

SUMMARY
The p53 transcription factor drives anti-proliferative gene expression programs in response to diverse stressors, including DNA damage and oncogenic signaling. Here, we seek to uncover new mechanisms through which p53 regulates gene expression using tandem affinity purification/mass spectrometry to identify p53-interacting proteins. This approach identified METTL3, an m6A RNA-methyltransferase complex (MTC) constituent, as a p53 interactor. We find that METTL3 promotes p53 protein stabilization and target gene expression in response to DNA damage and oncogenic signals, by both catalytic activity-dependent and independent mechanisms. METTL3 also enhances p53 tumor suppressor activity in in vivo mouse cancer models and human cancer cells. Notably, METTL3 only promotes tumor suppression in the context of intact p53. Analysis of human cancer genome data further supports the notion that the MTC reinforces p53 function in human cancer. Together, these studies reveal a fundamental role for METTL3 in amplifying p53 signaling in response to cellular stress.
Graphical Abstract
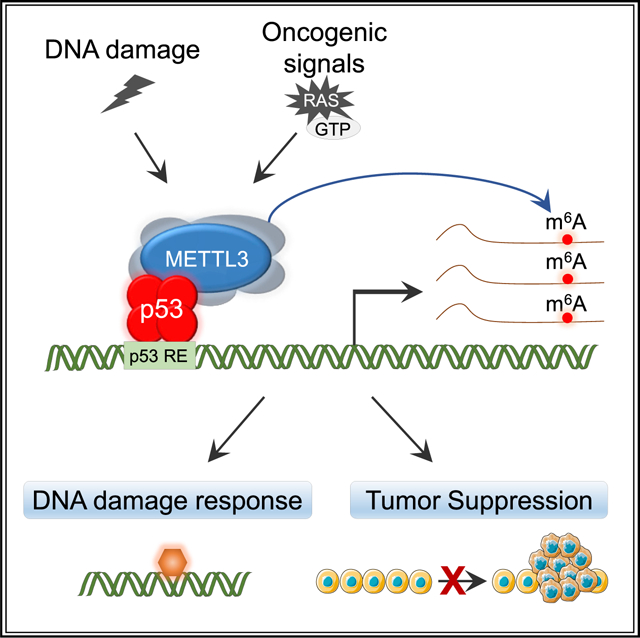
In brief
p53 is a transcriptional activator that suppresses tumorigenesis through the regulation of target genes with diverse biological functions. Raj et al. uncover METTL3, an RNA-methyltransferase, as a binding partner of p53 that reinforces p53 transcriptional activity and highlight a role for METTL3 in p53-mediated tumor suppression in mice and humans.
INTRODUCTION
The TP53 gene, which encodes the p53 protein, is mutated in over half of all human cancers, reflecting its fundamental role as a tumor suppressor (Kandoth et al., 2013). p53 is a transcription factor that integrates cellular stress cues such as DNA damage and oncogenic signaling to drive anti-proliferative or pro-apoptotic responses associated with tissue homeostasis and tumor suppression (Bieging et al., 2014; Kastenhuber and Lowe, 2017; Vousden and Prives, 2009). p53 activity as a transcriptional activator is critical for governing gene expression programs that limit tumorigenesis, as evidenced by the preponderance of TP53 mutations observed in sequences encoding the DNA-binding domain in human cancers and studies of transactivation-dead p53 mutant knock-in mice, which are highly tumor prone (Brady et al., 2011; Jiang et al., 2011; Mello et al., 2017b; Olivier et al., 2010). Despite this well-established role for p53 transcriptional activation function in tumor suppression, the mechanisms through which p53 regulates downstream gene expression programs remain only superficially understood. A complete understanding of the mechanisms of p53-regulated gene expression is of paramount importance for ultimately targeting this critical tumor suppressor in the clinic (Sabapathy and Lane, 2018).
p53 promotes specific gene expression programs through recruitment of the general transcriptional machinery via interaction with cofactors such as mediator subunits and TBP-associated factors (TAFs) to increase levels of transcriptional initiation (Espinosa et al., 2003; Haberle and Stark, 2018). p53 also recruits histone-modifying enzymes, including p300, CBP, and PCAF, to acetylate histones and remodel chromatin (Raj and Attardi, 2017). However, the mechanisms by which p53 regulates gene expression beyond effects on transcriptional initiation remain much less characterized. Recent studies have suggested that p53 acts at other levels of gene regulation. For example, p53 induces a splicing factor, ZMAT3, that modulates alternative splicing and is critical for tumor suppression (Bieging-Rolett et al., 2020). In response to ribosome dysfunction, p53 globally inhibits protein translation through the induction of a translational repressor, 4E-BP1 (Tiu et al., 2021). Elucidating and characterizing additional aspects of p53 activity will provide key insight into p53 function in tissue homeostasis and cancer suppression. The ability of eukaryotic gene expression to be regulated at multiple levels provides maximal capacity for fine-tuning in response to diverse signals such as DNA damage and oncogene expression (Vihervaara et al., 2018).
While the ultimate gene expression profile of a cell has long been known to be influenced by modulation of transcription, RNA splicing, and protein translation or stability, recent work has illuminated the importance of RNA modification in the regulation of gene expression. Specifically, N6-methyladenosine (m6A), the most abundant and prevalent internal modification in eukaryotic mRNAs, is installed on mRNAs co-transcriptionally, primarily by the METTL3-METTL14 methyltransferase writer complex. Depending on the context and the m6A readers expressed in a given setting, m6A modification can then affect gene expression through effects on RNA stability, subcellular localization, and translation. Importantly, m6A modification has been demonstrated to be critical for cell state transitions, such as during differentiation of stem cells and in response to a variety of stress signals, including DNA damage or heat shock (Shi et al., 2019). However, the factors that govern which transcripts are selected for m6A modification to drive specific cellular responses remain enigmatic.
Here, we sought to gain new insight into how p53 regulates gene expression programs in the context of stress signals using tandem affinity purification coupled with mass spectrometry to identify novel p53-interacting partners. To ensure that p53 pathways remain intact, we performed experiments using untransformed cells. We thus identified the METTL3 m6A RNA-methyltransferase as a p53-interacting protein. Remarkably, analysis of METTL3-deficient cells revealed that both p53 stabilization and p53 target gene induction by either DNA damage or oncogenic signals are significantly compromised by METTL3 deficiency. Interestingly, METTL3 binds to the p53 amino terminus, leading to p53 stabilization and co-transcriptional m6A modification of downstream components of the p53 pathway, thus reinforcing p53 function in response to DNA damage and in tumor suppression, both in mice and humans. Collectively, these studies reveal a fundamental role for METTL3 in amplifying p53 signaling, both through protein stabilization and epitranscriptome regulation.
RESULTS
METTL3 interacts with p53 and enhances p53 transcriptional activity
To identify novel p53-interacting partners, we expressed LAP (localization and affinity purification)-tagged p53 in untransformed Flp-In-3T3 fibroblasts and treated cells with the DNA double-strand break-inducer doxorubicin (dox), which activates p53 and triggers p53-dependent G1 arrest, providing a good model for understanding the biochemical basis for p53 action (Johnson et al., 2005). After p53-LAP purification and mass spectrometry, performed in triplicate, we identified numerous p53-associated proteins, including METTL3, the catalytic component of the core methyltransferase complex that installs N6-methyladenosine (m6A) on eukaryotic mRNAs (Shi et al., 2019), as a p53-interacting partner under DNA damage conditions (Figure 1A; Table S1). Individual immunoprecipitation (IP) assays showed that METTL3 interacts with LAP-tagged p53 in both untreated and dox-treated Flp-In-3T3s (Figure 1B). We confirmed the p53-METTL3 interaction by additional co-immunoprecipitation (coIP) assays after co-expressing hemagglutinin (HA)-tagged p53 and FLAG-tagged METTL3 in Flp-In-3T3 p53−/− cells (Figure S1A). We tested the ability of endogenous p53 and METTL3 to interact in primary mouse embryonic fibroblasts (MEFs) transformed by expressing the oncogenes E1A and HrasG12V, a model system we selected because of the well-characterized role for p53 in response to DNA damage and in transformation suppression (see below). Indeed, in this context, p53 interacted with METTL3 (Figure 1C). We next sought to establish whether the interaction between p53 and METTL3 is direct or could potentially be mediated by chromatin or a cellular protein. First, we found that the METTL3-p53 interaction is not indirectly mediated by DNA, as the interaction is still observed upon ethidium bromide or DNase treatment (Figures S1B and S1C). Second, we tested whether this interaction is direct, using glutathione S-transferase (GST) pull-down assays to assess the interaction between bacterially expressed, purified GST-p53 and in vitro-translated, HA-tagged METTL3, with His-tagged HDM2 as a positive control. We found that HA-METTL3 bound to GST-p53 but not GST alone, suggesting that METTL3 and p53 interact directly (Figures S1D and S1E).
Figure 1. METTL3 interacts with p53 and enhances p53 protein accumulation.
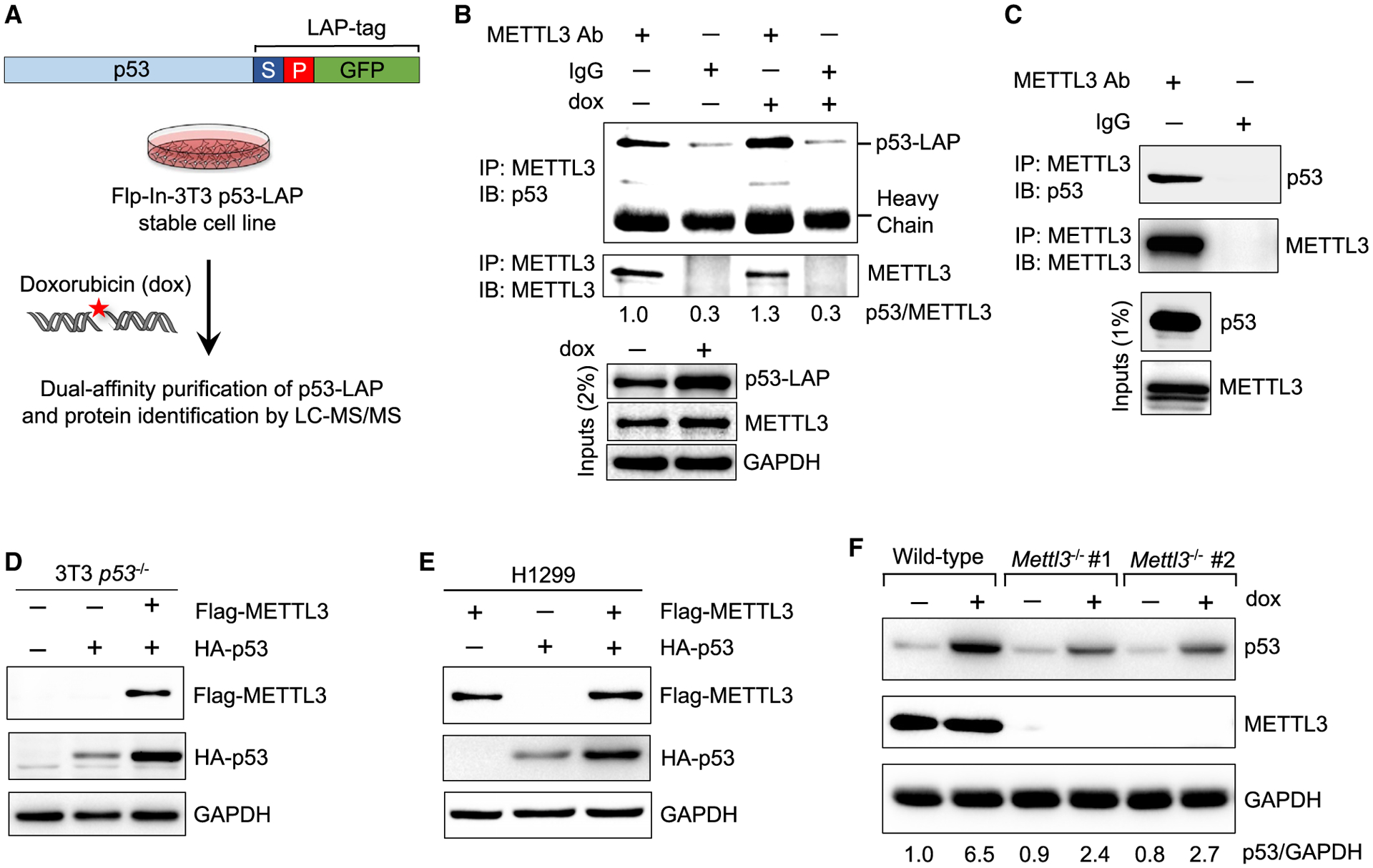
(A) Schematic of dual-affinity purification of LAP-tagged p53 protein in Flp-In-3T3 fibroblasts. Cells were treated with 0.2 μg/mL doxorubicin (dox) for 6 h, followed by dual-affinity purification of p53-bound protein complexes and protein identification by LC-MS/MS.
(B) Co-immunoprecipitation (coIP) and immunoblot assay to examine p53-LAP and endogenous METTL3 interaction in untreated and DNA damage-treated Flp-In-3T3 cells. Numbers indicate the amount of p53 co-precipitated relative to METTL3 (n = 2).
(C) CoIP and immunoblot assay to examine interaction of endogenous METTL3 and p53 in E1A;HRasG12V-expressing MEFs (n = 2).
(D) Immunoblot after transfection of FLAG-METTL3 and HA-p53 plasmids into Flp-In-3T3 p53−/− cells (n = 2).
(E) Immunoblot after co-transfection of FLAG-METTL3 and HA-p53 plasmids into H1299 cells (n = 3).
(F) p53 induction after 6 h dox (0.2 μg/mL) in wild-type (WT) and two different Mettl3−/− ES cell lines. Numbers indicate the amount of p53 relative to glycer-aldehyde 3-phosphate dehydrogenase (abbreviated GAPDH) loading control (n = 3). Representative immunoblots are shown in (B)–(F), and GAPDH serves as a loading control.
See also Figures S1 and S2A and Table S1.
Interestingly, upon co-expression of p53 and METTL3 in p53-deficient human or mouse cells, we noted that METTL3 enhances p53 protein levels, suggesting that METTL3 might affect p53 function (Figures 1D and 1E). To test this hypothesis, we assessed the consequences of METTL3 deficiency for p53 function. We first leveraged two different Mettl3-null embryonic stem (ES) cell lines and assessed how METTL3 loss affects p53 responses to DNA damage (Batista et al., 2014). Specifically, upon DNA damage signals, p53 protein levels rise due to enhanced stability, and p53 induces expression of a host of downstream target genes. We found that Mettl3 nullizygosity results in decreased p53 accumulation in response to DNA damage, which is accompanied by diminished induction of various p53 target genes, indicating that METTL3 enhances p53 protein accumulation and transcriptional activity in ES cells (Figures 1F and S2A).
To determine whether this response extends to other cell types, we next examined the tractable E1A;HRasG12V MEF model. E1A;HrasG12V MEFs expressing a control shRNA or shRNAs directed against Mettl3 to acutely knock down Mettl3, were either left untreated or treated for 6 h with dox to induce p53 accumulation and target gene activation (Figure 2A). As in ES cells, we found that Mettl3 knockdown results in reduced p53 accumulation in DNA damage-treated E1A;HRasG12V MEFs (Figure 2B). We then performed RNA sequencing (RNA-seq) to analyze global gene expression profiles in response to DNA damage in cells with Mettl3 expression or Mettl3 knock-down. Principal component analysis revealed that untreated and dox-treated samples clearly segregated, as expected (Figure 2C). The control and Mettl3 knockdown samples also clustered distinctly, with Mettl3 shRNA 1 and shRNA 2 samples showing a tight association. We next performed differential gene expression analysis to compare the expression patterns of control and Mettl3 knockdown samples under DNA damage conditions. Notably, some of the most statistically significant genes downregulated with Mettl3 knockdown included noncanonical p53 target genes found to be p53-bound and regulated in E1A;HRasG12V MEFs, such as Pltp, Mreg, Jag2, Kcnj4, and Ni-pal4 (Figure 2D; Table S2) (Valente et al., 2020). We next performed unbiased functional annotation analysis to reveal pathways associated with the METTL3-dependent DNA damage response. Interestingly, gene set enrichment analysis of genes downregulated after Mettl3 knockdown under DNA damage conditions revealed that the top pathway in numerous p53 chromatin immunoprecipitation (ChIP)-seq and gene expression data sets was the p53 pathway (Figures 2E, 2F, S2B, and S2C; Table S2). Indeed, analysis of the expression of genes known to be p53-bound and -regulated specifically in E1A;HrasG12V MEFs (Valente et al., 2020) underscored how profoundly Mettl3 knockdown compromises the DNA damage-induced expression but not the basal expression of these genes (Figures 2G and S2D). These results were supported by qRT-PCR analysis of individual p53 target genes (Figure S2E). Collectively, these findings indicate that METTL3 globally enforces robust p53 responses to DNA damage.
