

Abstract
T cell-B cell interaction is the key immune response to protect the host from severe viral infection. However, how T cells support B cells to exert protective humoral immunity in humans is not well understood. Here, we use COVID-19 as a model of acute viral infections and analyze CD4+ T cell subsets associated with plasmablast expansion and clinical outcome. Peripheral helper T cells (Tph cells; denoted as PD-1highCXCR5–CD4+ T cells) are significantly increased, as are plasmablasts. Tph cells exhibit “B cell help” signatures and induce plasmablast differentiation in vitro. Interestingly, expanded plasmablasts show increased CXCR3 expression, which is positively correlated with higher frequency of activated Tph cells and better clinical outcome. Mechanistically, Tph cells help B cell differentiation and produce more interferon γ (IFNγ), which induces CXCR3 expression on plasmablasts. These results elucidate a role for Tph cells in regulating protective B cell response during acute viral infection.
Keywords: PD-1highCXCR5–CD4+ peripheral helper T cells, Tph cells, IFNγ, CXCR3+ plasmablasts, T cell-B cell interactions
Graphical abstract
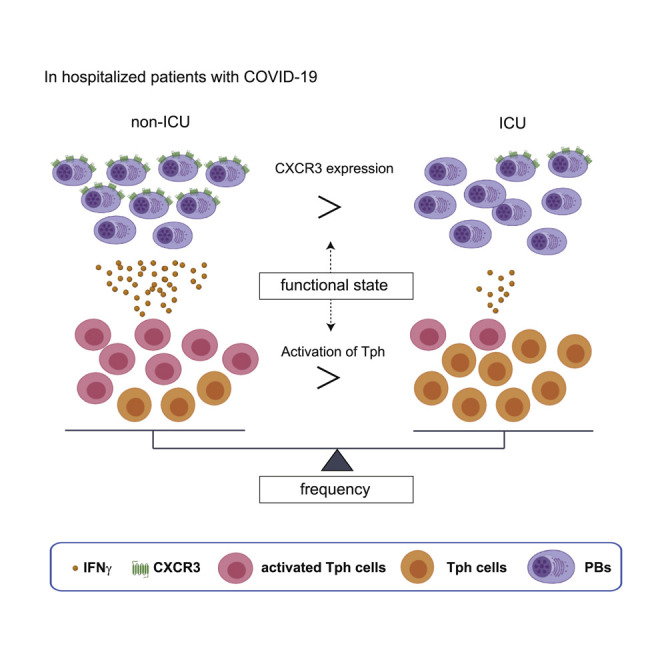
Asashima et al. report that peripheral helper T cells (denoted as PD-1highCXCR5–CD4+ T cells) are significantly increased in the acute phase of COVID-19, as are plasmablasts. Plasmablasts show increased CXCR3 expression, which is induced by IFNγ, and are positively correlated with activated Tph cells with better clinical outcome.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causes a wide spectrum of symptoms ranging from asymptomatic infections to acute respiratory distress syndrome (ARDS).1 , 2 COVID-19 is the clinical manifestation of SARS-CoV-2 infection, and it has become clear that a dysregulated immune response against SARS-CoV-2 in the early phase is central for disease severity.3 , 4 , 5 , 6 The generation of a robust antibody response is critical for clearing the virus, and the increase in plasmablasts, which can represent up to 30% of circulating B cells, is observed in a subset of patients with COVID-19 comparable to acute Ebola or dengue virus infections.7 , 8
During the early phase of viral infections, activated B cells differentiate into plasmablasts that migrate to target inflammatory tissues,9 indicating that the characteristics of plasmablasts are important in the disease course. These plasmablasts express chemoattractant receptors together with adhesion molecules and produce organ-specific protective antibodies for viral control.10 , 11 This rapid antibody response is critical as the early containment of virus reduces the risk of cytokine storm syndrome with excessive accumulation of immune cells in the lung parenchyma with ARDS.12 , 13 Recent studies suggested that the rapid generation of neutralizing antibodies is associated with protective immune response in COVID-19.14 , 15 Despite this important role of virus-specific antibodies, the control of expansion and chemoattractant receptors of plasmablasts in the early phase of human viral infections remains poorly understood.
During the adaptive immune response, T follicular helper (Tfh) cells play crucial roles in B cell differentiation.16 , 17 Circulating CXCR5+CD4+ T cells (cTfh) appear to represent the circulating compartment of Tfh cells.18 Intriguingly, while the plasmablast response correlates with the cTfh frequency in subjects recovered from COVID-19,19 that is not the case in the early phase of symptomatic patients with COVID-19.6 Moreover, Tfh-independent antibody responses are induced against both SARS-CoV-2 and influenza virus infections in mouse models.20 These data suggest that activated helper T cells other than Tfh cells indeed support the differentiation of B cells apart from germinal centers in the early phase of viral infections. However, what subset of T cells induce B cell differentiation in the early phase of human viral infections has not been elucidated.
To explore these mechanisms, we utilized immune profiles of COVID-19 as a model of acute viral infection in human and investigated the characteristics of B and T cells in patients with COVID-19 by integrating single cell RNA-seq (scRNA-seq) and flow cytometry datasets. Here, we report that PD-1highCXCR5–CD4+ T cells, so called peripheral helper T (Tph) cells,21 , 22 are significantly increased and positively correlated with the frequency of plasmablasts in peripheral blood in hospitalized patients with COVID-19. These Tph cells exhibit “B cell help” signatures to a similar degree as cTfh cells, but express higher inflammatory chemokine receptors, including CCR2 and CCR5, compared to cTfh cells. In vitro experiments indicate that activated PD-1highCXCR5– Tph cells exhibit higher IFNγ production than activated cTfh cells, which promote CXCR3 expression and differentiation of plasmablasts. Finally, we demonstrate that CXCR3+ plasmablasts are significantly increased in hospitalized patients with less severe disease without ICU admission, while they are decreased in patients admitted to ICU requiring mechanical ventilation. These findings provide a mechanism for the increase of plasmablasts apart from Tfh cells in the early phase of COVID-19, indicating that the induction of CXCR3+ plasmablasts by activated PD-1highCXCR5– Tph cells is important for disease control. These results elucidate a role for Tph cells in regulating antibody response associated with acute viral infection.
Results
PD-1highCXCR5–Tph cells are significantly increased in the early phase of COVID-19 and have a distinctive gene expression profile
Tfh cells provide essential B cell help, and circulating PD-1+CXCR5+ Tfh (cTfh) cells appear to represent circulating compartment of Tfh cells.18 , 23 , 24 As noted above, another T cell population expressing high levels of PD-1, but not CXCR5, was recently identified as playing a role in extra-follicular B cell differentiation in autoimmune disorders.21 , 22 In addition to an absence of germinal centers in lymphoid organs in the early phase of COVID-19,25 we hypothesized that PD-1highCXCR5− Tph cells may drive plasmablast expansion. We first categorized hospitalized patients with COVID-19 into 2 groups based on their admission to the intensive care unit (ICU): hospitalized patients without admission to the ICU (non-ICU subjects), and patients requiring mechanical ventilation in the ICU during hospitalization (ICU subjects) (Table S1). We evaluated CD45RA−CD4+ T cell subsets based on PD-1 and CXCR5 expression and found that PD-1highCXCR5− Tph cells were significantly increased in patients with COVID-19 and were positively correlated with the frequency of circulating plasmablasts (Figures 1A, 1B, and S1A). Samples were collected on the average of 11.8 days after first symptoms in patients with COVID-19, and there was no significant difference between two groups (non-ICU: 11.7 ± 10.1 days; ICU: 12.3 ± 10.5 days). In contrast, while PD1+CXCR5+ Tfh cells (both PD-1highCXCR5+ Tfh cells and PD-1intCXCR5+ Tfh cells) were also increased in the blood of patients with COVID-19, they were not correlated with plasmablasts in the early phase (Figures S1B–S1D). Moreover, the proportion of PD-1highCXCR5− Tph cells was not related to clinical characteristics (age, body mass index [BMI], and sex) (Figures S2A–S2C). These observations were replicated by reanalyzing flow cytometry data of patients with COVID-19 from a different study6 (Figures S3A–S3C).
Figure 1.

The characteristics of PD-1highCXCR5– Tph cells
(A) Representative data of PD-1highCXCR5– Tph cells in each group (left), the proportion of these T cells among healthcare workers (HCs) (n = 55), non-ICU patients with COVID-19 (non-ICU) (n = 56), and ICU patients (ICU) (n = 36). One-way ANOVA with Dunn’s multiple comparisons tests were performed (right). Data are represented as mean ± SEM.
(B) Correlation between PD-1highCXCR5– Tph cells and plasmablasts in patients (both non-ICU and ICU, n = 51). Linear regression is shown with 95% confidence interval (gray area). Correlation statistics is two-tailed Spearman’s rank correlation test.
