

Abstract
Rare coding variation has historically provided the most direct connections between gene function and disease pathogenesis. By meta-analyzing the whole-exomes of 24,248 cases and 97,322 controls, we implicate ultra-rare coding variants (URVs) in ten genes as conferring substantial risk for schizophrenia (odds ratios 3 – 50, P < 2.14 × 10−6), and 32 genes at a FDR < 5%. These genes have the greatest expression in central nervous system neurons and have diverse molecular functions that include the formation, structure, and function of the synapse. The associations of NMDA receptor subunit GRIN2A and AMPA receptor subunit GRIA3 provide support for the dysfunction of the glutamatergic system as a mechanistic hypothesis in the pathogenesis of schizophrenia. We observe an overlap of rare variant risk between schizophrenia, autism spectrum disorders (ASD)1, epilepsy and severe neurodevelopmental disorders (DD/ID)2, though in some shared genes different mutation types are implicated. Most genes described here however are not implicated in neurodevelopment and we demonstrate that genes prioritized from common variant analyses of schizophrenia are enriched in rare variant risk3, suggesting that common and rare genetic risk factors at least partially converge on the same underlying pathogenic biological processes. Even after excluding significantly associated genes, schizophrenia cases still carry a substantial excess of URVs, implying that more risk genes await discovery using this approach.
Introduction
Schizophrenia is a severe psychiatric disorder with signs and symptoms that include hallucinations, delusions, disorganized speech and behavior, diminished emotional expression, social withdrawal, and cognitive impairment. The disorder has a lifetime risk of ~0.7%, is often disabling, and reduces life expectancy by nearly 15 years4,5. Existing therapies largely address primarily positive symptoms (e.g., hallucinations and delusions) and response to existing antipsychotic medications is highly variable with ~30% of patients classified as treatment resistant6. The lack of progress in therapeutic development is in part a consequence of our limited understanding of the molecular etiology of psychiatric disorders6,7.
It is well-established that schizophrenia has a substantial genetic component with contributions from across the allele frequency spectrum8–11. As initially theorized, the high heritability, consistency of prevalence across populations and increasing risk observed for individuals in more densely affected families suggested that polygenic predisposition should play a dominant role in defining schizophrenia risk in the population4,12. This has been borne out by genome-wide association studies (GWAS) which have now, in a companion paper, identified 270 common (minor allele frequency [MAF] > 1%) risk loci of individually small effect (median odds ratio [OR] < 1.05)13. As a class of variation, common variants explain ~24% of the variance in disease liability14. Several rare (MAF < 0.1%) recurrent copy number variants (CNVs) have also been robustly associated with schizophrenia, as exemplified by the dramatically higher rates of schizophrenia in 22q11.2 deletion carriers10,15. This suggests a role for rare gene-disrupting mutations with much larger effects on individual risk (OR 2 – 60). Although the variants we have been able to implicate have large effects on risk in the individual, because they are rare they make only a small contribution to overall heritability in the population. Despite these successes in locus discovery, it remains challenging to move from individual associations to specific genes and disease mechanisms. Because causal variants in schizophrenia GWAS are predominantly non-coding, challenges related to fine-mapping and interpretation of intergenic and intronic elements limit our ability to confidently identify underlying genes, infer the mechanism by which they influence disease risk, and determine the direction of effect. CNVs of large effect, on the other hand, often disrupt hundreds of kilobases of the genome and multiple genes simultaneously, limiting our ability to derive clear functional insights10.
Analyzing rare coding variants offers a powerful complementary approach to identify genes in complex traits. Theory predicts that the forces of natural selection will tend to keep large effect risk variants at much lower frequencies in the population, especially in disorders such as schizophrenia that are associated with reduced fecundity16. However, most rare variants will have little or no functional consequence or impact on risk, posing a significant challenge in identifying those that are truly causal and complicating required analyses in which rare variants are tested as a group rather than individually. The most natural grouping for rare variants is within a gene, based on predicted functional consequence or evidence for deleteriousness16,17. Protein-truncating variants (PTVs) are among the most interpretable associations as they suggest that the effect on disease most commonly tracks with decreasing expression of the gene18. Earlier schizophrenia sequencing studies have established that ultra-rare and de novo mutations contribute to risk as a category, and have prioritized disease-relevant tissues and processes, specifically observing an enrichment in neuronal genes and synaptic processes9,11,19–23. Furthermore, these risk alleles are concentrated in genes with a near-complete depletion of protein-truncating variants in population studies, a result shared with other neurodevelopmental disorders9,11 and suggesting strong direct selection against such mutations. However, the analysis of URVs has had limited success in delivering individual gene discovery in schizophrenia because of power limitations, with only a single gene, SETD1A, identified as robustly associated16,21.
The Schizophrenia Exome Sequencing Meta-Analysis (SCHEMA) Consortium was formed as a global collaborative effort to analyze sequence data from many studies to advance gene discovery. Here, we generated, aggregated, harmonized variant identification, and meta-analyzed the exome sequences of 24,248 individuals with schizophrenia and 97,322 controls from seven continental populations. This analysis is, to our knowledge, one of the largest sequencing studies of a complex trait to date. As predicted by apparent rare variant burden in schizophrenia, increasing the sample size has led to the identification of 10 genes with URVs that confer substantial risk at exome-wide significance. Combining these findings with other large-scale sequencing studies, we find shared and distinct genetic signals between schizophrenia and other neurodevelopmental disorders. In tandem with a companion paper from the Psychiatric Genomics Consortium13, we provide evidence that common and ultra-rare coding variants identify an overlapping set of genes. Finally, we demonstrate that increased scale following this approach will uncover additional risk genes and help complete the genetic architecture of schizophrenia.
Results
Data description and quality control
We aggregated exome sequence data consisting of 24,248 individuals diagnosed with schizophrenia and 50,437 individuals without a known psychiatric diagnosis, recruited in eleven global collections that had previously contributed to common variant association efforts (Supplementary Methods, Figure 1A, Table S1). The sequence data for 7,979 cases had been previously presented in earlier publications9,11,19–22, while the remaining 16,269 cases are presented here for the first time. To ensure calibrated analyses, these samples were included in joint re-processing and variant calling using a standardized BWA-Picard-GATK pipeline as part of the larger Genome Aggregation Database (gnomAD) effort (Supplementary Methods); consequently, SCHEMA case-control samples with appropriate permissions are also included in the gnomAD v2 release24. After extracting SCHEMA samples from this callset, we performed quality control steps to ensure high quality of sequence data, exclude contaminated samples, identify parent-proband trios and other related individuals, and infer global ancestries (Supplementary Methods, Figure 1B, Figures S1–7, Table S2). We subsequently applied site- and genotype-level filters to generate a robust set of coding SNPs and indels for a well-matched case-control analysis (Supplementary Methods). Previous studies have shown that PTVs are concentrated in 3,063 genes under strong constraint in schizophrenia cases compared to controls11,25, and we replicated this result with consistent signals across our major cohorts (Pmeta = 7.6 × 10−35; OR = 1.26, 95% CI = 1.22 – 1.31, Figure 1C, Extended Data Figure 1).
Figure 1. Study design and analytic approach.
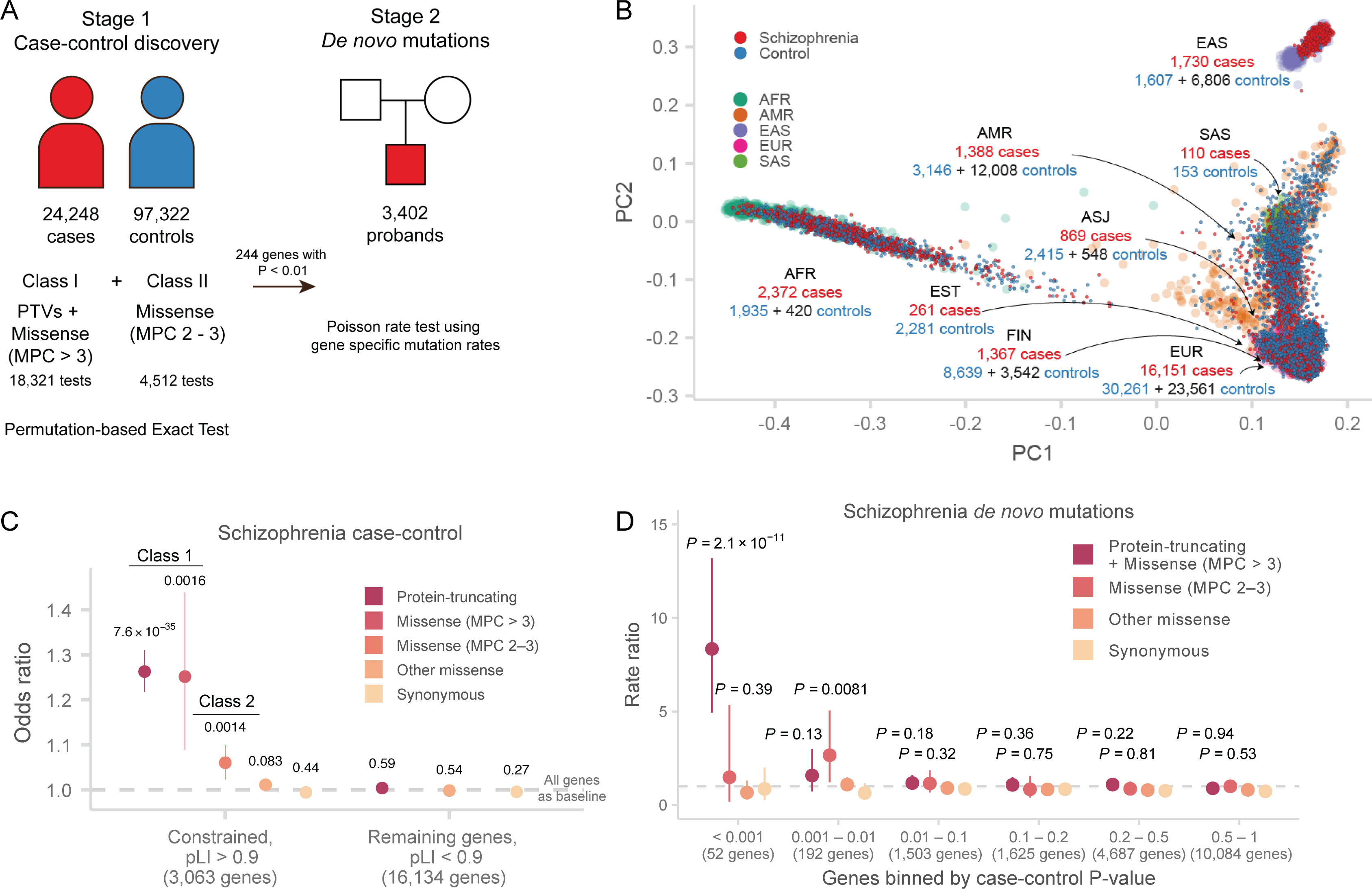
A: Study design. Case-control and parent-proband trio sample sizes, variant classes, and analytical methods are described. The case-control stage is shown on the left, and the de novo mutation stage is shown on the right.