Figure 2. METTL3 promotes p53 transcriptional programs in response to DNA damage.
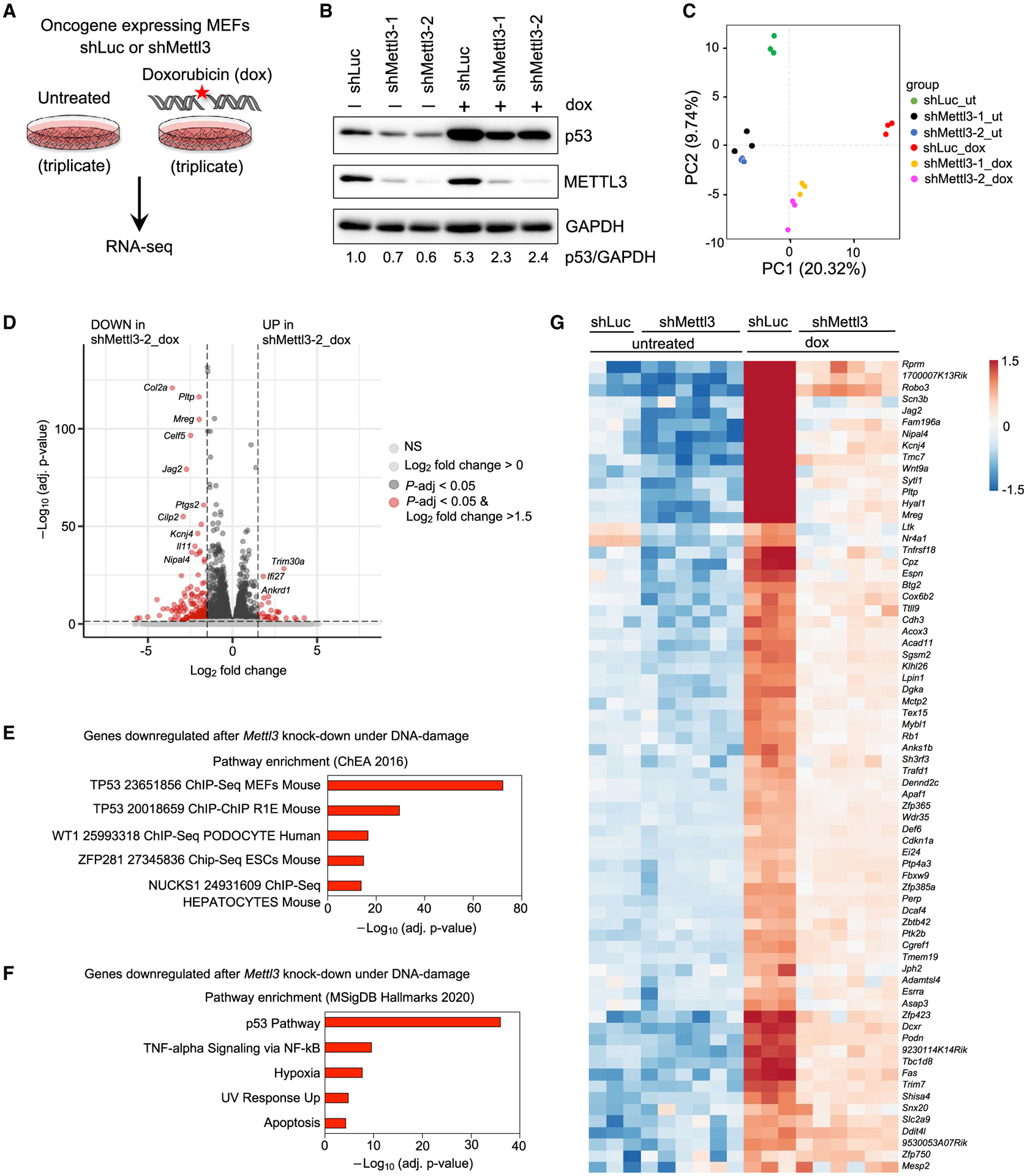
(A) E1A;HRasG12V-expressing WT MEFs transduced with shLuc or shMettl3 hairpins were either left untreated or treated with 0.2 μg/mL dox for 6 h followed by RNA-seq analysis.
(B) Immunoblots showing p53 and METTL3 protein levels in shLuc and shMettl3 shRNA-expressing cell lines. GAPDH serves as a loading control. Representative immunoblots are shown from three biological replicates, and two different MEF lines per genotype were used for this experiment.
(C) Principal component analysis of gene expression profiles of untreated and dox-treated shLuc and shMettl3 shRNA-expressing E1A;HRasG12V MEFs.
(D) Volcano plot showing differentially expressed genes after Mettl3 knockdown under DNA damage conditions. The horizontal dashed line represents the threshold of the adjusted p value of 0.05, while the vertical line represents the threshold of the log2 fold change of greater than 1.5.
(E and F) Functional annotation of genes downregulated in dox-treated shMettl3-2 MEFs relative to dox-treated shLuc MEFs, as identified by Enrichr analysis of ChEA 2016 (E) and MSigDB Hallmarks 2020 (F) data sets.
(G) Heat map showing expression patterns of p53-bound and -regulated target genes in RNA-seq analysis of untreated or dox-treated shLuc and shMettl3 E1A;HRasG12V MEFs.
METTL3 promotes p53 stability through interactions with the p53 amino terminus
We next investigated the molecular mechanisms by which METTL3 enhances p53 protein levels. p53 protein has a short half-life resulting from interaction with the MDM2 ubiquitin ligase, which targets p53 for ubiquitin-mediated proteolysis (Haupt et al., 1997; Kubbutat et al., 1997). In response to stress signals, such as DNA damage, MDM2 is displaced from p53, resulting in enhanced p53 protein stability and activation of p53 transcriptional programs (Momand et al., 1992; Oliner et al., 1993). We therefore sought first to assess whether METTL3 might affect p53 protein stability. We treated ES cells with dox to stabilize p53, then with cycloheximide to block translation, and interrogated how Mettl3 status affects p53 half-life. Interestingly, Mettl3 deficiency was characterized by a diminished p53 half-life, suggesting that METTL3 acts to enhance p53 protein stability (Figures 3A and 3B). To assess whether the enhanced p53 stabilization by METTL3 reflects differences in MDM2 binding, we performed coIP experiments to measure the MDM2-p53 interaction in the presence of Mettl3 or with Mettl3 knockdown. Intriguingly, with Mettl3 knockdown, we found that MDM2 interacts more strongly with p53 upon DNA damage than with Mettl3 expression (Figure 3C). These findings suggest that in response to DNA damage, MDM2 is more efficiently displaced from p53 when METTL3 is present than when it is absent.
Figure 3. METTL3 binds the p53 amino terminus and enhances p53 protein half-life.
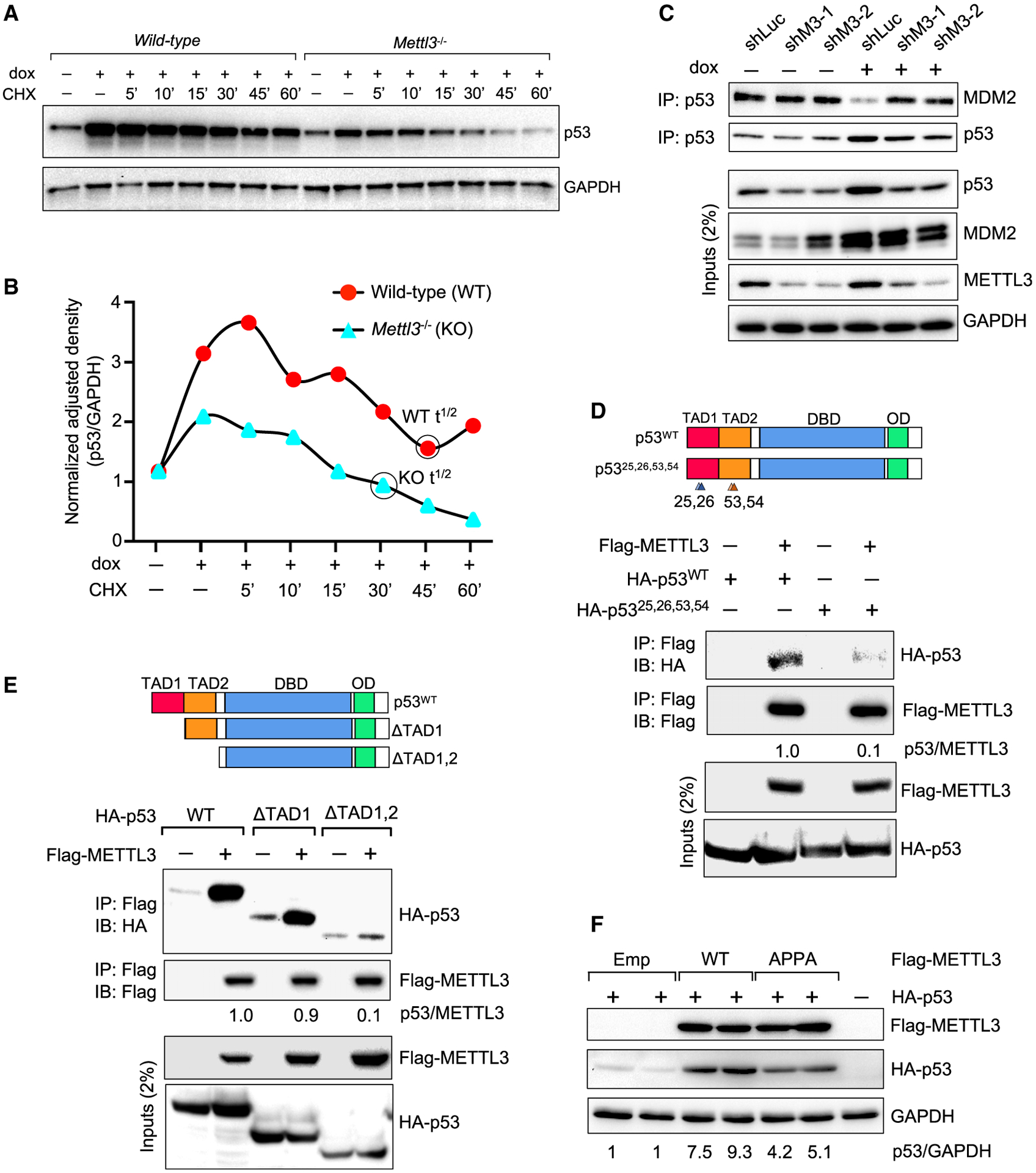
(A and B) Time course immunoblot analysis of p53 levels, normalized to GAPDH (in B), in WT and Mettl3−/− knockout (KO) mouse ES cells after treatment with 0.2 μg/mL dox and 100 μM cycloheximide (CHX) for indicated times (n = 2).
(C) CoIP and immunoblot assays to assess MDM2 pull-down with p53, in E1A;HRasG12V MEFs expressing control shLuc or shMettl3 RNAs, under basal and DNA damage conditions (6 h dox [0.2 μg/mL]). Representative immunoblots are shown from three biological replicates, and two different MEF lines per genotype were used.
(D) CoIP and immunoblot assays in 3T3 p53−/− cells to assess pull-down of HA-p53 WT or transcriptionally dead HA-p5325,26,53,54 (schematized at top) with FLAG-METTL3 (n = 2).
(E) CoIP and immunoblot assays in 3T3 p53−/− cells to assess pull-down of HA-p53 WT or HA-p53 deletion mutants (schematized at top) with FLAG-METTL3 (n = 3). In (D) and (E), numbers indicate the amount of HA-p53 co-immunoprecipitated relative to FLAG-METTL3.
(F) Immunoblot analysis to assess p53 levels upon co-transfection of HA-p53 plasmid and empty vector or vectors expressing FLAG-METTL3 WT or catalytically inactive FLAG-METTL3 (APPA) into p53−/− 3T3 cells. Numbers indicate HA-p53 levels relative to GAPDH. Representative immunoblots are shown and GAPDH serves as a loading control (n = 2).
This observation suggested the possibility that METTL3 and MDM2 might bind to similar regions of p53. We therefore tested whether METTL3 binds to the p53 amino terminus in a similar manner to MDM2. MDM2 binds to the two p53 transcriptional activation domains (TADs) that reside between residues 1–80 (Raj and Attardi, 2017). The interaction of MDM2 with p53 is, in fact, disrupted by mutations in the two TADs at residues L25,Q26 in TAD1 and F53,F54 in TAD2 (Mello et al., 2017b; Raj and Attardi, 2017). Notably, the ability of METTL3 to bind to p53 is reduced by ~50% with the p53L25Q,Q26S,F53Q,F54S mutant (Figure 3D). Moreover, deletion of both p53 TADs virtually abolished the interaction with METTL3 (Figure 3E). These observations, combined with our findings that p53 and METTL3 interact directly, suggest that METTL3 binds to the p53 TADs and support the idea that METTL3 might stabilize p53 by hindering MDM2 binding. If so, then the effects of METTL3 on p53 might be expected to be at least partially independent of its catalytic activity. To test this hypothesis, we used a METTL3 catalytic mutant (“APPA”; METTL3D395A,W398A) that lacks m6A methyltransferase activity (Bujnicki et al., 2002). Indeed, co-expression of this catalytically dead METTL3 mutant protein with p53 results in efficient enhancement of exogenous p53 stabilization, albeit somewhat less than with wild-type (WT) METTL3 (Figure 3F). Together, these findings reveal that METTL3 plays a catalytic activity-independent role to stabilize p53 but also suggest that METTL3 might exert a catalytic activity-dependent role in augmenting the p53 pathway.
METTL3 promotes m6A modification of select p53 pathway transcripts
To interrogate the catalytic activity-dependent role of METTL3 in the p53 pathway, we performed m6A-eCLIP-seq to identify METTL3-dependent m6A RNA modifications occurring after DNA damage. We utilized E1A;HRasG12V MEFs expressing control shRNAs or shRNAs directed against Mettl3 to acutely knock-down Mettl3, and we treated cells 6 h with dox before performing m6A-eCLIP-seq (Figure 4A). We first queried the site of m6A modifications in both samples and found that m6A modifications mapped primarily to the classical DRACH motif and were pre-dominantly localized to the 3′ UTR or last exon of the coding sequence of mRNAs (Figures 4B, 4C, and S3A–S3C). We next characterized the transcripts displaying METTL3-dependent m6A modification in the presence of DNA damage. After normalization of m6A IP signal to input to ensure analysis of differences in m6A modification between samples rather than differences in RNA levels (see STAR Methods), functional annotation analysis of the transcripts with enhanced METTL3-dependent m6A modification under DNA damaging agent conditions revealed various categories, including cell cycle, RNA processing, and transcriptional regulation by p53 (Figure 4D; Table S3). The overlap between the genes that undergo METTL3-dependent m6A modification under DNA damage conditions and 432 genes found previously to be bound and regulated by p53 upon DNA damage in MEFs (Kenzelmann Broz et al., 2013) was statistically significant (Fisher exact test, p = 1.0E-3). The subset of p53 target gene transcripts showing METTL3-dependent m6A modification upon DNA damaging agent treatment included several well-known p53 target genes (Neat1, Ptp4a3, Killer, Mdm2) (Basak et al., 2008; Mello et al., 2017a), as well as a number of tumor suppression-associated target genes we identified previously, including Tex15, Sulf2, Polk, and Slc19a2 (Bieging-Rolett et al., 2020) (Figures 4E and 4F). In contrast, no clear METTL3-dependent m6A modification was detected for the p53 transcript itself upon treating with DNA damage (data not shown).
Figure 4. METTL3-mediated m6A modification of p53 target gene transcripts under DNA damage.
