral infections. J. Immunol 198(5):1782–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobsen H, Klein SL. 2021. Sex differences in immunity to viral infections. Front. Immunol 12:720952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Griesbeck M, Ziegler S, Laffont S, Smith N, Chauveau L, et al. 2015. Sex differences in plasmacytoid dendritic cell levels of IRF5 drive higher IFN-α production in women. J. Immunol 195(11):5327–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fink AL, Engle K, Ursin RL, Tang WY, Klein SL. 2018. Biological sex affects vaccine efficacy and protection against influenza in mice. PNAS 115(49):12477–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guha-Sapir D, Schimmer B. 2005. Dengue fever: new paradigms for a changing epidemiology. Emerg. Themes Epidemiol 2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balogun MA, Vyse AJ, Hesketh LM, Kafatos G, Parry JV, Ramsay ME. 2009. Estimating hepatitis C infection acquired in England, 1986–2000. Epidemiol. Infect 137(9):1249–54 [DOI] [PubMed] [Google Scholar]
- 11.Siangphoe U, Archer KJ. 2015. Gene expression in HIV-associated neurocognitive disorders: a meta-analysis. J. Acquir. Immune Defic. Syndr 70(5):479–88 [DOI] [PubMed] [Google Scholar]
- 12.Potluri T, Fink AL, Sylvia KE, Dhakal S, Vermillion MS, et al. 2019. Age-associated changes in the impact of sex steroids on influenza vaccine responses in males and females. npj Vaccines 4(1):1–12. Erratum. 2019. npj Vaccines 4:35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rubtsova K, Marrack P, Rubtsov AV. 2015. Sexual dimorphism in autoimmunity. J. Clin. Investig 125(6):2187–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ngo ST, Steyn FJ, McCombe PA. 2014. Gender differences in autoimmune disease. Front. Neuroendocrinol 35(3):347–69 [DOI] [PubMed] [Google Scholar]
- 15.Arnold AP. 2020. Four core genotypes and XY* mouse models: update on impact on SABV research. Neurosci. Biobehav. Rev 119:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasidhar MV, Itoh N, Gold SM, Lawson GW, Voskuhl RR. 2012. The XX sex chromosome complement in mice is associated with increased spontaneous lupus compared with XY. Ann. Rheum. Dis 71(8):1418–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith-Bouvier DL, Divekar AA, Sasidhar M, Du S, Tiwari-Woodruff SK, et al. 2008. A role for sex chromosome complement in the female bias in autoimmune disease. J. Exp. Med 205(5):1099–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Youness A, Miquel CH, Guéry JC. 2021. Escape from X chromosome inactivation and the female predominance in autoimmune diseases. Int. J. Mol. Sci 22(3):1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plath K, Mlynarczyk-Evans S, Nusinow DA, Panning B. 2002. Xist RNA and the mechanism of X chromosome inactivation. Annu. Rev. Genet 36:233–78 [DOI] [PubMed] [Google Scholar]
- 20.Golden LC, Itoh Y, Itoh N, Iyengar S, Coit P, et al. 2019. Parent-of-origin differences in DNA methylation of X chromosome genes in T lymphocytes. PNAS 116(52):26779–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, et al. 2017. Landscape of X chromosome inactivation across human tissues. Nature 550:244–48. Erratum. 2018. Nature 555(7695):274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balaton BP, Brown CJ. 2021. Contribution of genetic and epigenetic changes to escape from X-chromosome inactivation. Epigenet. Chromatin 14:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mousavi MJ, Mahmoudi M, Ghotloo S. 2020. Escape from X chromosome inactivation and female bias of autoimmune diseases. Mol. Med 26(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, et al. 2018. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol 3(19):eaap8855 [DOI] [PubMed] [Google Scholar]