B: Principal components analysis of SCHEMA samples. 1000 Genomes samples with reported ancestry are plotted in the background, and SCHEMA samples are displayed in the foreground. For each global ancestry group, we report the number of cases and controls in the discovery data set in red and blue respectively, and the number of external controls in black. AFR: African, ASJ: Ashkenazi Jewish, AMR: Latin American, EAS: East Asian, EST: Estonian, FIN: Finnish, EUR: non-Finnish European, SAS: South Asian.
C: Case-control enrichment of ultra-rare protein-coding variants in genes intolerant of protein-truncating variants (n = 22,444 cases and n = 39,837 controls). Two-sided P values from logistic regression displayed are from comparing the burden of variants of the labeled consequence in cases compared to controls. By definition, MPC enrichment is only shown for pLI > 0.9 genes. The dot represents the odds ratio, and the bars represent the 95% CIs of the point estimates. pLI: probability of loss-of-function intolerant in the gnomAD database.
D: Enrichment of schizophrenia de novo mutations in P value bins derived from the Stage 1 (case-control) gene burden analysis (n = 3,402 schizophrenia trios). The one-sided enrichment P values displayed are calculated as a Poisson probability having equal or greater than the observed number of mutations given the baseline mutation rate. The relative rate is given by the ratio of observed to expected rate of de novo mutations. The dot represents the relative rate, and the bars represent the 95% CIs of the point estimates.
Analysis approach
To increase power for gene discovery, we incorporated variant counts from additional samples from non-psychiatric and non-neurological collections that were aggregated as part of the gnomAD consortium effort (Supplementary Methods)24. We attempted to control for technical and methodological batch effects that may arise from this approach in both variant calling and additionally via permutation testing described below. All samples in gnomAD and SCHEMA consortia were re-processed and joint called using the same pipeline, and the same variant filters were applied to arrive high-quality calls. Importantly, we restricted our analysis to coding exons with high-quality data across all major exome capture technologies, reducing any artifacts that may arise from coverage differences(Supplementary Methods, Figures S1–2). After incorporating variant counts from additional 46,885 gnomAD controls, our combined discovery data set is composed of 24,248 cases and 97,322 population controls (Figure 1A, 1B, Table S3).
Because only summary-level variant counts were available for the 46,885 external controls, we tested for an excess of disruptive variants per gene using a Fisher’s exact test in which statistical significance was determined by case-control permutations within each strata (Supplementary Methods, Table S3). As in other sequencing studies, we enriched for pathogenic variants by restricting our analysis to ultra-rare variants (defined as minor allele count [MAC] ≤ 5) that are also either PTVs (defined as stop-gained, frameshift, and essential splice donor or acceptor variants) or damaging missense variants as defined by the MPC pathogenicity score1,26(Supplementary Methods). We found that missense variants with MPC > 3 have a global signal on par with PTVs in schizophrenia, autism spectrum disorders, and severe neurodevelopmental disorders, while variants with MPC 2 – 3 has a significant but weaker signal than PTVs and were therefore analyzed separately (Figure 1C, Extended Data Figure 2, Extended Data Figure 3, Figures S8, Table S4, Supplementary Methods). Motivated by these observations, we performed a burden test of PTVs and MPC > 3 variants (Class I) to generate a P value for 18,321 protein-coding genes (Supplementary Methods). In the 4,512 genes with MPC 2 – 3 (Class II) variants, we perform an additional test aggregating these variants, and meta-analyze these gene statistics with Class I P values using a weighted Z-score method (Supplementary Methods). To ensure the robustness of the results generated by this approach, we observed the expected null distribution of P values in gene-based tests of synonymous variants in each strata and in the meta-analysis (Figure S9, S10). Additionally, we observed no inflation of synonymous P values using the Mantel-Haenszel test even after limiting our analysis to genes with larger total numbers of alleles (gene-wide MAC > 10, 50, or 100), where we had greater power to detect potential artifacts (Figure S11, S12).
Previous studies had integrated case-control and trio-based de novo mutations for gene discovery1,21, and to this end, we aggregated and re-annotated de novo mutations from 3,402 published parent-proband trios (Supplementary Methods). Despite the sizable number of trios, there were few de novo mutations for analysis with only 325 genes with one or more de novo PTV and only 449 with at least one Class I or Class II mutations. Using Poisson rate tests based on expected mutation rate27, we found these de novo mutations are enriched for the 244 genes with P < 0.01 in our case-control analysis (Figure S13, Table S5), with limited or no signal in the remaining genes in the genome (Figure 1D). The most striking enrichment was observed for the 52 genes with case-control P < 0.001 (Class I mutations: P = 2.1 × 10−11; Rate ratio = 8.3, 95% CI = 4.9 – 13), which provides additional reassurance of the robustness of our case-control gene results. Motivated by these observations, we calculated de novo Class I and II P values in the 244 genes with Pcase-control < 0.01 using the Poisson rate test and meta-analyzed them with our case-control test statistic using a weighted Z-score method to increase power (Supplementary Methods, Figure S13–S15).
Individual genes implicated by URVs
Combined, our meta-analysis of 24,248 cases, 97,322 controls, and de novo mutations from 3,402 trios implicates 10 genes in which ultra-rare coding variants are significantly associated with schizophrenia (P < 2.14 × 10−6 corresponding to 0.05/23,321 tests; Figure 2A, 2B). These top associations as a group are supported by complementary types of variation that include case-control PTVs, damaging missense variants, and de novo mutations (Table 1, Table S5). Although confidence intervals are wide, URVs in these genes appear to confer substantial risk, with odds ratio of PTVs and Class I variants ranging from 3 to 50. As expected, all ten genes are among the most constrained genes in the genome, with a substantial depletion of PTVs compared to chance expectation24. The annotated functions of these genes are diverse and include ion transport (CACNA1G, GRIN2A, and GRIA3), neuronal migration and growth (TRIO), transcriptional regulation (SP4, RB1CC1, and SETD1A), nuclear transport (XPO7), and ubiquitin ligation (CUL1, HERC1). We include a brief discussion of the known biological functions of these genes in the Supplementary Note. Beyond these ten genes, we identify 22 additional genes at a False Discovery Rate (FDR) < 5% (Figure 2A, Table S5). We observe notable deviation at the tail of the distribution beyond the associated genes, suggesting that more genes remain to be discovered (Figure 2B). We report all high-quality variants, relevant annotations, and gene-level results on a public browser at https://schema.broadinstitute.org.
Figure 2. Results from the meta-analysis of ultra-rare coding variants in 3,402 trios, 24,248 cases, and 97,322 controls.
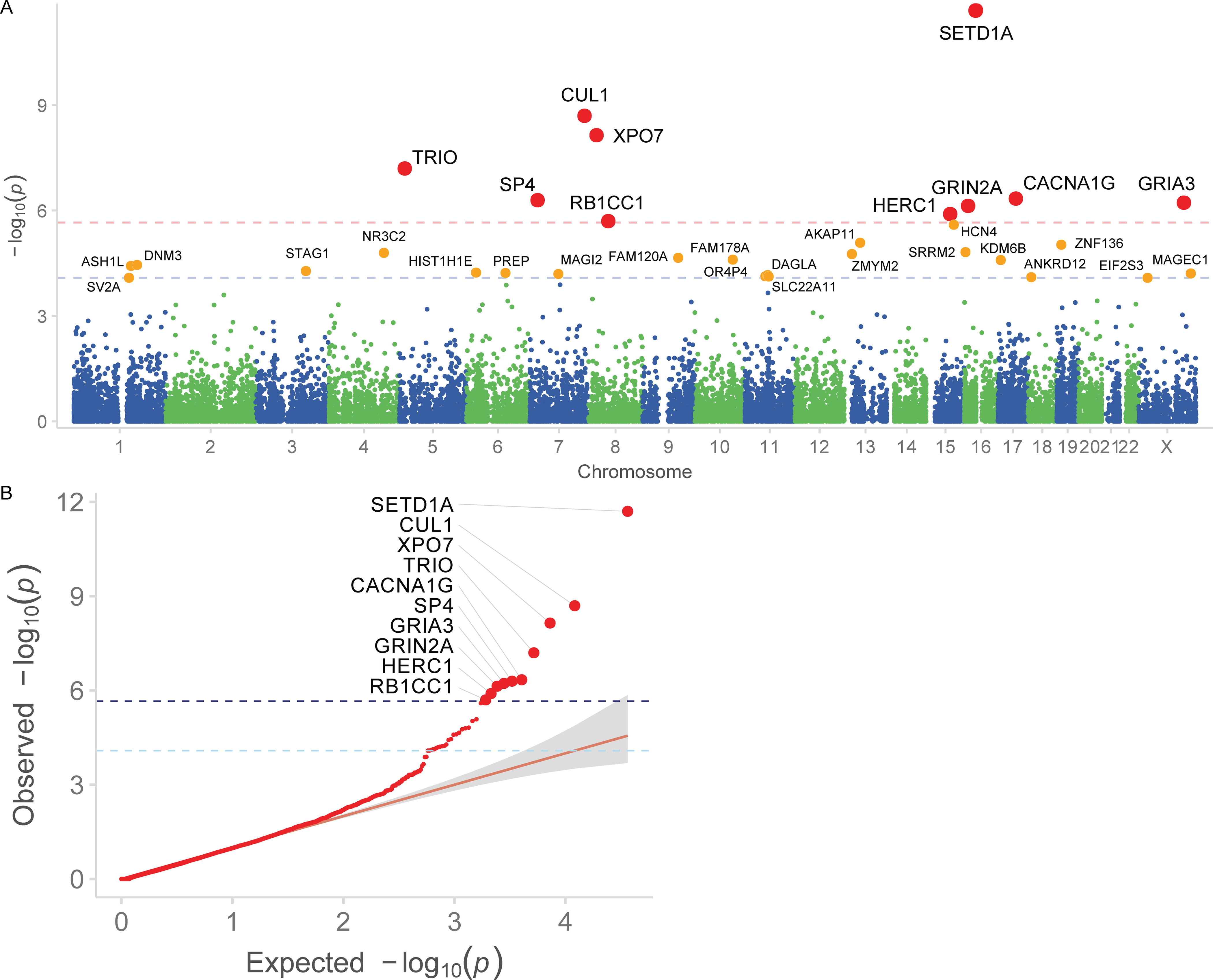
A: Manhattan plot. −log10 P values are plotted against the chromosomal location of each gene. The per-gene P values are calculated by meta-analyzing two-sided burden test P values from rare coding variants in 24,248 cases and 97,322 controls, and one-sided Poisson rate test P values from de novo mutations in 3,402 trios (see text and Supplementary Methods for more information). Genes reaching exome-wide significance (P < 2.14 × 10−6 corresponding to 0.05/23,321 tests) are in red, and genes significant at FDR < 5% are in orange. Red dashed line: P = 2.14 × 10−6; Blue dashed line: FDR < 5%, or P = 8.23 × 10-5.