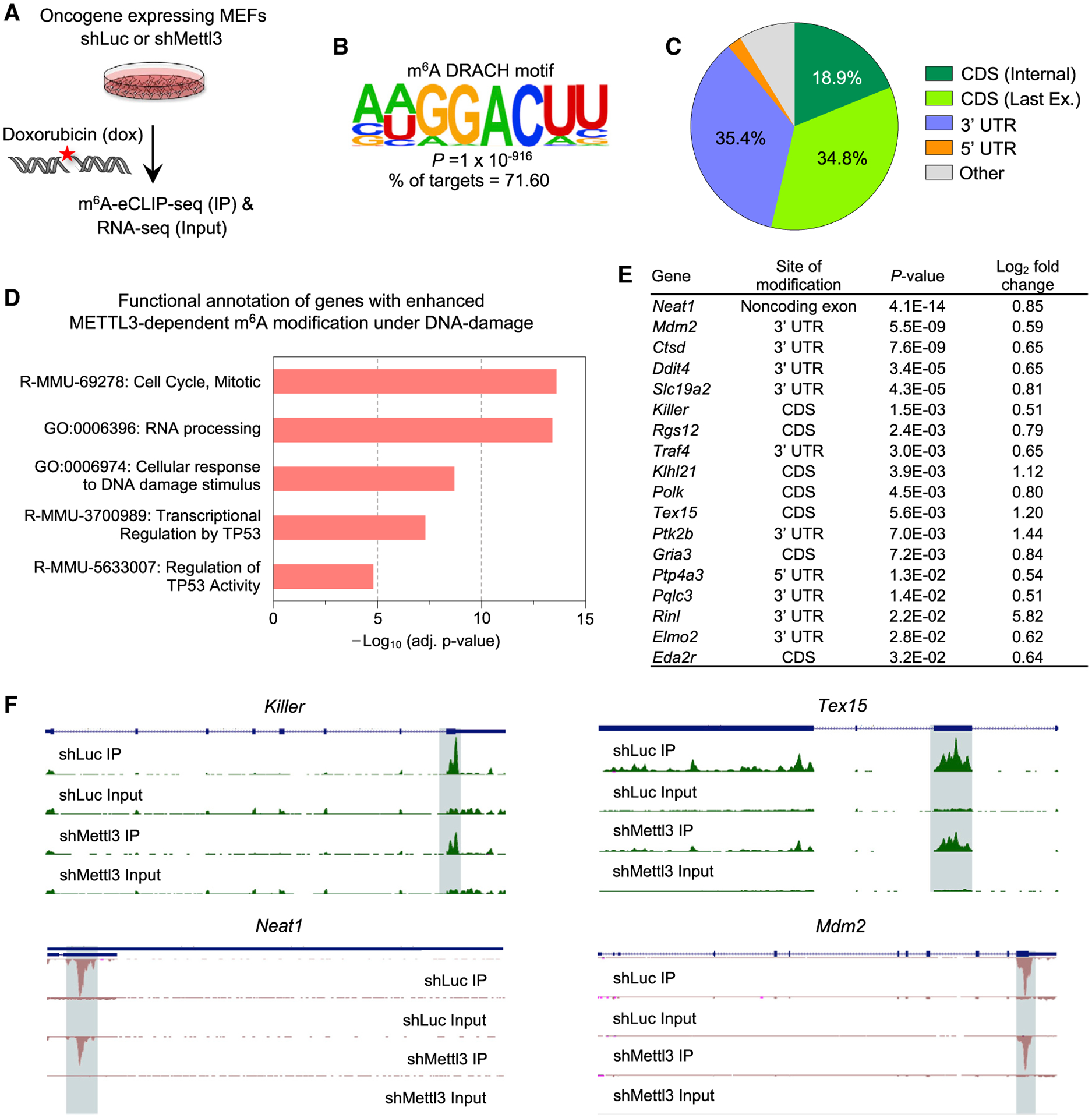
(A) E1A;HRasG12V MEFs transduced with shLuc or shMettl3 hairpins were treated with 0.2 μg/mL dox for 6 h followed by m6A-eCLIP-seq and RNA-seq analysis. m6A-IP reads were normalized to the total number of reads covering the m6A residue in the input sample.
(B) Identification of the known consensus m6A DRACH motif in mRNAs displaying m6A modification in the presence of DNA damage, by performing de novo motif search with the Hypergeometric Optimization of Motif EnRichment (HOMER) database (Heinz et al., 2010).
(C) Pie chart of the distribution of m6A peaks enriched upon DNA damage in E1A;HRasG12V MEFs transduced with the shLuc hairpin.
(D) Functional annotation analysis of METTL3-dependent m6A-modified gene transcripts enriched upon DNA damage, as identified by Metascape analysis.
(E) Table of p53 pathway transcripts with METTL3-dependent m6A modification under acute DNA damage showing the major site of modification, p values, and log2 fold change for enriched m6A peaks between dox-treated E1A;HRasG12V MEFs transduced with shLuc and shMettl3 hairpins.
(F) UCSC genome browser tracks showing RPM (reads per million) patterns of m6A-eCLIP-seq in Killer, Tex15, Neat1, and Mdm2 mRNAs in dox-treated E1A;HRasG12V MEFs transduced with either shLuc or shMettl3 RNAs, relative to input. Green and pink tracks are peaks on positive and negative DNA strands, respectively.
The METTL3-dependent modification of select p53 target gene transcripts upon DNA damage suggests that the p53-METTL3 interaction can augment the p53-dependent induction of some p53 target genes in part through co-transcriptional installation of m6A on transcripts. To elucidate the mechanism underlying this effect of METTL3, we tested whether METTL3 might associate with chromatin at p53 target genes. Using ChIP assays in MEFs, we found that METTL3 associates with the p53 binding sites of p53 target genes subject to m6A modification, including Killer and Mdm2, suggesting that METTL3 can associate with chromatin of p53 target genes to direct m6A modification (Figure 5A). This interaction is diminished in the absence of p53, underscoring the importance of p53 for METTL3 recruitment to these sites (Figure 5B). Together, these findings suggest that the METTL3 complex installs m6A marks on select p53-regulated mRNAs co-transcriptionally. Interestingly, METTL3 also localized to the p53 binding sites of genes whose transcripts were not found to be m6A modified (Cdkn1a, Trp53inp1, Noxa), suggesting that METTL3 exerts its catalytic activity-independent effects on p53 on DNA.
Figure 5. METTL3 associates with chromatin of p53 target genes.
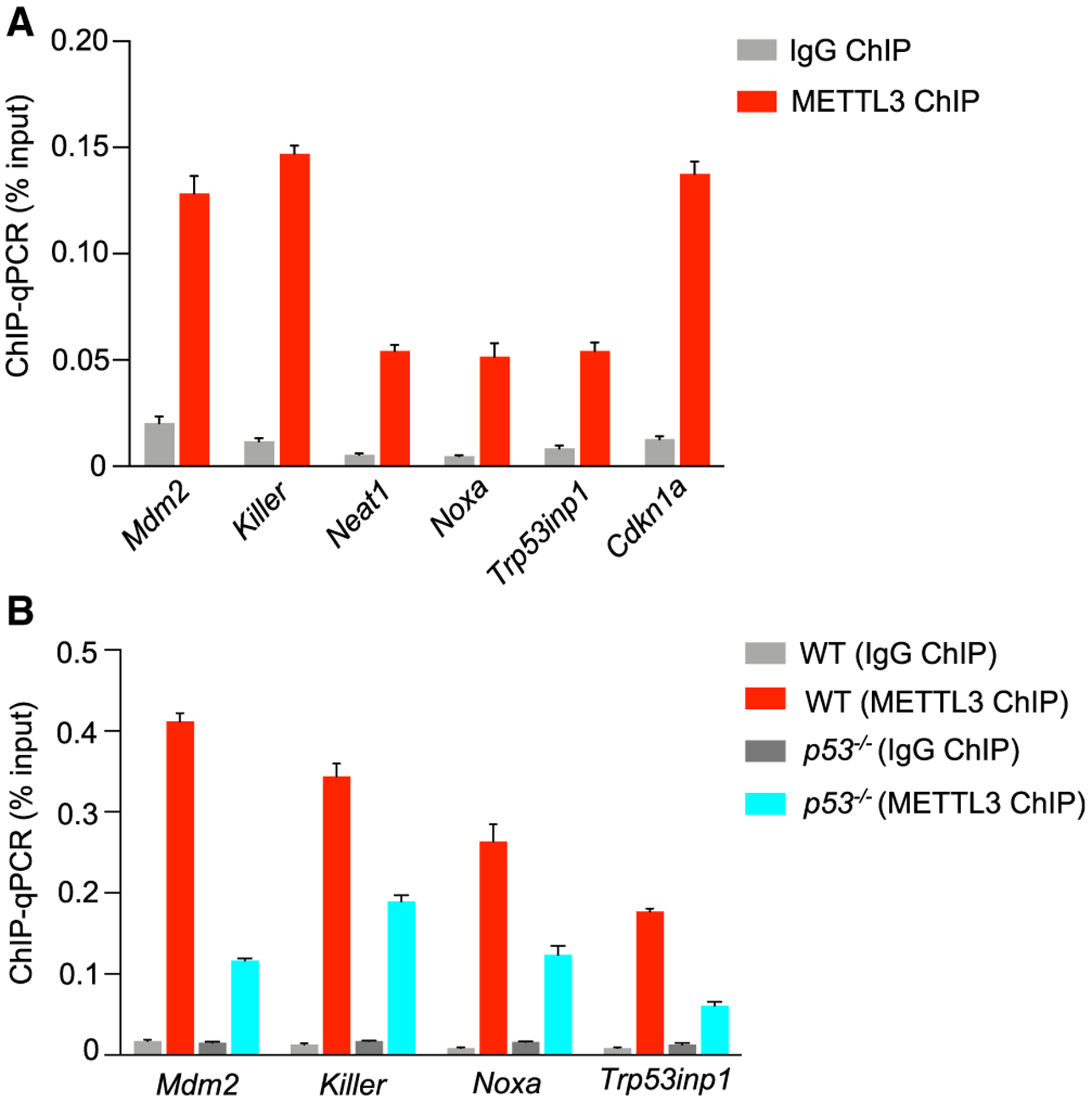
(A) ChIP analysis of METTL3 binding to p53 sites in p53 target gene loci, relative to input, in dox-treated E1A;HRasG12V MEFs.
(B) ChIP analysis of METTL3 binding to p53 sites in p53 target gene loci, relative to input, in dox-treated E1A;HRasG12V wild-type and p53−/− MEFs. In (A) and (B), IgG serves as a negative control antibody, and representative analysis from two biological replicates is shown.
M6A is well-understood to modulate both translation and RNA stability. To determine whether METTL3-mediated m6A modification of p53 target gene transcripts has a role in enhancing mRNA translation, we performed polysome fractionation of dox-treated WT or Mettl3-null ES cells followed by qRT-PCR analysis of m6A-modified (Mdm2, Killer) and unmodified p53 target gene transcripts (Cdkn1a, Noxa, Trp53inp1). We observed no discernable difference in the distribution of control housekeeping gene (β-Actin) or p53 target gene mRNAs in polysomes from WT and Mettl3-null ES cells. These findings suggest that METTL3-mediated m6A modification of p53 pathway transcripts does not impact translation of these mRNAs, but likely contributes to RNA stability (Figures S4A–S4F). Overall, our findings suggest that METTL3 exerts a dual effect on p53, primarily through stabilization of p53 and secondarily through m6A modification of select transcripts in the p53 pathway to enhance their expression.
METTL3 supports p53 in tumor suppression in mouse models
Beyond serving as a key sentinel to genotoxic damage, p53 plays a critical role in responses to oncogenic signals, as underscored by its frequent mutation in human cancer (Bieging et al., 2014). In response to oncogene expression, p53 suppresses transformation in vitro and tumorigenesis in vivo. To determine whether METTL3 might also contribute to p53 tumor-suppressive function, we first utilized E1A;HRasG12V MEFs, a classical transformation model in which p53 potently suppresses transformation, as described above (Bieging-Rolett et al., 2020). We knocked down Mettl3 expression in E1A;HRasG12V MEFs using shRNAs. Analysis of these cells revealed that attenuated METTL3 protein expression enhanced their clonogenic potential, a cardinal feature of transformed cells, supporting a role for METTL3 in suppressing transformation (Figure 6A). In contrast, Mettl3 knockdown in E1A;HRasG12V p53 null MEFs did not enhance clonogenic potential, indicating that the role for METTL3 in transformation suppression is specifically in the context of intact p53 (Figures S5A and S5B). We next assessed the consequences of Mettl3 knockdown for tumor growth by subcutaneous injection of oncogene-expressing cells into immunocompromised mice. Tumor growth in this context is significantly enhanced by p53 deficiency (Jiang et al., 2011); similarly, we found that Mettl3 knockdown with either of two shRNAs dramatically enhanced tumor growth in vivo in p53-proficient MEFs but not in p53-null MEFs, suggesting a role for METTL3 in tumor suppression (Figure 6B). The tumor growth with Mettl3 knockdown was not as great as with p53 nullizygosity, consistent with the observation that Mettl3 deficiency dampens but does not eliminate p53 pathway activity. Moreover, expression of Mettl3 shRNAs in E1A;HRasG12V;p53−/− cells did not enhance tumor growth, indicating again that METTL3 tumor-suppressive activity is seen specifically in the context of WT p53. Interestingly, Mettl3 ablation with the more potent shRNA even inhibited colony growth (Figure S5B).
Figure 6. METTL3 supports p53-mediated tumor suppression in mouse models.
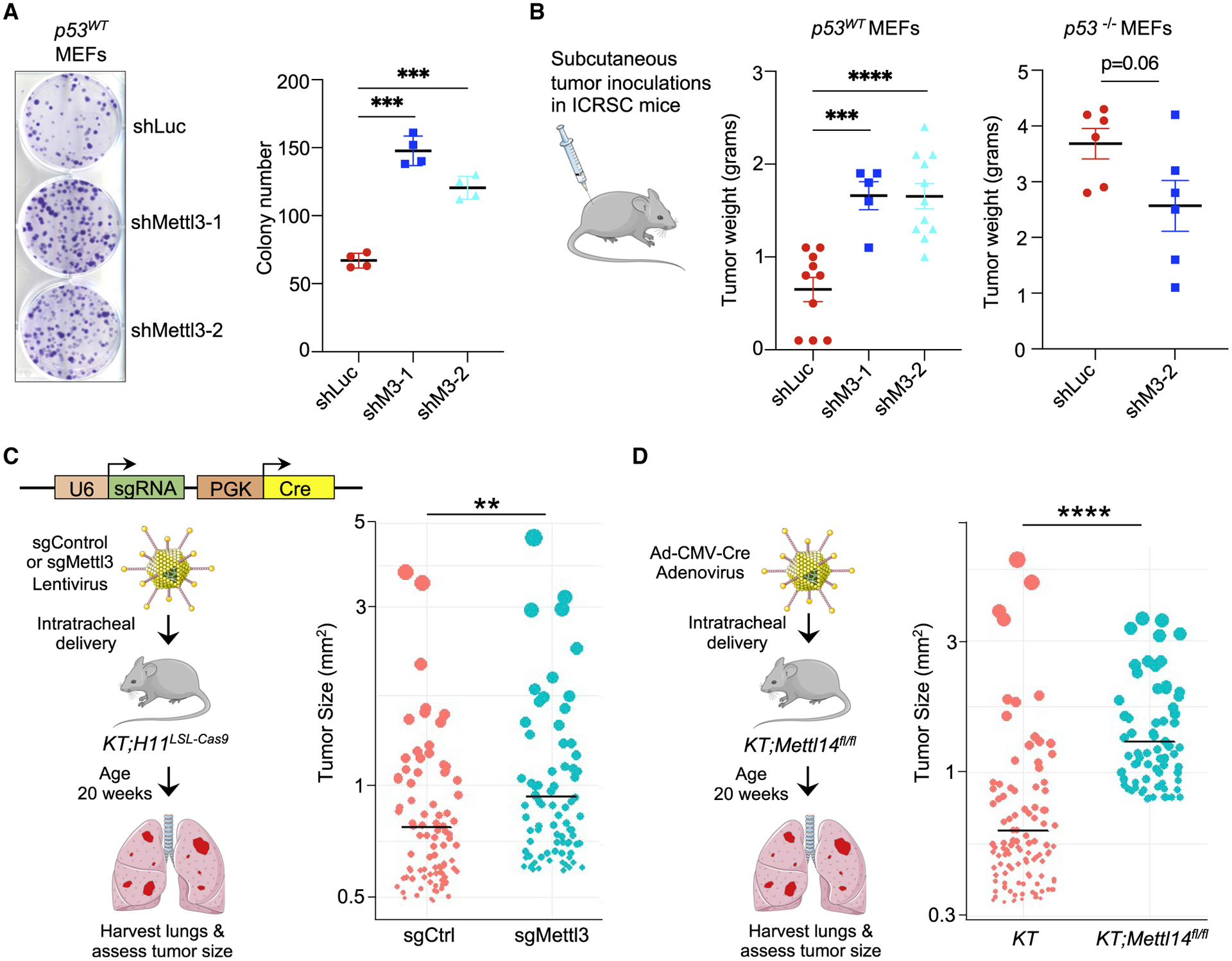
(A) Low-density plating assay to assess clonogenic potential of E1A;HRasG12V-expressing WT MEFs transduced with either shLuc or shMettl3 RNAs. (Left) Representative crystal violet-stained colonies are shown. (Right) Average colony number (n = 4, with triplicate samples, using two different MEF lines per genotype).
(B) (Left) Average weight of tumors from E1A;HRasG12V p53WT MEF transduced with either shLuc or shMettl3 RNAs after growth in vivo. (Right) Average weight of E1A;HRasG12V p53−/− MEF tumors with shLuc or shMettl3 expression after growth in vivo. In (A) and (B), bar shows mean ± SEM.
