

Abstract
G protein coupled receptors (GPCRs) are membrane bound proteins that are ubiquitously expressed in many cell types and take part in mediating multiple signaling pathways. GPCRs are dynamic proteins and exist in an equilibrium between an ensemble of conformational states such as inactive and fully active states. This dynamic nature of GPCRs is one of the factors that confers their basal activity even in the absence of any ligand mediated activation. Ligands selectively bind and stabilize a subset of the conformations from the ensemble leading to a shift in the equilibrium towards the inactive or the active state depending on the nature of the ligand. This ligand-selective effect is achieved through allosteric communication between the ligand binding site and G protein or β-arrestin coupling site. Similarly, the G protein coupling to the receptor exerts the allosteric effect on the ligand binding region leading to increased binding affinity for agonists and decreased affinity for antagonists or inverse agonists. In this review, we enumerate the current state of our understanding of the mechanism of allosteric communication in GPCRs with a specific focus on the critical role of computational methods in delineating the residues involved in allosteric communication. Analyzing allosteric communication mechanism using molecular dynamics simulations have revealed (i) a structurally conserved mechanism of allosteric communication that regulates the G protein coupling, (ii) a rational structure-based approach to designing selective ligands and (iii) an approach to designing allosteric GPCR mutants that are either ligand and G protein or β-arrestin selective.
Keywords: GPCRs, allosteric communication, ligand efficacy, molecular dynamics, Allosteer
Graphical Abstract
Introduction
Allosteric communication in GPCRs
Membrane bound G-protein coupled receptors (GPCRs) are seven helical proteins that are expressed ubiquitously in multiple cells and hence form the largest family of drug targets. Upon activation by a ligand, GPCRs couple to one or more of the members of the G protein families and/or β-arrestin families. The multitude of structures of GPCRs and their complexes with the G proteins and β-arrestins have opened up numerous opportunities for designing high potency ligands for a given GPCR[1–3]. GPCRs are inherently dynamic proteins in an equilibrium between an ensemble of conformations[4,5]. Although we know from multiple GPCR structures that many of the GPCR ligands bind to the extracellular region of the receptor, we now have examples of GPCR structures where ligand binds in the intracellular region or in the crevices between transmembrane (TM) helices leading to a number of targetable sites [6]. Immaterial of where the ligands bind in the GPCR, the perturbation caused by ligand binding at one site is relayed to the distant intracellular G protein or β-arrestin coupling site causing a shift in the equilibrium of conformations and changes in the receptor dynamics. Exactly which amino acid residues are involved in this allosteric communication continues to be a critical research endeavor in many laboratories. The results of these studies will collectively reveal if there is a common mechanism of allosteric communication in GPCRs even if the strength of such communication will vary depending on the nature of the ligand, the GPCR and the G protein or β-arrestin bound. Knowledge of the residues involved in allosteric communication will also allow us to engineer GPCR mutants that would bias the receptor towards one signaling pathway over the other. In the same vein, many disease-associated mutations are located in GPCRs[7,8] and these mutations can lead to gain or loss of function of the receptors. Identifying the residues involved in allosteric communication and their overlap with disease associated mutations would allow us to annotate the functional role of these mutations at a protein level and open up therapeutic opportunities. In this review we will illustrate how to integrate different computational methods at the sequence, structure and dynamics level to identify possible allosteric communication pathways between distant structural regions of GPCRs. Our focus is on elucidating the mechanistic insights on allosteric communication in GPCRs that we have gathered from computational methods. It should be noted that most of these computational methods are generalizable and hence applicable for any protein or protein complexes.
There are multiple terms related to allostery in proteins that needs to be explained prior to beginning this review. The binding site of the endogenous ligands that activate or deactivate the GPCRs is known as the orthosteric site. On the other hand, there are known small molecules, peptides or nanobodies that exert a positive or negative effect on the binding and potency/efficacy of the agonist or antagonist and such molecules are called allosteric modulators. While the agonist or antagonist bind to the orthosteric site, the modulators typically bind to allosteric sites that are distant from the orthosteric site. Here are some excellent reviews published in 2020 on allosteric molecules[9–12]. Allosteric communication in GPCRs falls into two types:
The communication between residue patches in distant sites of the GPCR that occur concurrently in time leading to large scale conformational transitions. Such processes involve longer time scale (typically hundreds of microseconds to milliseconds) that is measurable by NMR measurements.
The statistical spatial correlation or mutual information in the covariance of cartesian coordinates of atoms or torsion angle distributions of residue patches located in distant sites is another type of allosteric communication. Such spatially correlated dynamics of GPCRs is the type of correlated movement that leads to lowering of entropy of the system and hence stabilization of a given conformational state. Molecular dynamics simulations that map the spatiotemporal dynamics of residues at an atomic level over a shorter time scale (lower microseconds) is suitable for mapping the spatial correlation of GPCR dynamics.
Here we cover computational methods that delineate spatially correlated conformational changes, the type of correlated movement that leads to stabilization of a conformational state of a GPCR. The strength of the spatial correlated movement can be modulated by the type of ligand bound to the GPCR[13]. For example, an antagonist or inverse agonist bound GPCR shows high level of spatial correlation in residue movements that reduces the overall entropy of the system and stabilizes the inactive state[14–16]. However, when bound to an agonist the GPCR becomes more flexible with increased entropy resulting from reduced correlated movement among distant residues[14]. This was shown to be true for the time correlated movements of labeled residues by NMR[17,18]. Throughout this review, we have diligently cited the most relevant current publications (and reviews if the number of primary publications is high). Please pardon us if we have inadvertently omitted any relevant literature.
GPCRs exhibit a continuum of conformation states:
GPCRs are dynamic proteins and exist in an equilibrium among multiple conformational states even in the absence of agonist binding. The well characterized conformational states through crystallography[1–3,19], and electron microscopy[20–22] studies are antagonist or inverse agonist bound inactive state, agonist bound active-intermediate state and agonist and G protein or β-arrestin bound fully active state. Each of these distinct functional states is not characterized by a single snapshot but rather, constitutes an ensemble of receptor conformational states[23]. The structural characteristics of these functional states are typified by inter-residue distances located in the intracellular regions of TM3 and TM6 as well as inter-residue distances between TM3 and TM7. However, it should be noted that these are just one type of measure to distinguish these states and in no way a complete one. As shown in Fig. 1, analysis of these distances in various three-dimensional structures of inactive, intermediate and fully active state structures shows that there is no clear distinction between the defined inactive or active intermediate states. This suggests that GPCRs exist in a continuum of states and each state is tuned to G protein or β-arrestin coupling to different levels. There are NMR[17,18,24–29], DEER[30–33], FRET[34–37] and other experimental studies[38–40] that clearly illustrate that ligand selectively bind and stabilize specific receptor conformations [40–43] that shifts the equilibrium and the relative population of the various conformational states. Multiple studies have also shown that other factors such as lipid components of cell membrane as well as cations such as Na+[44], Ca2+ and Mg2+ can affect the conformation equilibrium of GPCRs and hence their activity[29,45–48]. The changes effected by ligands, G proteins, β-arrestins, lipids, divalent cations on GPCR conformations and hence its activity is through allosteric communication. In summary, it is clear that GPCRs are dynamic proteins and its activity emerges from a continuum of conformation states each with different level activity towards a specific agonist binding and a specific effector protein (G protein or β-arrestin) coupling. In the next section we outline briefly, the experimental evidence for allosteric communication.