- 25.Itoh Y, Golden LC, Itoh N, Matsukawa MA, Ren E, et al. 2019. The X-linked histone demethylase Kdm6a in CD4+ T lymphocytes modulates autoimmunity. J. Clin. Investig 129(9):3852–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cook KD, Shpargel KB, Starmer J, Whitfield-Larry F, Conley B, et al. 2015. T follicular helper cell-dependent clearance of a persistent virus infection requires T cell expression of the histone demethylase UTX. Immunity 43(4):703–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walport LJ, Hopkinson RJ, Vollmar M, Madden SK, Gileadi C, et al. 2014. Human UTY(KDM6C) is a male-specific Nϵ-methyl lysyl demethylase. J. Biol. Chem 289(26):18302–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le Coz C, Trofa M, Syrett CM, Martin A, Jyonouchi H, et al. 2018. CD40LG duplication-associated autoimmune disease is silenced by nonrandom X-chromosome inactivation. J. Allergy Clin. Immunol 141(6):2308–11.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Syrett CM, Kramer MC, Basu A, Atchison ML, Anguera MC. 2016. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. PNAS 113(14):E2029–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. 2007. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol 179(9):6352–58 [DOI] [PubMed] [Google Scholar]
- 31.Aarts SABM, Seijkens TTP, van Dorst KJF, Dijkstra CD, Kooij G, Lutgens E. 2017. The CD40–CD40L dyad in experimental autoimmune encephalomyelitis and multiple sclerosis. Front. Immunol 8:1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pyfrom S, Paneru B, Knox JJ, Cancro MP, Posso S, et al. 2021. The dynamic epigenetic regulation of the inactive X chromosome in healthy human B cells is dysregulated in lupus patients. PNAS 118(24):e2024624118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fang H, Disteche CM, Berletch JB. 2019. X inactivation and escape: epigenetic and structural features. Front. Cell Dev. Biol 7:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Syrett CM, Paneru B, Sandoval-Heglund D, Wang J, Banerjee S, et al. 2019. Altered X-chromosome inactivation in T cells may promote sex-biased autoimmune diseases. JCI Insight. 4(7):e126751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zito A, Davies MN, Tsai PC, Roberts S, Andres-Ejarque R, et al. 2019. Heritability of skewed X-inactivation in female twins is tissue-specific and associated with age. Nat. Commun 10(1):5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ozbalkan Z, Bagişlar S, Kiraz S, Akyerli CB, Ozer HTE, et al. 2005. Skewed X chromosome inactivation in blood cells of women with scleroderma. Arthritis Rheum. 52(5):1564–70 [DOI] [PubMed] [Google Scholar]
- 37.Chabchoub G, Uz E, Maalej A, Mustafa CA, Rebai A, et al. 2009. Analysis of skewed X-chromosome inactivation in females with rheumatoid arthritis and autoimmune thyroid diseases. Arthritis Res. Ther 11(4):R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brix TH, Knudsen GPS, Kristiansen M, Kyvik KO, Orstavik KH, Hegedüs L. 2005. High frequency of skewed X-chromosome inactivation in females with autoimmune thyroid disease: a possible explanation for the female predisposition to thyroid autoimmunity. J. Clin. Endocrinol. Metab 90(11):5949–53 [DOI] [PubMed] [Google Scholar]
- 39.Santiwatana S, Mahachoklertwattana P, Limwongse C, Khlairit P, Pongratanakul S, et al. 2018. Skewed X chromosome inactivation in girls and female adolescents with autoimmune thyroid disease. Clin. Endocrinol 89(6):863–69 [DOI] [PubMed] [Google Scholar]
- 40.Moulton VR. 2018. Sex hormones in acquired immunity and autoimmune disease. Front. Immunol 9:2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bupp MRG, Jorgensen TN. 2018. Androgen-induced immunosuppression. Front. Immunol 9:794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson MS, Su MA. 2016. AIRE expands: new roles in immune tolerance and beyond. Nat. Rev. Immunol 16(4):247–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dragin N, Bismuth J, Cizeron-Clairac G, Biferi MG, Berthault C, et al. 2016. Estrogen-mediated downregulation of AIRE influences sexual dimorphism in autoimmune diseases. J. Clin. Investig 126(4):1525–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu P, Conley B, Nelson JS, Free M, Martin A, et al. 2016. Sex bias in CNS autoimmune disease mediated by androgen control of autoimmune regulator. Nat. Commun 48(7):829–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohammad I, Starskaia I, Nagy T, Guo J, Yatkin E, et al. 2018. Estrogen receptor contributes to T cell–mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci. Signal 11(526):eaap9415 [DOI] [PubMed] [Google Scholar]