(C) Principal-component analysis (PCA) of RNA-seq transcriptomes (n = 3, patients with COVID-19). Based on the expressions of PD-1 and CXCR5, six subsets were evaluated.
(D) Heatmap of Tfh-related genes.21
(E) Clustered heatmap of 100 genes that were differentially expressed (left column) in PD-1highCXCR5– Tph cells compared with cTfh cells (PD-1highCXCR5+ Tfh cells) (|log2FC| > 1, FDR < 0.05). The right column shows the log2FC.
(F) Representative data of CCR5 and CCR2 expression on PD-1highCXCR5– Tph cells compared with PD-1highCXCR5+ Tfh cells.
See also Figures S1–S3.
To further characterize these T helper populations, transcriptional profiles of six subsets of memory CD4+ T cells (CD45RA–CD4+ T cells), categorized by PD-1 and CXCR5 expression levels, were examined by bulk RNA sequencing (RNA-seq) (Figure 1C). Principal-component analysis (PCA) placed Tph cells at a distinctive position relative to the other subsets. Tph cells express Tfh-related genes (MAF, TIGIT, SLAMF6, and IL21),21 which are important for Tfh functions (Figure 1D). PD-1intCXCR5+ Tfh cells also expressed these genes but had less ICOS expression than PD-1highCXCR5+ Tfh cells, suggesting that PD-1highCXCR5+ Tfh cells are more activated cTfh cells.16 We identified 100 genes that were differentially expressed in PD-1highCXCR5− Tph cells compared with PD-1highCXCR5+ Tfh cells (|log2FC| > 1, FDR < 0.05) (Figure 1E). PD-1highCXCR5− Tph cells expressed activation markers such as HLA-DRB1 and pathways associated with “cell adhesion molecules,” “adheres junctions,” and “cytokine-cytokine receptor interaction” were enriched in Tph cells (Figures S2D and S2E). In addition, Tph cells showed marked upregulation of tissue-resident chemokine receptors, including CCR2, CCR5, and CX3CR1 (Figure 1F), and Th1-like signatures (CXCR3, TBX21, and STAT1) (Figure S2F). Thus, Tph cells exhibit “B cell help” signatures to a similar degree as cTfh cells but with a distinctive gene expression profile.
Activated PD-1highCXCR5− Tph cells are significantly increased in non-ICU patients and induce plasmablasts in vitro
To further evaluate the specific gene signatures of PD-1highCXCR5− Tph cells, we compared them with the other five subsets in memory CD4+ T cells. CXCR6, LAG3, and PRR5L were significantly upregulated, while CHD7, ZBTB20, ZNF251, GRK25, and GPRASP1 were significantly downregulated in PD-1highCXCR5− Tph cells (Figures 2A and 2B). Flow cytometry analysis also showed the same trend for upregulation of LAG3 and CXCR6 (Figures S4A and S4B). By interrogating these gene lists with CD4+ T cell clusters from our single-cell RNA-seq (scRNA-seq) dataset,5 we found that dividing CD4+ T cells best fit with these signatures (Figure 2C). We previously reported that dividing CD4+ T cells share the characteristics of HLA-DR+CD38+ activated T cells,5 and indeed, more than half of the activated CD4+ T cells fell into the PD-1highCXCR5− Tph cell compartment (Figure 2D). Of note, this activated state of PD-1highCXCR5− Tph cells was significantly increased in the less severely ill non-ICU patients compared with ICU patients (Figure 2E). We confirmed that these observations were not confounded by known risk factors for disease severity such as age, BMI, and sex (Figures S4C–S4E). While there was no difference in the proportion of PD-1highCXCR5− Tph cells between non-ICU and ICU patients at baseline, the expansion of Tph cells was accelerated at the later phase in the more severely ill ICU patients, and the difference reached significance at 2 weeks after the baseline measurement (Figure S4F). In contrast, the higher activation state of PD-1highCXCR5− Tph cells in non-ICU compared with ICU patients at baseline became less significant over time (Figure S4G). These differences were not attributed to the start date of treatment (p = 0.4337; Mann-Whitney test).
Figure 2.

Activated PD-1highCXCR5– Tph cells are significantly increased in non-ICU patients with COVID-19
(A) Venn diagrams showing the overlapped genes among those significantly upregulated (log2FC > 1, FDR < 0.05) (left) and downregulated (log2FC < −1, FDR < 0.05) (right) in PD-1highCXCR5– Tph cells compared with five subsets.
(B) Heatmap of PD-1highCXCR5– Tph cell-related genes (selected in A).
(C) Heatmap of PD-1highCXCR5– Tph cell-related genes (selected in A) among each T cell cluster of our scRNA-seq dataset reported.5
(D) Representative data for each T cell subset among HLA-DR+CD38+CD45RA–CD4+ T cells (left), and their proportions were evaluated by one-way ANOVA with Dunn’s multiple comparisons tests (right). COVID-19 samples that have more than 5% of HLA-DR+CD38+ T cells were evaluated (n = 11).
(E) Representative data of HLA-DR+CD38+ activated cells in PD-1highCXCR5– Tph cells between non-ICU and ICU patients (left). The proportions of activated PD-1highCXCR5– Tph cells were evaluated (non-ICU; n = 56, ICU; n = 36) by two-tailed unpaired Student’s t test (right).
(F and G) Each T cell subset and autologous CD20+CD27+ B cells were co-cultured (n = 7, patients with COVID-19). Representative data of CD27highCD138+ plasma cells after co-culture (F, left) and the proportion of plasma cells (F, right). IgG concentrations in supernatants were evaluated (G). PD-1int/−CXCR5– T cells indicate both PD-1intCXCR5– T cells (subset iii) and PD-1−CXCR5– T cells (v).
Data are represented as mean ± SEM (D–G).
See also Figure S4.
In the light of cTfh cells being implicated in the later phase of anti-SARS-CoV-2 antibody production,19 , 26 , 27 we examined whether PD-1highCXCR5− Tph cells promote B cell differentiation in vitro. As well as cTfh cells, PD-1highCXCR5− Tph cells helped differentiation of memory B cells into plasmablasts and initiated immunoglobulin G (IgG) production (Figures 2F and 2G). These data suggest that the activated PD-1highCXCR5− Tph cells can support B cell differentiation and that their early induction was associated with better clinical outcome.
The proportion of CXCR3+ plasmablasts is positively correlated with that of activated PD-1highCXCR5- Tph cells in the early phase of COVID-19
Next, we evaluated B cell signatures and confirmed that the frequency of plasmablasts is significantly increased in the peripheral blood of patients with COVID-193 , 28 (Figure 3A). Notably, there was no significant difference in the proportion of plasmablasts between non-ICU and ICU groups in hospitalized patients. To further understand the characteristics of B cells in patients with COVID-19, we analyzed our scRNA-seq data5 and subclustered B cells (total 13,550 cells from 31 samples) into eight clusters (Figures 3B, S5A, and S5B). These samples were collected on the average of 11.7 days after first symptom, and there was no difference between the two groups in patients with COVID-19 (non-ICU: 11.4 ± 5.9 days, ICU: 12.3 ± 5 days), which was the same as flow cytometry analysis. We identified naive B cells (MS4A1 + IGHD +); germinal center (GC)-like B cells (MS4A1 + NEIL1 +); intermediate memory B cells (IGHD + CD27 +); memory B cells (MS4A1 + CD27 +); and two plasma cell clusters, plasmablasts (MZB1 + CD38 +) and Ki67+ plasmablasts (MZB1 + CD38 + MKI67 +), in accordance with a previous report.29 Additionally, we were able to identify two more clusters, namely, FCRL5+ B cells (MS4A1 + FCRL5 +) and CD1c+ B cells (MS4A1 + CD1C +). FCRL5+ B cells also express higher levels of ITGAX and ZEB2 than other B cells, and this cluster was similar with atypical B cells or double-negative (DN) cells30 , 31 , 32 (Figure S5C). CD1c+ B cells express higher levels of CD52, which implies that their gene expression signatures resemble marginal zone-like B cells.33 , 34 We found that the proportions of two plasmablast clusters within total B cells were not different between non-ICU and ICU subjects, while GC-like B cells was significantly decreased in ICU patients compared with healthy controls (Figure S5D). With regard to B cell receptor (BCR) sequences, both plasmablast clusters expressed IgG, indicating that they had undergone class switching (Figure S6A). While there was no significant difference in the clonal diversity or the frequency of unmutated clones between the two clusters (Figures S6B and S6C), Ki67+ plasmablasts showed a significantly lower frequency of somatic hypermutations compared with the other plasmablasts (Figure S6D).
Figure 3.

The divergent immunological features of B cells in non-ICU and ICU patients with COVID-19
(A) Representative data of CD19+CD27+CD38+ plasmablasts (left). Plasmablasts between HCs (n = 15) and non-ICU (n = 31) and ICU patients with COVID-19 (n = 20) were evaluated by one-way ANOVA with Dunn’s multiple comparisons tests (right).