Figure 1:
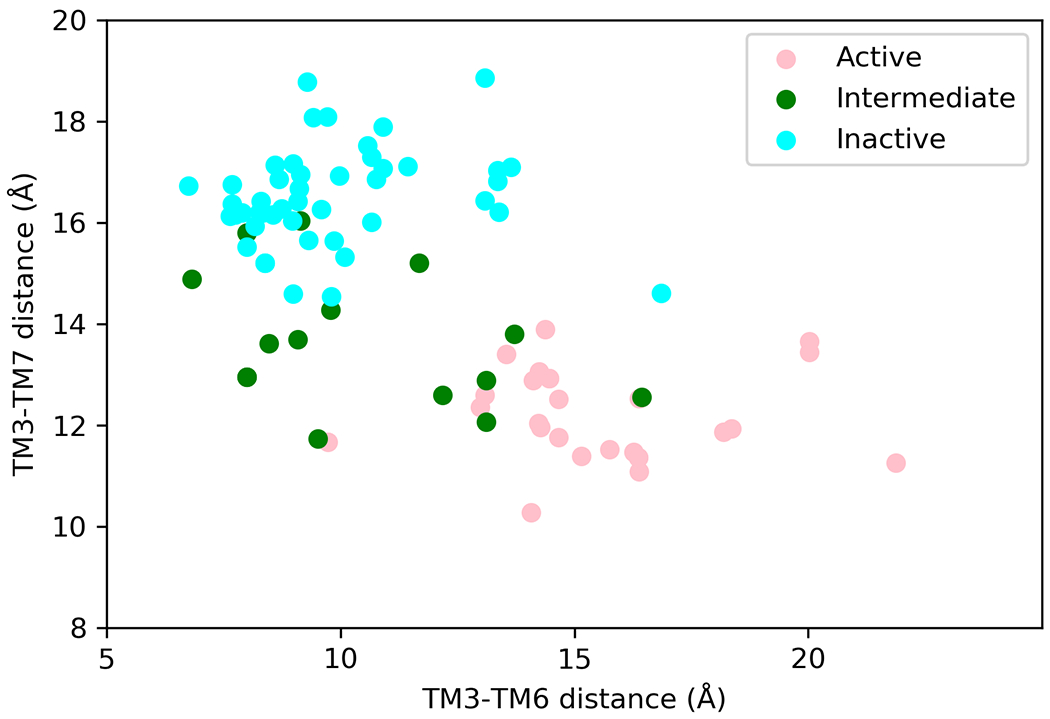
Three dimensional structures show that GPCRs exist in a continuum of states with overlap between inactive, intermediate and fully active conformations. Comparison of the inter-residue distances between residues 3.50 on TM3 and 6.34 on TM6 on x-axis and residues 3.50 and 7.53 on the y-axis. These residue numbers follow the commonly used Ballesteros-Weinstein residue numbering scheme for class A GPCRs. We used several crystal structures of class A GPCRs, in the inactive, intermediate and fully active states of class A GPCRs. Only one structure for each conformational state of a given class A GPCR was selected for this plot. The protein data bank identification codes for the three-dimensional structures used here are: 6G79, 6WHA, 5TUD, 6BQG, 6D9H, 5WF6, 6OIJ, 4MQT, 6K42, 6IBL, 6OS1,7C6A, 6WWZ, 5XR8, 6PT0, 6LFO, 6VMS, 6LW5, 7CFN, 6LI3, 5T04, 6B73, 5C1M, 6FK8, 6AK3, 7D7M, 4IAR, 6A94, 6BQH, 5UEN, 6WJC, 5ZKC, 4U15, 5DSG, 6OL9, 6KUW, 4BVN, 6PS2, 4ZUD, 5VBL, 6I9K, 6GPX, 5UIW, 6QZH, 5LWE, 5U09, 5ZTY, 6LFL, 3ODU, 6CM4, 3PBL, 5WIU, 6IGK, 6KO5, 7BR3, 3RZE, 4Z36, 6ME2, 6ME6, 6HLP, 5ZBQ, 4N6H, 4DJH, 4DKL, 5DHH, 2Z73, 6TOS, 5WQC, 6TPK, 6D27, 5X33, 3V2Y, 4IB4, 6RZ5, 6RZ7, 5TZR, 6LI0, 6W25, 1U19, 4XNV, 4PXZ, 3VW7, 5NDD, 5ZKQ, 5XSZ, 6RNK, 6IIU.
Evidence for Allosteric communication in GPCRs:
Experimental binding measurements using purified GPCRs in nanodiscs and detergent micelles have shown that agonist bound receptor exhibit stronger coupling strength to the G proteins than in the absence of the agonist[40,49]. Similarly, the G protein or nanobody sensors coupled receptor also showed increase in the agonist affinity to the receptor[49–51]. A crystal structure study of a constitutively active mutant of neurotensin receptor 1 showed opening of the TM6 even in the absence of a ligand[52]. A recent study on the basal activity of β2AR using pressure-resolved DEER[30], showed a finite population of the active state even in the absence of the agonist. Taken together it is clear that there is two-way allosteric communication between the ligand binding sites and the G protein or β-arrestin coupling sites in GPCRs.