B: Q-Q plot. Observed −log10 P values are plotted against expectation given a uniform distribution. The per-gene P values are calculated by meta-analyzing two-sided burden test P values from rare coding variants in 24,248 cases and 97,322 controls, and one-sided Poisson rate test P values from de novo mutations in 3,402 trios (see text and Supplementary Methods for more information). Genes reaching exome-wide significance are plotted with a larger size. The direction of effect is indicated by the color of each point. The gray shaded area indicates the 95% CI under the null. Dark blue dashed line: P = 2.14 × 10−6; Light blue dashed line: FDR < 5%.
The identification of individual genes provides support for more specific mechanistic hypotheses underlying schizophrenia pathogenesis. Developed from neuropharmacological and neuropathological observations, the glutamatergic hypothesis postulates that the hypofunction of glutamatergic signaling through NMDA receptors is a possible mechanism of disease28 (Supplementary Note). Here, we find that PTV and damaging missense variants in NMDA receptor subunit GRIN2A confer substantial risk for schizophrenia (P = 7.37 × 10−7; Class I [PTV and MPC > 3] OR 24.1, 95% CI 5.36 – 221; Class II [MPC > 2] OR 2.37, 95% CI 1.1 – 4.92). Schizophrenia GWAS also identified a common variant at GRIN2A (OR = 1.057, P = 1.57 × 10−10), providing an allelic series in which different perturbation of gene function results in severity of disease risk (Figure 3A)8.The NMDA receptor changes in composition during prenatal to postnatal neurodevelopment with GRIN2A predominantly expressed during late childhood and adolescence, recapitulating expected epidemiological observations on schizophrenia age-of-onset (Supplementary Methods, Figure 3B)29. We additionally find that risk URVs in AMPA receptor subunit GRIA3 confer substantial risk (P = 5.98 × 10−7; Class I [PTV and MPC > 3] OR 20.1 95% CI 4.28 – 188; Table 1). Combined, our results from exome sequencing support the dysregulation of the glutamatergic system as a mechanistic hypothesis for the development of schizophrenia, and that the specific identification of genes by coding variation may provide new avenues of understanding disease pathogenesis.
Figure 3. Biological insights from exome sequence data.
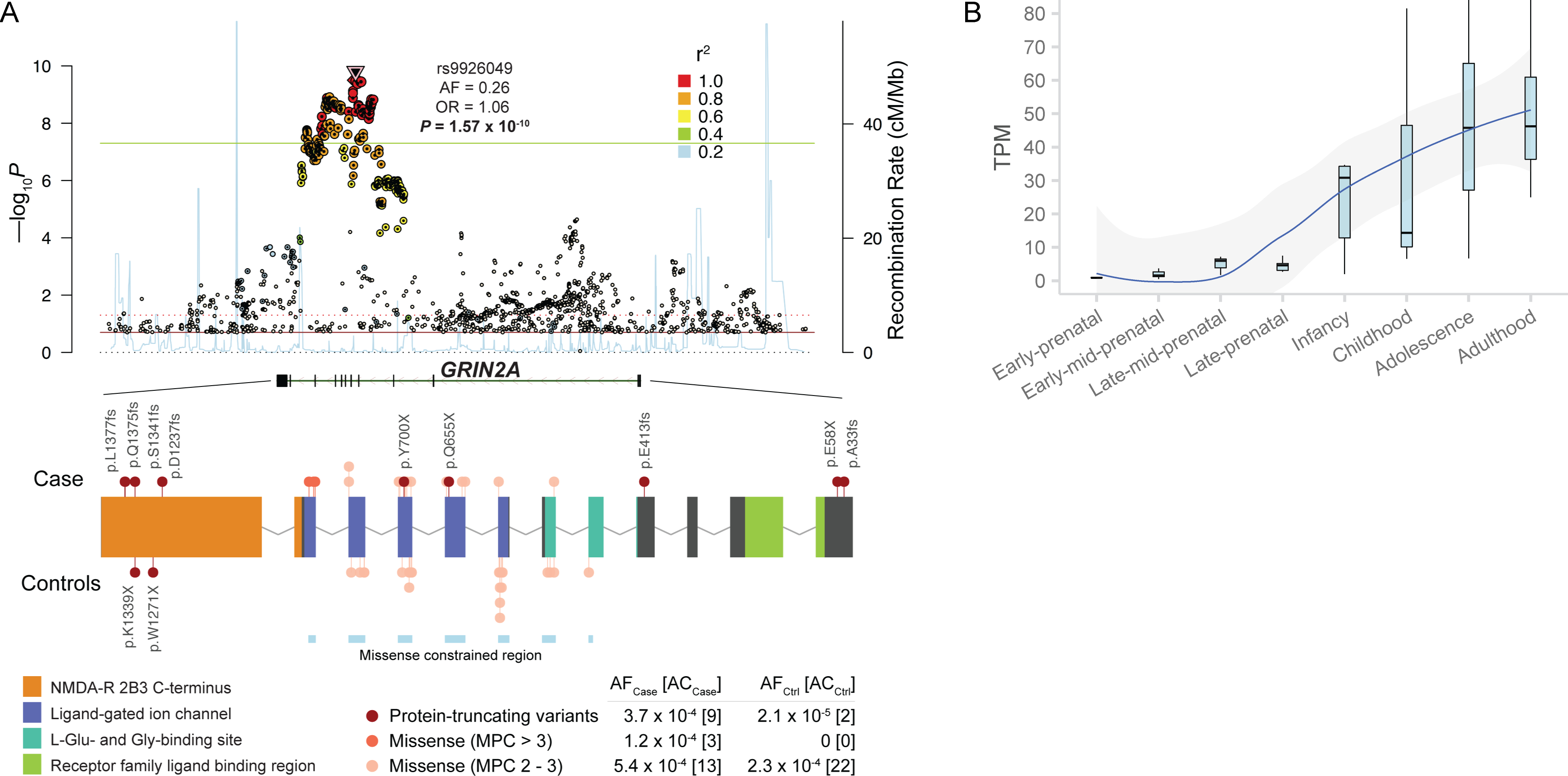
A: Common and rare allelic series at NMDA receptor subunit GRIN2A. The Locus Zoom plot (top) displays the common variant (GWAS) association of the gene3. The two-sided P values of each SNP from the GWAS meta-analysis are shown along the y-axis. The color of each dot corresponds to the LD with the index SNP, and the properties of the index SNP are displayed. The gene plot (bottom) displays the protein-coding variants that contribute to the exome signal in GRIN2A. Variants discovered in cases are plotted above the gene, and those from control are plotted below. Each variant is colored based on inferred consequence, and the protein domains and missense constrained regions of the gene are also labelled26,43. The frequencies and counts in cases and controls are displayed for each variant class. AF: allele frequency, AC: allele count.
B: Temporal expression of GRIN2A in the human brain (n = 42 samples). We show GRIN2A expression in four prenatal and four postnatal periods derived from whole-brain tissue in BrainSpan29. The expression values plotted are in transcript-per-million (TPM). In the box plot, the lower hinge is the 25% quantile, the middle line is the median, the upper hinge is the 75% quantile, the lower whisker extends to the smallest observation greater than or equal to the lower hinge − 1.5 * IQR, and the upper whisker extends to the largest observation less than or equal to the upper hinge + 1.5 * IQR.
Shared genes with GWAS loci
Pathway analyses of common variants have prioritized disease-relevant tissues and cell types, and in some cases, independently recapitulating known biology8,30,31. To derive insights from global patterns of rare coding variants, we tested for an excess burden of URVs in schizophrenia cases compared to controls in 1,732 broadly-defined gene sets from databases of biological pathways (e.g. Gene Ontology, REACTOME, KEGG) and experimental data (Supplementary Methods)11. We observed significant enrichment of URVs in 33 gene sets (P < 2.9 × 10−5) that recapitulated consistent and overlapping cellular compartments and biological processes, including definitions of the postsynaptic density (human cortex biopsy post-synaptic density; P = 1.2 × 10−12), chromatin modification (GO:0016568; P = 1.8 × 10−12), regulation of ion transmembrane transport (GO:0034765; P = 6.7× 10−7), axon guidance (P = 5.4 × 10−6), voltage-gated cation channel activity (GO:0022843; P = 8.1 × 10−6), and synaptic transmission (GO:0007268; P = 1.79 × 10−5) (Table S6, Figure S16). Because of the clear synaptic signal, we investigated in the refined synaptic ontology defined by the SynGO consortium32, and found consistent enrichment for postsynaptic components and processes (GO:0098794; P = 3.9 × 10−6; Table S7). These global observations are consistent with the known functions of the individual risk genes now implicated by rare variation (Supplementary Note). Following earlier reports studying heritability enrichment in GTEx tissues8,31, we found that genes with the highest specific expression in brain regions showed the strongest enrichment of risk URVs, most significantly in the human frontal cortex (P = 1.63 × 10−8) and with limited signal in the other tissue types (Extended Data Figure 4, Table S8, S9). To further deconvolute this signal, we investigated which single cell types in the mouse nervous system show the highest specific expression for the 32 (FDR < 5%) schizophrenia risk genes (Supplementary Methods)33,34. Here, we found widespread enrichments across central nervous system neurons with limited to no signal in glial cells and peripheral nervous system neurons (Table S10, Figure S17). Thus, at a high level, global analysis of ultra-rare protein-coding variation independently recapitulated known biology related to schizophrenia pathogenesis, including processes, cellular components, and tissues previously implicated by common variant analyses.