(B) Uniform manifold approximation and projection (UMAP) representation of subclustered B cells from HCs (n = 13) and COVID-19 samples (n = 18 from 10 patients).
(C) Heatmap of chemoattractant receptors36 among HCs and non-ICU and ICU patients with COVID-19 in clusters of both plasmablasts and Ki67+ plasmablasts. Average expression per subject is shown.
(D) Representative data of CXCR3 expression on CD19+CD27+CD38+ plasmablasts in patients with COVID-19 (left). CXCR3+ plasmablasts between non-ICU (n = 31) and ICU (n = 20) patients with COVID-19 were evaluated by two-tailed unpaired Student’s t test (right). FMO, fluorescence minus one.
(E) Correlation between activated PD-1highCXCR5– Tph cells and CXCR3+ plasmablasts (both non-ICU and ICU, n = 51). Linear regression is shown with 95% confidence interval (gray area). Correlation statistics is two-tailed Spearman’s rank correlation test.
Data are represented as mean ± SEM (A and D).
See also Figures S5–S7.
To further examine the characteristics of each cluster identified by scRNA-seq, we assessed the expression of genes that are associated with B cell function36 (Figure S5E). It was of interest that the expression level of CXCR3, the chemoattractant receptor, was upregulated in plasmablast clusters in viral-infected subjects, specifically in the Ki67+ plasmablast cluster. In contrast, the homing receptors35 known to guide immune cells to lymph nodes (CCR7, SELL) were expressed in healthy control plasmablasts (Figures 3C and 3D). Additionally, we observed a positive correlation between activated PD-1highCXCR5− Tph cells and CXCR3+ plasmablasts (Figure 3E). We also evaluated the relationship between activated PD-1high/intCXCR5+ Tfh cells and CXCR3+ plasmablasts, which did not have a significant positive correlation (r = 0.2592, p = 0.0662).
To elucidate the functional readouts with regard to PD-1highCXCR5− Tph cells and CXCR3+ plasmablasts, we evaluated anti-SARS-CoV-2 antibodies. The proportion of CXCR3+ plasmablasts was positively correlated with the level of SARS-CoV-2 neutralizing antibodies (Figure S7A), and moreover, antibody titers were more correlated with the frequencies of activated PD-1highCXCR5− Tph cells than those of activated PD-1+CXCR5+ Tfh cells (Figures S7B–S7D).
Taken together, increased CXCR3 expression was one of the features in plasmablasts, and their frequency was positively correlated with that of activated PD-1highCXCR5− Tph cells, which might be beneficial in preventing severe viral expansion especially in the early phase.
CXCR3 expression on plasmablasts is induced by higher IFNγ from PD-1highCXCR5− Tph cells
To evaluate how CXCR3+ plasmablasts can be induced by PD-1highCXCR5− Tph cells, we examined their cytokine productions. Stimulation with anti-CD3/CD28 antibodies induced greater IFNγ and interleukin-10 (IL-10) production from PD-1highCXCR5− Tph cells (Figure 4A), which is of interest as IFNγ is known to upregulate CXCR3 expression during B cell differentiation.37 We confirmed that PD-1highCXCR5− Tph cells produced IL-21 and CXCL13 to the same degree as PD-1highCXCR5+ Tfh cells (Figure 4B). We also observed that CXCR3 expression on plasmablasts was upregulated by IFNγ in a dose-dependent manner, while IL-10 was not (Figures 4C and 4D). In fact, the addition of anti-IFNγ blocking antibodies diminished Tph cell-mediated differentiation of CXCR3+ plasmablasts in vitro (Figure 4E). Moreover, other chemoattractant receptors such as CCR2 were upregulated besides CXCR3, but CXCR4 was not (Figure 4F). These data indicate that IFNγ produced by PD-1highCXCR5− Tph cells induces CXCR3 expression on plasmablasts.
Figure 4.

CXCR3 expression on plasmablasts is induced by via IFNγ from PD-1highCXCR5– Tph cells
(A) T cells (n = 5, patients with COVID-19) were stimulated with anti-CD3/28 for 48 h, then cytokine productions were measured (IFNγ, IL-10) by ELISA.
(B) IL-21 and CXCL13 levels in the supernatants of co-cultures were measured by ELISA (n = 7, patients with COVID-19).
(C and D) CD20+CD27+ memory B cells (n = 5, healthy controls) were cultured with CD40L, IL-21, and IL-10 or different concentrations of IFNγ for 7 days (n = 5, healthy controls). Representative histogram for CXCR3 expression on plasma cells (C, left) and CXCR3 gMFI was evaluated (C, right). After 7 days in culture, CD19+CD27+CD138+ plasma cells were sorted, and CXCR3 expression was measured by qPCR (D).
(E) Representative data of CXCR3 expression on CD27highCD138+ plasma cells after co-culture with Tph cells (n = 5, patients with COVID-19) with anti-human IFNγ antibodies (anti-IFNγ Abs) or IgG isotype controls (IgG control) (E, top). CXCR3+CD27highCD138+ plasma cells (bottom) were evaluated by Wilcoxon matched-pairs signed rank test (E, bottom).
(F) After 7 days culture of CD20+CD27+ memory B cells (n = 5, healthy donors) with various conditions, gene expressions of CD19+CD27highCD138+ plasma cells were measured by qPCR (n = 5, healthy controls).
Data were evaluated by one-way ANOVA with Tukey’s multiple comparisons tests (A–D and F). Data are represented as mean ± SEM (A–D and F).
Discussion
PD-1highCXCR5− Tph cells were first described in the synovial fluid of rheumatoid arthritis and the peripheral blood of patients with systemic lupus erythematosus (SLE),21 , 38 indicating an important role in autoimmune diseases with tissue-specific, ectopic antibody production. Here, we analyzed T cells and B cells in patients with COVID-19 and demonstrated that in the early phase of COVID-19, PD-1highCXCR5− Tph cells, exhibiting “B cell help” signatures and promoting B cell differentiation in vitro, were significantly increased in hospitalized patients, with COVID-19 correlating with frequencies of plasmablasts. Furthermore, PD-1highCXCR5− Tph cells produced more IFNγ, which induced CXCR3 on plasmablasts, and they were correlated with increases in activated Tph cells and anti-SARS-CoV-2 neutralizing antibodies, which were significantly decreased in ICU patients on mechanical ventilation. These results elucidate a role for Tph cells in promoting CXCR3+ plasmablasts by secretion of IFNγ and in regulating antibody responses associated with viral infection.
Our data demonstrate that not only cTfh cells but also Tph cells play an important role in supporting the expansion of plasmablasts during acute viral infection. One of the common features between the early phase of COVID-19 and chronic autoimmune conditions such as SLE is type-1 IFN (IFN-I) signatures.5 , 39 , 40 IFN-I downregulates CXCR5 expression and upregulates PD-1 expression in human T cells in vitro.41 , 42 Intriguingly, many differentially expressed genes on Tph cells such as CXCR6, LAG3, ZBTB20, and CHD7 seem to be regulated by IFN-I in human T cells,42 indicating that IFN-I might be contributing to Tph cell differentiation.
The antibody responses against infected bacteria are established by day 3 through 5 weeks.43 However, GC formation was observed approximately 1 month after infection; therefore, antibody responses at the early phase were not GC dependent. In fact, antibodies with low frequencies of somatic hypermutation are detected in the acute viral infections before GC formation and play protective roles against viral infection.13 , 44 , 45 Similarly, in blood from patients with COVID-19, earlier antibody responses are likely to be associated with better recovery.14 , 15 , 46 Our findings that the rapid induction of CXCR3+ plasmablasts was linked with both a better outcome of disease and the production of protective antibodies support these data. Additionally, we demonstrated that PD-1highCXCR5− Tph cells in the activated state support the induction of CXCR3+ plasmablasts, and these T cells were significantly increased in less severe, non-ICU patients at earlier stages. The significant difference in the frequency of activated PD-1highCXCR5− Tph cells between non-ICU and ICU patients waned over time, indicating that the prompt induction of activated PD-1highCXCR5− Tph cells might be critical for the promotion of CXCR3+ plasmablasts and the production of subsequent neutralizing antibodies to effectively remove virus linked with better clinical outcomes. These data provide evidence for the function of activated Tph cells by providing a protective role in the early phase of hospitalized patients with COVID-19, whereas the stronger and delayed/prolonged activation of T cells is known to be associated with worse clinical outcomes.6 Not only sex differences3 but also genetic backgrounds are reported to be related to T cell activation,47 , 48 and further analyses combined with these factors will lead to the more precise identification of patients at higher risk and are thus expected to be valuable for the development of personalized treatments.