(C) (Left) Scheme for in vivo tumor experiment with CRISPR-Cas9 system. Lentiviral vectors expressing Cre recombinase and Mettl3 or control sgRNA were delivered intratracheally into KT; H11LSL-Cas9 mice (KT = KrasLSL-G12D/+;Rosa26LSL-tdTomato/LSL-tdTomato), and tumor size of all lung tumors was assessed after 20 weeks. (Right) Graph showing sizes of top 10% of all tumors assessed in each group (n = 12 control and 11 sgMettl3 mice; n = 907 control and 826 sgMettl3 tumors).
(D) (Left) Scheme for in vivo tumor experiment. Ad-CMV-Cre adenovirus was delivered intratracheally into KT;Mettl14fl/fl or control KT mice (KT = KrasLSL-G12D/+;Rosa26LSL-tdTomato/LSL-tdTomato) and tumor size of all lung tumors was assessed after 20 weeks. (Right) Graph showing sizes of top 10% of all tumors assessed in each group (n = 12 KT and 10 KT;Mettl14fl/fl mice; n = 1,075 KT and 708 KT;Mettl14fl/fl tumors). In (C) and (D), each dot represents a tumor, with area proportional to the tumor size. Bar shows the mean for each group. p values were determined by unpaired, two-tailed Student’s t test; **p < 0.01, ***p < 0.001, ****p < 0.0001.
See also Figure S5.
To assess the impact of METTL3 deficiency more broadly on cancer development, we employed an autochthonous mouse lung adenocarcinoma (LUAD) model in which we could perform targeted gene inactivation using CRISPR-Cas9-mediated gene editing. In this model, lentiviral-Cre instillation induces oncogenic KrasG12D, Cas9, and tdTomato expression as well as delivering sgRNAs targeting Mettl3 (sgMettl3) or a non-targeting control sgRNA (sgCtrl). Importantly, p53 deficiency in this model is known to enhance tumor growth (Bieging-Rolett et al., 2020). Tumor quantification in this model is assessed by measuring the range of hundreds of individual tumor sizes within H&E-stained histological lung sections. Analysis of tumor sizes from 12 sgCtrl and 11 sgMettl3 mice revealed that Mettl3 ablation by CRISPR-Cas9 resulted in larger tumors than in control mice, suggesting a role for Mettl3 in LUAD suppression (Figure 6C). To complement these experiments, we leveraged Mettl14 conditional knockout mice (Weng et al., 2018) to address the role of METTL14 in tumor suppression, as METTL14 and METTL3 form the stable heterodimeric core of the methyltransferase complex (MTC). In this model, adenoviral-Cre instillation induces oncogenic KrasG12D and tdTomato reporter expression (denoted as KT), with concomitant Cre-mediated inactivation of Mettl14. Interestingly, KT;Mettl14fl/fl mice developed significantly larger tumors than KT mice (Figures 6D and S5C). Together, these findings suggest that Mettl3 and Mettl14 both have tumor suppressor activity in lung adenocarcinoma in vivo, in which p53 has an established role in suppressing cancer.
METTL3 reinforces p53 activity in human cells
We next sought to interrogate the importance of the p53/methyltransferase axis in tumor suppression in humans. We first used A549 human LUAD cells with intact p53 to assess whether METTL3 expression affects p53 protein levels. Indeed, similar to our observations in mouse cells, we observed that expression of FLAG-METTL3 strongly enhanced endogenous p53 protein levels (Figure 7A). We next assessed the functional conservation of the METTL3-p53 tumor-suppressive pathway by performing clonogenic assays. As expected, A549 cells in which we engineered a TP53 deletion formed colonies more efficiently than p53-expressing A549 cells (Figure S5D). We then expressed a scrambled control shRNA or either of two different shRNAs to knock down METTL3 in p53-expressing A549 cells (Figure 7B). We found that METTL3 knockdown with either shRNA enhanced transformation potential relative to the control shRNA in low-density plating and soft-agar assays, indicating that METTL3 suppresses transformation in human LUAD cells (Figures 7C and S5E). Notably, as we observed with p53-null oncogene-expressing MEFs, METTL3 knockdown in TP53 knockout (TP53−/−) A549 cells did not enhance transformation (Figures 7D and S5F), indicating that METTL3 also specifically suppresses transformation in the context of an intact p53 pathway in human cells. Interestingly, we again observed that METTL3 knockdown in TP53−/− A549 cells dramatically impaired colony growth (Figure 7D).
Figure 7. METTL3 MTC and p53 operate in a common tumor-suppressive pathway in human cancers.
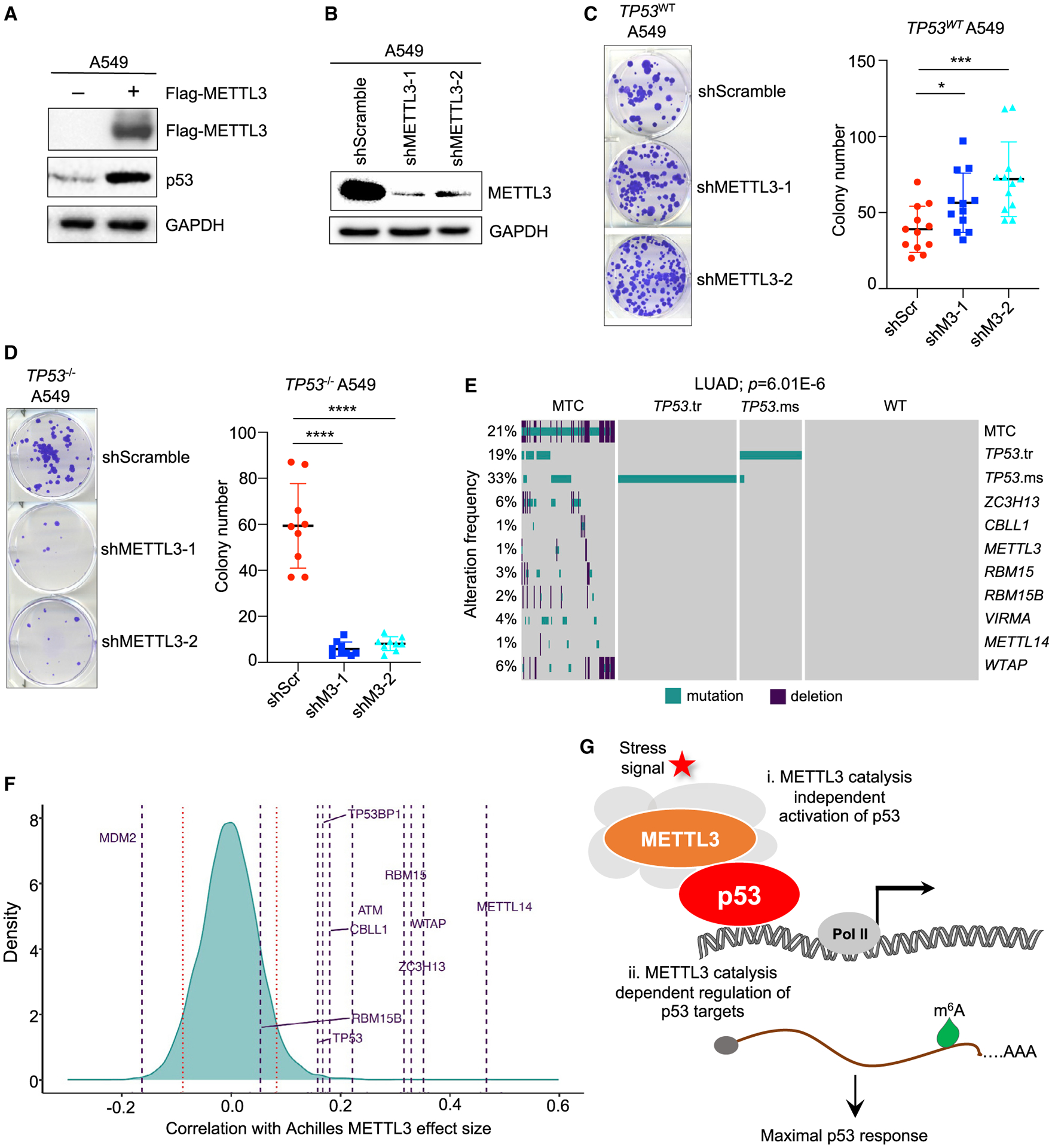
(A) Immunoblotting for endogenous p53 after FLAG-METTL3 plasmid transfection into A549 cells (n = 3).
(B) Immunoblotting for METTL3 in A549 cells transduced with either shScramble (control) or either of two shMETTL3 RNAs (n = 2). In (A) and (B), GAPDH serves as a loading control.
(C and D) Low-density plating assay to assess clonogenic potential of TP53WT (C) and TP53−/− A549 (D) cells transduced with lentiviruses expressing either shScramble or shMETTL3 RNAs. (Left) Representative crystal violet-stained colonies are shown. (Right) Average colony number (n = 3, with triplicate samples). Bar shows mean ± SEM. p values were determined by unpaired, two-tailed Student’s t test; *p < 0.05, ***p < 0.001, ****p < 0.0001.
(E) Oncoplot showing alteration frequencies of TP53 and METTL3 MTC components in human LUAD. TP53.tr refers to truncation mutations, while TP53.ms refers to missense mutations in TP53. p value shows significance of DISCOVER test unadjusted and adjusted for multiple testing.
(F) Density distribution of Pearson correlations between METTL3 Achilles scores. Horizontal lines represent genes of interest, including MTC components, TP53, and regulators of the p53 pathway. Red bars represent the 5th and 95th quantiles of the distribution.
(G) Proposed model of METTL3-MTC regulation of the p53 pathway to potentiate full p53 responses to stress signals. In response to stress signals, METTL3 as part of the MTC (gray shading), stabilizes p53 protein in an m6A catalysis-independent manner (i), and METTL3 regulates the expression of select p53 pathway transcripts by governing their m6A-modification (ii).
See also Figures S6 and S7.
TP53 and METTL3 operate in a common pathway in human cancers
We next explored the role of the p53-METTL3 interaction in human cancer development using TCGA data. METTL3 is part of a multi-protein MTC that not only includes METTL14 but also WTAP, RBM15, RBM15B, VIRMA, CBLL1, and ZC3H13 (Zaccara et al., 2019). We thus sought to identify point mutations and deletions in METTL3 complex components in human cancers and assess whether there is a mutually exclusive relationship with TP53 mutations. Indeed, examination of the mutational spectrum in the full complement of MTC components in various carcinomas, including lung adenocarcinoma, breast, and head and neck cancers, revealed a mutually exclusive pattern of mutations with TP53, suggesting that when constituents of the MTC are mutated, there is less selective pressure to mutate TP53 (Figures 7E and S6A–S6C). We found further that, as with TP53 mutation, MTC mutations compromise expression of various p53 target genes in a range of human cancer types, albeit not to the full extent seen with the TP53 mutation (Figures S6D and S6E). These data suggest that the MTC augments the p53 transcriptional program in humans.
To test the functional significance of MTC activity in the p53 pathway, we leveraged cancer dependency map (DepMap) data to assess the impact of METTL3 knockout on the proliferation of human cancer cell lines of different TP53 statuses (DepMap: https://depmap.org/portal/) (Meyers et al., 2017). DepMap profiles hundreds of cancer cell lines for sensitivity to genetic perturbations via RNAi or CRISPR, followed by measurement of proliferation to provide a gene effect score, termed the Achilles score, that reflects the essentiality of individual genes for proliferation. If two genes have similar dependency scores across cell lines, they are termed co-dependent, which suggests that these genes are in the same pathway. Notably, we found that the most statistically significant METTL3 co-dependency with all genes was with TP53 (Figure S7A). Moreover, the highest Pearson correlation between Achilles scores with METTL3 knockout includes not only other MTC components, as expected, but also TP53 and other positive regulators of the p53 pathway, TP53BP1 and ATM, further supporting the idea that the MTC is a component of the p53 pathway (Figure 7F). Conversely, METTL3 showed a negative correlation with MDM2, as expected for a negative regulator of p53. TP53 and its regulators showed similar co-dependencies with other MTC components such as WTAP (Figure S7B). Interestingly, the Achilles scores for METTL3 are significantly lower in TP53 mutant cell lines than in cell lines harboring WT TP53 (Figure S7C), reminiscent of the reduced proliferation seen in oncogene-expressing, p53-deficient MEFs and A549 cells upon Mettl3/METTL3 knockdown (Figures 7D and S5B). Collectively, our findings support the idea that the METTL3 MTC and p53 operate in a common tumor-suppressive pathway in various human cancers, with the MTC acting to promote full p53 activity.
DISCUSSION
Here, we identify METTL3 as a novel p53-interacting partner that plays a fundamental role in augmenting p53 activity at two levels. The primary effect of METTL3 is through binding to the amino terminus of p53 to enhance its half-life through a catalytic activity-independent mechanism. METTL3 can also promote p53 activity by installing the m6A modification on select p53 target gene transcripts in acute DNA damage responses and in tumor suppression to ensure their potent expression (Figure 7G). The amplification of p53 activity by METTL3 is important for transformation suppression in mouse and human cells and LUAD suppression in mouse models. Moreover, analyses of DepMap and human cancer genome data provide genetic support for the importance of the p53-MTC axis in tumor suppression. Overall, the participation of the METTL3 complex in p53 programs ensures that a robust p53 response can be induced and provides the potential for fine-tuning p53 activity.
Recruitment of the MTC by p53 and other sequence-specific transcription factors allows for co-transcriptional m6A modification that can provide an additional level of regulation for gene expression programs. Such a role for METTL3 and the MTC in increasing the activity of sequence-specific transcription factors has been reported, albeit through distinct mechanisms. For example, in acute myeloid leukemia (AML), METTL3 is recruited by the CEBPZ transcription factor to the promoters of oncogenes to drive m6A modification of emerging transcripts, a signal that ultimately augments translation of these transcripts to sustain AML cells (Barbieri et al., 2017). In human pluripotent stem cells, in response to differentiation cues, the SMAD2/3 transcription factors recruit the MTC to RNAs to facilitate co-transcriptional m6A modification, leading to destabilization of specific transcripts, such as Nanog, to drive the exit from pluripotency and promote differentiation (Bertero et al., 2018). Recruitment of the MTC to actively expressed genes in ES cells can also occur through METTL14 binding to H3K36me3, a mark of transcription elongation, to drive co-transcriptional m6A modification of nascent mRNAs, also leading to destabilization of pluripotency gene mRNAs (Huang et al., 2019). Thus, METTL3 can be recruited to DNA by transcriptional regulators to promote m6A modification. Similarly, our findings suggest that METTL3 is recruited to p53-bound sites in chromatin to co-transcriptionally install m6A marks on p53 target gene messages. Recruitment of the MTC by specific transcription factors may help to explain which transcripts are selected for m6A modification in a given setting, a point that has remained enigmatic.
m6A modification on RNA by writers such as the MTC affects gene expression through a variety of mechanisms mediated by different m6A reader proteins (Shi et al., 2019). Although m6A marks can be read by YTHDF2/3 reader proteins to trigger mRNA destabilization, they can also be recognized by IGF2BP1–3 to promote mRNA stabilization (Du et al., 2016; Huang et al., 2018; Shi et al., 2017; Wang et al., 2014). m6A modification can also be recognized by the YTHDC1 nuclear reader to regulate splicing and RNA export (Xiao et al., 2016). In addition, m6A can affect translation through diverse mechanisms (Shi et al., 2019). For example, METTL3-driven m6A modification on the 5′ UTR triggers binding to multiple subunits of the eukaryotic initiation factor 3 (eIF3) complex, while m6A marks in the coding sequence (CDS) and 3′ UTR result in YTHDF1/3 and eIF3 recruitment to promote translation (Meyer et al., 2015; Shi et al., 2017; Wang et al., 2015). Interestingly, METTL3 also promotes translation by acting as a reader: it can bind to an m6A mark in the 3′ UTR and interact with translation initiation machinery components such as eIF3H at the 5′ end of the transcript via RNA circularization, thus leading to enhanced translation of m6A-modified mRNAs (Choe et al., 2018; Lin et al., 2016). In this capacity, METTL3 catalytic activity is dispensable. Our initial findings suggest that METTL3 may promote stabilization of p53 target gene mRNAs rather than enhanced translation of these mRNAs, but future analyses will be required to further elaborate these mechanisms, including which readers are involved in sensing these modified transcripts.