To evaluate the overlap of schizophrenia associations from common variants and ultra-rare coding variant analyses, we jointly analyzed our results with the largest GWAS of schizophrenia to date, which identified common variant associations at 270 distinct loci from the analysis of 69,369 cases and 236,642 controls13. Statistical fine-mapping prioritized the likely underlying protein-coding gene at 64 of these associations (Table S11, Figure S18), and we found a case-control enrichment of URVs in these genes (Pmeta = 3.9 × 10−4; ORClass I = 1.46, 1.2 – 1.77 95% CI; Figure 4A, Table S12). Beyond the statistical enrichment, GRIN2A and SP4, two of the ten significant rare variant genes, had clear associations in schizophrenia GWAS (Figure 3A, Figure 4B). Furthermore, FAM120A and STAG1 resided in more complex GWAS-associated regions containing multiple genes but were prioritized among their neighbors as FDR < 5% in our sequencing study (Figure 4C, 4D). Combined, these results suggest there is at least partial convergence in the genes and biological processes implicated by common and ultra-rare genetic variation, and that ultra-rare coding variants can be leveraged to prioritize genes within GWAS loci.
Figure 4. Shared genetic signal with schizophrenia GWAS.
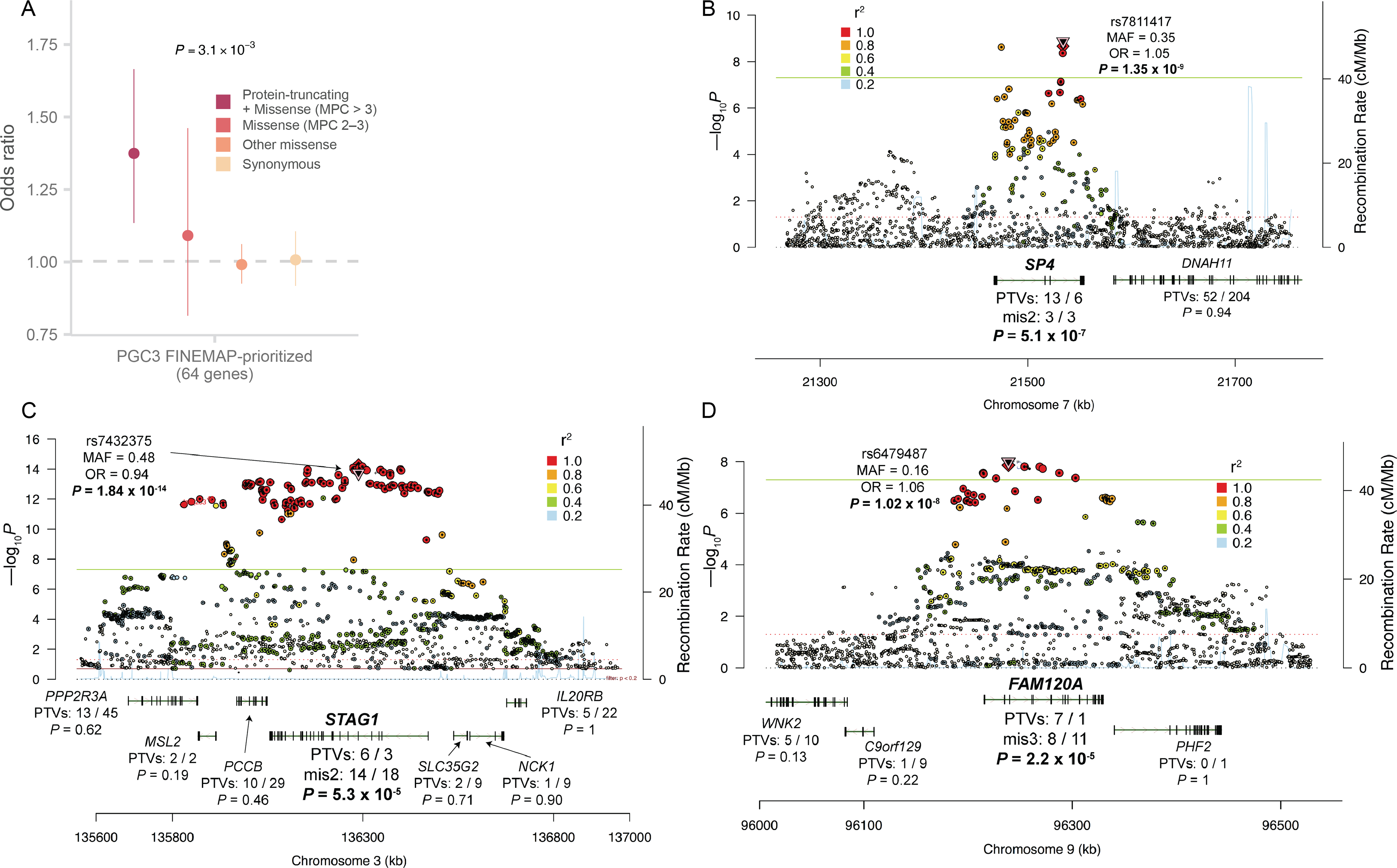
A: Case-control enrichment of ultra-rare protein-coding variants in genes prioritized from fine-mapping of the PGC schizophrenia GWAS (n = 22,444 cases and n = 39,837 controls)13. The reported P value is from applying the Fisher’s combined probability method on the two-sided P values of Class I and Class II variants. The dot represents the odds ratio, and the bars represent the 95% CIs of the point estimates.
B, C, D: Prioritization of GWAS loci using exome data. The Locus Zoom plot of three GWAS loci is displayed. The two-sided P values of each SNP from the GWAS meta-analysis are shown along the y-axis. Below, for each gene in or adjacent to the region, we show the case-control counts of PTVs in the exome data, along with the two-sided burden test meta-analysis P values. SP4, STAG1 and FAM120A are highlighted as the only genes with notable signals in the exome data within each locus.
Shared and distinct genes with DD/IDs
Exome sequencing studies of autism spectrum disorders (ASD) and severe neurodevelopmental disorders (DD/ID) have leveraged ultra-rare coding variants to identify risk genes. These studies have established that the genetic signals were concentrated in constrained genes and shared between the two disorders35,36. Most recently, the analysis of de novo mutations from 31,058 DD/ID trios implicated 299 genes, while the analysis of 11,986 ASD cases identified 102 genes at FDR < 10% (Table S11)1,37. We found a significant excess of URVs in schizophrenia cases compared to controls in the 299 DD/ID-associated genes (Pmeta = 1.5 × 10−14; ORClass I = 1.44, 1.3 – 1.6 95% CI), and in the 102 ASD-associated genes (Pmeta = 3.7 × 10−7; ORClass I = 1.45, 1.23 – 1.72 95% CI; Figure 5A; Table S12). Thus, some schizophrenia rare variant risk appears to be shared with other neurodevelopmental disorders.
Figure 5. Shared genetic signal between schizophrenia and other neurodevelopmental disorders.
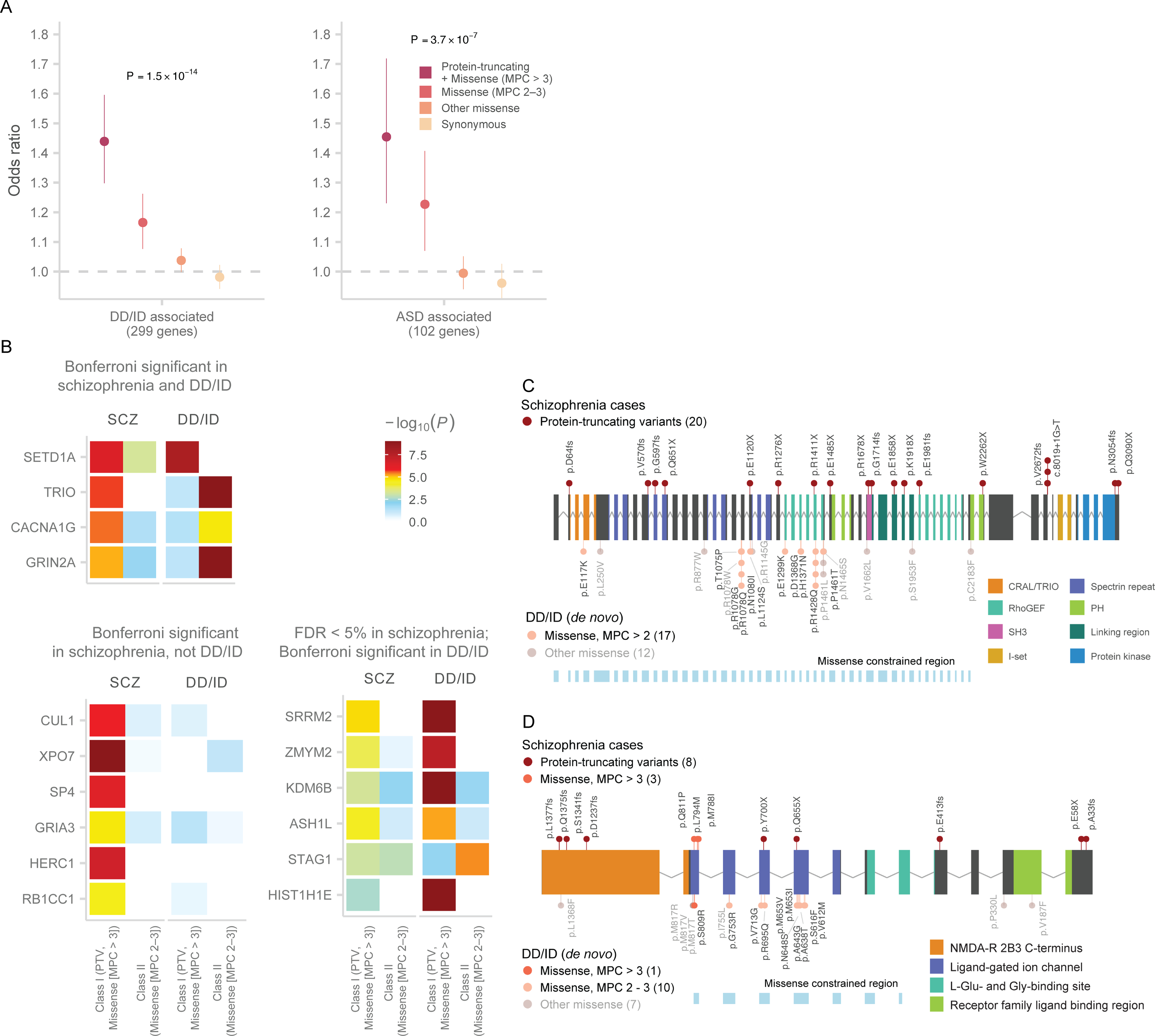
A: Case-control enrichment of ultra-rare protein-coding variants in DD/ID and ASD-associated genes (n = 22,444 cases and n = 39,837 controls). We test for the burden of schizophrenia URVs in genes identified in the most recent exome sequencing studies of ASD and DD/ID1,37. The reported P value is from applying the Fisher’s combined probability method on the two-sided P values of Class I and Class II variants. The dot represents the odds ratio, and the bars represent the 95% CIs of the point estimates.