Tph cells are implicated in the extra-follicular B cell responses,22 and a recent study reported that extra-follicular B cell activation was observed in severe patients with COVID-19,49 which is seemingly contradictory to our results. However, the categorization of disease severity was different between these studies. The previous study defined the severity based on the hospitalization so that all non-hospitalized patients (i.e., outpatients) were grouped as mild and all hospitalized patients as severe. In contrast, we studied only hospitalized patients and divided inpatients into either a non-ICU cohort or an ICU cohort with severe disease requiring mechanical ventilation or death. Thus, while these clinical categories have some degree of variability, our results nevertheless support previous findings that extra-follicular features are detected in hospitalized patients.
The temporal dynamics of T cell-B cell interaction is crucial to understand immune response during the disease progression of COVID-19. We demonstrated a relationship between PD-1highCXCR5− Tph cells and CXCR3+ plasmablasts in the acute phase, especially within 14 days after symptom onset. In contrast, Tfh cells play a central role during the recovery phase of COVID-19.19 , 27 Recently, Koutsakos et al.26 identified a correlation between antibody titers and the proportion of cTfh1 cells in patients with COVID-19. However, the dynamics of rapid expansion of plasmablasts, peaking between 1 and 2 weeks from symptom onset, and the gradual increase of cTfh1 cells peaking in 3–4 weeks implicates that T cells other than cTfh1 cells support the rapid expansion of plasmablasts at the earlier phase. Indeed, the PD-1highCXCR5+ T cells with the highest CXCR3 expression (subset 2 in Figure 1A), which correspond to most of cTfh1 cells, were not positively correlated with plasmablast expansion (Figure S3C). Thus, our findings suggest that PD-1highCXCR5− Tph cells play a major role in the rapid expansion of plasmablasts in the acute phase of hospitalized patients with COVID-19. Subsequently, the transition of “B cell help” T cell population from Tph cells to Tfh cells could shift T cell-B cell interaction from extra-follicular to a GC response that governs long-lasting humoral immunity.
In summary, our data implicate PD-1highCXCR5− Tph cells as triggering the induction of CXCR3+ plasmablasts via IFNγ in the early phase of acute viral infection by using COVID-19 as an in vivo model of human viral infection. These data provide a potential framework for assessing immune response to pathogenic virus and other IFN-I-inducing viral infections. Moreover, these data shed light as to how T cells can drive B cell differentiation in acute viral infections and provide potential insights into the role of PD-1highCXCR5− Tph cells on a variety of immune-mediated diseases with the possible contribution of aberrant T-B interaction, including chronic autoimmune diseases.
Limitations of the study
Although we performed a bidirectional analysis between T cells and B cells in blood, we could not directly assess the lymphoid tissue samples. Moreover, given that the definition of Tph cells is based on PD1highCXCR5− memory CD4+ T cells, the functional overlaps between Tph cells and PD-1high Th1 cells are not clear. To decipher the relationship among PD-1highCXCR5− Tph cells, cTfh1 cells, and PD-1highTh1 cells from the perspective of “B cell help” functions, the spatial interactions between those T cells and B cells at tissue sites can be assessed in future studies. Another limitation is that we could not evaluate the antigen specificity of Tph cells because of limited samples and cell numbers from patients with COVID-19 who have vaccine- and past-infection-free status. The use of a SARS-CoV-2-specific tetramer with genotyping would have been useful and will be performed in future investigations. In addition, both Tph cells and plasmablasts are small subsets in total lymphocytes, and the assessment of absolute population numbers would have been more helpful. Although our finding highlights the potential role of CXCR3+ plasmablasts during acute viral infection, the functional differences between CXCR3+ and CXCR3− plasmablasts were not addressed, which is challenging in human but can be clarified by in vivo models with Cxcr3 conditional knockout mice. Finally, our correlative data between Tph cells and plasmablasts may reflect the general kinetics of acute viral response and does not necessarily imply that these cells are interdependent. To clarify those causal interactions between Tph cells and plasmablasts in vivo, further investigation using murine models is warranted.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BV605 anti-human CD3 (clone UCHT1) | Biolegend | 300480 |
| BV785 anti-human CD4 (clone SK3) | Biolegend | 344642 |
| APCFire750 anti-human CD8 (clone SK1) | Biolegend | 344746 |
| BV421 anti-human CD197(CCR7) (clone G043H7) | Biolegend | 353208 |
| Alexa Fluor 700 anti-human CD45RA (clone HI100) | BD Biosciences | 560673 |
| PE-CF594 anti-human CD25 (clone BC96) | BD Biosciences | 567489 |
| PE anti-human PD-1 (cline EH12.2H7) | Biolegend | 329906 |
| APC anti-human TIM3 (cline F38-2E2) | Biolegend | 345012 |
| BV711 anti-human CD38 (clone HIT2) | Biolegend | 303528 |
| BB515 anti-human HLA-DR (clone G46-6) | BD Biosciences | 564516 |
| BB700 anti-human CXCR5 (clone RF8B2) | BD Biosciences | 566469 |
| PE-Cy7 anti-human CD127 (clone HIL-7R-M21) | BD Biosciences | 560822 |
| BV785 anti-human CD19 (clone SJ25C1) | Biolegend | 302240 |
| BV421 anti-human CD138 (clone MI15) | Biolegend | 356516 |
| Alexa Fluor 700 anti-human CD20 (clone 2H7) | Biolegend | 302322 |
| Alexa Fluor 647 anti-human CD27 (clone M-T271) | Biolegend | 356434 |
| PE/Dazzle594 ant-human IgD (clone IA6-2) | Biolegend | 348240 |
| BB700 anti-human CD183/CXCR3 (clone 1C6/CXCR3) | BD Biosciences | 566532 |
| PE-Cy7 anti-human CD86 (clone IT2.2) | Biolegend | 305422 |
| APC/Fire750 anti-human IgM (clone MHM-88) | Biolegend | 314546 |
| BV605 anti-human CD24 (clone ML5) | Biolegend | 311124 |
| Alexa Fluor 647 anti-human CXCR5 (clone RF8B2) | BD Biosciences | 558113 |
| BV421 anti-human CD192(CCR2) (clone K036C2) | Biolegend | 357209 |
| FITC anti-human CD195(CCR5) (clone J418F1) | Biolegend | 359119 |
| Alexa Fluor 647 anti-human CD45RA (clone HI100) | BD Biosciences | 560673 |
| Alexa Fluor 488 anti-human CD183/CXCR3 (clone 1C6/CXCR3) | BD Biosciences | 558047 |
| BV421 anti-human CD27 (clone M-T271) | Biolegend | 356418 |
| BV510 anti-human CD20 (clone 2H7) | Biolegend | 302340 |
| BV711 anti-human CD138 (clone MI15) | Biolegend | 356522 |
| PE-Cy7 anti-human CD183/CXCR3 (clone 1C6/CXCR3) | BD Biosciences | 560831 |
| BV421 anti-human CX3CR1 (clone 2A9-1) | Biolegend | 341619 |
| BV421 anti-human LAG3 (clone 11C3C65) | Biolegend | 369314 |
| FITC anti-human CD186(CXCR6) (clone K041E5) | Biolegend | 356019 |
| FITC anti-human CD3 (clone UCHT1) | BD Biosciences | 561806 |
| PE-Cy7 anti-human CD4 (clone OKT4) | Biolegend | 317414 |
| Ultra-LEAF™ Purified anti-human IFN-γ Antibody (clone B27) | Biolegend | 506532 |
| mouse IgG1 k isotype controls (clone MOPC-21) | Biolegend | 400102 |
| Purified NA/LE Mouse Anti-Human CD3 (clone UCHT1) | BD Biosciences | 555329 |
| Purified NA/LE Mouse Anti-Human CD28 | BD Biosciences | 555725 |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI 1640 medium | Gibco | 11875-085 |
| Fetal Bovine Serum | Gemini | 100-106 |
| L-glutamine (200mM) | Gibco | 25030-081 |
| Pen Strep | Gibco | 15140-122 |
| Enterotoxin Type B from Staphylococcus aureus | LIST BIOLOGICAL LABS INC | 122 |
| LPS from E. Coli, TLR4 ligand | NOVUS Biologicals | NBP2-25295 |
| MEGACD40L Protein | Enzo | ALX-522-110-C010 |
| Recombinant Human IL-10 Protein | R&D | 217-IL-010/CF |
| Recombinant Human IL-21 Protein | R&D | 8879-IL-050/CF |
| Recombinant Human IFN-gamma Protein | R&D | 285-IF-100/CF |
| Human TruStan FcX | Biolegend | 422302 |
| Live/Dead Fixable Aqua | Thermofisher | L34966 |
| Live/Dead Fixable Near-ID | Thermofisher | L34976 |
| Critical commercial assays | ||
| Human B cell isolation kit | Stemcell Technologies | 19054 |
| RNeasy Micro Kit | QIAGEN | 74034 |
| SuperScript IV VILO Master Mix | Thermofisher | 11756050 |
| TaqMan Fast Univ PCR Master Mix | Applied Biosystems | 4352046 |