- 46.Vasanthakumar A, Chisanga D, Blume J, Gloury R, Britt K, et al. 2020. Sex-specific adipose tissue imprinting of regulatory T cells. Nature 579(7800):581–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walecki M, Eisel F, Klug J, Baal N, Paradowska-Dogan A, et al. 2015. Androgen receptor modulates Foxp3 expression in CD4+CD25+Foxp3+ regulatory T-cells. Mol. Biol. Cell 26(15):2845–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Russi AE, Ebel ME, Yang Y, Brown MA. 2018. Male-specific IL-33 expression regulates sex-dimorphic EAE susceptibility. PNAS 115(7):E1520–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roubinian JR, Papoian R, Talal N. 1977. Androgenic hormones modulate autoantibody responses and improve survival in murine lupus. J. Clin. Investig 59(6):1066–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yesilova Z, Ozata M, Kocar IH, Turan M, Pekel A, et al. 2000. The effects of gonadotropin treatment on the immunological features of male patients with idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab 85(1):66–70 [DOI] [PubMed] [Google Scholar]
- 51.Kocar IH, Yesilova Z, Özata M, Turan M, Sengül A, Özdemir I. 2000. The effect of testosterone replacement treatment on immunological features of patients with Klinefelter’s syndrome. Clin. Exp. Immunol 121(3):448–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilhelmson AS, Lantero Rodriguez M, Stubelius A, Fogelstrand P, Johansson I, et al. 2018. Testosterone is an endogenous regulator of BAFF and splenic B cell number. Nat. Commun 9(1):2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, et al. 2013. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339(6123):1084–88 [DOI] [PubMed] [Google Scholar]
- 54.Peckham H, de Gruijter NM, Raine C, Radziszewska A, Ciurtin C, et al. 2020. Male sex identified by global COVID-19 meta-analysis as a risk factor for death and ITU admission. Nat. Commun 11(1):6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takahashi T, Ellingson MK, Wong P, Israelow B, Lucas C, et al. 2020. Sex differences in immune responses that underlie COVID-19 disease outcomes: overview of the study design. Nature 588(7837):315–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, et al. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271–80.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sungnak W, Huang N, Bécavin C, Berg M, Queen R, et al. 2020. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med 26(5):681–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stelzig KE, Canepa-Escaro F, Schiliro M, Berdnikovs S, Prakash YS, Chiarella SE. 2020. Estrogen regulates the expression of SARS-CoV-2 receptor ACE2 in differentiated airway epithelial cells. Am.J. Physiol. Cell. Mol. Physiol 318(6):L1280–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clinckemalie L, Spans L, Dubois V, Laurent M, Helsen C, et al. 2013. Androgen regulation of the TMPRSS2 gene and the effect of a SNP in an androgen response element. Mol. Endocrinol 27(12):2028–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mikkonen L, Pihlajamaa P, Sahu B, Zhang FP, Jänne OA. 2010. Androgen receptor and androgen-dependent gene expression in lung. Mol. Cell. Endocrinol 317(1–2):14–24 [DOI] [PubMed] [Google Scholar]