The METTL3 complex is critical for responses to extracellular cues, either to promote cell fate transitions or homeostasis. For example, METTL3 is required in mouse and human ES cells to restrict self-renewal and promote differentiation through down-regulation of core pluripotency factor transcripts such as Nanog (Batista et al., 2014). Similarly, METTL3-mediated m6A modification in human hematopoietic stem cells is critical for differentiation in vivo and for inhibiting self-renewal in glioblastoma stem cells (Cui et al., 2017; Lee et al., 2019). Intriguingly, several studies have established a similar role of p53 in opposing stemness and promoting differentiation: p53 loss in mice triggers an expansion of both normal and cancer stem cells, and p53 restricts cellular reprogramming in iPS cell generation (Kaiser and Attardi, 2018; Krizhanovsky and Lowe, 2009). Beyond modulating cell state, METTL3 can also ensure homeostasis by promoting resolution to cellular stress. For example, in response to UV, METTL3/METTL14 localizes to DNA damage sites to transiently induce m6A RNA modification and promote DNA repair through recruitment of DNA polymerase (Xiang et al., 2017). METTL3 also plays a critical role in the resolution of heat shock responses by inducing m6A modification on Hsp70 mRNA, leading to its destabilization (Knuckles et al., 2017). Our findings similarly underscore the importance of METTL3 in promoting responses to signals, specifically for ensuring a robust p53 response to DNA damage or oncogene expression.
m6A modification and the MTC have a highly context-dependent role in cancer (Huang et al., 2020). Although METTL3 has been reported to be an oncogene in numerous cancers, such as AML, colon cancer, NSCLC (non-small-cell lung cancer), bladder cancer, and ovarian cancer (Barbieri et al., 2017; Huang et al., 2020; Vu et al., 2017), it has been shown to be a tumor suppressor in other settings, such as renal cell carcinoma, glioblastoma, and endometrial cancer (Cui et al., 2017; Li et al., 2017b; Liu et al., 2018). Similarly, METTL14 has been reported to promote some cancer types yet perform a tumor-suppressive role in hepatocellular carcinoma (HCC), glioblastoma (GBM), and skin cancer development (Cui et al., 2017; Huang et al., 2020; Yang et al., 2021). Our findings suggest that the MTC can be tumor suppressive in the context of intact p53, where it bolsters p53 activity. Interestingly, our clonogenic assays in mouse and human cells suggest that METTL3 may support tumor growth in the absence of p53, as knockdown is detrimental for cell growth in this context. This difference in METTL3 action in the context of active or deficient p53 provides one potential explanation for observed differences in the role of the MTC in cancer development. Importantly, these findings also provide initial critical insight into when it would be beneficial to therapeutically target METTL3 in human cancer. Specifically, our observations suggest that METTL3 inhibitors would be most efficacious in cancers with p53 pathway deficiency, either through direct TP53 mutation or through other alterations in the p53 pathway. Indeed, the METTL3 inhibitor STM2457 has efficacy in AML, a cancer in which the p53 pathway is often inactivated through overexpression of negative regulators of p53, such as MDM2 and MDM4 (Carvajal et al., 2018; Tan et al., 2014; Yankova et al., 2021). Further analysis of the precise contexts in which METTL3 promotes or suppresses tumor development will be critical for better understanding pathways to tumorigenesis and for ultimately designing therapeutic interventions based on this pathway.
Limitations of the study
Our work presents a highly novel role for METTL3 in the p53 transcriptional program that is enforced primarily by protein-protein interaction between p53 and METTL3. Although we have shown that the METTL3-p53 interaction is direct and does not require DNA, the role of RNA in mediating the interaction is still unclear and could be important, as p53 has been shown previously to bind to other RNA species (Riley and Maher, 2007; Tournillon et al., 2017; Yoshida et al., 2004). In addition, although our data suggest that MDM2 is not efficiently displaced from p53 by DNA damage in the absence of METTL3, whether MDM2 and METTL3 directly compete for p53 binding remains unclear. Finally, our data suggest that p53 pathway status is one factor that dictates whether METTL3 is an oncogene or tumor suppressor. However, our analysis was limited to the studies conducted in clonogenic and allograft tumor assays. It will be interesting to assess this more widely with multiple cancer types using in vivo mouse models where p53 status is clear. This insight would have deep therapeutic implications.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the lead contact, Laura D. Attardi (attardi@stanford.edu).
Materials Availability
Plasmids and cell lines generated by this study are available upon request.
Data and Code Availability
RNA-seq and m6A-eCLIP-seq data have been deposited at GEO and are publicly available as of the date of publication. The paper analyzes previously published, publicly available datasets. Accession numbers are listed in the key resources table. The Achilles DepMap dataset (DepMap: CCLE_Depmap_21Q4 release) is available from the DepMap portal, DepMap: https://depmap.org/portal/download/. TCGA gene expression data was downloaded from NCI Genomic Data Commons: gdc.cancer.gov. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
All code to reproduce the figures has been deposited at GitHub (GitHub: https://github.com/CancerCompBioLab/Mettl3_paper) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p53 antibody (CM5) | Leica (Novocastra) | NCL-L-p53-CM5p; RRID: AB_563933 |
| p53 antibody (1C12) | Cell Signaling Technology | 2524; RRID: AB_331743 |
| METTL3 polyclonal antibody | Abclonal | A8370; RRID: AB_2770344 |
| METTL3 monoclonal antibody | Abcam | ab195352; RRID: AB_2721254 |
| HA antibody | Thermo Fisher Scientific | 71–5500; RRID: AB_2533988 |
| FLAG antibody | Sigma-Aldrich | F1804; RRID: AB_262044 |
| MDM2 antibody (2A10) | Abcam | ab16895; RRID: AB_2143534 |
| GAPDH antibody | Fitzgerald | 10R-G109A; RRID: AB_1285808 |
| N6-Methyladenosine (m6A) (D9D9W) antibody | Cell Signaling Technology | 56593; RRID: AB_2799515 |
| Goat anti-rabbit HRP antibody | Jackson ImmunoResearch | 111-035-144; RRID: AB_2307391 |
| Goat anti-mouse HRP antibody | Jackson ImmunoResearch | 115-035-003; RRID: AB_10015289 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG, light chain specific | Jackson ImmunoResearch | 115-035-174; RRID: AB_2338512 |
| Peroxidase IgG Fraction Monoclonal Mouse Anti-Rabbit IgG, light chain specific | Jackson ImmunoResearch | 211-032-171; RRID: AB_2339149 |
| Bacterial and virus strains | ||
| Ad-Cre or Ad5CMVCre | University of Iowa | VVC-U of Iowa-5 |
| Lenti-U6-sgMettl3/Cre | This paper | N/A |
| Lenti-U6-sgNeoR1/Cre | Laboratory of Dr. Monte Winslow | N/A |
| E.coli BL21 expressing GST-0 | Laboratory of Dr. Hua Lu and Shelya Zeng | N/A |
| E.coli BL21 expressing GST-p53 | Laboratory of Dr. Hua Lu and Shelya Zeng | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 2000 | Thermo Fisher Scientific | 11668019 |
| Doxorubicin hydrochloride | Sigma-Aldrich | D1515 |
| Puromycin dihydrochloride | Sigma-Aldrich | P7255 |
| Cycloheximide | Sigma-Aldrich | C7698 |
| Glutathione Sepharose 4B | Cytiva | 17-0756-01 |
| GST-0 | This paper | N/A |
| GST-p53 | This paper | N/A |
| 6X-His-HDM2 | Laboratory of Dr. Hua Lu and Shelya Zeng | N/A |
| Critical commercial assays | ||
| Clarity Western ECL Substrate | Bio-Rad | 1705060 |
| ECL Prime Western Blotting System | Millipore-Sigma | GERPN2232 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | 23227 |
| TRIzol reagent | Thermo Fisher | 15596026 |
| PowerUP SYBR green master mix | Thermo Fisher | 4367660 |
| M-MLV Reverse Transcriptase | Thermo Fisher | 28025013 |
| iScript Supermix cDNA synthesis kit | Bio-Rad | 1708841 |
| Gateway BP Clonase II Enzyme Mix | Thermo Fisher | 11789-020 |
| Gateway LR Clonase II Enzyme Mix | Thermo Fisher | 11791-020 |
| Q5 Site-directed mutagenesis kit | NEB | E0554S |
| TNT SP6 High-Yield Wheat Germ Protein Expression System | Promega | L3260 |
| Deposited data | ||
| M6A-eCLIP-seq and RNA-seq | This paper | GEO:GSE169343 |
| shLuc and shMettl3 MEF RNA-seq | This paper | GEO:GSE193341 |
| Mendeley Data | This paper | Mendeley Data: https://dx.doi.org/10.17632/gdpmnddt7v.1 |
| Mouse p53 ChIP-seq | Kenzelmann-Broz et al. (2013) | GEO:GSE46240 |
| E1A;HRasG12V p53WT and p53 null MEF RNA-seq | Valente et al. (2020) | GEO:GSE136355 |
| Achilles DepMap (CCLE_Depmap_21Q4 release) | Meyers et al. (2017) | DepMap: http://depmap.org |
| Scripts used in the study | This paper | GitHub: https://github.com/CancerCompBioLab/Mettl3_paper |
| Experimental models: Cell lines | ||
| Mouse: E1A;HRasG12V p53WT MEFs | Laboratory of Dr. Laura Attardi | N/A |
| Mouse: E1A;HRasG12V p53−/− MEFs | Laboratory of Dr. Laura Attardi | N/A |
| Mouse: E1A;HRasG12V p53WT MEFs shLuc | This paper | N/A |
| Mouse: E1A;HRasG12V p53WT MEFs shMettl3-1 | This paper | N/A |
| Mouse: E1A;HRasG12V p53WT MEFs shMettl3-2 | This paper | N/A |
| Mouse: E1A;HRasG12V p53−/− MEFs shLuc | This paper | N/A |
| Mouse: E1A;HRasG12V p53−/− MEFs shMettl3-1 | This paper | N/A |
| Mouse: E1A;HRasG12V p53−/− MEFs shMettl3-2 | This paper | N/A |
| Mouse: Flp-In™-3T3 | Thermo Fisher | R76107 |
| Mouse: Flp-In-3T3 p53-LAP | This paper | N/A |
| Mouse: Flp-In-3T3 p53−/− | This paper | N/A |
| Mouse: Embryonic Stem cells Wild-type | Laboratory of Dr. Howard Y. Chang | N/A |
| Mouse: Embryonic Stem cells Mettl3−/− | Laboratory of Dr. Howard Y. Chang | N/A |
| Human: A549 lung cancer cell line: TP53WT | Stanford University School of Medicine laboratories | N/A |
| Human: A549 lung cancer cell line: TP53−/− | This manuscript | N/A |
| Human: H1299 lung cancer cell line | Stanford University School of Medicine laboratories | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: ICR-Scid: IcrTac:ICR-Prkdcscid | Taconic Biosciences | ICRSC-M |
| Mouse: KrasLSL-G12D mice: B6.129S4-Krastm4Tyj/J | Laboratory of Dr. Tyler Jacks | N/A |
| Mouse: Rosa26LSL-tdTomato | Madisen et al. (2010) | N/A |
| Mouse: H11LSL-Cas9: B6;129-Igs2tm1(CAG-cas9+)Mmw/J | Chiou et al. (2015); Laboratory of Dr. Monte Winslow | 026816 |
| Mouse: Mettl14fl/fl | Wang et al. (2018); Stanford University School of Medicine laboratories | N/A |
| Oligonucleotides | ||
| See Table S4 for primers | This paper | N/A |
| Recombinant DNA | ||
| pENTR p53 | This paper | N/A |
| pG-LAP7/puro destination vector | Laboratory of Dr. Peter Jackson | N/A |
| pG-LAP7/puro p53 (p53-LAP) | This paper | N/A |
| pENTR Mettl3 | This paper | N/A |
| pG-LAP2 Mettl3WT (FLAG-METTL3 WT) | This paper | N/A |
| pG-LAP2 Mettl3APPA (FLAG-METTL3 APPA) | This paper | N/A |
| pCS2-HA destination vector | Laboratory of Dr. Peter Jackson | N/A |
| pCS2-HA-Mettl3 (HA-METTL3) | This paper | N/A |
| pcDNA3.1–3XHA-p53 | Laboratory of Dr. Laura Attardi | N/A |
| pcDNA3.1–3XHA-p53 Δ1–42 | This paper | N/A |
| pcDNA3.1–3XHA-p53 Δ1–61 | This paper | N/A |
| pX330 p53 | Addgene | 59910 |
| shRNA vectors with shRNAs targeting Luciferase, Mettl3, METTL3 | Laboratory of Dr. Pedro Batista | N/A |
| Lenti-U6-sgMettl3 | This paper | N/A |
| Lenti-U6-sgNeoR1 | Laboratory of Dr. Monte Winslow | N/A |
| pX458 TP53 series | This paper | N/A |
| Software and algorithms | ||
| Enrichr | Chen et al. (2013), Kuleshov et al. (2016) | https://maayanlab.cloud/Enrichr/ |
| Metascape | Zhou et al. (2019) | http://metascape.org/gp/index.html#/main/step1 |
| Byonic | Protein Metrics | https://proteinmetrics.com/resources/byonic-advanced-peptide-and-protein-identification-software/ |
| Image Lab | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| NDPview 2.0 | Hamamatsu | https://www.hamamatsu.com/us/en/product/type/U12388-01/index.html |
| QPath | Univ of Edinburgh | https://qupath.github.io/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Umi_tools (v0.5.1) | Smith et al. (2017) | https://github.com/CGATOxford/UMI-tools |
| Cutadapt (v2.7) | Martin (2011) | https://github.com/marcelm/cutadapt |
| STAR (v2.6.0c) | Dobin et al. (2013) | https://github.com/alexdobin/STAR |
| CLIPper | Lovci et al. (2013) | https://github.com/YeoLab/clipper |
| GENCODE (release M21) | Frankish et al. (2019) | https://www.gencodegenes.org/human/ |
| HOMER (v3.1) | Heinz et al. (2010) | http://homer.ucsd.edu/homer/motif/motifDatabase.html |
| limma (v3.46) | Ritchie et al. (2015) | https://bioconductor.org/packages/release/bioc/html/limma.html |
| genefu (v2.22.1) | Gendoo et al. (2016) | http://bioconductor.org/packages/release/bioc/html/genefu.html |
| DISCOVER package | Canisius et al. (2016) | https://ccb.nki.nl/software/discover/ |
| DEseq2 | Love et al. (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Fastp | Chen et al. (2018) | https://github.com/OpenGene/fastp |
| Featurecounts (1.5.3) | Liao et al. (2014) | https://rnnh.github.io/bioinfo-notebook/docs/featureCounts.html |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
ICR SCID male mice were obtained from Taconic Biosciences (Cat # ICRSC-M). KrasLSL-G12D (K), Rosa26LSL-Tomato (T), H11LSL-Cas9 (Cas9) and Mettl14fl/fl mice have been described previously (Chiou et al., 2015; Jackson et al., 2001; Madisen et al., 2010; Wang et al., 2018). All mice were maintained under pathogen-free conditions at the Stanford animal care facility. All experiments were approved by Administrative Panel on Laboratory Animal Care at Stanford University.
Subcutaneous tumor assays
Subcutaneous tumor studies were performed as described previously (Jiang et al., 2011). Briefly, 1 × 106 shLuc or shMettl3 E1A;HRasG12V MEFs were subcutaneously injected into the flanks of Scid mice, and tumors were weighed at least 21 days post-injection.
Mouse lung adenocarcinoma assay
Lentiviral particles were produced and titered as described previously (Rogers et al., 2017). Lung tumors were initiated by intratracheal infection of mice as described previously (DuPage et al., 2009) using lentiviral- or adenoviral-Cre vectors. Lung tumors were induced by intratracheal administration of 90,000 particles of Lenti-U6-sgRNA/PGK-Cre virus in 6–12 week old KrasLSL-G12D (K); Rosa26LSL-Tomato (T); H11LSL-Cas9 (C) mice or Ad5-CMV-Cre (Univ. of Iowa) virus in KrasLSL-G12D (K); Rosa26LSL-Tomato (T);Mettl14fl/fl mice as previously described (DuPage et al., 2009). Lungs were harvested at 20 weeks after inoculation and tumor burden was assessed by histology as indicated.
Cell culturing and drug treatments
Mouse embryonic fibroblasts, Flp-In-3T3, H1299, and A549 cells were maintained in Dulbecco’s Modified Eagle Medium (Gibco) supplemented with 10% Fetal Calf Serum (FCS), 1% penicillin/streptomycin, 50 μg/mL gentamicin and incubated at 37°C in a carbon dioxide incubator. Mouse ES cells were cultured as previously described (Batista et al., 2014). Doxorubicin (Sigma, Cat # D1515) treatment was done at a concentration of 0.2 μg/ml for 6 hours. Cycloheximide (Sigma, Cat # C7698) was used at 100 μM for the indicated period of time. Protein extracts were treated with DNase I (Invitrogen, Cat # 18047019) at 40 U/ml for 1 hr at 4°C. Ethidium bromide was added at 10 μg/ml to protein extracts prior to and during IP and to the wash buffer.