B: Heatmap displaying the strength of association for schizophrenia-associated genes in our discovery data set and in genes implicated by de novo mutations in trios diagnosed with DD/ID. We display three groups of genes: Bonferroni significant in schizophrenia and DD/ID, Bonferroni significant only in schizophrenia, and FDR < 5% in schizophrenia and Bonferroni significant in DD/ID. The degree of association from each sequencing study is displayed as the color corresponding to −log10 P values in that study. The two-sided case-control burden test P value is reported for schizophrenia, while one-sided P value from the de novo enrichment using the Poisson rate test is reported for DD/ID. Results are further stratified to tests of Class I (PTV and MPC > 3) and Class II (missense [MPC 2 – 3]) variants.
C: Allelic series in TRIO between schizophrenia and DD/ID risk variants. The gene plot displays the protein-coding variants that contribute to the exome signal in TRIO. Variants discovered in schizophrenia cases are plotted above the gene, and missense de novo mutations from DD/ID probands are plotted below. Each variant is colored based on inferred consequence, and the protein domains of the gene are also labelled. The variant counts are displayed for each variant class.
D: Allelic series in GRIN2A. See D for description.
With 31,058 trios, the scale of gene discovery in severe DD/ID provided sufficient power to evaluate the individual schizophrenia risk genes associated in our study for a role in broader neurodevelopmental disorders. Nine of the ten schizophrenia genes showed limited de novo PTV signal in DD/ID, with a combined 8 de novo PTVs observed in these genes (Xexp = 4.98; PPois = 0.13; Figure 5B; Table S13). SETD1A had a significant de novo PTV signal in DD/ID (Xobs = 8, Xexp = 0.41; P = 1.3 × 10−8), supporting an earlier report that described SETD1A as a gene associated with both schizophrenia and broader neurodevelopmental disorders21. We also observed a missense signal in SETD1A in our study (Table 1; Figure S19). Extending this analysis to the additional 22 FDR < 5% genes, we found that six genes (STAG1, ASH1L, ZMYM2, KDM6B, SRRM2, and HIST1H1E) were significantly associated with DD/ID in addition to schizophrenia (Figure 5B; Table S13). Among these FDR < 5% genes, ASH1L, KDM6B and NR3C2 were associated with ASD 1 (Table S13). Broadly speaking, while PTV mutations in certain genes are joint risk factors for schizophrenia and DD/ID, the majority of schizophrenia associations reported here appear to have little or no role in DD/ID despite the enormous power of published DD/ID studies to date.
Notably, three of the ten risk genes for schizophrenia (TRIO, GRIN2A, and CACNA1G) were associated with risk of severe DD/IDs exclusively through de novo missense mutations that cluster within each gene (Figure 5B; Table S13), while the schizophrenia signal was largely driven by PTVs. De novo missense mutations in TRIO significantly disrupted the exons preceding or containing the RhoGEF domain (Figure 5C)37,38, and de novo missense mutations in GRIN2A cluster at the base of the ion channel with the most mutations in the exon encoding for the pore of the complex (Figure 5D). STAG1, which had a common and rare variant signal in schizophrenia (Figure 4D), was associated with DD/ID primarily through de novo missense mutations (Figure 5B; Table S13). These observations suggest schizophrenia and childhood onset neurodevelopmental disorders share some genes and biological processes, but that at least in some cases, the severity or the nature of the functional impairment differs between disorders.
We explored what properties may differ between schizophrenia- and DD/ID-associated risk genes, and hypothesized that DD/ID genes were under stronger evolutionary constraint with a bias towards prenatal expression when compared to schizophrenia genes. While schizophrenia genes (FDR < 5%) were under substantial genic constraint compared to expectation (Figure S20; M-W U test; P = 2.9 × 10−7; Supplementary Methods), they are significantly less constrained than DD/ID-associated genes (M-W U test; P = 3.5 × 10−5). Furthermore, schizophrenia genes as a group did not show pre- or postnatal bias in brain expression (P = 0.21; Figure S21), while DD/ID-associated genes were overwhelmingly prenatal in expression (P = 7.5 × 10−20). Indeed, individual genes like SETD1A, TRIO, and SP4 exhibited prenatal expression while GRIN2A and GRIA3 showed postnatal expression (Figure S22). These observations offer the possibility that certain properties may differentiate genes for adult psychiatric disorders and more severe DD/IDs.
Contribution of ultra-rare PTVs to risk
Efforts in the past decade are beginning to generate a more comprehensive view of the genetic architecture for schizophrenia, composed of common variants of small effects, large CNVs with elevated frequencies driven by genomic instability, and now, URVs of large effect implicating individual genes (Figure 6A)8,10. Because schizophrenia as a trait is under strong selection39–41, we expect that URVs of large effect to be frequently de novo or of very recent origin and contribute to risk in only a fraction of diagnosed patients. We quantified the contribution of PTVs to risk first in our full schizophrenia data set, and then partitioned the de novo and inherited contributions in 2,304 parent-proband trios. We restrict these analyses to the 3,063 PTV-intolerant (pLI > 0.9) genes in which schizophrenia risk URVs are concentrated. We observed 0.057 (0.049 – 0.065 95% CI) extra singleton PTV variants per individual in cases compared to controls, suggesting ~5.7% of cases carried a PTV relevant to disease risk. In the 2,304 trios, 0.0394 (0.014 – 0.065 95% CI; 74%) extra singleton PTV variants were inherited per proband, and 0.0121 (0.0022 – 0.02 95% CI; or 26%) extra de novo PTV mutations in constrained genes were identified in cases compared to controls. In contrast, DD/ID probands have 0.111 (0.103 – 0.119 95% CI) extra de novo PTV mutations in constrained genes, while ASD individuals have 0.0478 (0.0387 – 0.0568 95% CI) extra de novo PTV mutations (Figure S23; Supplementary Methods). In the ten schizophrenia-associated genes, 7 de novo mutations and 13 transmitted variants are observed in 2,304 trios, suggesting that 0.86% of patients are carriers and ~35% of variants are de novo. Finally, the genome-wide signal in constrained genes (pLI > 0.9: OR = 1.26, P = 7.6 × 10−35) remains significant even after excluding the 32 FDR < 5% genes (OR = 1.23, P = 4.3 × 10−27; Figure 6B, Table S4), reaffirming the genetic heterogeneity underlying schizophrenia risk and suggesting that the majority of schizophrenia risk genes in which rare variants confer risk remain to be discovered.
Figure 6. The contributions of ultra-rare PTVs to schizophrenia risk.
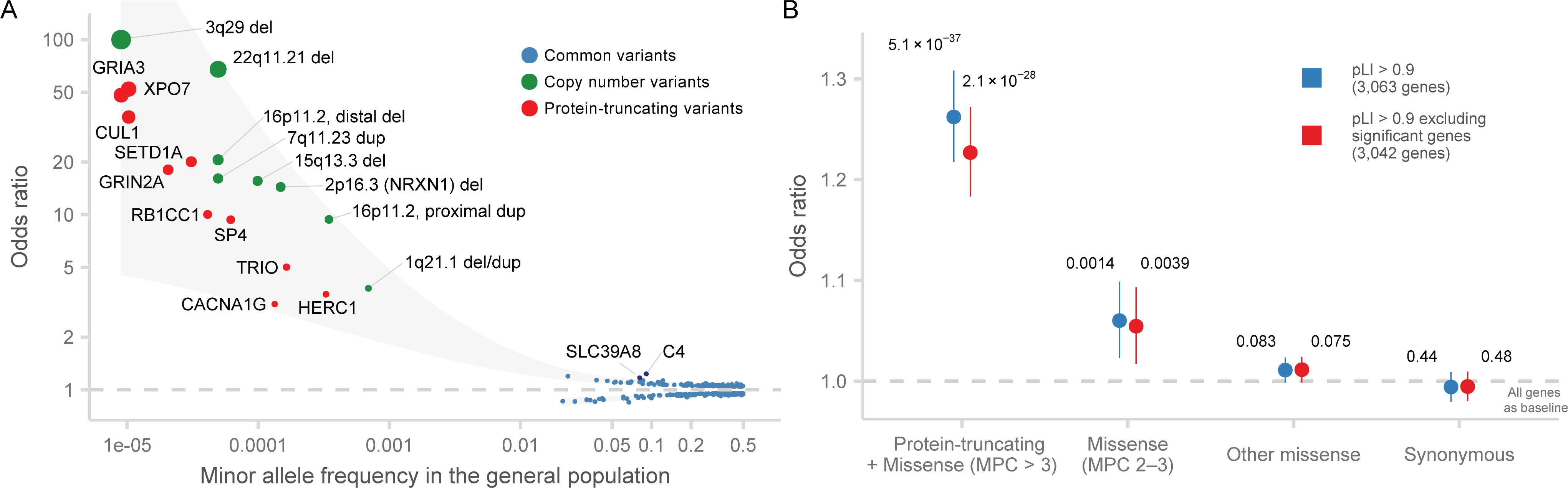
A: Genetic architecture of schizophrenia. Significant genetic associations for schizophrenia from the most recent GWAS, CNV, and sequencing studies are displayed. The in-sample odds ratio is plotted against the minor allele frequency in the general population. The color of each dot corresponds to the source of the association, and the size of the dot to the odds ratio. The shaded area represented the loess-smoothed lines of the upper and lower bounds of the point estimates.