Allosteric Communication and Ligand Efficacy:
Based on their effect on receptor activity, ligands of GPCRs can be broadly classified as agonists, partial agonists, antagonists and inverse agonists. These broad definitions are based on the measured efficacy of the ligand. What are the atomic level features of the ligand:GPCR complexes and the ligand:GPCR:effector protein complexes that contribute to the ligand efficacy? The strength of the allosteric communication from the ligand binding site to the effector coupling site in the GPCR and the reverse allostery are critical factors in determining the molecular efficacy of ligands. As described in the section above FRET, NMR, DEER based studies have shown evidence for ligand specific conformational changes in the receptor that dictates the coupling strength to the transducer proteins. Partial agonists binding to β2AR have been shown to alter the balance of relative population of the active and inactive states of the receptor[34]. However, in the adenosine receptor A2AR, partial agonists have been shown to stabilize distinct receptor active states[43]. Using single molecule spectroscopy, it was shown that when the Gs protein is bound to β2AR in the presence of partial agonists, it has higher affinity for GDP than in the presence of full agonists[34]. Single molecule FRET and other FRET sensor-based techniques studying the effect of several agonists on β2-adrenergic receptor (β2AR) provide an estimate of the relative population of the different conformational states and an estimate of molecular efficacy[34,53]. Isogai et al [54] studied turkey β1AR in the presence of six different ligands (two agonists – isoproterenol and dobutamine; 4 antagonists – atenolol, alprenolol, carvedilol and cyanopindolol) and in the apo form using backbone labeled valines in NMR. The authors found that upon ligand binding there are concerted changes in the intracellular side of TM5, which correlated linearly with ligand efficacy for the G protein pathway. They also showed that the binding of a nanobody produced strong chemical shifts of residues throughout the receptor including in the extracellular region, indicative of important connections in allosteric signal transmission networks.
This clearly highlights the need that in order to understand the molecular origins of ligand efficacy one needs to probe the residues involved in the two-way allosteric communication between the ligand binding site and the effector protein coupling site. Determination of the strength of the allosteric communication for different ligands and different effector protein bound complexes would aid in determining the atomic level contribution to molecular efficacy of ligands and to molecular ligand bias factor. As detailed in the next section, computational methods play a crucial role in determining the residues involved in allosteric communication and the relative strength of allosteric communication in different ligand bound GPCR states.
Computational Methods to delineate the residues involved in Allosteric Communication in GPCR Signaling
Computational methods based on amino acid sequence analysis, structure-based analysis and dynamics-based analysis including molecular dynamics have been developed and applied to study allosteric communication in GPCRs.
Sequence based analysis of residues involved in Allosteric Communication:
Amino acid sequence based Evolutionary analysis methods using multiple sequence alignments of GPCRs have been used to delineate residues with functional significance in GPCRs[55–58]. Using multiple sequence alignment of a subset of class A GPCRs, Madubashi et al used evolutionary trace analysis that ranks the relative importance of each residue position based on the number of branches that are above the residue position in the phylogenetic tree. The top ranking residue positions were used to identify residues that are proposed to play an important role in the GPCR function[55]. Many of these residues form communities of residues that are neither in the ligand binding site not in the G protein coupling region. These communities of residues were shown to cause functional defects upon mutation[55,57]. Subsequent advances in the evolutionary trace method involved calculating the propensity of covariation or correlated mutation using the mutual information. Again, the covariation analysis was done using the multiple sequence alignment of selected GPCRs. This method was used for identifying residues involved in allosteric communication in dopamine D2 receptor function[59]. Thus, the sequence-based information can be used to calculate covariation information and identify the network of residues involved in intramolecular allosteric communication. However, a minor drawback is that the residue communities identified to be involved in allosteric communication through these analyses were disjointed and did not show a pathway of connected residues from the ligand binding site to the effector protein coupling site. Additionally, the sequence covariation information is limited since it does not differentiate the residue positions that could be involved in maintaining structural stability of GPCRs from those that are involved in modulating receptor activity or both these functions.
Static three-dimensional structure-based analysis of Allosteric Communication in GPCRs:
Using graph theoretical properties, a protein structure can be cast into a network model. There is tremendous amount of work in this field covered by detailed reviews[60–62]. Here we discuss this method only as applied to understanding GPCR allostery. Using the network model derived from the static three-dimensional structures of GPCRs and applying normal mode analysis, residues at a distance that are highly interconnected (high edge strengths in the network model) were identified. Starting from three dimensional structures of a GPCR in different conformational states, the differences in the inter-residue edge strengths for the network models derived in the fully active versus inactive states of GPCRs were delineated[63]. Further development of this class of methods was done with the purpose of extending the single structure analysis to a dynamic structure network. This advancement was based on perturbing the static network model using the Elastic Network or Gaussian Network approach to generate a dynamic network of the GPCR structures. Using mutual information and cross-correlations in the Cartesian coordinates of the Cα atoms of each residue, calculated from the dynamic network, a community of residues involved in allosteric communication were derived. Application of the dynamic network methods to several class A GPCRs have provided explanations of the roles of mutations that activate or deactivate the receptor, affect ligand binding, and receptor dimerization[64–66]. Such dynamic network-based analysis has the advantage of being faster than running molecular dynamics simulations. However, the perturbations induced in the protein network model are stochastic and not suitable for deriving thermodynamic properties of the systems. More importantly, the Gaussian network uses a coarse grain model of the GPCR using only the Cα atoms for each residue and the functional motions of the GPCR that happens at an atomic scale are lost. The atomic level details on the allosteric communication mechanism derived from molecular dynamics simulation techniques are detailed in the next section.
Residues involved in allosteric communication in GPCRs derived from molecular dynamics simulations combined with graph theory network models
The dynamic couplings in residue motions within the GPCR or within the GPCR:G protein complex, at an atomic level, form one of the fundamental components that initiate, propagate and effect allosteric couplings. All-atom Molecular dynamics simulations is a well-established computational technique that is routinely used to probe and track atomic level motion in low microseconds time scale. The trajectories derived using molecular dynamics simulations on GPCRs provide the building maps of dynamic coupling. In this section we enumerate how the molecular dynamics simulation trajectories have been harnessed to derive the mechanism of allosteric communication in GPCRs and to enumerate the residues involved in such communication. We will also show the caveats in the methods and future developments needed in this area.
In 2014, we developed a generalizable method, Allosteer [14–16,67], that uses molecular dynamics simulation trajectories to calculate the allosteric communication pathways in proteins. In the Allosteer method we seek to study the equilibrium properties of two distant domains in a protein via the statistical correlation in their dynamics. Please note that this method, as of now, does not uncover any time related correlated movements that occur in longer time scales leading to conformational transitions typically observed in NMR. In brief, the procedure to calculate allosteric pathways from the extracellular region of the GPCRs, passing through the ligand binding site to the G protein coupling region is as follows: In the first step we calculate the mutual information for every pair of dihedral angles in the GPCR using the first and second order entropies. Further, for the pairs of residues that show high mutual information in their torsion angle distributions we use graph theory to calculate the shortest allosteric communication pathways with maximum total mutual information of residues in the pathways. Overlapping pathways are clustered to form allosteric communication pipelines[14]. The strength of the allosteric communication pipelines is the number of overlapping pathways that are clustered in the pipeline. The network of residues that make up the allosteric pipelines modulate the coupling strength of different G proteins and β-arrestins to the GPCR. We observed that the strongest allosteric communication pipelines in GPCRs emanate from the extracellular regions through the agonist binding site terminating in the spatially distant G-protein coupling site. Mutation of the residues in the allosteric communication pipelines have been shown to increase or decrease the protein activity in multiple proteins that include GPCRs[14–16,52,67,68], kinases[69,70], phosphatases[71] and DNA repair proteins[72]. This approach can be used on any protein system without any prior knowledge on the allosteric communication in the protein under study.