Plasmids
Construction of pG-LAP7/puro p53 (p53-LAP) expression plasmid
Gateway entry vector for p53 was created by BP recombination between pDONR221 and PCR amplified p53 cDNA fragment. Flp-In system compatible C-terminally LAP-tagged p53 (p53-LAP) was generated by LR recombination between p53 entry vector (pENTR p53) and pG-LAP7/puro destination vector (gift from Peter Jackson, Stanford University).
Construction of pG-LAP2 Mettl3 (Flag-METTL3) expression plasmid
Gateway entry vector for Mettl3 was created by BP recombination between pDONR221 and PCR amplified Mettl3 cDNA fragment. Flp-In system compatible N-terminally Flag-tagged METTL3 (Flag-METTL3) was generated by LR recombination between Mettl3 entry vector and pG-LAP2/puro destination vector (gift from Peter Jackson lab, Stanford University). Q5 site-directed mutagenesis (NEB E0554S) kit was used to generate the Flag-METTL3 APPA mutant expression construct.
Cloning of p53 deletion mutants in pcDNA3.1–3XHA-p53 backbone
Mutant p53 cDNAs were generated by polymerase chain reaction using pcDNA3.1–3XHA-p53 vector that contains N-terminal HA tagged full-length wild-type p53 as a template. To generate p53 ΔTAD1 (Δ1–42), the pair of primers used were: forward primer 5’-TTTTGGCGCGCCGATCTGTTGCTGCCCCAG-3’; and reverse primer: 5’-TTTTTTAATTAATCAGTCTGAGTCAGGCCC-3’. To generate p53 ΔTAD1,2 (Δ1–61), the pair of primers used were: forward primer 5’-TTTTGGCGCGCC CGAGTGTCAGGAGCTCCT-3’; and reverse primer 5’-TTTTTTAATTAATCAGTCTGAGTCAGGCCC-3’.
Construction of pCS2-HA-Mettl3 expression plasmid
Gateway entry vector for Mettl3 were created by BP recombination between pDONR221 and PCR amplified Mettl3 cDNA fragment. LR recombination between Mettl3 entry vector (pENTR Mettl3) and pCS2-HA destination vector (gift from Peter Jackson, Stanford University) was performed using Gateway LR Clonase II enzyme mix (Thermo Fisher Scientific, 11791100) according to manufacturer’s instructions.
Construction of Lenti-U6-sgMettl3/Cre vector
We generated lentiviral vectors carrying PGK-Cre as well as an sgRNA targeting Mettl3 or control sgRNA targeting Neo. Lenti-U6-sgRNA/Cre vectors containing each sgRNA were generated as described previously (Chiou et al., 2015; Rogers et al., 2017). Briefly, Q5 site-directed mutagenesis (NEB E0554S) kit was used to insert Mettl3 or control sgRNAs into the parental lentiviral vector containing the U6 promoter as well as PGK-Cre.
Construction of pX458 TP53 vector
We generated pX458 TP3 vector by cloning sgRNAs targeting human TP53 into the pX458 plasmid (Addgene Plasmid #48138). Guides were designed using the design tool at https://chopchop.cbu.uib.no/. The pX458 plasmid was digested using BbsI and a pair of partially complementary annealed oligos containing overhangs from BbsI site and TP53 sgRNA sequence were cloned into the vector. The oligo sequences used were: 5′-CACCGCCATTGTTCAATATCGTCCG-3′ and 5’-AAACCGGACGATATTGAACAATGGC-3’; 5’-CACCGCGACGCTAGGATCTGACTG-3’ and 5’-AAACCAGTCAGATCCTAGCGTCGC-3’; 5’-CACCGCCCGGACGATATTGAACAA-3’ and 5’-AAACTTGTTCAATATCGTCCGGGC-3’.
Construction of Flp-In 3T3 p53-LAP, Flp-In 3T3 p53−/− and A549 TP53−/− cell lines
We used Flp-In™−3T3 cells (Thermo Fisher Scientific, Cat # R76107) to construct a cell line stably expressing C-terminally LAP-tagged wild-type p53 cDNA. Gateway entry vector for p53 was created by BP recombination between pDONR221 and PCR amplified p53 cDNA fragment. Flp-In system compatible C-terminally LAP-tagged p53 (p53-LAP) was generated by LR recombination between p53 entry vector and pG-LAP7/puro destination vector (gift from Peter Jackson, Stanford University). Flp-In 3T3 cells stably expressing p53-LAP were generated by co-transfecting 0.4 μg of the preceding vector with 3.6 μg of pOG44, followed by selection with 4 μg/ml puromycin. Flp-In 3T3 p53−/− cell line was generated by Crispr/Cas9 by co-transfecting Flp-In 3T3 cells with pX330 p53 plasmid (gift from Tyler Jacks, Addgene Plasmid #59910) expressing Cas9 and sgRNA targeting mouse p53 and pmaxGFP plasmid (Lonza). Two days post transfection, the GFP positive population was sorted by FACS and clonally expanded. Individual cell clones were screened for p53 deletion using standard PCR and TIDE analysis. Loss of p53 protein expression was confirmed by immunoblotting. A549 TP53−/− cells were generated using pX330-based plasmid, pX458 (gift from Feng Zhang, Addgene Plasmid #48138) expressing Cas9, GFP and sgRNA targeting human TP53. A549 cells were co-transfected with three pX458 TP53 plasmids expressing three distinct sgRNAs targeting TP53. Two days post transfection, the GFP positive population was sorted by FACS and clonally expanded. Individual cell clones were screened for p53 loss by immunoblotting.
Lentiviral shRNA cell lines
Mouse and human shMettl3 constructs were a gift from Pedro Batista, NIH. Lentivirus carrying shMettl3 were produced by co-transfecting 293T cells with 150 ng of pCMV-VSV-G, 350 ng of pCMV-dR8.2 dvpr, and 500 ng of shMettl3 plasmids. Media was replaced 16 hr after transfection to remove transfection reagent, and virus was harvested at 48 hr post-transfection. Virus was then filtered with a 0.45 μm PVDF filter (SLHV013SL, Millipore) and mixed with polybrene (TR-1003-G, Millipore). Wild-type or p53 null E1A;HRasG12V-expressing MEFs (Bieging-Rolett et al., 2020) or A549 cells were infected for 48 hr, followed by selection with 2 μg/ml puromycin. Knock-down of METTL3 expression was confirmed by immunoblotting using anti-METTL3 (Abcam, ab195352, 1:1000).
METHOD DETAILS
Tandem Affinity Purification
After constructing Flp-In-3T3 expressing p53-LAP, we grew large scale cultures in roller bottles and affinity purified protein complexes for mass spectrometry. The cultures were either left untreated or treated with 0.2 μg/ml dox for 6 hours. Large-scale preparations of whole-cell lysates were subjected to dual-affinity purification, first with anti-GFP antibody-coupled beads to pull-down p53-LAP complexes. We then employed PreScission™ protease, which cleaves at a unique site between the GFP and S-tags and performed a second round of affinity purification using a Protein S Agarose column that binds the S-tag. The bound p53-S-tag and any interacting proteins that were co-purified were eluted off the beads under denaturing conditions and run on a gradient gel, which was stained with Coomassie blue, and each lane was cut into 8 discrete bands, which were submitted for mass spectrometric protein identification. A 10 ml packed cell volume was re-suspended with 20 mL of LAP-resuspension buffer (300 mM KCl, 50 mM HEPES-KOH [pH 7.4], 1 mM EGTA, 1 mM MgCl2, 10% glycerol, 0.5 mM DTT), and protease inhibitors (Thermo Fisher Scientific, PI88266), lysed by gradually adding 0.6 mL 10% NP-40 to a final concentration of 0.3%, then incubated on ice for 10 min. The lysate was first centrifuged at 14,000 rpm (27,000 g) at 4°C for 30 min, and the resulting supernatant was centrifuged at 43,000 rpm (100,000 g) for 1 hr at 4°C to further clarify the lysate. High speed spin supernatant was mixed with 0.5 mL of GFP-coupled beads and rotated for 1 hr at 4°C to capture GFP-tagged proteins, and washed five times with 1 mL LAP200N buffer (200 mM KCl, 50 mM HEPES-KOH [pH 7.4], 1 mM EGTA, 1 mM MgCl2, 10% glycerol, 0.5 mM DTT, protease inhibitors, and 0.05% NP-40). After re-suspending the beads with 1 mL LAP200N buffer lacking DTT and protease inhibitors, the GFP tag was cleaved by adding 5 mg of PreScission protease and rotating tubes at 4°C for 16 hours. All subsequent steps until the cutting of bands from protein gels were performed in a laminar flow hood to prevent keratin contamination. PreScission protease-eluted supernatant was added to 100 mL of S-protein agarose (EMD Millipore, 69704–3) to capture S-tagged protein. After washing three times with LAP200N buffer lacking DTT and twice with LAP100 buffer (100 mM KCl, 50 mM HEPES-KOH [pH 7.4], 1mM EGTA, 1mM MgCl2, and 10% glycerol), purified protein complexes were eluted with 50 μL of 2X LDS buffer and boiled at 95°C for 3 min. 5% of the total eluate was run on a gradient gel and silver-stained as quality control. Samples were then run on Bolt Bis-Tris Plus Gels (Thermo Fisher Scientific, NW04120BOX) in Bolt MES SDS Running Buffer (Thermo Fisher Scientific, B000202). Gels were fixed in 100 mL of fixing solution (50% methanol, 10% acetic acid in Optima LC/MS grade water (Thermo Fisher Scientific, W6–1) at room temperature, and stained with Colloidal Blue Staining Kit (Thermo Fisher Scientific, LC6025). After the buffer was replaced with Optima water, the bands were cut into eight pieces, followed by washing twice with 500 μL of 50% acetonitrile in Optima water. LC-MS/MS was performed by in-gel tryptic digestion of the gel bands followed by protein identification on a high performance Thermo Scientific Orbitrap Fusion™ Tribrid™ mass spectrometer as described below.
Mass Spectrometry
Samples were processed for mass spectrometry by Stanford University Mass Spectrometry Facility. In a typical experiment, protein gel bands were first diced into 1 mm cubes and reduced with 5 mM DTT, 50 mM ammonium bicarbonate. After removal of residual solvent, proteins were alkylated using 10 mM acrylamide in 50 mM ammonium bicarbonate for 30 min at room temperature. Digestion was performed using Trypsin/LysC (Promega, Cat # V5071) in the presence of 0.02% Protease Max (Promega, Cat # V2071) overnight at 37°C. The following day, solid particulate was condensed by centrifugation and peptides extracted by adding 60% acetonitrile, 39.9% water, 0.1% formic acid and incubating for 15 min. Extracted peptides were dried in a speed vac and then reconstituted in 12.5 μl reconstitution buffer (2% acetonitrile with 0.1% Formic acid) and 3 μl of it was injected on the instrument.
Mass spectrometry experiments were performed using an Orbitrap Fusion™ Tribrid™ mass spectrometer (Thermo Scientific, San Jose, CA) with liquid chromatography performed using an Acquity M-Class UPLC (Waters Corporation, Milford, MA). For a typical LC MS experiment, a pulled-and-packed fused silica C18 reverse phase column was used, with Dr. Maisch 1.8-micron C18 beads as the packing material and a length of ~25 cm. A flow rate of 450 nL/min was used with a mobile phase A of aqueous 0.2% formic acid and mobile phase B of 0.2% formic acid in acetonitrile. Peptides were directly injected onto the analytical column using a gradient (3–45% B, followed by a high-B wash) for 80 min. The mass spectrometer was operated in a data dependent fashion, with MS1 survey spectra collected in the orbitrap and MS2 fragmentation using CID for in the ion trap.
For data analysis, the .RAW data files were processed using Byonic (Protein Metrics, San Carlos, CA) to identify peptides and infer proteins. Proteolysis was assumed to be tryptic in nature and allowed for up to two missed cleavage sites. Precursor mass accuracies were held within 12 ppm, with MS/MS fragments held to a 0.4 Da mass accuracy. Proteins were held to a false discovery rate of 1%, using standard approaches (Elias and Gygi, 2007). Spectral counts from Byonic output were normalized by calculating NSAF values (Zybailov et al., 2006) and bait-prey interactions were scored based on large numbers of unrelated affinity pull-downs in mouse cell lines(Li et al., 2017a).
Co-immunoprecipitation assays
Cells were harvested by scraping method using cell scrapers (Corning, Cat # 3010) and lysed with ice cold NP-40 lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 0.5 mM EDTA, 10% glycerol) containing protease inhibitors (Roche cOmplete™, Cat # 11 697 498 001). Protein was quantitated using the bicinchoninic acid protein assay (BCA) kit (Pierce, Cat # 23227). 1–2 mg total protein was used for each immunoprecipitation reaction (IP) reaction in 500 μl final volume. Lysates were first pre-cleared using 50% slurry of BSA blocked Protein A sepharose beads (GE, Cat # 17–0780-01) by incubating for 30 minutes at 4°C. For METTL3 IPs, the pre-cleared lysates were incubated with 1–2 μg METTL3 polyclonal antibody (Abclonal, A8370) overnight at 4°C on a nutator to allow protein complexes to form. For p53 IPs, pre-cleared lysates were incubated with 2–4 μl p53 polyclonal antibody (Leica Biosystems, NCL-L-p53-CM5p) overnight at 4°C on a nutator to allow p53 protein complexes to form. The day after, immune complexes were retrieved with 50 μl of 50% slurry of BSA blocked Protein A Sepharose beads for 4 hours at 4°C. Post the incubation, the beads were washed 3 times using 0.1% NP-40 containing wash buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.1% NP-40, 0.5 mM EDTA, 10% glycerol). The immobilized immunoprecipitated complexes were eluted by boiling the sepharose beads in 2X SDS sample buffer. For Flag-HA CoIPs, lysates were pre-cleared using Protein A/G magnetic beads (Thermo Fisher Scientific, Cat # 26162) for 30 minutes at 4°C. The pre-cleared lysates were incubated with 25 μl Flag M2 magnetic beads (Millipore, Cat # M8823) for 4 hours at 4°C to immunoprecipitate Flag-tagged METTL3. Beads were washed 4 times using 1% Triton X-100 containing wash buffer (10 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA). Flag-protein complexes were eluted using Flag peptide (Millipore, Cat # F3290) at 150 μg/ml by incubating the beads at room temperature for 30 minutes. The eluates were resolved on a 10% SDS-PAGE gel and the proteins were electroblotted onto PVDF membranes (Millipore, Immobilon-P, Cat # IPVH20200) for probing with following primary and secondary antibodies: anti-p53 (gift from Helin K, Univ. of Copenhagen, clone AI25, 1:500), anti-METTL3 (Abclonal, A8370, 1:500), anti-MDM2 (Abcam, ab16895, 1:500), anti-Flag (Sigma, F1804, 1:1000) and anti-HA (Thermo Fisher Scientific, 71–5500, 1:500), peroxidase Affinipure goat anti-mouse IgG, light chain specific (Jackson ImmunoResearch, 115–035-174, 1:5,000), peroxidase IgG fraction monoclonal mouse anti-rabbit IgG, light chain specific (Jackson ImmunoResearch, 211–032-171, 1:5,000). Inputs represent 2–5% of the lysate subjected to immunoprecipitation.
GST pull-down assays
BL21 competent E.coli expressing plasmids encoding GST only or GST-tagged p53 were grown on LB-Amp plates. Isolated colonies were inoculated in LB media and grown overnight at 37°C with agitation. Starter cultures were diluted 1:200 and incubated at 37°C with agitation until an optical density of 0.5 was reached. Protein expression was induced with 1 mM IPTG (Sigma-Aldrich, Cat # 16758) for 5 hours. Cultures were centrifuged at 5000 rpm at 4°C for 15 minutes to pellet the cells. Pellets were resuspended in ice-cold GST-lysis buffer (1x PBS, 0.1% NP-40, 10% glycerol, 1 mM DTT) containing protease inhibitors (Roche cOmplete™, Cat # 11 697 498 001) and sonicated for 3 cycles of 30 pulses, where each pulse was at 40% amplitude and lasted 1 second, with 2 seconds between each pulse. The samples were centrifuged at 10,000 rpm at 4°C for 20 minutes. The supernatant was incubated with 50% slurry of Glutathione Sepharose 4B beads (Cytiva, Cat # 17–0756-01) in GST-lysis buffer for 15 minutes at room temperature. The beads were pelleted via centrifugation in a swinging bucket rotor at 1500 rpm at 4°C for 1 minute. The beads were washed 3x at 4°C with light agitation, first using GST-lysis buffer, followed by GST-lysis buffer + 500 mM NaCl, then followed by GST-lysis buffer. Beads were resuspended in BC-100 buffer (20 mM Tris pH 7.9, 100 mM KCl, 0.2 mM EDTA, 15% glycerol, 10 mM β-mercaptoethanol, 0.5 mM DTT) containing protease inhibitors (Roche cOmplete™, Cat # 11 697 498 001) for storage at 4°C. GST proteins were analyzed by SDS-PAGE and Coomassie blue staining.