- 61.Qiao Y, Wang XM, Mannan R, Pitchiaya S, Zhang Y, et al. 2020. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS2. PNAS 118(1):e2021450118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, et al. 2020. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181(5):1016–35.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, et al. 2020. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369(6504):718–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berghöfer B, Frommer T, Haley G, Fink L, Bein G, Hackstein H. 2006. TLR7 ligands induce higher IFN-α production in females. J. Immunol 177(4):2088–96 [DOI] [PubMed] [Google Scholar]
- 65.Hagen SH, Henseling F, Hennesen J, Savel H, Delahaye S, et al. 2020. Heterogeneous escape from X chromosome inactivation results in sex differences in type I IFN responses at the single human pDC level. Cell Rep. 33(10):108485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laffont S, Rouquié N, Azar P, Seillet C, Plumas J, et al. 2014. X-chromosome complement and estrogen receptor signaling independently contribute to the enhanced TLR7-mediated IFN-α production of plasmacytoid dendritic cells from women. J. Immunol 193(11):5444–52 [DOI] [PubMed] [Google Scholar]
- 67.Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, et al. 2020. Autoantibodies against IFNs in patients with life-threatening COVID-19. Science 370(6515):eabd4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bastard P, Orlova E, Sozaeva L, Lévy R, James A, et al. 2021. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J. Exp. Med 218(7):e20210554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klein SL, Flanagan KL. 2016. Sex differences in immune responses. Nat. Rev. Immunol 16(10):626–38 [DOI] [PubMed] [Google Scholar]
- 70.Hewagama A, Patel D, Yarlagadda S, Strickland FM, Richardson BC. 2009. Stronger inflammatory/ic T-cell response in women identified by microarray analysis. Genes Immun. 10(5):509–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang MA, Rego D, Moshkova M, Kebir H, Chruscinski A, et al. 2012. Peroxisome proliferator-activated receptor (PPAR)α and -γ regulate IFNγ and IL-17A production by human T cells in a sex-specific way. PNAS 109(24):9505–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roberts CW, Walker W, Alexander J. 2001. Sex-associated hormones and immunity to protozoan parasites. Clin. Microbiol. Rev 14(3):476–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takahashi T, Iwasaki A. 2021. Sex differences in immune responses. Science 371(6527):347–48 [DOI] [PubMed] [Google Scholar]
- 74.Mathew D, Giles JR, Baxter AE, Oldridge DA, Greenplate AR, et al. 2020. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 369(6508):eabc8511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liao M, Liu Y, Yuan J, Wen Y, Xu G, et al. 2020. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med 26(6):842–44 [DOI] [PubMed] [Google Scholar]
- 76.Long QX, Liu BZ, Deng HJ, Wu GC, Deng K, et al. 2020. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat. Med 26(6):845–48 [DOI] [PubMed] [Google Scholar]
- 77.Wu J, Liang B, Chen C, Wang H, Fang Y, et al. 2021. SARS-CoV-2 infection induces sustained humoral immune responses in convalescent patients following symptomatic COVID-19. Nat. Commun 12(1):1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tang J, Ravichandran S, Lee Y, Grubbs G, Coyle EM, et al. 2021. Antibody affinity maturation and plasma IgA associate with clinical outcome in hospitalized COVID-19 patients. Nat. Commun 12(1):1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaneko N, Kuo HH, Boucau J, Farmer JR, Allard-Chamard H, et al. 2020. Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell 183(1):143–57.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dimitrijević M, Arsenović-Ranin N, Kosec D, Bufan B, Nacka-Aleksić M, et al. 2020. Sex differences in Tfh cell help to B cells contribute to sexual dimorphism in severity of rat collagen-induced arthritis. Sci. Rep 10(1):1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Greenfield A, Carrel L, Pennisi D, Philippe C, Quaderi N, et al. 1998. The UTX gene escapes X inactivation in mice and humans. Hum. Mol. Genet 7(4):737–42 [DOI] [PubMed] [Google Scholar]