B: Case-control enrichment of ultra-rare protein-coding variants in genes intolerant of protein-truncating variants after excluding schizophrenia-associate genes (n = 22,444 cases and n = 39,837 controls). We perform the test with all constrained genes (pLI > 0.9) and after excluding all schizophrenia-associated genes with FDR < 5%. Two-sided P values from logistic regression displayed are from comparing the burden of variants of the labeled consequence in cases compared to controls. The dot represents the odds ratio, and the bars represent the 95% CIs of the point estimates.
Discussion
In one of the largest exome sequencing studies to date, we identify genes in which disruptive coding variants confer substantial risk for schizophrenia at exome--wide significance. This effort required re-processing a decade of sequence data, harmonization of variant calling and quality control, inclusion of external controls, and integration of PTV, damaging missense, and de novo variants. Global, collaborative efforts such as this provide a template for tackling the genetic contributions in other complex diseases.
Genome-wide analyses recapitulated known biological processes and reaffirm that schizophrenia risk genes are involved in the postsynaptic density and broader synaptic function, and enriched in expression in neuronal tissues. Furthermore, the identification of specific genes supports more specific mechanistic hypotheses. The association of PTVs in the NMDA receptor subunit GRIN2A to schizophrenia risk provides genetic support for the dysregulation of glutamatergic signaling as a possible mechanism of disease. A natural dose-response curve occurs at this gene in which common regulatory variants modestly influence disease risk and PTV and predicted damaging missense variants increase risk more substantially. Interestingly, the NMDA receptor is composed of two GRIN2 units (GRIN2A and/or GRIN2B) along with two constitutive GRIN1 units, and GRIN2A increases dramatically in expression later in childhood and adolescence, mimicking the age of onset of disease for schizophrenia. De novo mutations in GRIN2B conversely are associated with more severe disorders of neurodevelopment that manifest in childhood, including intellectual disability and autism42. Such findings provide a unique opportunity to identify experiments of nature which help to build and support mechanistic hypotheses that may lead to a better understanding of disease biology.
Joint analysis with genetic data from DD/ID and ASD consortia have provided evidence for shared genes between neuropsychiatric and broader neurodevelopmental disorders. Indeed, seven of the 32 FDR < 5% genes are also associated with DD/ID, providing additional confidence in those associations. The shared genes suggest that there is at least some contribution from early brain developmental processes that predisposes to schizophrenia. Despite this sharing, PTVs in 9 of the 10 most confidently associated genes are associated with schizophrenia and not for DD/ID, which may provide avenues for identifying disease-specific processes. Of further interest, we observe allelic series in GRIN2A, TRIO, and CACNA1G in which PTVs increase schizophrenia risk and de novo missense mutations confer strong DD/ID risk. De novo missense mutations in these genes clustered in specific domains and are associated with more severe neurodevelopmental, syndromic disorders with cognitive impairment, suggesting an alternate or gain-of-function effect. Analyses estimating relative penetrance for different phenotypes will increase in power as consortium efforts studying specific diseases and biobank efforts continue to grow, all of which would be fruitful in informing what is shared and distinct across disorders.
We show for the first time that common regulatory variants from GWAS and ultra-rare coding variants disrupt an overlapping set of genes, including an allelic series in four genes in which common variants and rare coding variants increase risk to varying degrees. Combined, these results suggest that exome sequencing identifies some common, shared underlying biology that is dysregulated across the allele frequency spectrum, rather than syndromic forms of disease with unrelated biology regulated by common variation. Furthermore, because of this sharing, coding variants can help refine and fine-map common variant associations like at the STAG1 and FAM120A loci. As common and rare variant association studies continue to grow, we can better determine the actual degree of overlap of genes that are regulated by both types of variation. Ultimately, the emerging evidence of an overlap between common and ultra-rare variation gives confidence that the integration of results from sequencing consortia with the GWAS efforts will have significant value for identifying specific genes beyond what any single strategy can achieve on its own.
A decade of genotyping and sequencing studies now establish specific genetic contributions from common variants, copy number variants, and ultra-rare coding variants as conferring risk for schizophrenia. Despite this progress, it is clear that we are still in the early stages of gene discovery13. The vast majority of risk alleles, their direction and magnitude of effect, mode of action, and responsible genes are yet to be discovered. These emerging genetic findings will serve in part to direct and motivate mechanistic studies that begin to unravel disease biology. The success of common variant association studies, and now exome sequencing, suggest concrete progress towards understanding the causes of human complex traits and diseases, and provide a clear roadmap towards understanding the genetic architecture of schizophrenia.
Extended Data
Extended Data Figure 1. Schizophrenia case-control enrichment in constrained genes (pLI > 0.9) in different SCHEMA cohorts (n = 22,444 cases and n = 39,837 controls).
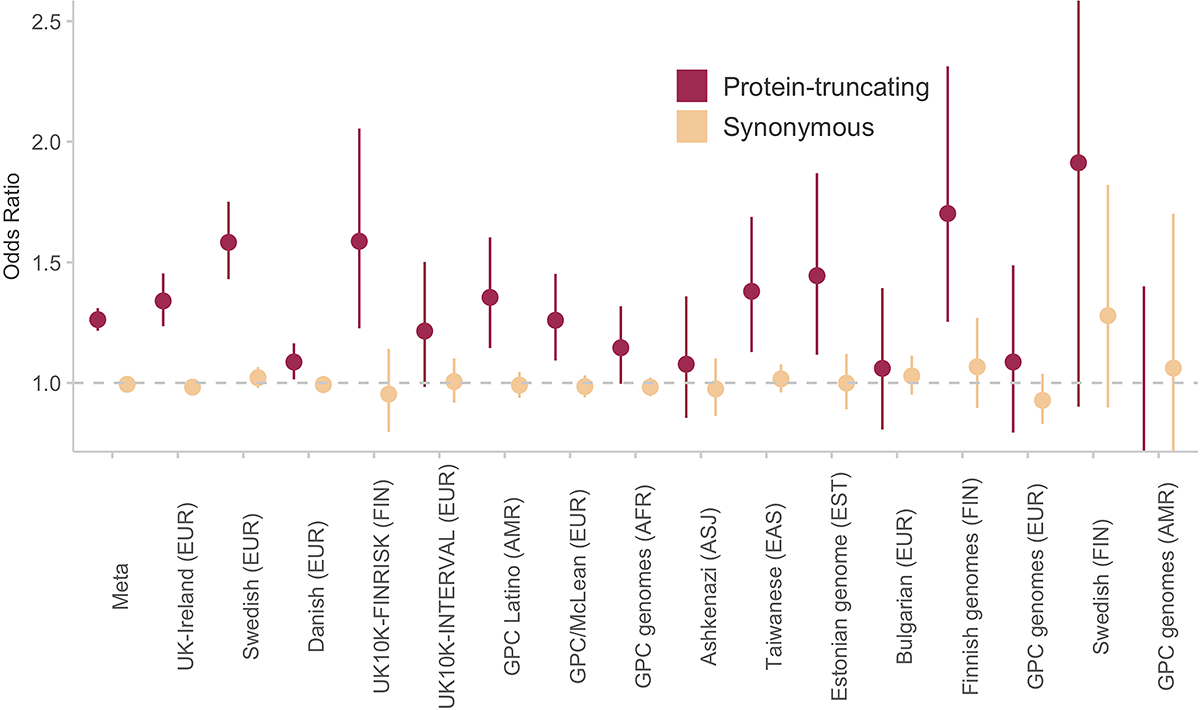
The odds ratio and standard error of PTVs and Synonymous variants are provided for each cohort. The meta-analyzed odds ratio and standard error is calculated using inverse-variance. PTVs show consistent signals across the different cohorts, and synonymous variants do not deviate from expectation. Bars represent the 95% CIs of the point estimates.
Extended Data Figure 2. Schizophrenia case-control enrichment in constrained genes (pLI > 0.9) stratified by different variant annotations and inferred consequences (n = 22,444 cases and n = 39,837 controls).
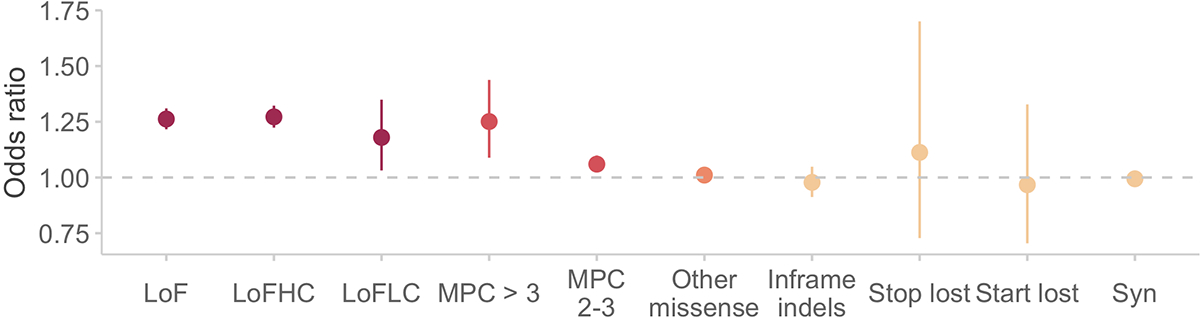
LoF: all loss-of-function or PTVs; LoFHC: high-confidence LOFTEE PTVs; LoFLC: low-confidence based on LOFTEE; MPC > 3: missense variants with MPC > 3; MPC 2 – 3: missense variants with MPC 2 – 3; Other missense: missense variants with MPC < 2; Syn: synonymous variants. The dot represents the odds ratio, and the bars represent the 95% CIs of the point estimates.