GST-p53 and HDM2 interaction was tested by mixing 2.5 μL of GST only or GST-p53 beads with 3 μL of purified 6xHis-HDM2 (gift from Hua Lu and Shelya Zheng, Tulane University). Reaction volume was made up to 50 μL with GST-lysis buffer and reactions were incubated with gentle shaking at room temperature for 45 minutes. The beads were washed 3x with GST-lysis buffer with light vortexing between each wash. Protein complexes were eluted from the beads by boiling in 1X SDS loading buffer at 95°C for 5 minutes. Samples were analyzed by immunoblotting using anti-MDM2 (Abcam, ab16895, 1:1000). To examine the interaction between GST-p53 and METTL3, the same protocol was performed except replacing HDM2 with either 2.5 μL or 5 μL of in vitro translated HA-METTL3. HA-METTL3 was generated by in vitro translation reaction using pCS2-HA-Mettl3- plasmid and a coupled transcription/translation system (Promega, L3260) according to manufacturer’s instructions. Briefly, 6 μg of pCS2-HA-Mettl3 plasmid was added to 30 μL of TNT master mix and the final volume was made up to 50 μL with nuclease-free water. Reactions were incubated at room temperature for 2 hours and extracts were analyzed by immunoblotting using anti-METTL3 (Abcam, ab195352, 1:1000).
Immunoblotting
Protein was extracted using NP-40 lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5 mM EDTA, and 10% glycerol) containing protease inhibitors (Roche cOmplete™, Cat # 11 697 498 001). Protein was quantitated using the BCA kit (Pierce, Cat # 23227). 20 μg of protein was resolved on a 10% SDS-PAGE gel, electroblotted onto PVDF membranes (Millipore, Immobilon-P, Cat # IPVH20200) and blocked in 5% non-fat dry milk prepared in TBS with 0.1% Tween-20 (TBST). Three washes were performed in TBST, and the following primary and secondary antibodies were used: rabbit anti-p53 (Leica Biosystems, NCL-L-p53-CM5p, 1:5000), rabbit anti-METTL3 (Abclonal, A8370, 1:1000), rabbit anti-METTL3 (Abcam, ab195352, 1:1000), mouse anti-MDM2 (Abcam, ab16895, 1:1000), mouse anti-Flag (Sigma, F1804, 1:1000), rabbit anti-HA (Thermo Fisher Scientific, 71–5500, 1:500), mouse anti-p53 (Cell Signaling Technology, 2524, 1:1000), mouse anti-GAPDH (Fitzgerald, 10R-G109A, 1:10,000), peroxidase Affinipure goat anti-rabbit IgG (H+L) (Jackson ImmunoResearch, 111–035-144, 1:5,000), and peroxidase Affinipure goat anti-mouse IgG (H+L) (Jackson ImmunoResearch, 115–035-003, 1:5,000). Immunodetection was performed using ECL™ Prime (Millipore-Sigma, Cat# GERPN2232) or Clarity™ Western ECL substrate (Bio-Rad, Cat# 1705060).
qRT-PCR
RNA extraction was performed using TRIzol reagent (Thermo Fisher Scientific, Cat #15596018) according to the manufacturer’s protocol. RNA (2–5 μg) was treated with DNAse I (Thermo Fisher Scientific, Cat # AM1906) according to the manufacturer’s instructions. Reverse transcription was conducted with M-MLV reverse transcriptase (Thermo Fisher Scientific, Cat # 28025) and random primers (Thermo Fisher Scientific, Cat # 48190). 1 μg of total RNA was used for cDNA synthesis. cDNA was diluted 1:5 in nuclease-free water and stored at −80°C until used. Quantitative PCR was performed in triplicate using PowerUP SYBR green master mix (Thermo Fisher Scientific, Cat # A25743) and a 7900HT Fast Real-Time PCR machine (Applied Biosystems). Expression analysis was performed using specific primers for each gene (Table S4). The mean of housekeeping gene β-Actin was used as an internal control to normalize the variability in expression levels. All qRT-PCR performed using PowerUP SYBR Green was conducted at 50°C for 2 min, 95°C for 10 min, and then 40 cycles of 95°C for 15 s and 60°C for 1 min. Melt curve analysis was done to verify the specificity of the reaction. Samples were quantified using a standard curve.
ChIP
Analysis of METTL3 chromatin binding was done in wild-type and p53−/− E1A;HRasG12V-expressing MEFs. MEFs were seeded at 7 × 106 cells per 10 cm dish, one day prior to the ChIP experiment. After treatment with 0.2 μg/ml doxorubicin for 6h, cells were harvested to prepare chromatin for immunoprecipitation using METTL3 polyclonal antibodies (Abclonal, Cat # A8370). To prepare chromatin for immunoprecipitation, proteins were cross-linked to DNA by adding 1% formaldehyde (Sigma, Cat # F8775) and incubating at room temperature for 10 mins. The cross-linking reaction was quenched by adding glycine at a final concentration of 0.125 M and swirling gently to mix. The medium in the plates was removed and cells were washed with cold 1X PBS. PBS was aspirated and 5 mL cold Farnham lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40) containing protease inhibitors (Roche cOmplete™, Cat # 11 697 498 001) was added. Cells were scraped off the plate with a cell scraper and transferred into 15-ml conical tubes on ice. Cells were pelleted at 2,000 rpm for 5 mins at 4°C. Cells were placed on ice and supernatant was carefully removed. Cell pellets were snapfrozen in liquid nitrogen and stored at −80°C.
For further processing, each pellet was resuspended in 1 mL Farnham lysis buffer on ice and mixed gently by flicking the tube. The lysate was passed through a 21-gauge needle 20 times, avoiding air bubbles. The crude nuclear preparation was collected by centrifuging at 2,000 rpm for 5 minutes at 4°C. The supernatant was discarded, and the pellet was resuspended in 200 μL RIPA buffer (1X PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease inhibitors. Samples were sonicated to shear the chromatin using a Bioruptor 300 (Diagenode) with a circulating water bath, according to the manufacturer’s instructions, at the medium setting (14 cycles of 30 seconds ON, 50 seconds OFF, 4°C). The sonicated lysates were centrifuged at 14,000 rpm in a microfuge for 15 minutes at 4°C, and the supernatant was collected. Antibodies were pre-coupled to Protein A/G magnetic beads (Thermo Fisher Scientific, Cat # 26162), according to the bead manufacturer’s instructions. The bead-antibody complex was incubated with the chromatin preparation by mixing 100 μL antibody-coupled beads to each 200 μL chromatin preparation and incubating on a rotator for one hour at room temperature, followed by one hour at 4°C. Beads containing immuno-bound chromatin were collected by placing the microfuge tube on a magnet rack and discarding the supernatant. Washes were performed two times with low-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl at pH 8.1, 150 mM NaCl), three times with high-salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl at pH 8.1, 500 mM NaCl), and four times with LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholic acid, 1 mM EDTA, 10 mM Tris at pH 8.1). 1 ml TE buffer (1M Tris-HCL at pH 8.0, 0.5 M EDTA at pH 8.0) was added to the washed beads. After 1 minute of mixing on the rotator, the tubes were placed on the magnet rack to collect the beads, and the supernatant was discarded. Beads were resuspended in 200 μL IP elution buffer (1% SDS, 0.1 M NaHCO3). To elute the immuno-bound chromatin from the beads, the beads were incubated in a 65°C water bath for 1 hour, with vortexing every 15 minutes. Beads were centrifuged at 14,000 rpm in a microfuge at room temperature for 3 minutes, and the supernatant was collected. Supernatant containing protein-DNA complexes was incubated in a 65°C water bath overnight to complete the reversal of the formaldehyde cross-links. To prepare a control reverse cross-link chromatin library, a sample of sheared chromatin representing at least 1×106 cells was incubated in a 65°C water bath overnight to reverse the cross-links. Volumes were made up to 200 μL with 1X PBS, 20 μL of Proteinase K (Invitrogen 25530) was added, and samples were incubated at room temperature for 2 minutes. 4 μL of RNase A (Thermo Scientific EN0531) was added, followed by 5 volumes of Qiagen buffer PB from the QIAquick PCR purification kit (Qiagen, Cat #28104), and samples were incubated at room temperature for 20 minutes. DNA in the reverse-cross-linked samples was recovered by column chromatography using the Qiagen Qiaquick PCR purification kit, according to the manufacturer’s instructions. Chromatin-immunoprecipitated DNA was analyzed by quantitative PCR with primers flanking various p53 binding sites (Table S4) using PowerUP SYBR green master mix (Thermo Fisher Scientific, Cat # A25743) and a 7900HT Fast Real-Time PCR machine (Applied Biosystems). ChIP-qPCR data were analyzed by the percent input method of analysis in which signals obtained from the ChIP were divided by signals obtained from an input sample.
RNA-seq library preparation and data analysis
RNA was extracted from cell lines using TRIzol reagent (Thermo Fisher Scientific, Cat #15596018) according to the manufacturer’s protocol. RNA quality was assessed using a BioAnalyzer (2100 Bioanalyzer Instrument, Agilent). Novogene generated and sequenced RNA-seq libraries using the NEBNext Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA) following manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Library quality was assessed on the Agilent Bioanalyzer 2100 system. Raw data (raw reads) of FASTQ format were firstly processed through fastp (Chen et al., 2018). In this step, clean data (clean reads) were obtained by removing reads containing adapter and poly-N sequences and reads with low quality from raw data. At the same time, Q20, Q30 and GC content of the clean data were calculated. All the downstream analyses were based on the clean data with high quality. Reference genome and gene model annotation files were downloaded from genome website browser (NCBI/UCSC/Ensembl) directly. Paired-end clean reads were aligned to the reference genome using the Spliced Transcripts Alignment to a Reference (STAR) software, which is based on a previously undescribed RNA-seq alignment algorithm that uses sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure. STAR exhibits better alignment precision and sensitivity than other RNA-seq aligners for both experimental and simulated data. FeatureCounts was used to count the read numbers mapped of each gene (Liao et al., 2014). And then RPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. RPKM, Reads Per Kilobase of exon model per Million mapped reads, considers the effect of sequencing depth and gene length for the reads count at the same time, and is currently the most commonly used method for estimating gene expression levels (Mortazavi et al., 2008). Differential expression analysis between two conditions/groups (three biological replicates per condition) was performed using DESeq2 R package (Love et al., 2014). DESeq2 provides statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting P values were adjusted using the Benjamini and Hochberg’s approach for controlling the False Discovery Rate (FDR). Genes with an adjusted P value < 0.05 found by DESeq2 were assigned as differentially expressed. Pathway analysis was performed using Enrichr (Enrichr: http://amp.pharm.mssm.edu/Enrichr/) (Chen et al., 2013; Kuleshov et al., 2016).
m6A-eCLIP-seq
m6A-eCLIP was performed at Eclipse BioInnovations Inc, San Diego CA as described below. The procedure started by isolating mRNAs through poly(A) selection using oligo (dT) beads (Thermo Fisher Scientific) followed by magnetic bead separation from 50 μg of total RNA. The mRNA was DNase treated at 37°C for 10 minutes then immediately sheared into 100–200nt fragments by heating at 95°C for 12 minutes in 1x Turbo DNase buffer (Thermo Fisher Scientific). An anti-m6A antibody (CST) was added, and samples were UV-C crosslinked using a UVP CL-1000 crosslinker at 2 rounds of 150 mJ/cm2 using 254nm wavelength. The antibody-RNA complexes were then coupled overnight to protein G beads (CST). Following overnight coupling, library preparation (including adapter ligations, SDS-PAGE electrophoresis and nitrocellulose membrane transfer, reverse transcription, and PCR amplification) was performed as previously described for standard eCLIP (Van Nostrand et al., 2016), with the 30–110 kDa region size-selected by cutting from the membrane (corresponding to RNA fragments cross-linked to antibody heavy and light chains). 10 ng of fragmented mRNA was run as an RNA-seq input control, starting with FastAP treatment as described (Van Nostrand et al., 2016). The final library’s shape and yield was assessed by Agilent TapeStation.
The eCLIP cDNA adapter contains a sequence of 10 random nucleotides at the 5′ end. This random sequence serves as a unique molecular identifier (UMI) (Kivioja et al., 2011) after sequencing primers are ligated to the 3′ end of cDNA molecules. Therefore, eCLIP reads begin with the UMI and, in the first step of analysis, UMIs were pruned from read sequences using umi_tools (v0.5.1) (Smith et al., 2017). UMI sequences were saved by incorporating them into the read names in the FASTQ files to be utilized in subsequent analysis steps. Next, 3′-adapters were trimmed from reads using cutadapt (v2.7) (Martin, 2011) and reads shorter than 18 bp in length were removed. Reads were then mapped to a database of mouse repetitive elements and rRNA sequences compiled from Dfam (Hubley et al., 2016) and Genbank (Benson et al., 2014). All non-repeat mapped reads were mapped to the mouse genome (mm10) using STAR (v2.6.0c) (Dobin et al., 2013). PCR duplicates were removed using umi_tools (v0.5.1) by utilizing UMI sequences from the read names and mapping positions. Peaks were identified within m6A-eCLIP samples using the peak caller CLIPper (GitHub: https://github.com/YeoLab/clipper) (Lovci et al., 2013). For each peak, IP versus input fold enrichment values were calculated as a ratio of counts of reads overlapping the peak region in the IP and the input samples (read counts in each sample were normalized against the total number of reads in the sample after PCR duplicate removal). The RPM value for a particular region was calculated by counting the number of reads in that region and dividing that by the “per million” scaling factor, which is defined as the total number of reads in the sample divided by 1,000,000. The log2 fold changes for a peak are calculated as follows: Log2 (RPM in IP in peak region / RPM in input in peak region). Log2 fold change values (RPM in IP in peak region / RPM in input in peak region) were calculated for individual peaks in each m6A-IP to normalize the IP reads to the total number of reads covering the m6A residue. A p-value was calculated for each peak by the Yates’ Chi-Square test, or Fisher Exact Test if the observed or expected read number was below 5. Peaks were annotated using transcript information from GENCODE (Release M21) (Frankish et al., 2019) with the following priority hierarchy to define the final annotation of overlapping features: protein coding transcript (CDS, UTRs, intron), followed by non-coding transcripts (exon, intron).
Significance of the difference in IP enrichment, i.e. IP/input ratios, between shLuc and shMettl3-2 samples treated with dox, was used to identify genes that were most enriched for a specific m6A modification under DNA damage in a METTL3-dependent manner. For each peak called in IP library of shLuc_dox, we calculated enrichment and p-values relative to normalized counts of reads overlapping these peaks in shMettl3-2_dox. Significance of the difference in IP enrichment was evaluated using the Yates’ Chi-Squared Test. Since RPM normalization can result in numerically very small values, prior to performing statistical tests the normalized values were scaled in order to increase power. Scaling factors were chosen as an inverse count of mapped reads in the deepest library out of IP and input libraries in samples in the comparison, which had an effect of scaling RPM normalized values corresponding to a read count of 1 in the deepest library to also be equal to 1. Finally, resulting p-values were adjusted for multiple testing using the Benjamini-Hochberg technique.
Cutoffs for significance were chosen as follows:
IP/input ratios were ≥ 1 (log2 scale) in either the shLuc_dox or the shMettl3-2_dox
Difference of IP/input ratios between shLuc_dox and shMettl3-2_dox was ≥ 0.5 on log2 scale
Adjusted p-value ≤ 0.05
Using above-described statistical method, we identified 1215 unique genes with enhanced METTL3-dependent m6A modification under DNA damage (Table S3). Pathway analysis of these genes was performed using Metascape (v3.5).