| Taqman qPCR Probe (CXCR3) | Thermofisher | Hs01847760_s1 |
| Taqman qPCR Probe (CXCR4) | Thermofisher | Hs00607978_s1 |
| Taqman qPCR Probe (CCR2) | Thermofisher | Hs00356601_m1 |
| Taqman qPCR Probe (B2M) | Thermofisher | Hs00187842_m1 |
| IgG (Total) Human Uncoated ELISA Kit | Thermofisher | 88-50550-88 |
| Human CXCL13/BLC/BCA-1 DuoSet ELISA kit | R&D | DY801 |
| Human IFNg DuoSet ELISA kit | R&D | DY285 |
| Human IL-10 DuoSet ELISA kit | R&D | DY217B |
| Human IL-21 DuoSet ELISA kit | R&D | DY8879-05 |
| Human SARS-CoV-2 Spike (Trimer) IgM ELISA Kit | Thermofisher | BMS2325 |
| Human SARS-CoV-2 Spike (Trimer) IgG ELISA Kit | Thermofisher | BMS2324 |
| Anti-SARS-CoV-2 S-RBD protein Human IgG ELISA Kit | Proteintech | KE30003 |
| Anti-SARS-CoV-2 S-RBD protein Human IgM ELISA Kit | Proteintech | KE30004 |
| LEGENDplex™ SARS-CoV-2 Neut. Ab Assay | Biolegend | 741127 |
| SMART-Seq v4 Ultra Low Input RNA Kit | Takara/Clontech | 634898 |
| Nextera XT DNA Library Preparation kit | Illumina | FC-131-1024 |
| Deposited data | ||
| RNA-seq data | This paper | GSE 214624 |
| Software and algorithms | ||
| R Statistical Computing Software (v3.6) | The R Foundation | https://www.r-project.org/ |
| Seurat (v3.2.0) | Stuart et al.50 | https://satijalab.org/seurat/ |
| IMGT/GENE-DB (v3.1.26) | Giudicelli et al.51 | https://www.imgt.org/IMGTindex/IMGTgene-db.php |
| IgBLAST (v.1.15.0) | Ye et al.,52 | https://www.ncbi.nlm.nih.gov/igblast/ |
| Change-O (v.1.0.0) | Gupta et al.53 | http://clip.med.yale.edu/changeo/ |
| SHazaM (v1.0.2.999) | Yaari et al.54 | https://shazam.readthedocs.io/en/stable/ |
| Alakazam (v1.0.2.999) | Gupta et al.53 | https://alakazam.readthedocs.io/en/stable/ |
| PRINSEQ++ (v1.2) | Cantu et al.55 | https://github.com/Adrian-Cantu/PRINSEQ-plus-plus |
| STAR (v2.7.1) | Dobin et al.56 | https://github.com/alexdobin/STAR |
| RSEM (v1.3.0) | Li and Dewey.57 | https://github.com/deweylab/RSEM |
| DESeq2 (v1.34.0) | Love et al.58 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GraphPad Prism version 7 | GraphPad | https://www.graphpad.com/guides/prism/8/user-guide/tips_for_using_prism.htm |
| Enrichr | Kuleshov et al.59 | https://maayanlab.cloud/Enrichr/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tomokazu S. Sumida (tomokazu.sumida@yale.edu).
Materials availability
This study did not generate new unique reagents.
Experimental models and subject details
Ethics Statement
This study was approved by the Institutional Review Board at the Yale School of Medicine (FWA00002571, Protocol IDs: 2000027690 and 2000027291REG). Informed consent was obtained from all enrolled patients, healthcare workers, and healthy donors.
Patients and samples
Adult patients (≥18 years old) admitted to Yale-New Haven Hospital, positive for SARS-CoV-2 by RT-PCR from nasopharyngeal and/or oropharyngeal swabs, and able to provide informed consent (surrogate consent accepted) were eligible (Table S1). Individuals with pregnant patients, patients with background hematological abnormalities, patients with autoimmune diseases and patients with a history of organ transplantation and on immunosuppressive agents, were excluded from this study.
For the characterization of T cells and B cells, the flow data deposited in the Yale IMPACT Biorepository study were analyzed as described elsewhere.3 , 28 All the patients were admitted between 30 March and 27 May 2020 and hospitalized. No samples had histories of prior COVID-19 infection nor vaccinations and only the baseline data were analyzed except for the analysis of time kinetics. All the experiments were performed on fresh peripheral blood mononuclear cells (PBMCs), and samples were drawn on the average of 11.8 days after first symptoms. 92 hospitalized patients with COVID-19 and 64 COVID-19-uninfected healthcare workers (HCs) were enrolled. Patients with COVID-19 who required admission to the ICU had been classified as “ICU” and 27.8% of them were expired. The other patients classified as “non-ICU” were all discharged without ICU admission. For the patients who were 90 years-old or older, their ages were protected health information, and ‘90’ was put as the surrogate value for the analyses. HCs were all negative in both PCR and serology tests.
Single cell RNA-seq (scRNA-seq) was performed on cryopreserved PBMC samples of 10 patients with COVID-19 following the same criteria as above and 13 age- and sex-matched controls. All the samples were drawn on the average of 11.7 days after first symptoms. Control samples were already collected before the first report of COVID-19 in 2018. From eight of ten patients with COVID-19, PBMC samples from two different time points had been analyzed. Four patients had been classified as “ICU”, who required admission to the ICU with mechanical ventilation, and the other six patients classified as “non-ICU” who were hospitalized, but not admitted to ICU and all discharged, and the same criteria as flow data. We have described the full cohort elsewhere.5
All the other experiments which include in vitro experiments and bulk RNA-seq were performed with fresh PBMCs at the baseline. Patients were admitted between 21st July 2020 and 24th Sep 2021 and all were immunologically naive except blocking experiments (Figure 4E), which had history of prior SARS-CoV-2 vaccinations.
Method details
Peripheral blood mononuclear cells isolation
PBMCs were prepared from whole blood by Ficoll gradient centrifugation (Histopaque (Sigma) in the Yale IMPACT Biorepository study and Lymphoprep (Stemcell) in other experiments). The PBMC layer was collected into a new 50-mL tube and washed twice with PBS to remove any remaining Lymphoprep/Histopaque. As for scRNA-seq and flow cytometry, the pelleted cells were treated with ACK buffer for red cell lysis. All the other experiments including bulk RNA-seq samples were processed without lysis buffer.
Flow cytometry and sorting
In the Yale IMPACT Biorepository study, the staining was performed mainly in two separate panels for (1) T cell surface staining and (2) B cell surface staining. PBMCs were plated at 1-2 × 106 cells in a 96-well U-bottom plate, and resuspended in Live/Dead Fixable Aqua (ThermoFisher) for 20 min at 4 °C. Following a wash, cells were then blocked with Human TruStan FcX (BioLegend) for 10 min at room temperature. Cocktails of desired staining antibodies were directly added to this mixture for 30 min at room temperature. Before analysis, cells were washed and resuspended in 100 μL of 4% paraformaldehyde for 30 min at 4 °C. We have described the detailed methods elsewhere.3 Data acquisition had been done, dividing into several times (T cell panel; 32 batches, B cell panel; 19 batches) by the specific flow cytometry (Attune NXT; Thermofisher). They did not contain any specific experiment batches with significant outliers (more than mean +2SD or less than mean – 2SD) than the others. For other experiments, freshly isolated PBMCs were stained with cocktails of desired staining antibodies for 30 min at 4°C. Specific T cell and B cell subsets were sorted on a Sony MA900 cell sorter.
T-B-cell co-culture experiments
Co-culture experiments were performed referenced as described previously.21 In brief, sorted T cell populations (5000-20000 cells) from PBMCs of patients with COVID-19 were co-cultured with autologous CD20+CD27+ memory B cells at a ratio of 1:3 in 200 μL of RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 nM L-glutamine, and 100 U/ml penicillin, 100 μg/mL streptomycin (Gibco), stimulated with SEB (1 μg/mL) and LPS (1 μg/mL) for 7 days. Supernatants were collected, and total IgG (Invitrogen), and cytokine/chemokine levels (R&D systems) were measured by ELISA. Some experiments added anti-human IFNγ antibodies (5 μg/mL) or mouse IgG1 k isotype controls on day 0 and 4 for the neutralization of IFNγ bioactivities. Cells were harvested and analyzed by flow cytometry, with plasma cells defined as CD27highCD138+ cells.
B cell differentiation in vitro
We extracted PBMCs from healthy volunteers. After the isolation of CD19+ B cells from PBMCs using Human B cell isolation kit (Stemcell Technologies), CD20+CD27+ memory B cells were sorted on a FACS Aria (BD Biosciences) and stimulated with CD40L (0.05 μg/mL), IL-21 (20 ng/mL) and other cytokines in culture medium the same as above. After 7 days, CD27highCD138+ plasma cells were sorted for gene expression analysis by qPCR.