Using Allosteer on microseconds-long molecular dynamics simulation trajectories on different conformational states of β2AR, we elucidated[14] the allosteric communication pipelines in three different conformational states of β2AR: 1) the inverse agonist-bound inactive state; 2), the agonist-bound intermediate state; and 3), the agonist- and G-protein-bound fully active state. We observed that the inactive state of β2AR with antagonist bound showed strong allosteric communication compared with the agonist bound intermediate state and agonist and G protein bound fully active state of β2AR. The strength of the allosteric communication pipelines from the extracellular domain to the intracellular domain is weakened when an agonist binds to β2AR. Thus, agonist binding leads to decoupling the extracellular domain from the intracellular domain of the receptor and making the receptor more dynamic compared with the other states. This was shown experimentally to be true using NMR studies[17].
In the same lines of the Allosteer method, subsequent studies on GPCRs used the mutual information in Cartesian coordinates of residues instead of the torsion angles of the residues as in the Allosteer method. They calculated the residues with high mutual information and identified communities of residues that relay the allosteric information[73–76]. Other studies[77] have delineated the allosteric network of residues that mediate the ligand binding to the effector coupling regions based on analysis of inter-residue distances that showed concurrent rearrangement. Such analysis is system specific and requires a priori knowledge on residues whose movements need to be analyzed.
Relative strength of the allosteric communication pipelines to G protein versus β-arrestin coupling sites correlates with ligand bias factor
GPCR agonists that elicit differential responses when coupled to G-proteins or β-arrestins are called “biased agonists” and this phenomenon is called “biased signaling”. It is widely appreciated that the ligand bias factor is influenced by structural factors[78], as well as cell and tissue-specific effects[79]. The intrinsic structural factors of the GPCR-G-protein or GPCR:β-arrestin complexes are important factors that contribute to ligand bias. We postulated that the relative strength of the allosteric communication between the ligand binding site and G protein or β-arrestin coupling site is an intrinsic structure factor that contributes to ligand bias. In our recent work[67], we calculated the ratio of the strength of the allosteric communication pipelines from the extracellular region passing through the residues in the ligand binding site to the residues in the G-protein and β-arrestin coupling interface in the GPCRs. We calculated this ratio for an agonist of interest with respect to a reference agonist. We called this as the “molecular ligand bias” since this ratio represents the atomic level contribution to ligand bias. The calculated molecular ligand bias showed a qualitative correlation with the experimentally measured ligand bias factor for several ligands in three different class A GPCRs, angiotensin 2 receptor 1, κ-opioid receptor (here we used a homology model as the active state crystal structure of the κ-opioid receptor was unavailable then), β2-adrenergic receptor. This correlation demonstrates that the strength of allosteric communication plays an important role in ligand bias and that computational methods play a critical role in estimating the molecular level ligand bias. We also showed that the allosteric network of residues located in the allosteric communication pipelines can influence the conformations of the residues in the ligand binding sites. We termed these residues in the ligand binding site as “functional hotspots” [67]. Knowledge of the functional hotspot residues in the ligand binding site will greatly aid design of biased ligands as well as subtype-selective ligands [80].
The synergy of iterative computational-experimental cycles in identifying allosteric network of residues
The most exciting and nerve-wracking moment and at the same time providing a true test for the computational methods is when we make predictions that get tested subsequently by experiments. An elegant study by Chen et al[38] showed through a series of iterative predictions and experiments that modifying and optimizing the residues located in the allosteric pipeline could stabilize a ligand specific conformation state, such as inactive or active state of the receptor. Such engineered mutations also shift the receptor response to ligand binding. For example, they engineered mutations in the allosteric communication pipelines through rational design that would enhance the allosteric coupling of the agonist-bound ligand site with the active state and not with the inactive conformation. Such mutations were shown to stabilize the G protein bound active state conformation and thereby the enhanced agonist binding affinity.
Using Allosteer, we recently predicted the residues involved in allosteric communication from the extracellular regions to the Gq and β-arrestin2 coupling sites in the angiotensin 2 receptor 1. The residues predicted to be involved in allosteric communication to the Gq or β-arrestin2 coupled sites were tested for their effect upon mutation to alanine, while existing alanine residues were replaced with glycine using BRET-based biosensors in cells[81–84]. A majority of residues involved in allosteric communication to the Gq coupling site were located in TM5 and TM6, followed by ICL2, TM3 and TM4, while residues that communicate to the β-arrestin2 coupling sites are not only numerous, but also distributed more widely across the AT1R structure[85].
Conclusions
What we have learned about Allostery in GPCRs so far and what lies ahead:
The experimental and computational studies on GPCR allostery have shown that:
There is allosteric communication between the ligand binding site and G protein or β-arrestin coupling sites in GPCRs. The strength of the allosteric communication varies depending on the type of ligand and the effector protein bound to the receptor.
Inverse agonist bound receptor shows strong allosteric communication that leads to lower entropy of the whole receptor and hence less flexible receptor.
Agonist binding to the receptor weakens the allosteric communication between the extracellular and intracellular domains of the GPCR. This results in overall higher entropy and flexibility of the receptor that probably enables opening of the intracellular region to allow G protein or β-arrestin coupling.
Both agonist and G protein bound fully active states of GPCRs show stronger allosteric communication than the state with just the agonist bound.
The residues involved in allosteric communication across several class A GPCRs in their antagonist or inverse agonist bound inactive states are located in the same structural position in the GPCRs structure although they are not conserved across even same subfamily of class A GPCRs[15].
The relative strength of allosteric communication from the ligand binding site to the G protein and β-arrestin coupling sites is an important structural factor contributing to the molecular ligand bias.
Using the allosteric network of residues delineated using computational methods one could engineer GPCR mutants that show relatively stronger biased coupling to G protein or β-arrestin.
The Allosteer method can be used to identify residues in the ligand binding sites whose conformations are influenced by residues in the allosteric communication pipelines. We termed these residues as functional hotspots[67]. Knowledge of the functional hotspot residues in the ligand binding site will greatly aid design of biased ligands.