- 82.Klein SL, Pekosz A, Park HS, Ursin RL, Shapiro JR, et al. 2020. Sex, age, and hospitalization drive antibody responses in a COVID-19 convalescent plasma donor population. J. Clin. Investig 130(11):6141–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang S, Cowley LA, Liu XS. 2019. Sex differences in cancer immunotherapy efficacy, biomarkers, and therapeutic strategy. Molecules 24(18):3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Klein SL, Morgan R. 2020. The impact of sex and gender on immunotherapy outcomes. Biol. Sex Differ 11(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stewart TJ, Abrams SI. 2008. How tumours escape mass destruction. Oncogene 27:5894–903 [DOI] [PubMed] [Google Scholar]
- 86.Wei SC, Duffy CR, Allison JP. 2018. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 8(9):1069–86 [DOI] [PubMed] [Google Scholar]
- 87.Haslam A, Prasad V. 2019. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open 2(5):e192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Conforti F, Pala L, Bagnardi V, De Pas T, Martinetti M, et al. 2018. Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta-analysis. Lancet Oncol. 19(6):737–46 [DOI] [PubMed] [Google Scholar]
- 89.Wallis CJD, Butaney M, Satkunasivum K, Freedland J, Patel SP, et al. 2019. Association of patient sex with efficacy of immune checkpoint inhibitors and overall survival in advanced cancers: a systematic review and meta-analysis. JAMA Oncol. 5(4):529–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Das S, Johnson DB. 2019. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 7:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ye Y, Jing Y, Li L, Mills GB, Diao L, et al. 2020. Sex-associated molecular differences for cancer immunotherapy. Nat. Commun 11(1):1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang S, Zhang J, He Z, Wu K, Liu X-S. 2019. The predictive power of tumor mutational burden in lung cancer immunotherapy response is influenced by patients’ sex. Int. J. Cancer 145(10):2840–49 [DOI] [PubMed] [Google Scholar]
- 93.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, et al. 2019. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med 381(16):1535–46 [DOI] [PubMed] [Google Scholar]
- 94.Arnaud-Coffin P, Maillet D, Gan HK, Stelmes J-J, You B, et al. 2019. A systematic review of adverse events in randomized trials assessing immune checkpoint inhibitors. Int. J. Cancer 145(3):639–48 [DOI] [PubMed] [Google Scholar]
- 95.Triggianese P, Novelli L, Galdiero MR, Chimenti MS, Conigliaro P, et al. 2020. Immune checkpoint inhibitors-induced autoimmunity: the impact of gender. Autoimmun. Rev 19(8):102590. [DOI] [PubMed] [Google Scholar]
- 96.Danlos FX, Voisin AL, Dyevre V, Michot JM, Routier E, et al. 2018. Safety and efficacy of anti-programmed death 1 antibodies in patients with cancer and pre-existing autoimmune or inflammatory disease. Eur. J. Cancer 91:21–29 [DOI] [PubMed] [Google Scholar]
- 97.Collins M, Michot JM, Danlos FX, Mussini C, Soularue E, et al. 2017. Inflammatory gastrointestinal diseases associated with PD-1 blockade antibodies. Ann. Oncol 28(11):2860–65 [DOI] [PubMed] [Google Scholar]
- 98.de Malet A, Antoni G, Collins M, Soularue E, Marthey L, et al. 2019. Evolution and recurrence of gastrointestinal immune-related adverse events induced by immune checkpoint inhibitors. Eur. J. Cancer 106:106–14 [DOI] [PubMed] [Google Scholar]
- 99.Zhang ML, Neyaz A, Patil D, Chen J, Dougan M, Deshpande V. 2020. Immune-related adverse events in the gastrointestinal tract: diagnostic utility of upper gastrointestinal biopsies. Histopathology 76(2):233–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ramos-Casals M, Maria A, Suárez-Almazor ME, Lambotte O, Fisher BA, et al. 2019. Sicca/Sjögren’s syndrome triggered by PD-1/PD-L1 checkpoint inhibitors: data from the International ImmunoCancer Registry (ICIR). Clin. Exp. Rheumatol 37(3 Suppl. 118):114–22 [PubMed] [Google Scholar]