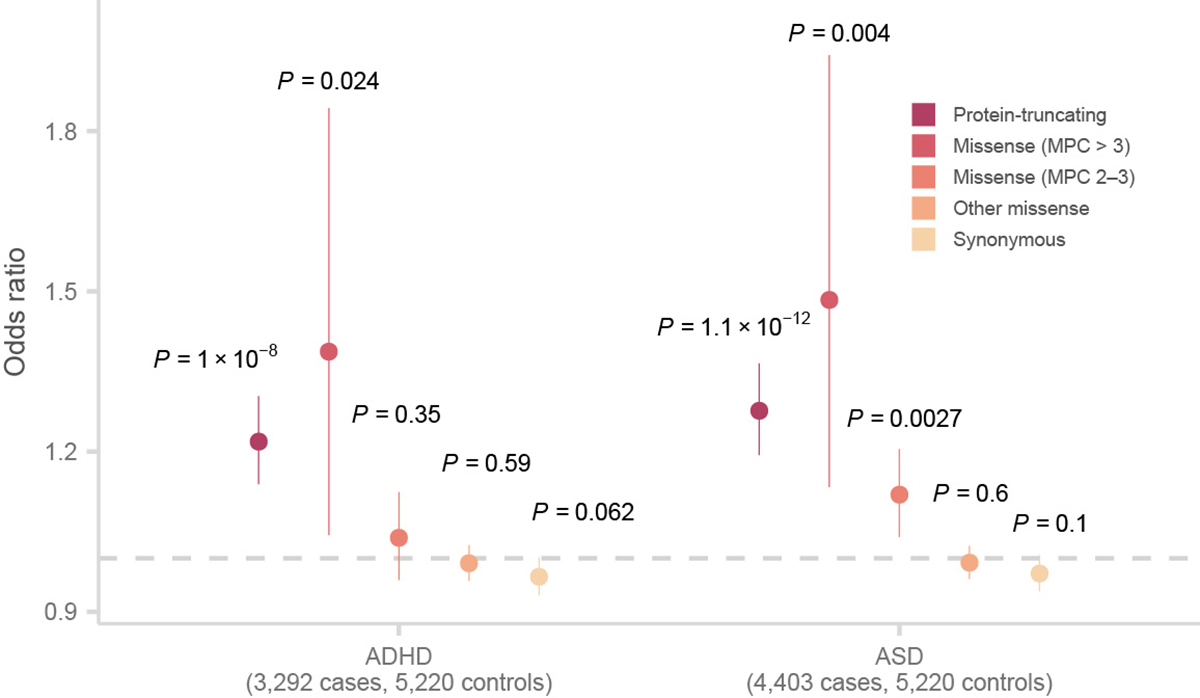
Extended Data Figure 3. Enrichment of URVs in n = 4,403 ASD and n = 3,292 ADHD cases compared to n = 5,220 controls stratified by variant annotation and consequences in constrained genes (pLI > 0.9).
Two-sided P values from logistic regression displayed are from comparing the burden of variants of the labeled consequence in cases compared to controls. The dot represents the odds ratio, and the bars represent the 95% CIs of the point estimates.
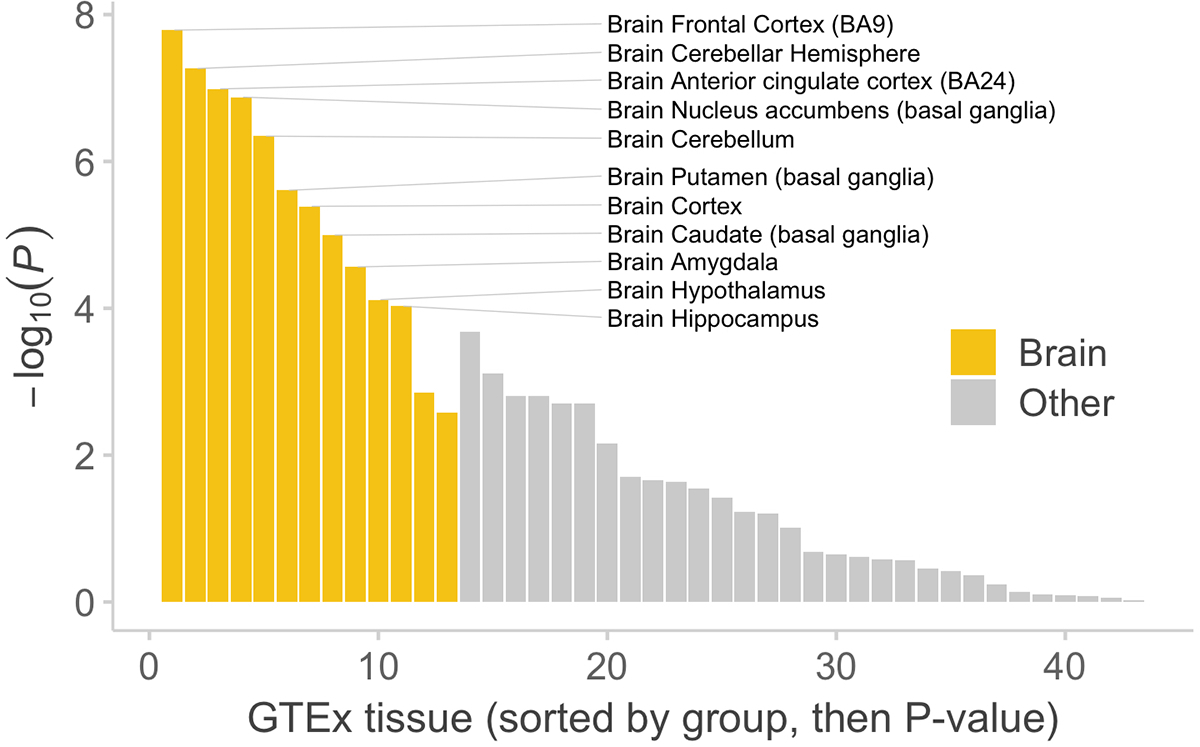
Extended Data Figure 4. Schizophrenia case-control gene set enrichment in brain and non-brain GTEx tissues.
We test for the burden of rare PTVs in genes with the strongest specific expression in that tissue type relative to other tissues as defined in 31. Gene set burden statistics are calculated using a logistic regression model of rare variants from n = 22,444 cases and n = 39,837 controls. We report two-sided P values. Each bar is a different tissue in GTEx, grouped by whether it is part of the central nervous system and sorted by P value (Table S8).
Extended Data Table 1.
Case-control and de novo counts of the ten Bonferroni significant genes in the main analysis.
| Gene Symbol | Case PTV | Ctrl PTV | Case mis3 | Ctrl mis3 | Case mis2 | Ctrl mis2 | De novo PTV | De novo mis3 | De novo mis2 | P value | Q value | OR (PTV) | OR (Class I) | OR (Class II) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SETD1A | 15 | 3 | 3 | 4 | 11 | 10 | 3 | 2.00E-12 | 3.62E-08 | 20.1 (5.68–108) | 10.3 (4.12–29.3) | 4.42 (1.7–11.6) | ||
| CUL1 | 8 | 1 | 2 | 0 | 7 | 16 | 3 | 2.01E-09 | 1.82E-05 | 36.1 (5.01–1570) | 44.2 (6.42–1880) | 1.76 (0.611–4.51) | ||
| XPO7 | 12 | 1 | 1 | 1 | 10 | 32 | 1 | 7.18E-09 | 4.34E-05 | 52.2 (7.84–2190) | 28.1 (6.46–253) | 1.25 (0.55–2.62) | ||
| TRIO | 18 | 16 | 0 | 0 | 24 | 102 | 2 | 6.35E-08 | 2.88E-04 | 5.02 (2.47–10.4) | 5.02 (2.47–10.4) | 0.944 (0.579–1.48) | ||
| CACNA1G | 10 | 13 | 8 | 4 | 55 | 134 | 1 | 4.57E-07 | 1.54E-03 | 3.09 (1.21–7.63) | 4.25 (2.07–8.78) | 1.68 (1.21–2.31) | ||
| SP4 | 13 | 6 | 3 | 3 | 0 | 2 | 1 | 5.08E-07 | 1.54E-03 | 9.37 (3.38–29.7) | 7.59 (3.2–19.3) | 0 (0–21.4) | ||
| GRIA3 | 5 | 0 | 3 | 2 | 10 | 24 | 1 | 1 | 5.98E-07 | 1.55E-03 | Inf (4.73-Inf) | 20.1 (4.28–188) | 1.67 (0.714–3.63) | |
| GRIN2A | 9 | 2 | 3 | 0 | 13 | 22 | 7.37E-07 | 1.67E-03 | 18.1 (3.74–172) | 24.1 (5.36–221) | 2.37 (1.1–4.92) | |||
| HERC1 | 28 | 32 | 0 | 0 | 2 | 8 | 1.26E-06 | 2.54E-03 | 3.51 (2.04–6.03) | 3.51 (2.04–6.03) | 1 (0.104–5.03) | |||
| RB1CC1 | 9 | 4 | 0 | 0 | 0 | 0 | 2 | 2.00E-06 | 3.63E-03 | 10 (2.89–43.9) | 10 (2.89–43.9) | 0 (0-Inf) |
Case-control counts displayed are the total counts for variants with minor allele count <= 5. PTV: protein-truncating variant, mis3: missense variants with MPC > 3, mis2: missense variants with MPC 2 – 3; Q value: adjusted P value after FDR adjustment; Class I: PTV and missense variants (MPC > 3); Class II: missense variants (MPC 2 – 3). Two-sided gene P values for Class I and Class II variants are calculated using the permuted Fisher’s exact test. Gene P values for de novo mutations are calculated using a one-sided Poisson rate test. The meta-analysis gene P value is calculated from the weighted Z-score method.