Multiple three-dimensional structures of GPCRs show ligand binding in multiple regions of the GPCR structure. While most of the ligands for which we have structures bind in the extracellular region of the receptor, some ligands have been shown to bind to the intracellular region and in the extramembrane part of the transmembrane region of the GPCR (Fig. 2). Additionally, in the past decade, positive and negative allosteric modulators targeting GPCRs have shown great promise due to some of their desirable pharmacological properties. Allosteric modulators have been shown to enhance the subtype selectivity of orthosteric ligands to closely related GPCRs[86,87]. Other studies detailed in these fine reviews have shown that allosteric modulators show bias to specific signaling pathways (G protein or β-arrestin) and/or enhance the ligand bias of the orthosteric ligands[87,88]. Allosteric modulators bind to multiple structural regions of GPCRs highlighting the presence of druggable binding sites across GPCR structures. However, there is sparse information on the mechanisms by which the allosteric communication occurs in these systems where ligand binds in different structural regions, or when allosteric modulators are bound to the receptor in addition to the orthosteric ligands. We speculate that there should be allosteric communication pipelines from these ligand binding sites to the G protein coupling sites. Additionally, the changes in the mechanism of allosteric communication in GPCRs when both an allosteric modulator and an orthosteric ligand are bound needs to be understood. These pipelines from various structural regions of GPCRs can be used to identify other putative ligand binding sites in GPCRs. GPCR mutants have been designed to bind to a ligand of choice[89]. These mutations that are located in the ligand binding site reshape the binding site to enhance binding to the desired ligand. The residues in the allosteric communication pipelines that are distant from the ligand binding site and the G protein or β-arrestin coupling site can be used to design GPCR mutants that confer specificity to bind to either a ligand of choice or to a transducer protein of choice.
Figure 2:
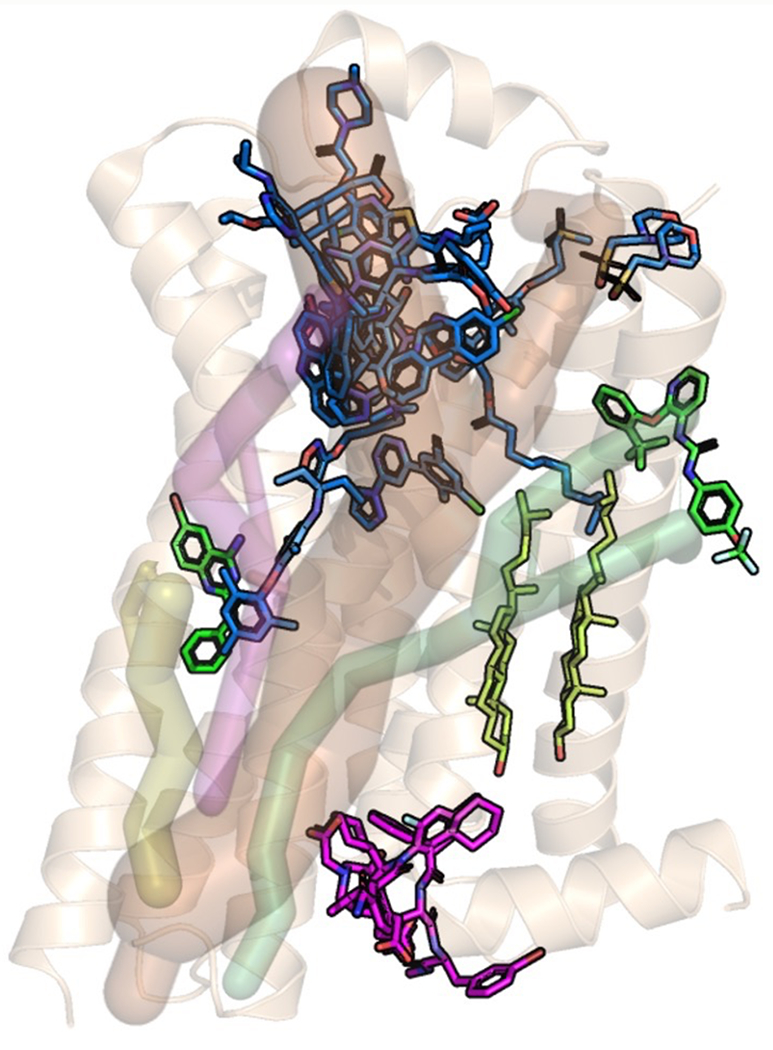
Positions of ligands extracted from three dimensional structures of a few class A GPCRs that show diverse ligand binding sites. These structures were overlaid on the crystal structure conformation of the inactive state β2-adrenergic receptor (pdb ID:6OBA); the color codes are: blue — ligands binding to the extracellular loops and N terminus, and orthosteric site; green — ligands binding in the middle and outside of the transmembrane domain; magenta — ligands binding to the intracellular part of the transmembrane domain or near the G protein interface, yellowish green — cholesterol; We have shown ligands from the following crystal structures: pdb IDs: 4Z35, 5CGC, 4OO9, 4OR2, 4K5Y, 4ZJ8, 4XEE, 2RH1, 3PWH, 4RWS, 3VW7, 4MQT, 4XNV, 4PHU, 5T1A, 5X7D, and 5UIG. The shaded pipes shown in the figure are the speculated allosteric communication pipelines to different ligand binding regions in the GPCR structure. To date there is data to demonstrate allosteric communication pipelines to the orthosteric ligand binding site. There is no data yet, showing any of the other possible allosteric communication pipelines.
Acknowledgements:
The support for the work was provided by NIH R01GM117923 and R01GM097261. We thank Dr. Supriyo Bhattacharya for insightful discussions. We thank Mr. Keith Le for generating the data for Figure 1.
Abbreviations:
- GPCR
G protein coupled receptor
- TM
transmembrane
- β2AR
human β2 adrenergic receptor
- β1AR
turkey β1 adrenergic receptor
Footnotes
Conflicts of Interest: The authors declare no competing interests.