- 101.Lidar M, Giat E, Garelick D, Horowitz Y, Amital H, et al. 2018. Rheumatic manifestations among cancer patients treated with immune checkpoint inhibitors. Autoimmun. Rev 17(3):284–89 [DOI] [PubMed] [Google Scholar]
- 102.Muir CA, Clifton-Bligh RJ, Long GV, Scolyer RA, Lo SN, et al. 2021. Thyroid immune-related adverse events following immune checkpoint inhibitor treatment. J. Clin. Endocrinol. Metab 106(9):e3704–13 [DOI] [PubMed] [Google Scholar]
- 103.Álvarez-Sierra D, Marín-Sánchez A, Ruiz-Blázquez P, de Jesús Gil C, Iglesias-Felip C, et al. 2019. Analysis of the PD-1/PD-L1 axis in human autoimmune thyroid disease: insights into pathogenesis and clues to immunotherapy associated thyroid autoimmunity. J. Autoimmun 103:102285. [DOI] [PubMed] [Google Scholar]
- 104.Kotwal A, Gustafson MP, Bornschlegl S, Kottschade L, Delivanis DA, et al. 2020. Immune checkpoint inhibitor-induced thyroiditis is associated with increased intrathyroidal T lymphocyte subpopulations. Thyroid. 30(10):1440–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yasuda Y, Iwama S, Sugiyama D, Okuji T, Kobayashi T, et al. 2021. CD4+ T cells are essential for the development of destructive thyroiditis induced by anti–PD-1 antibody in thyroglobulin-immunized mice. Sci. Transl. Med 13(593):eabb7495 [DOI] [PubMed] [Google Scholar]
- 106.de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. 2019. A systematic review and meta-analysis of endocrine-related adverse events associated with immune checkpoint inhibitors. Horm. Metab. Res 51(3):145–56 [DOI] [PubMed] [Google Scholar]
- 107.Di Dalmazi G, Ippolito S, Lupi I, Caturegli P. 2019. Hypophysitis induced by immune checkpoint inhibitors: a 10-year assessment. Expert Rev. Endocrinol. Metab 14(6):381–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.von Euw E, Chodon T, Attar N, Jalil J, Koya RC, et al. 2009. CTLA4 blockade increases Th17 cells in patients with metastatic melanoma. J. Transl. Med 7:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Perez-Ruiz E, Minute L, Otano I, Alvarez M, Ochoa MC, et al. 2019. Prophylactic TNF blockade uncouples efficacy and toxicity in dual CTLA-4 and PD-1 immunotherapy. Nature 569(7756):428–32 [DOI] [PubMed] [Google Scholar]
- 110.Schneider AH, Kanashiro A, Dutra SGV, Souza RN, Veras FP, et al. 2019. Estradiol replacement therapy regulates innate immune response in ovariectomized arthritic mice. Int. Immunopharmacol 72:504–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bluestone JA, Anderson M, Herold KC, Stamatouli AM, Quandt Z, et al. 2018. Collateral damage: insulin-dependent diabetes induced with checkpoint inhibitors. Diabetes 67(8):1471–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ma C, Hodi FS, Giobbie-Hurder A, Wang X, Zhou J, et al. 2019. The impact of high-dose glucocorticoids on the outcome of immune-checkpoint inhibitor–related thyroid disorders. Cancer Immunol. Res 7(7):1214–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Abdel-Rahman O 2018. Does a patient’s sex predict the efficacy of cancer immunotherapy? Lancet Oncol. 19(6):716–17 [DOI] [PubMed] [Google Scholar]
- 114.Natl. Inst. Health Off. Res. Wom. Health. 2018. History of women’s participation in clinical research. Office of Research on Women’s Health. https://orwh.od.nih.gov/toolkit/recruitment/history [Google Scholar]
- 115.Natl. Inst. Health. 2015. Consideration of sex as a biological variable in NIH-funded research. Notice NOT-OD-15–102, Natl. Inst. Health, Bethesda, MD. https://grants.nih.gov/grants/guide/notice-files/not-od-15-102.html [Google Scholar]
- 116.Tannenbaum C, Schwarz JM, Clayton JA, De Vries GJ, Sullivan C. 2016. Evaluating sex as a biological variable in preclinical research: The devil in the details. Biol. Sex Differ 7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Woitowich NC, Woodruff TK. 2019. Implementation of the NIH sex-inclusion policy: attitudes and opinions of study section members. J. Women’s Heal 28(1):9–16 [DOI] [PubMed] [Google Scholar]