T cell stimulation in vitro
We extracted PBMCs from patients with COVID-19. Each subset of memory CD4+ T cells based on the expression levels of PD-1 and CXCR5 were stimulated with anti-CD3/CD28 (each 1 μg/mL) for 48 h and cytokine levels were measured by ELISA (all from R&D systems) according to the manufacturer’s instructions. 1% Triton X-100 for 60 min at room temperature was added before ELISA to reduce risk from any potential virus in the supernatant.60
Quantitative PCR
Total RNA was extracted using RNeasy Micro Kit (QIAGEN) according to the manufacturer’s instructions. cDNA was synthesized with SuperScript IV VILO Master Mix (Thermofisher). cDNAs were amplified with Taqman probes (Taqman Gene Expression Arrays) and TaqMan Fast Advanced Master Mix on a StepOne Real-Time PCR System (Applied Biosystems) according to the manufacturer’s instructions. The RNA expression was measured relative to B2M expression.
The measurement of anti-SARS-CoV-2 antibodies
Sera was collected from patients with COVID-19 and ELISA assays (Proteintech) were performed. In brief, sera were diluted with 1:100 and added to the RBD S-RBD pre-coated plate. After 30 min incubation at room temperature, wells were washed and horseradish peroxidase-conjugated anti-human IgG or IgM were added. After 30 min incubation, wells were washed, followed by TMB reagents. Reagents including sulfuric acid were used to stop reactions and the color intensities were evaluated at 450nm with the correction wavelength set at 630nm. For the evaluation of neutralizing antibodies, sera were diluted with 1:100 and beads-based SARS-CoV-2 neutralizing antibody assay (Biolegend) was used, following the protocol.
Single cell RNA-seq data processing
A PBMC scRNA-seq dataset which had been previously performed and reported by us5 was reanalyzed. In brief, single cell barcoding of PBMC and library construction had been performed using the 10x Chromium NextGEM 5 prime kit according to manufacturer’s instructions. Libraries had been sequenced on an Illumina Novaseq 6000 platform. Raw reads had been demultiplexed and processed using Cell Ranger (v3.1) mapping to the GRCh38 (Ensembl 93) reference genome. Resulting gene-cell matrices had been analyzed using the package Seurat50 , 61 in the software R (v3.6.2) including integration of data, clustering, multiplet identification and cell type annotation. We have described the detailed methods elsewhere.5 PBMCs were processed with four different experiments, and we could not find critical batch differences among experiments. The annotated R object was used for sub-clustering of B cells.
The three cell populations, "Memory B cells", "Naive B cells " and "Plasma cells" in total PBMCs were re-clustered to obtain a finer cell type granularity. To remove batch- and single-donor effects, we integrated all 31 samples of these populations into one dataset using reference-based anchor finding and integration workflow. We chose 2 healthy donor samples (C27 and C32) and 2 COVID-19 samples (NS1B and TS3A), which have enough B cell numbers, as references for anchor finding and integration. The top 2000 variable genes were selected, and integration anchors were determined by “FindIntegrationAnchors” without k.filter for low cell numbers in some samples. These anchors were used to integrate the data using the “IntegrateData” function with top 30 dimensions and scaled. The top 17 PCs were used for data integration and downstream steps, along with a clustering resolution of 0.4. Cluster-specific gene expression profiles were established using the “FindAllMarkers” per cluster and per subset to annotate the clusters. Doublet clusters were determined by co-expression of heterogeneous lineage markers (e.g., MS4A1 and CD3). These clusters were removed prior to finalizing the UMAPs.
B cell receptor repertoire analysis
B cell receptor (BCR) V(D)J repertoire data processing, clonal clustering, and unmutated germline ancestor sequence reconstruction was previously performed.5 Briefly, V(D)J genes aligned to the IMGT/GENE-DB v3.1.2651 germline reference database using IgBLAST v.1.15.0.52 Cells with multiple IGH V(D)J sequences were assigned to the most abundant IGH V(D)J sequence by UMI count, and ties were broken by the first identified heavy chain. Non-functional sequences were removed. V(D)J sequences within each patient were grouped into clonal clusters by first partitioning based on common IGHV gene annotations, IGHJ gene annotations, and junction lengths. Within these groups, sequences differing from one another by a length normalized Hamming distance of 0.15 within the junction region were defined as clones by single-linkage clustering using Change-O v.1.0.0.53 Germline sequences were reconstructed for each clone with the D segment and N/P regions masked (replaced with “N” nucleotides) using the CreateGermlines.py function within Change-O v.1.0.0.
Somatic hypermutation frequency was calculated using SHazaM v1.0.2.99954 as the frequency of non-ambiguous nucleotide differences along the IGHV gene segment (IMGT positions 1-312) between each sequence and its inferred germline ancestor. To identify unmutated B cell clones of different cell types and isotypes, B cell clones were separated by cell type and isotype and considered “unmutated” if the median somatic hypermutation frequency of their constituent sequences was <1%. This cutoff was also used in.5 , 62 To quantify B cell clonal diversity, we calculated Simpson’s diversity within each patient for plasmablast subsets using the alphaDiversity function of Alakazam v1.0.2.999.53 To account for differences in sequence depth, the number of sequences within each patient were down-sampled to the same number of sequences, and the mean of 100 such re-sampling repetitions was reported. Only patients with at least 30 B cells were included in diversity calculations. All statistical analyses of BCR sequences were performed with R (v3.6.1).
Bulk RNA-seq
cDNA and library preparation and sequencing
RNAs were isolated using RNeasy Plus Micro Kit (QIAGEN) and cDNAs were generated using the SMART-Seq v4 Ultra Low Input RNA Kit for sequencing (Takara/Clontech). Barcoded libraries were generated by the Nextera XT DNA Library Preparation kit (Illumina) and sequenced with a 2x100 bp paired-end protocol on the HiSeq 4000 Sequencing System (Illumina).
Bulk RNA-seq data analysis
Low quality ends (less than phred score = 30) and short read length (minimum length = 30) was trimmed using PRINSEQ++55 (version1.2). Trimmed reads were aligned to the hg38 genome reference using STAR56 (v2.7.1), and subsequently RSEM (RNA-Seq by Expectation Maximization)57 was used to count reads mapping to the genes from Ensembl release 93. We applied limma to model each gene as a linear combination of donor-specific effects. Top 1000 genes by variance were analyzed for PCA. Heat maps show row-normalized relative gene expression z-scores across columns. Pairwise differential expression was performed using the R package DESeq2.58 The cutoff value to select differentially expressed genes is provided in each figure legend.
Pathway analysis
The differentially expressed genes upregulated in PD-1highCXCR5−CD4+ T cells compared with PD-1highCXCR5+CD4+ T cells were inputted to Enrichr59 to calculate enrichment of pathway-associated terms.
Quantification and statistical analysis
All statistical analyses were performed using R or GraphPad Prism 7 (GraphPad Software). Detailed information about statistical analysis, including tests and values used, is provided
in the figure legends.
Acknowledgments
We would like to thank all the hospital staff who helped care for the patients and obtain samples; all the members of the YALE IMPACT research team who obtained data, P.T.H. Giang and D. Dimitri for the organization of additional sample collection for revised experiments, Dr. K. O’Connor for feedback and discussions, Dr. P. Coish for his proofreading, Drs. L. Devine and C. Wang for assistance with cell sorting, Drs. G. Wang and C. Castaldi at the Yale Center for Genome Analysis for support with 10x Genomics library preparation and sequencing, and M. Zhang for preparation of bulk RNA-seq libraries and sequencing. D.A.H., A.I., S.H.K., R.R.M., and A.C.S. thank the HIPC Consortium for valuable input. This study was supported by grants to H.A. from Daiichi Sankyo Foundation of Life Science and Uehara Memorial Foundation for his scholarship; to D.A.H. from the National Institutes of Health (NIH) (U19 AI089992, R25 NS079193, P01 AI073748, U24 AI11867, R01 AI22220, UM 1HG009390, P01 AI039671, P50 CA121974, and R01 CA227473), the National Multiple Sclerosis Society (NMSS) (CA 1061- A-18 and RG-1802-30153), the Nancy Taylor Foundation for Chronic Diseases, and Erase MS; to N.K. from NIH (R01HL127349, R01HL141852, and U01HL145567); to K.B.H. and S.H.K. from NIH (R01AI104739); to A.I. from NIH (R01AI157488 and R01NS111242); and to A.C.S. from NIH (K24AG042489). RNA-seq service was conducted at the Yale Center for Genome Analysis and the Yale Stem Cell Center Genomics Core facility, with the latter supported by the Connecticut Regenerative Medicine Research Fund and the Li Ka Shing Foundation.
Author contributions
Overall study design, H.A., D.A.H., and T.S.S.; biospecimen collection/processing, H.A., S.M., M.C., W.E.R., P.W., J.K., C.L., K.R., O.C., A.U., B.E., R.R.M., A.I., A.C.S., and T.S.S.; data analysis, H.A., W.E.R., K.B.H., I.C., S.C., N.L., L.G., S.H.K., and T.S.S.; original draft writing, H.A.; supervising the study, D.A.H. and T.S.S.; reviewing and editing the manuscript, all authors.