References:
- 1.Wang J, Hua T & Liu Z-J (2020) Structural features of activated GPCR signaling complexes. Curr Opin Struct Biol 63, 82–89. [DOI] [PubMed] [Google Scholar]
- 2.Sutkeviciute I & Vilardaga J-P (2020) Structural insights into emergent signaling modes of G protein–coupled receptors. J Biol Chem 295, 11626–11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Congreve M, de Graaf C, Swain NA & Tate CG (2020) Impact of GPCR Structures on Drug Discovery. Cell 181, 81–91. [DOI] [PubMed] [Google Scholar]
- 4.Wingler LM & Lefkowitz RJ (2020) Conformational Basis of G Protein-Coupled Receptor Signaling Versatility. Trends Cell Biol 30, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weis WI & Kobilka BK (2018) The Molecular Basis of G Protein–Coupled Receptor Activation. Annu Rev Biochem 87, 897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Yu Z, Xiao W, Lu S & Zhang J (2020) Allosteric binding sites at the receptor–lipid bilayer interface: novel targets for GPCR drug discovery. Drug Discov Today. [DOI] [PubMed] [Google Scholar]
- 7.O’Hayre M, Vázquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S & Gutkind JS (2013) The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev Cancer 13, 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schöneberg T & Liebscher I (2021) Mutations in G Protein–Coupled Receptors: Mechanisms, Pathophysiology and Potential Therapeutic Approaches. Pharmacol Rev 73, 89–119. [DOI] [PubMed] [Google Scholar]
- 9.Changeux J-P & Christopoulos A (2016) Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 166, 1084–1102. [DOI] [PubMed] [Google Scholar]
- 10.Skiba MA & Kruse AC (2020) Autoantibodies as Endogenous Modulators of GPCR Signaling. Trends Pharmacol Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizuno H & Kihara Y (2020) Druggable Lipid GPCRs: Past, Present, and Prospects. In pp. 223–258. [DOI] [PubMed] [Google Scholar]
- 12.Singh KD & Karnik SS (2021) Current Trends in GPCR Allostery. J Membr Biol. [DOI] [PubMed] [Google Scholar]
- 13.Kim TH, Chung KY, Manglik A, Hansen AL, Dror RO, Mildorf TJ, Shaw DE, Kobilka BK & Prosser RS (2013) The Role of Ligands on the Equilibria Between Functional States of a G Protein-Coupled Receptor. J Am Chem Soc 135, 9465–9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhattacharya S & Vaidehi N (2014) Differences in Allosteric Communication Pipelines in the Inactive and Active States of a GPCR. Biophys J 107, 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaidehi N & Bhattacharya S (2016) Allosteric communication pipelines in G-protein-coupled receptors. Curr Opin Pharmacol 30, 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhattacharya S, Salomon-Ferrer R, Lee S & Vaidehi N (2016) Conserved Mechanism of Conformational Stability and Dynamics in G-Protein-Coupled Receptors. J Chem Theory Comput 12, 5575–5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, Thian FS, Kobilka TS, Shaw DE, Mueller L, Prosser RS & Kobilka BK (2013) The Dynamic Process of β2-Adrenergic Receptor Activation. Cell 152, 532–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, Lerch MT, Kobilka TS, Thian FS, Hubbell WL, Prosser RS & Kobilka BK (2015) Structural Insights into the Dynamic Process of β 2 -Adrenergic Receptor Signaling. Cell 161, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gusach A, Maslov I, Luginina A, Borshchevskiy V, Mishin A & Cherezov V (2020) Beyond structure: emerging approaches to study GPCR dynamics. Curr Opin Struct Biol 63, 18–25. [DOI] [PubMed] [Google Scholar]
- 20.Glukhova A, Draper-Joyce CJ, Sunahara RK, Christopoulos A, Wootten D & Sexton PM (2018) Rules of Engagement: GPCRs and G Proteins. ACS Pharmacol Transl Sci 1, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.García-Nafría J & Tate CG (2020) Cryo-Electron Microscopy: Moving Beyond X-Ray Crystal Structures for Drug Receptors and Drug Development. Annu Rev Pharmacol Toxicol 60, 51–71. [DOI] [PubMed] [Google Scholar]
- 22.García-Nafría J & Tate CG (2019) Cryo-EM structures of GPCRs coupled to Gs, Gi and Go. Mol Cell Endocrinol 488, 1–13. [DOI] [PubMed] [Google Scholar]
- 23.Lee S, Nivedha AK, Tate CG & Vaidehi N (2019) Dynamic Role of the G Protein in Stabilizing the Active State of the Adenosine A2A Receptor. Structure 27, 703–712.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye L, Neale C, Sljoka A, Lyda B, Pichugin D, Tsuchimura N, Larda ST, Pomès R, García AE, Ernst OP, Sunahara RK & Prosser RS (2018) Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat Commun 9, 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, McFarland A, Madsen JJ, Aalo E & Ye L (2021) The Potential of 19F NMR Application in GPCR Biased Drug Discovery. Trends Pharmacol Sci 42, 19–30. [DOI] [PubMed] [Google Scholar]
- 26.Frei JN, Broadhurst RW, Bostock MJ, Solt A, Jones AJY, Gabriel F, Tandale A, Shrestha B & Nietlispach D (2020) Conformational plasticity of ligand-bound and ternary GPCR complexes studied by 19F NMR of the β1-adrenergic receptor. Nat Commun 11, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Didenko T, Liu JJ, Horst R, Stevens RC & Wüthrich K (2013) Fluorine-19 NMR of integral membrane proteins illustrated with studies of GPCRs. Curr Opin Struct Biol 23, 740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma X, Hu Y, Batebi H, Heng J, Xu J, Liu X, Niu X, Li H, Hildebrand PW, Jin C & Kobilka BK (2020) Analysis of β 2 AR-G s and β 2 AR-G i complex formation by NMR spectroscopy. Proc Natl Acad Sci 117, 23096–23105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones AJY, Gabriel F, Tandale A & Nietlispach D (2020) Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches. Molecules 25, 4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lerch MT, Matt RA, Masureel M, Elgeti M, Kumar KK, Hilger D, Foys B, Kobilka BK & Hubbell WL (2020) Viewing rare conformations of the β 2 adrenergic receptor with pressure-resolved DEER spectroscopy. Proc Natl Acad Sci 117, 31824–31831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altenbach C, Kusnetzow AK, Ernst OP, Hofmann KP & Hubbell WL (2008) High-resolution distance mapping in rhodopsin reveals the pattern of helix movement due to activation. Proc Natl Acad Sci 105, 7439–7444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wingler LM, Elgeti M, Hilger D, Latorraca NR, Lerch MT, Staus DP, Dror RO, Kobilka BK, Hubbell WL & Lefkowitz RJ (2019) Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 176, 468–478.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Eps N, Altenbach C, Caro LN, Latorraca NR, Hollingsworth SA, Dror RO, Ernst OP & Hubbell WL (2018) G i - and G s -coupled GPCRs show different modes of G-protein binding. Proc Natl Acad Sci 115, 2383–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, Zhou Z, Perez-Aguilar JM, Hauge M, Mathiasen S, Javitch JA, Weinstein H, Kobilka BK & Blanchard SC (2017) Single-molecule analysis of ligand efficacy in β2AR–G-protein activation. Nature 547, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandhu M, Touma AM, Dysthe M, Sadler F, Sivaramakrishnan S & Vaidehi N (2019) Conformational plasticity of the intracellular cavity of GPCR–G-protein complexes leads to G-protein promiscuity and selectivity. Proc Natl Acad Sci, 201820944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halls ML & Canals M (2018) Genetically Encoded FRET Biosensors to Illuminate Compartmentalised GPCR Signalling. Trends Pharmacol Sci 39, 148–157. [DOI] [PubMed] [Google Scholar]