Supplementary Material
Acknowledgements
We would like to thank the patients and families who participated in our studies in the past two decades, without whom our research and findings would not be possible. Research reported in this publication was supported by the National Institute of Mental Health, and the National Human Genome Research Institute of the National Institutes of Health under award numbers: U01MH10564, U01MH105578, U01MH105666, U01MH109539, R01MH085548, R01MH085521, and U54HG003067. We would also like to acknowledge the generous support from the Stanley Family Foundation, Kent and Elizabeth Dauten, and The Dalio Foundation who have enabled us to rapidly expand our data generation collections with the goal of moving towards better treatments for schizophrenia and other psychiatric disorders. We wish to acknowledge all of the research participants in the BRIDGES cohort. This work was supported by NIMH (R01 MH094145; Michael Boehnke and Richard M.Myers, PIs and U01 MH105653 (Michael Boehnke, PI). The collection and storage of cases and controls from the Centre for Addiction and Mental Health (CAMH) in Toronto and from the Institute of Psychiatry, Psychology and Neuroscience (IoPPN), King’s College London in London, U.K. was supported by funding from GlaxoSmithKline. CAMH was supported by the Canadian Institutes of Health Research (MOP-172013, PI John B. Vincent, CAMH). IoPPN was supported by funding from the National Institute for Health Research(NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London (IoPPN). The views expressed are those of the author(s) and not necessarily those of the UK NHS, the NIHR or the UK Department of Health. Case and control collection was supported by Heinz C. Prechter Bipolar Research Fund at the University of Michigan Depression Center to Melvin G. McInnis. Data and biomaterials were collected for the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD), a multi-center, longitudinal project selected from responses to RFP #NIMH-98-DS-0001, “Treatment for Bipolar Disorder” which was led by Gary Sachs and coordinated by Massachusetts General Hospital in Boston, MA with support from 2N01 MH080001-001. The Genomic Psychiatric Cohort (GPC) was supported by NIMH (U01 MH105641 (PI Carlos Pato), R01 MH085548 (PIs Carlos Pato and Michele Pato), R01MH104964 (PIs Carlos Pato and Michele Pato). The MCTFR study was supported through grants from the National Institutes of Health DA037904, DA024417, DA036216, DA05147, AA09367, DA024417,HG007022, and HL117626. The work at Cardiff University was supported by Medical Research Council Centre Grant No. MR/L010305/1 and Program Grant No. G0800509.
Footnotes
Competing Interests
M.J.D. is a founder of Maze Therapeutics and RBNC Therapeutics. B.M.N. is a member of the scientific advisory board at Deep Genomics and RBNC Therapeutics, Member of the scientific advisory committee at Milken and a consultant for Camp4 Therapeutics, Merck and Biogen. A.P. is a member of Astra Zeneca’s Genomics Advisory Board. M.C.O, M.J.O, and J.T.W. are supported by a collaborative research grant from Takeda Pharmaceuticals. D.S.P. was an employee of Genomics plc, all analyses reported in this paper were performed as part of D.S.P.’s employment at the Massachusetts General Hospital and Broad Institute. The remaining authors declare no competing interests.
Ethics declarations
Written IRB approvals and study consent forms from each of the sample contributing organizations were sent to the Broad Institute of Harvard and M.I.T. before samples were sequenced and analyzed. All relevant ethical guidelines have been followed, and any necessary IRB and/or ethics committee approvals have been obtained. All ethical approvals are on file at the Massachusetts General Brigham (MGB), formerly Partners, IRB office amended to protocol #2014P001342, title: “Molecular Profiling of Psychiatric Disease” and undergoes annual continuing review by the Mass General Brigham Human Research Committee (MGBHRC) Institutional Review Board (IRB) of Mass General Brigham (Mass General Brigham IRB, Mass General Brigham, 399 Revolution Drive, Suite 710, Somerville, MA 02145). All necessary patient/participant consent has been obtained and the appropriate institutional forms have been archived.
Code availability
Software and code used are described throughout the Supplementary Methods of the manuscript. In brief, for sequence data generation, we used GATK v3.4 and v3.6, Picard version 1.1431, and VerifyBamID version 1.0.0. Sample, variant QC, and analyses were performed using Hail 0.1 and 0.2 (https://hail.is/), with functions and arguments referred to in the Supplementary Methods. Wrappers and methods using Hail code can be found at https://github.com/TarjinderSingh/hailutils. Additional (basic) processing and visualization was performed using base R (v3.6) with tidyverse libraries (https://www.tidyverse.org/packages/).
Data availability
We describe all datasets in the manuscript or Supplementary Information. We provide summary-level data at the variant and gene level in an online browser for viewing and download (https://schema.broadinstitute.org). There are no restrictions on the aggregated data released on the browser. For contributing data sets that are permitted to be distributed at the individual level, we have deposited, or are currently depositing, the data in a public repository (the database of Genotypes and Phenotypes [dbGAP] and/or the European Genome-phenome Archive [EGA]) and provide the accessions in Table S1. Whole Exome Sequence data generated under this study are currently hosted on and shared with the collaborating study groups via the controlled access Terra platform (https://app.terra.bio/). The Terra environment, created by the Broad Institute, contains a rich system of workspace functionalities centered on data sharing and analysis. Requests for access to the controlled datasets are managed by data custodians of the SCHEMA consortium and the Broad Institute and sent to sample contributing investigators for approval.
References
- 1.Satterstrom FK et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell (2020) doi: 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplanis J et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature (2020) doi: 10.1038/s41586-020-2832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schizophrenia Working Group of the Psychiatric Genomics Consortium, Ripke S, Walters JTR & O’Donovan MC Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv (2020) doi: 10.1101/2020.09.12.20192922. [DOI] [Google Scholar]
- 4.McGrath J, Saha S, Chant D & Welham J Schizophrenia: A Concise Overview of Incidence, Prevalence, and Mortality. Epidemiol. Rev 30, 67–76 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Hjorthøj C, Stürup AE, McGrath JJ & Nordentoft M Years of potential life lost and life expectancy in schizophrenia: a systematic review and meta-analysis. The Lancet Psychiatry 4, 295–301 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Lehman AF et al. Practice guideline for the treatment of patients with schizophrenia, second edition. Am. J. Psychiatry 161, 1–56 (2004). [PubMed] [Google Scholar]
- 7.Hyman SE Revolution stalled. Sci. Transl. Med. 4, 155cm11 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genovese G et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 19, 1433–1441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshall CR et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27–35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh T et al. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat. Genet. 1–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottesman II & Shields J A polygenic theory of schizophrenia. Proc. Natl. Acad. Sci. U. S. A. 58, 199–205 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. Submitted (2020).
- 14.Loh P-R et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 47, 1385–1392 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karayiorgou M et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc. Natl. Acad. Sci. U. S. A. 92, 7612–7616 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuk O et al. Searching for missing heritability: Designing rare variant association studies. Proceedings of the National Academy of Sciences 111, E455–E464 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S, Abecasis GR, Boehnke M & Lin X Rare-variant association analysis: study designs and statistical tests. Am. J. Hum. Genet. 95, 5–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rivas MA et al. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 348, 666–669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fromer M et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purcell SM et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh T et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 19, 571–577 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howrigan DP et al. Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nat. Neurosci. 23, 185–193 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gulsuner S et al. Genetics of schizophrenia in the South African Xhosa. Science 367, 569–573 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lek M et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samocha KE et al. Regional missense constraint improves variant deleteriousness prediction. bioRxiv 148353 (2017) doi: 10.1101/148353. [DOI] [Google Scholar]
- 27.Samocha KE et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu W, MacDonald ML, Elswick DE & Sweet RA The glutamate hypothesis of schizophrenia: evidence from human brain tissue studies. Ann. N. Y. Acad. Sci. 1338, 38–57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang HJ et al. Spatio-temporal transcriptome of the human brain. Nature 478, 483–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Psychiatric Genetics Consortium. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. (2015) doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koopmans F et al. SynGO: An Evidence-Based, Expert-Curated Knowledge Base for the Synapse. Neuron 103, 217–234.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeisel A et al. Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skene NG et al. Genetic identification of brain cell types underlying schizophrenia. Nat. Genet. 50, 825–833 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Rubeis S et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaplanis J et al. Integrating healthcare and research genetic data empowers the discovery of 49 novel developmental disorders. bioRxiv 797787 (2019) doi: 10.1101/797787. [DOI] [Google Scholar]
- 38.Barbosa S et al. Opposite Modulation of RAC1 by Mutations in TRIO Is Associated with Distinct, Domain-Specific Neurodevelopmental Disorders. Am. J. Hum. Genet. (2020) doi: 10.1016/j.ajhg.2020.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haukka J, Suvisaari J & Lönnqvist J Fertility of patients with schizophrenia, their siblings, and the general population: a cohort study from 1950 to 1959 in Finland. Am. J. Psychiatry 160, 460–463 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Laursen TM & Munk-Olsen T Reproductive patterns in psychotic patients. Schizophr. Res. 121, 234–240 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Power RA et al. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. Arch. Gen. Psychiatry 70, 22–30 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Endele S et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 42, 1021–1026 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Finn RD et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We describe all datasets in the manuscript or Supplementary Information. We provide summary-level data at the variant and gene level in an online browser for viewing and download (https://schema.broadinstitute.org). There are no restrictions on the aggregated data released on the browser. For contributing data sets that are permitted to be distributed at the individual level, we have deposited, or are currently depositing, the data in a public repository (the database of Genotypes and Phenotypes [dbGAP] and/or the European Genome-phenome Archive [EGA]) and provide the accessions in Table S1. Whole Exome Sequence data generated under this study are currently hosted on and shared with the collaborating study groups via the controlled access Terra platform (https://app.terra.bio/). The Terra environment, created by the Broad Institute, contains a rich system of workspace functionalities centered on data sharing and analysis. Requests for access to the controlled datasets are managed by data custodians of the SCHEMA consortium and the Broad Institute and sent to sample contributing investigators for approval.