Declaration of interests
D.A.H. has received research funding from Bristol-Myers Squibb, Novartis, Sanofi, and Genentech. He has been a consultant for Bayer Pharmaceuticals, Bristol Myers Squibb, Compass Therapeutics, EMD Serono, Genentech, Juno Therapeutics, Novartis Pharmaceuticals, Proclara Biosciences, Sage Therapeutics, and Sanofi Genzyme. Further information regarding funding is available at https://openpaymentsdata.cms.gov/physician/166753/general-payments. N.K. reports personal fees from Boehringer Ingelheim, Third Rock, Pliant, Samumed, NuMedii, Indalo, Theravance, LifeMax, Three Lake Partners, and RohBar in the last 36 months and Equity in Pliant. N.K. is also a recipient of a grant from Veracyte and non-financial support from Miragen. In addition, N.K. has patents on New Therapies in Pulmonary Fibrosis and ARDS (unlicensed) and Peripheral Blood Gene Expression as biomarkers in IPF (licensed to biotech), all outside the submitted work. S.H.K. receives consulting fees from Northrop Grumman. K.B.H. receives consulting fees from Prellis Biologics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111895.
Supplemental information
Data and code availability
Single-cell RNA-seq data have been deposited at GEO database under accession code: GSE155224. Bulk RNA-seq data are also available under accession code: GSE214624. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Wu Z., McGoogan J.M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese center for disease control and prevention. JAMA. 2020;323:1239–1242. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 2.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., et al. Novel coronavirus from patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi T., Ellingson M.K., Wong P., Israelow B., Lucas C., Klein J., Silva J., Mao T., Oh J.E., Tokuyama M., Lu P., Venkataraman A., Park A., Liu F., Meir A., Sun J., Wang E.Y., Casanovas-Massana A., Wyllie A.L., Vogels C.B.F., Earnest R., Lapidus S., Ott I.M., Moore A.J., Yale IMPACT Research Team. Shaw A., Fournier J.B., Odio C.D., Farhadian S., Dela Cruz C., Grubaugh N.D., Schulz W.L., Ring A.M., Ko A.I., Omer S.B., Iwasaki A. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nature. 2020;588:315–320. doi: 10.1038/s41586-020-2700-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giamarellos-Bourboulis E.J., Netea M.G., Rovina N., Akinosoglou K., Antoniadou A., Antonakos N., Damoraki G., Gkavogianni T., Adami M.E., Katsaounou P., et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27:992–1000.e3. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Unterman A., Sumida T.S., Nouri N., Yan X., Zhao A.Y., Gasque V., Schupp J.C., Asashima H., Liu Y., Cosme C., et al. Single-cell multi-omics reveals dyssynchrony of the innate and adaptive immune system in progressive COVID-19. Nat. Commun. 2022;13:440. doi: 10.1038/s41467-021-27716-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathew D., Giles J.R., Baxter A.E., Oldridge D.A., Greenplate A.R., Wu J.E., Alanio C., Kuri-Cervantes L., Pampena M.B., D’Andrea K., et al. Deep immune profiling of COVID-19 patients reveals patient heterogeneity and distinct immunotypes with implications for therapeutic interventions. Science. 2020;369:eabc8511. doi: 10.1126/science.abc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McElroy A.K., Akondy R.S., Davis C.W., Ellebedy A.H., Mehta A.K., Kraft C.S., Lyon G.M., Ribner B.S., Varkey J., Sidney J., et al. Human Ebola virus infection results in substantial immune activation. Proc. Natl. Acad. Sci. USA. 2015;112:4719–4724. doi: 10.1073/pnas.1502619112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wrammert J., Onlamoon N., Akondy R.S., Perng G.C., Polsrila K., Chandele A., Kwissa M., Pulendran B., Wilson P.C., Wittawatmongkol O., et al. Rapid and massive virus-specific plasmablast responses during acute dengue virus infection in humans. J. Virol. 2012;86:2911–2918. doi: 10.1128/JVI.06075-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alon R., Sportiello M., Kozlovski S., Kumar A., Reilly E.C., Zarbock A., Garbi N., Topham D.J. Leukocyte trafficking to the lungs and beyond: lessons from influenza for COVID-19. Nat. Rev. Immunol. 2021;21:49–64. doi: 10.1038/s41577-020-00470-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kunkel E.J., Butcher E.C. Plasma-cell homing. Nat. Rev. Immunol. 2003;3:822–829. doi: 10.1038/nri1203. [DOI] [PubMed] [Google Scholar]
- 11.Nutt S.L., Hodgkin P.D., Tarlinton D.M., Corcoran L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015;15:160–171. doi: 10.1038/nri3795. [DOI] [PubMed] [Google Scholar]
- 12.Lerner R.A. Rare antibodies from combinatorial libraries suggests an S.O.S. component of the human immunological repertoire. Mol. Biosyst. 2011;7:1004–1012. doi: 10.1039/c0mb00310g. [DOI] [PubMed] [Google Scholar]
- 13.Murin C.D., Wilson I.A., Ward A.B. Antibody responses to viral infections: a structural perspective across three different enveloped viruses. Nat. Microbiol. 2019;4:734–747. doi: 10.1038/s41564-019-0392-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lucas C., Klein J., Sundaram M.E., Liu F., Wong P., Silva J., Mao T., Oh J.E., Mohanty S., Huang J., et al. Delayed production of neutralizing antibodies correlates with fatal COVID-19. Nat. Med. 2021;27:1178–1186. doi: 10.1038/s41591-021-01355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khoury D.S., Cromer D., Reynaldi A., Schlub T.E., Wheatley A.K., Juno J.A., Subbarao K., Kent S.J., Triccas J.A., Davenport M.P. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 2021;27:1205–1211. doi: 10.1038/s41591-021-01377-8. [DOI] [PubMed] [Google Scholar]
- 16.Ueno H. T follicular helper cells in human autoimmunity. Curr. Opin. Immunol. 2016;43:24–31. doi: 10.1016/j.coi.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Crotty S. T follicular helper cell Biology : a decade of discovery and diseases. Immunity. 2019;50:1132–1148. doi: 10.1016/j.immuni.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morita R., Schmitt N., Bentebibel S.E., Ranganathan R., Bourdery L., Zurawski G., Foucat E., Dullaers M., Oh S., Sabzghabaei N., et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34:108–121. doi: 10.1016/j.immuni.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Juno J.A., Tan H.X., Lee W.S., Reynaldi A., Kelly H.G., Wragg K., Esterbauer R., Kent H.E., Batten C.J., Mordant F.L., et al. Humoral and circulating follicular helper T cell responses in recovered patients with COVID-19. Nat. Med. 2020;26:1428–1434. doi: 10.1038/s41591-020-0995-0. [DOI] [PubMed] [Google Scholar]
- 20.Chen J.S., Chow R.D., Song E., Mao T., Israelow B., Kamath K., Bozekowski J., Haynes W.A., Filler R.B., Menasche B.L., et al. High-affinity, neutralizing antibodies to SARS-CoV-2 can be made without T follicular helper cells. Sci. Immunol. 2022;7:eabl5652. doi: 10.1126/sciimmunol.abl5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rao D.A., Gurish M.F., Marshall J.L., Slowikowski K., Fonseka C.Y., Liu Y., Donlin L.T., Henderson L.A., Wei K., Mizoguchi F., et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542:110–114. doi: 10.1038/nature20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshitomi H., Ueno H. Shared and distinct roles of T peripheral helper and T follicular helper cells in human diseases. Cell. Mol. Immunol. 2021;18:523–527. doi: 10.1038/s41423-020-00529-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Locci M., Havenar-Daughton C., Landais E., Wu J., Kroenke M.A., Arlehamn C.L., Su L.F., Cubas R., Davis M.M., Sette A., et al. Human circulating PD-1+CXCR3−CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. 2013;39:758–769. doi: 10.1016/j.immuni.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ueno H., Banchereau J., Vinuesa C.G. Pathophysiology of T follicular helper cells in humans and mice. Nat. Immunol. 2015;16:142–152. doi: 10.1038/ni.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaneko N., Kuo H.H., Boucau J., Farmer J.R., Allard-Chamard H., Mahajan V.S., Piechocka-Trocha A., Lefteri K., Osborn M., Bals J., et al. Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell. 2020;183:143–157.e13. doi: 10.1016/j.cell.2020.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koutsakos M., Rowntree L.C., Hensen L., Chua B.Y., van de Sandt C.E., Habel J.R., Zhang W., Jia X., Kedzierski L., Ashhurst T.M., et al. Integrated immune dynamics define correlates of COVID-19 severity and antibody responses. Cell Rep. Med. 2021;2:100208. doi: 10.1016/j.xcrm.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gong F., Dai Y., Zheng T., Cheng L., Zhao D., Wang H., Liu M., Pei H., Jin T., Yu D., Zhou P. Peripheral CD4+ T cell subsets and antibody response in COVID-19 convalescent individuals. J. Clin. Invest. 2020;130:6588–6599. doi: 10.1172/JCI141054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J.Y., Wang X.M., Xing X., Xu Z., Zhang C., Song J.W., Fan X., Xia P., Fu J.L., Wang S.Y., et al. Single-cell landscape of immunological responses in patients with COVID-19. Nat. Immunol. 2020;21:1107–1118. doi: 10.1038/s41590-020-0762-x. [DOI] [PubMed] [Google Scholar]