- 37.Gupte TM, Ritt M, Dysthe M, Malik RU & Sivaramakrishnan S (2019) Minute-scale persistence of a GPCR conformation state triggered by non-cognate G protein interactions primes signaling. Nat Commun 10, 4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen K-YM, Keri D & Barth P (2020) Computational design of G Protein-Coupled Receptor allosteric signal transductions. Nat Chem Biol 16, 77–86. [DOI] [PubMed] [Google Scholar]
- 39.Lane JR, May LT, Parton RG, Sexton PM & Christopoulos A (2017) A kinetic view of GPCR allostery and biased agonism. Nat Chem Biol 13, 929–937. [DOI] [PubMed] [Google Scholar]
- 40.Yao XJ, Velez Ruiz G, Whorton MR, Rasmussen SGF, DeVree BT, Deupi X, Sunahara RK & Kobilka B (2009) The effect of ligand efficacy on the formation and stability of a GPCR-G protein complex. Proc Natl Acad Sci 106, 9501–9506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghanouni P, Gryczynski Z, Steenhuis JJ, Lee TW, Farrens DL, Lakowicz JR & Kobilka BK (2001) Functionally Different Agonists Induce Distinct Conformations in the G Protein Coupling Domain of the β2Adrenergic Receptor. J Biol Chem 276, 24433–24436. [DOI] [PubMed] [Google Scholar]
- 42.Yao X, Parnot C, Deupi X, Ratnala VRP, Swaminath G, Farrens D & Kobilka B (2006) Coupling ligand structure to specific conformational switches in the β2-adrenoceptor. Nat Chem Biol 2, 417–422. [DOI] [PubMed] [Google Scholar]
- 43.Ye L, Van Eps N, Zimmer M, Ernst OP & Scott Prosser R (2016) Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268. [DOI] [PubMed] [Google Scholar]
- 44.Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V & Stevens RC (2014) Allosteric sodium in class A GPCR signaling. Trends Biochem Sci 39, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White KL, Eddy MT, Gao Z-GG, Han GW, Lian T, Deary A, Patel N, Jacobson KA, Katritch V & Stevens RC (2018) Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 26, 259–269.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jaakola V-P, Lane JR, Lin JY, Katritch V, IJzerman AP & Stevens RC (2010) Ligand Binding and Subtype Selectivity of the Human A 2A Adenosine Receptor. J Biol Chem 285, 13032–13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacques F & Yahi N (2015) Brain Lipids in Synaptic Function and Neurological Disease. In 1 st, pp. 163–181. Elsevier. [Google Scholar]
- 48.Prasanna X, Jafurulla M, Sengupta D & Chattopadhyay A (2016) The ganglioside GM1 interacts with the serotonin 1A receptor via the sphingolipid binding domain. Biochim Biophys Acta - Biomembr 1858, 2818–2826. [DOI] [PubMed] [Google Scholar]
- 49.DeVree BT, Mahoney JP, Vélez-Ruiz GA, Rasmussen SGF, Kuszak AJ, Edwald E, Fung J-JJ, Manglik A, Masureel M, Du Y, Matt RA, Pardon E, Steyaert J, Kobilka BK & Sunahara RK (2016) Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 535, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warne T, Edwards PC, Doré AS, Leslie AGW & Tate CG (2019) Molecular basis for high-affinity agonist binding in GPCRs. Science (80- ) 364, 775–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Livingston KE, Mahoney JP, Manglik A, Sunahara RK & Traynor JR (2018) Measuring ligand efficacy at the mu-opioid receptor using a conformational biosensor. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krumm BE, Lee S, Bhattacharya S, Botos I, White CF, Du H, Vaidehi N & Grisshammer R (2016) Structure and dynamics of a constitutively active neurotensin receptor. Sci Rep 6, 38564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kum K, Paulekas S, Sadler F, Gupte TM, Ritt M, Dysthe M, Vaidehi N & Sivaramakrishnan S (2021) β2-adrenoceptor ligand efficacy is tuned by a two-stage interaction with the Gαs C-terminus. Communicated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Isogai S, Deupi X, Opitz C, Heydenreich FM, Tsai C-J, Brueckner F, Schertler GFX, Veprintsev DB & Grzesiek S (2016) Backbone NMR reveals allosteric signal transduction networks in the β1-adrenergic receptor. Nature 530, 237–241. [DOI] [PubMed] [Google Scholar]
- 55.Madabushi S, Gross AK, Philippi A, Meng EC, Wensel TG & Lichtarge O (2004) Evolutionary Trace of G Protein-coupled Receptors Reveals Clusters of Residues That Determine Global and Class-specific Functions. J Biol Chem 279, 8126–8132. [DOI] [PubMed] [Google Scholar]
- 56.Schöneberg T, Hofreiter M, Schulz A & Römpler H (2007) Learning from the past: evolution of GPCR functions. Trends Pharmacol Sci 28, 117–121. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez GJ, Yao R, Lichtarge O & Wensel TG (2010) Evolution-guided discovery and recoding of allosteric pathway specificity determinants in psychoactive bioamine receptors. Proc Natl Acad Sci 107, 7787–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schönegge A-M, Gallion J, Picard L-P, Wilkins AD, Le Gouill C, Audet M, Stallaert W, Lohse MJ, Kimmel M, Lichtarge O & Bouvier M (2017) Evolutionary action and structural basis of the allosteric switch controlling β2AR functional selectivity. Nat Commun 8, 2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sung Y-M, Wilkins AD, Rodriguez GJ, Wensel TG & Lichtarge O (2016) Intramolecular allosteric communication in dopamine D2 receptor revealed by evolutionary amino acid covariation. Proc Natl Acad Sci 113, 3539–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Collier G & Ortiz V (2013) Emerging computational approaches for the study of protein allostery. Arch Biochem Biophys 538, 6–15. [DOI] [PubMed] [Google Scholar]
- 61.Wang J, Jain A, McDonald LR, Gambogi C, Lee AL & Dokholyan NV. (2020) Mapping allosteric communications within individual proteins. Nat Commun 11, 3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Di Paola L & Giuliani A (eds.) (2021) Allostery Springer US, New York, NY. [Google Scholar]
- 63.Gadiyaram V, Dighe A, Ghosh S & Vishveshwara S (2021) Network Re-Wiring During Allostery and Protein-Protein Interactions: A Graph Spectral Approach. In pp. 89–112. [DOI] [PubMed] [Google Scholar]
- 64.Jiang Y, Yuan Y, Zhang X, Liang T, Guo Y, Li M & Pu X (2016) Use of network model to explore dynamic and allosteric properties of three GPCR homodimers. RSC Adv 6, 106327–106339. [Google Scholar]
- 65.Fanelli F, Felline A, Raimondi F & Seeber M (2016) Structure network analysis to gain insights into GPCR function. Biochem Soc Trans 44, 613–618. [DOI] [PubMed] [Google Scholar]
- 66.Woods KN, Pfeffer J, Dutta A & Klein-Seetharaman J (2016) Vibrational resonance, allostery, and activation in rhodopsin-like G protein-coupled receptors. Sci Rep 6, 37290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nivedha AK, Tautermann CS, Bhattacharya S, Lee S, Casarosa P, Kollak I, Kiechle T & Vaidehi N (2018) Identifying Functional Hotspot Residues for Biased Ligand Design in G-Protein-Coupled Receptors. Mol Pharmacol 93, 288–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee S, Bhattacharya S, Tate CG, Grisshammer R & Vaidehi N (2015) Structural dynamics and thermostabilization of neurotensin receptor 1. J Phys Chem B 119, 4917–4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma N, Lippert LG, Devamani T, Levy B, Lee S, Sandhu M, Vaidehi N & Sivaramakrishnan S (2018) Bitopic Inhibition of ATP and Substrate Binding in Ser/Thr Kinases through a Conserved Allosteric Mechanism. Biochemistry 57, 6387–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee S, Devamani T, Song HD, Sandhu M, Larsen A, Sommese R, Jain A, Vaidehi N & Sivaramakrishnan S (2017) Distinct structural mechanisms determine substrate affinity and kinase activity of protein kinase Cα. J Biol Chem 292, 16300–16309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tautermann CS, Binder F, Büttner FH, Eickmeier C, Fiegen D, Gross U, Grundl MA, Heilker R, Hobson S, Hoerer S, Luippold A, Mack V, Montel F, Peters S, Bhattacharya S, Vaidehi N, Schnapp G, Thamm S & Zeeb M (2019) Allosteric Activation of Striatal-Enriched Protein Tyrosine Phosphatase (STEP, PTPN5) by a Fragment-like Molecule. J Med Chem 62, 306–316. [DOI] [PubMed] [Google Scholar]
- 72.Bhargava R, Sandhu M, Muk S, Lee G, Vaidehi N & Stark JM (2018) C-NHEJ without indels is robust and requires synergistic function of distinct XLF domains. Nat Commun 9, 2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kapoor A, Martinez-Rosell G, Provasi D, de Fabritiis G & Filizola M (2017) Dynamic and Kinetic Elements of μ-Opioid Receptor Functional Selectivity. Sci Rep 7, 11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meral D, Provasi D & Filizola M (2018) An efficient strategy to estimate thermodynamics and kinetics of G protein-coupled receptor activation using metadynamics and maximum caliber. J Chem Phys 149, 224101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miao Y & McCammon JA (2018) Mechanism of the G-protein mimetic nanobody binding to a muscarinic G-protein-coupled receptor. Proc Natl Acad Sci 115, 3036–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miao Y & McCammon JA (2016) Graded activation and free energy landscapes of a muscarinic G-protein–coupled receptor. Proc Natl Acad Sci 113, 12162–12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Suomivuori C-M, Latorraca NR, Wingler LM, Eismann S, King MC, Kleinhenz ALW, Skiba MA, Staus DP, Kruse AC, Lefkowitz RJ & Dror RO (2020) Molecular mechanism of biased signaling in a prototypical G protein–coupled receptor. Science (80- ) 367, 881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shukla AK, Singh G & Ghosh E (2014) Emerging structural insights into biased GPCR signaling. Trends Biochem Sci 39, 594–602. [DOI] [PubMed] [Google Scholar]
- 79.Luttrell LM, Maudsley S & Bohn LM (2015) Fulfilling the Promise of “Biased” G Protein–Coupled Receptor Agonism. Mol Pharmacol 88, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Suno R, Lee S, Maeda S, Yasuda S, Yamashita K, Hirata K, Horita S, Tawaramoto MS, Tsujimoto H, Murata T, Kinoshita M, Yamamoto M, Kobilka BK, Vaidehi N, Iwata S & Kobayashi T (2018) Structural insights into the subtype-selective antagonist binding to the M2 muscarinic receptor. Nat Chem Biol 14, 1150–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Namkung Y, LeGouill C, Kumar S, Cao Y, Teixeira LB, Lukasheva V, Giubilaro J, Simões SC, Longpré J-M, Devost D, Hébert TE, Piñeyro G, Leduc R, Costa-Neto CM, Bouvier M & Laporte SA (2018) Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci Signal 11, eaat1631. [DOI] [PubMed] [Google Scholar]
- 82.Zimmerman B, Beautrait A, Aguila B, Charles R, Escher E, Claing A, Bouvier M & Laporte SA (2012) Differential -Arrestin-Dependent Conformational Signaling and Cellular Responses Revealed by Angiotensin Analogs. Sci Signal 5, ra33–ra33. [DOI] [PubMed] [Google Scholar]
- 83.Namkung Y, Radresa O, Armando S, Devost D, Beautrait A, Le Gouill C & Laporte SA (2016) Quantifying biased signaling in GPCRs using BRET-based biosensors. Methods 92, 5–10. [DOI] [PubMed] [Google Scholar]
- 84.Namkung Y, Le Gouill C, Lukashova V, Kobayashi H, Hogue M, Khoury E, Song M, Bouvier M & Laporte SA (2016) Monitoring G protein-coupled receptor and β-arrestin trafficking in live cells using enhanced bystander BRET. Nat Commun 7, 12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nivedha AK, Cao Y, Bhattacharya S, Lee S, Laporte SA & Vaidehi N (2021) Allosteric Communication by Biased Agonists Elicit Distinct Molecular Signatures in Angiotensin II Type 1 Receptor. Communicated. [Google Scholar]
- 86.Foster DJ & Conn PJ (2017) Allosteric Modulation of GPCRs: New Insights and Potential Utility for Treatment of Schizophrenia and Other CNS Disorders. Neuron 94, 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thal DM, Glukhova A, Sexton PM & Christopoulos A (2018) Structural insights into G-protein-coupled receptor allostery. Nature 559, 45–53. [DOI] [PubMed] [Google Scholar]
- 88.Slosky LM, Bai Y, Toth K, Ray C, Rochelle LK, Badea A, Chandrasekhar R, Pogorelov VM, Abraham DM, Atluri N, Peddibhotla S, Hedrick MP, Hershberger P, Maloney P, Yuan H, Li Z, Wetsel WC, Pinkerton AB, Barak LS & Caron MG (2020) β-Arrestin-Biased Allosteric Modulator of NTSR1 Selectively Attenuates Addictive Behaviors. Cell 181, 1364–1379.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Urban DJ & Roth BL (2015) DREADDs (Designer Receptors Exclusively Activated by Designer Drugs): Chemogenetic Tools with Therapeutic Utility. Annu Rev Pharmacol Toxicol 55, 399–417. [DOI] [PubMed] [Google Scholar]