- 30.Kim C.C., Baccarella A.M., Bayat A., Pepper M., Fontana M.F. FCRL5+ memory B cells exhibit robust recall responses. Cell Rep. 2019;27:1446–1460.e4. doi: 10.1016/j.celrep.2019.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pérez-Mazliah D., Ndungu F.M., Aye R., Langhorne J. B-cell memory in malaria: myths and realities. Immunol. Rev. 2020;293:57–69. doi: 10.1111/imr.12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenks S.A., Cashman K.S., Zumaquero E., Marigorta U.M., Patel A.V., Wang X., Tomar D., Woodruff M.C., Simon Z., Bugrovsky R., et al. Distinct effector B cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity. 2018;49:725–739.e6. doi: 10.1016/j.immuni.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riedel R., Addo R., Ferreira-Gomes M., Heinz G.A., Heinrich F., Kummer J., Greiff V., Schulz D., Klaeden C., Cornelis R., et al. Discrete populations of isotype-switched memory B lymphocytes are maintained in murine spleen and bone marrow. Nat. Commun. 2020;11:2570. doi: 10.1038/s41467-020-16464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanz I., Wei C., Jenks S.A., Cashman K.S., Tipton C., Woodruff M.C., Hom J., Lee F.E.H. Challenges and opportunities for consistent classification of human B cell and plasma cell populations. Front. Immunol. 2019;10:2458. doi: 10.3389/fimmu.2019.02458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pattanapanyasat K., Khowawisetsut L., Chuansumrit A., Chokephaibulkit K., Tangnararatchakit K., Apiwattanakul N., Techasaensiri C., Thitilertdecha P., Sae-Ung T., Onlamoon N. B cell subset alteration and the expression of tissue homing molecules in dengue infected patients. J. Biomed. Sci. 2018;25:64. doi: 10.1186/s12929-018-0467-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glass D.R., Tsai A.G., Oliveria J.P., Hartmann F.J., Kimmey S.C., Calderon A.A., Borges L., Glass M.C., Wagar L.E., Davis M.M., Bendall S.C. An integrated multi-omic single-cell atlas of human B cell identity. Immunity. 2020;53:217–232.e5. doi: 10.1016/j.immuni.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muehlinghaus G., Cigliano L., Huehn S., Peddinghaus A., Leyendeckers H., Hauser A.E., Hiepe F., Radbruch A., Arce S., Manz R.A. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood. 2005;105:3965–3971. doi: 10.1182/blood-2004-08-2992. [DOI] [PubMed] [Google Scholar]
- 38.Bocharnikov A.V., Keegan J., Wacleche V.S., Cao Y., Fonseka C.Y., Wang G., Muise E.S., Zhang K.X., Arazi A., Keras G., et al. PD-1hiCXCR5– T peripheral helper cells promote B cell responses in lupus via MAF and IL-21. JCI Insight. 2019;4:e130062. doi: 10.1172/jci.insight.130062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conigliaro P., Perricone C., Benson R.A., Garside P., Brewer J.M., Perricone R., Valesini G. The type I IFN system in rheumatoid arthritis. Autoimmunity. 2010;43:220–225. doi: 10.3109/08916930903510914. [DOI] [PubMed] [Google Scholar]
- 40.Muskardin T.L.W., Niewold T.B. Type I interferon in rheumatic diseases. Nat. Rev. Rheumatol. 2018;14:214–228. doi: 10.1038/nrrheum.2018.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmitt N., Liu Y., Bentebibel S.E., Munagala I., Bourdery L., Venuprasad K., Banchereau J., Ueno H. The cytokine TGF-β2 co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFHcells. Nat. Immunol. 2014;15:856–865. doi: 10.1038/ni.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sumida T.S., Dulberg S., Schupp J.C., Lincoln M.R., Stillwell H.A., Axisa P.P., Comi M., Unterman A., Kaminski N., Madi A., et al. Type I interferon transcriptional network regulates expression of coinhibitory receptors in human T cells. Nat. Immunol. 2022;23:632–642. doi: 10.1038/s41590-022-01152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cunningham A.F., Gaspal F., Serre K., Mohr E., Henderson I.R., Scott-Tucker A., Kenny S.M., Khan M., Toellner K.M., Lane P.J.L., MacLennan I.C.M. Salmonella induces a switched antibody response without germinal centers that impedes the extracellular spread of infection. J. Immunol. 2007;178:6200–6207. doi: 10.4049/jimmunol.178.10.6200. [DOI] [PubMed] [Google Scholar]
- 44.Kalinke U., Bucher E.M., Ernst B., Oxenius A., Roost H.P., Geley S., Kofler R., Zinkernagel R.M., Hengartner H. The role of somatic mutation in the generation of the protective humoral immune response against vesicular stomatitis virus. Immunity. 1996;5:639–652. doi: 10.1016/s1074-7613(00)80277-0. [DOI] [PubMed] [Google Scholar]
- 45.Kalinke U., Oxenius A., Lopez-Macias C., Zinkernagel R.M., Hengartner H. Virus neutralization by germ-line vs. hypermutated antibodies. Proc. Natl. Acad. Sci. USA. 2000;97:10126–10131. doi: 10.1073/pnas.97.18.10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y., Tong X., Li Y., Gu B., Yan J., Liu Y., Shen H., Huang R., Wu C. A comprehensive, longitudinal analysis of humoral responses specific to four recombinant antigens of SARS-CoV-2 in severe and non-severe COVID-19 patients. PLoS Pathog. 2020;16:e1008796. doi: 10.1371/journal.ppat.1008796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kofler D.M., Severson C.A., Mousissian N., De Jager P.L., Hafler D.A. The CD6 multiple sclerosis susceptibility allele is associated with alterations in CD4+ T cell proliferation. J. Immunol. 2011;187:3286–3291. doi: 10.4049/jimmunol.1100626. [DOI] [PubMed] [Google Scholar]
- 48.Karakas Celik S., Cakmak Genc G., Dursun A. A bioinformatic approach to investigating cytokine genes and their receptor variants in relation to COVID-19 progression. Int. J. Immunogenet. 2021;48:211–218. doi: 10.1111/iji.12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woodruff M.C., Ramonell R.P., Nguyen D.C., Cashman K.S., Saini A.S., Haddad N.S., Ley A.M., Kyu S., Howell J.C., Ozturk T., et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 2020;21:1506–1516. doi: 10.1038/s41590-020-00814-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., III, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giudicelli V., Chaume D., Lefranc M.P. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res. 2005;33:D256–D261. doi: 10.1093/nar/gki010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ye J., Ma N., Madden T.L., Ostell J.M. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41:W34–W40. doi: 10.1093/nar/gkt382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gupta N.T., Vander Heiden J.A., Uduman M., Gadala-Maria D., Yaari G., Kleinstein S.H. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics. 2015;31:3356–3358. doi: 10.1093/bioinformatics/btv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yaari G., Vander Heiden J.A., Uduman M., Gadala-Maria D., Gupta N., Stern J.N.H., O’Connor K.C., Hafler D.A., Laserson U., Vigneault F., Kleinstein S.H. Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front. Immunol. 2013;4:358. doi: 10.3389/fimmu.2013.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cantu V.A., Sadural J., Edwards R. PRINSEQ++, a multi-threaded tool for fast and efficient quality control and preprocessing of sequencing datasets. PeerJ Prepr. 2019;7:e27553v1. doi: 10.7287/peerj.preprints.27553v1. [DOI] [Google Scholar]
- 56.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Remy M.M., Alfter M., Chiem M.N., Barbani M.T., Engler O.B., Suter-Riniker F. Effective chemical virus inactivation of patient serum compatible with accurate serodiagnosis of infections. Clin. Microbiol. Infect. 2019;25 doi: 10.1016/j.cmi.2018.10.016. 907.e7-907907.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nielsen S.C.A., Yang F., Jackson K.J.L., Hoh R.A., Röltgen K., Jean G.H., Stevens B.A., Lee J.Y., Rustagi A., Rogers A.J., et al. Human B cell clonal expansion and convergent antibody responses to SARS-CoV-2. Cell Host Microbe. 2020;28:516–525.e5. doi: 10.1016/j.chom.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO database under accession code: GSE155224. Bulk RNA-seq data are also available under accession code: GSE214624. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.