Polysome fractionation assay
Mouse embryonic stem cells were seeded at a density of 1.5 million in a 10 cm plate and incubated for 24 h at 37°C. Cells were then treated with 0.2 μg/mL doxorubicin for 8 hours. Following this, cells were treated with 100 μg/mL of cycloheximide (CHX) and incubated for 5 min. Cells were then washed with 1X PBS plus CHX and harvested in 300 μL of Polysome Lysis Buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 15 mM MgCl2, 1 mM DTT, 8% glycerol, 1% Triton X-100, 100 μg/ml CHX (Sigma-Aldrich, C7698–1G), 200 U/mL SUPERase RNase Inhibitor (Thermo Fisher, AM2696), 20 U/ml Turbo DNAse (Thermo Fisher, AM2238), and 1X Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher, 78442)). Samples were centrifuged at 1,300×g for 5 min and 10,000×g for 5 min to remove cell debris. Samples were then layered onto a 15–45% sucrose gradient containing 20 mM Tris-HCl pH 7.5, 100 mM NaCl, 15 mM MgCl2, and 100 μg/ml CHX. The 15–45% gradients were formed on a BioComp Model 108 Gradient Master. Layered gradients were ultracentrifuged with a Beckman SW41-Ti rotor at 100,000xg for 2.5h at 4C. After centrifugation, gradients were fractioned on a Density Gradient Fractionation System (Brandel, BR-188) at a flow rate of 1.5 mL/min. To each fraction, 100 μL of 10% SDS and 100 μL of 3M NaOAc, pH 5.5, was added. For normalization in RT-qPCR, 5 pg of in vitro transcribed Renilla luciferase mRNA was spiked into each individual fraction. RNA was then extracted by mixing samples with Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1) (Thermo Fisher, AM9722), incubating at 65°C, 5 min, and subsequent centrifugation at 21,000×g for 10 min at room temperature. The aqueous phase was isolated and mixed with an equal volumes of 100% ethanol. RNA was then further purified using the RNA Clean & Concentrator-5 kit (Zymo Research, R1013). Next, 100 ng of RNA was used for cDNA synthesis with the iScript Supermix (Bio-rad, 1708841). cDNA was diluted 10-fold in nuclease-free water and stored at −80°C until used. Quantitative PCR was performed in triplicate using PowerUP SYBR green master mix (Thermo Fisher Scientific, Cat # A25743) and a 7900HT Fast Real-Time PCR machine (Applied Biosystems). Expression analysis was performed using specific primers for each gene (Table S4). For analysis, the candidate transcript qPCR Ct value in each fraction was normalized to that of the spike-in in vitro transcribed Renilla luciferase RNA. Normalized Ct values were converted from log to linear space and then normalized to the sum total across all fractions for a given sample.
Low plating assays
E1A;HRasG12V MEFs or A549 cells were plated on six-well plates at 150 cells per well in triplicate and grown for 10–12 days. Cells were fixed with 10% formalin and stained with 0.1% crystal violet stain. Plates were scanned, and colonies were counted manually.
Immunohistochemistry
Hematoxylin and eosin (H&E) staining on paraffin embedded lung tissues were performed using standard protocols. A NanoZoomer 2.0-RS (Hamamatsu) or Aperia AT2 (Leica) slide scanner was used for imaging.
Mutual Exclusivity, Differential Expression Analysis and Achilles Perturbation Data
TCGA gene expression data was downloaded from NCI Genomic Data Commons: gdc.cancer.gov. Whole Exome Sequencing annotations (MAF file) was downloaded from (Ellrott et al., 2018). Gene expression raw counts were normalized using voom from limma package. Limma was used to calculate the TP53 targets differential expression between TP53 truncated tumors and TP53 wild-type, and between MTC altered (mutated, amplified or deleted) tumors and MTC wild-type for each tumor type individually (Ritchie et al., 2015). Summary fold changes were obtained using the function combine.est from genefu package (Gendoo et al., 2016).
The Discover test (Canisius et al., 2016) was used to calculate mutual exclusivity across MTC genes (mutated or deleted) and TP53 (missense or truncated mutations) across all TCGA cancer tissues individually and combined (adjusted by tissue).
Achilles perturbation scores and CCLE mutations were downloaded from DepMap site (DepMap: depmap.org, version 21Q4) (Meyers et al., 2017). Two sample Wilcoxon test was used to compare METTL3 Achilles score and mutation profiles (hotspot mutations as defined in DepMap). The METTL3 Achilles scores were scaled such that the mean is 0 and the standard deviation is 1. Scaled scored were calculated using the formula: scaled score = (actual score-mean (actual score))/sd(actual score). The Pearson correlation coefficient was used to calculate the correlation across Achilles scores.
QUANTIFICATION AND STATISTICAL ANALYSIS
The Benjamini-Hochberg approach was used for controlling the FDR in RNA-seq differential gene expression analysis. In m6A-eCLIP-seq analysis, the significance of the difference in IP enrichment between two samples was evaluated for each m6A peak using the Yates’ Chi-Squared Test, and p-values were adjusted for multiple testing using the Benjamini-Hochberg approach. The DISCOVER method was used to test for mutual exclusivity. Two sample Wilcoxon signed-rank test was used to compare the METTL3 Achilles score and mutation profiles across the DepMap. The Pearson correlation coefficient was used to calculate the correlation across Achilles scores. The unpaired two-tailed Student’s t-test was used for all the other statistical analyses. Error bars represent standard deviation or standard error of the mean (see figure legends). Significance was defined as a p-value ≤0.05, unless otherwise stated. Details and significance values can be found in the figure legends.
Supplementary Material
Highlights.
Affinity purification of p53 reveals METTL3 as a novel p53-interacting partner
METTL3 enhances p53 protein stability and transcriptional programs with stress cues
METTL3 promotes m6A modification of select p53 target gene RNAs upon DNA damage
METTL3 enhances p53-mediated tumor suppression in mouse and human cancers
ACKNOWLEDGMENTS
This work was supported by an NIH R35 grant (CA197591) and the Virginia and D.K. Ludwig Fund for Cancer Research to L.D.A. H.Y.C. is an investigator of the HHMI. P.K.J. was supported by an NIH R01 grant (CA250534). M.B. is an NYSCF Robertson Investigator and was supported by an NIH R01 (HD086634). C.C. is supported by the NIH through the NIH Director’s Pioneer Award (DP1-CA238296). We thank Pedro Batista for the kind gift of the shMettl3 constructs, Grace Li for support with coIP assays, and Pauline Chu for support with histology.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2022.04.010.
DECLARATION OF INTERESTS
H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio, and an advisor of 10x Genomics, Arsenal Biosciences, and Spring Discovery. H.Y.C. is an advisory board member of Molecular Cell. C.C. is an advisor and equity holder of GRAIL/Illumina, Ravel Biotechnology, and DeepCell, and an advisor to NanoString and Genentech. The remaining authors declare no competing interests.
REFERENCES
- Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millán-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, et al. (2017). Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basak S, Jacobs SB, Krieg AJ, Pathak N, Zeng Q, Kaldis P, Giaccia AJ, and Attardi LD (2008). The metastasis-associated gene Prl-3 is a p53 target involved in cell-cycle regulation. Mol. Cell 30, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. (2014). m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, and Sayers EW (2014). GenBank. Nucleic Acids Res. 43, D30–D35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, Kadiwala J, Hubner NC, de Los Mozos IR, Sadée C, et al. (2018). The SMAD2/3 interactome reveals that TGFbeta controls m(6)A mRNA methylation in pluripotency. Nature 555, 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging KT, Mello SS, and Attardi LD (2014). Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging-Rolett KT, Kaiser AM, Morgens DW, Boutelle AM, Seoane JA, Van Nostrand EL, Zhu C, Houlihan SL, Mello SS, Yee BA, et al. (2020). Zmat3 is a key splicing regulator in the p53 tumor suppression program. Mol. Cell 80, 452–469.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. (2011). Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujnicki JM, Feder M, Radlinska M, and Blumenthal RM (2002). Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J. Mol. Evol 55, 431–444. [DOI] [PubMed] [Google Scholar]
- Canisius S, Martens JW, and Wessels LF (2016). A novel independence test for somatic alterations in cancer shows that biology drives mutual exclusivity but chance explains most co-occurrence. Genome Biol. 17, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal LA, Neriah DB, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T, Narayanagari SR, Wheat JC, Todorova TI, Mitchell K, et al. (2018). Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou SH, Winters IP, Wang J, Naranjo S, Dudgeon C, Tamburini FB, Brady JJ, Yang D, Grüner BM, Chuang CH, et al. (2015). Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 29, 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, Du P, Kim W, Tang S, Sliz P, et al. (2018). mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561, 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun G, Lu Z, Huang Y, Yang CG, et al. (2017). m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 18, 2622–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, Ma J, and Wu L (2016). YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4–NOT deadenylase complex. Nat. Commun 7, 12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc 4, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa JM, Verdun RE, and Emerson BM (2003). p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol. Cell 12, 1015–1027. [DOI] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendoo DM, Ratanasirigulchai N, Schröder MS, Paré L, Parker JS, Prat A, and Haibe-Kains B (2016). Genefu: an R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 32, 1097–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle V, and Stark A (2018). Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol 19, 621–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, and Oren M (1997). Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, and Chen J (2020). m(6)A modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell 37, 270–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al. (2018). Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol 20, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, Auer F, et al. (2019). Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 567, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubley R, Finn RD, Clements J, Eddy SR, Jones TA, Bao W, Smit AF, and Wheeler TJ (2016). The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–D89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, and Tuveson DA (2001). Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Brady CA, Johnson TM, Lee EY, Park EJ, Scott MP, and Attardi LD (2011). Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc. Natl. Acad. Sci. USA 108, 17123–17128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TM, Hammond EM, Giaccia A, and Attardi LD (2005). The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat. Genet 37, 145–152. [DOI] [PubMed] [Google Scholar]
- Kaiser AM, and Attardi LD (2018). Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 25, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenhuber ER, and Lowe SW (2017). Putting p53 in context. Cell 170, 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, and Attardi LD (2013). Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 27, 1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, and Taipale J (2011). Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 9, 72–74. [DOI] [PubMed] [Google Scholar]
- Knuckles P, Carl SH, Musheev M, Niehrs C, Wenger A, and Bühler M (2017). RNA fate determination through cotranscriptional adenosine methylation and microprocessor binding. Nat. Struct. Mol. Biol 24, 561–569. [DOI] [PubMed] [Google Scholar]
- Krizhanovsky V, and Lowe SW (2009). Stem cells: the promises and perils of p53. Nature 460, 1085–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, and Vousden KH (1997). Regulation of p53 stability by Mdm2. Nature 387, 299–303. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44. W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Bao S, Qian Y, Geula S, Leslie J, Zhang C, Hanna JH, and Ding L (2019). Stage-specific requirement for Mettl3-dependent m(6)A mRNA methylation during haematopoietic stem cell differentiation. Nat. Cell Biol 21, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Ding S, Feng N, Mooney N, Ooi YS, Ren L, Diep J, Kelly MR, Yasukawa LL, Patton JT, et al. (2017a). Drebrin restricts rotavirus entry by inhibiting dynamin-mediated endocytosis. Proc. Natl. Acad. Sci. USA 114, E3642–E3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Tang J, Huang W, Wang F, Li P, Qin C, Qin Z, Zou Q, Wei J, Hua L, et al. (2017b). The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget 8, 96103–96116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lin S, Choe J, Du P, Triboulet R, and Gregory RI (2016). The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, et al. (2018). m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol 20, 1074–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovci MT, Ghanem D, Marr H, Arnold J, Gee S, Parra M, Liang TY, Stark TJ, Gehman LT, Hoon S, et al. (2013). Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat. Struct. Mol. Biol 20, 1434–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. j 17, 3. [Google Scholar]
- Mello SS, Sinow C, Raj N, Mazur PK, Bieging-Rolett K, Broz DK, Imam JFC, Vogel H, Wood LD, Sage J, et al. (2017a). Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev. 31, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello SS, Valente LJ, Raj N, Seoane JA, Flowers BM, McClendon J, Bieging-Rolett KT, Lee J, Ivanochko D, Kozak MM, et al. (2017b). A p53 super-tumor suppressor reveals a tumor suppressive p53-Ptpn14-yap axis in pancreatic cancer. Cancer Cell 32, 460–473.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, and Jaffrey SR (2015). 5′ UTR m(6)A promotes Cap-independent translation. Cell 163, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, and Levine AJ (1992). The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, and Wold B (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. [DOI] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, and Vogelstein B (1993). Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362, 857–860. [DOI] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, and Hainaut P (2010). TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol 2, a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj N, and Attardi LD (2017). The transactivation domains of the p53 protein. Cold Spring Harb. Perspect. Med 7, a026047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley KJ, and Maher LJ 3rd. (2007). p53 RNA interactions: new clues in an old mystery. RNA 13, 1825–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers ZN, McFarland CD, Winters IP, Naranjo S, Chuang CH, Petrov D, and Winslow MM (2017). A quantitative and multiplexed approach to uncover the fitness landscape of tumor suppression in vivo. Nat. Methods 14, 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabapathy K, and Lane DP (2018). Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol 15, 13–30. [DOI] [PubMed] [Google Scholar]
- Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, and He C (2017). YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 27, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wei J, and He C (2019). Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan BX, Khoo KH, Lim TM, and Lane DP (2014). High Mdm4 levels suppress p53 activity and enhance its half-life in acute myeloid leukaemia. Oncotarget 5, 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiu GC, Kerr CH, Forester CM, Krishnarao PS, Rosenblatt HD, Raj N, Lantz TC, Zhulyn O, Bowen ME, Shokat L, et al. (2021). A p53-dependent translational program directs tissue-selective phenotypes in a model of ribosomopathies. Dev. Cell 56, 2089–2102.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournillon AS, López I, Malbert-Colas L, Findakly S, Naski N, Olivares-Illana V, Karakostis K, Vojtesek B, Nylander K, and Fåhraeus R (2017). p53 binds the mdmx mRNA and controls its translation. Oncogene 36, 723–730. [DOI] [PubMed] [Google Scholar]
- Valente LJ, Tarangelo A, Li AM, Naciri M, Raj N, Boutelle AM, Li Y, Mello SS, Bieging-Rolett K, DeBerardinis RJ, et al. (2020). p53 deficiency triggers dysregulation of diverse cellular processes in physiological oxygen. J. Cell Biol 219. e201908212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara A, Duarte FM, and Lis JT (2018). Molecular mechanisms driving transcriptional stress responses. Nat. Rev. Genet 19, 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, and Prives C (2009). Blinded by the light: the growing complexity of p53. Cell 137, 413–431. [DOI] [PubMed] [Google Scholar]
- Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. (2017). The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med 23, 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Yue M, Wang J, Kumar S, Wechsler-Reya RJ, Zhang Z, Ogawa Y, Kellis M, Duester G, and Zhao JC (2018). N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci 21, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, et al. (2018). METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 22, 191–205. e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al. (2017). RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al. (2016). Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61, 507–519. [DOI] [PubMed] [Google Scholar]
- Yang Z, Yang S, Cui YH, Wei J, Shah P, Park G, Cui X, He C, and He YY (2021). METTL14 facilitates global genome repair and suppresses skin tumorigenesis. Proc. Natl. Acad. Sci. USA 118. e2025948118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, Pilka ES, Aspris D, Leggate D, Hendrick AG, et al. (2021). Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Izumi H, Torigoe T, Ishiguchi H, Yoshida T, Itoh H, and Kohno K (2004). Binding of RNA to p53 regulates its oligomerization and DNA-binding activity. Oncogene 23, 4371–4379. [DOI] [PubMed] [Google Scholar]
- Zaccara S, Ries RJ, and Jaffrey SR (2019). Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol 20, 608–624. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nature communications 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, and Washburn MP (2006). Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res 5, 2339–2347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and m6A-eCLIP-seq data have been deposited at GEO and are publicly available as of the date of publication. The paper analyzes previously published, publicly available datasets. Accession numbers are listed in the key resources table. The Achilles DepMap dataset (DepMap: CCLE_Depmap_21Q4 release) is available from the DepMap portal, DepMap: https://depmap.org/portal/download/. TCGA gene expression data was downloaded from NCI Genomic Data Commons: gdc.cancer.gov. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
All code to reproduce the figures has been deposited at GitHub (GitHub: https://github.com/CancerCompBioLab/Mettl3_paper) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p53 antibody (CM5) | Leica (Novocastra) | NCL-L-p53-CM5p; RRID: AB_563933 |
| p53 antibody (1C12) | Cell Signaling Technology | 2524; RRID: AB_331743 |
| METTL3 polyclonal antibody | Abclonal | A8370; RRID: AB_2770344 |
| METTL3 monoclonal antibody | Abcam | ab195352; RRID: AB_2721254 |
| HA antibody | Thermo Fisher Scientific | 71–5500; RRID: AB_2533988 |
| FLAG antibody | Sigma-Aldrich | F1804; RRID: AB_262044 |
| MDM2 antibody (2A10) | Abcam | ab16895; RRID: AB_2143534 |
| GAPDH antibody | Fitzgerald | 10R-G109A; RRID: AB_1285808 |
| N6-Methyladenosine (m6A) (D9D9W) antibody | Cell Signaling Technology | 56593; RRID: AB_2799515 |
| Goat anti-rabbit HRP antibody | Jackson ImmunoResearch | 111-035-144; RRID: AB_2307391 |
| Goat anti-mouse HRP antibody | Jackson ImmunoResearch | 115-035-003; RRID: AB_10015289 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG, light chain specific | Jackson ImmunoResearch | 115-035-174; RRID: AB_2338512 |
| Peroxidase IgG Fraction Monoclonal Mouse Anti-Rabbit IgG, light chain specific | Jackson ImmunoResearch | 211-032-171; RRID: AB_2339149 |
| Bacterial and virus strains | ||
| Ad-Cre or Ad5CMVCre | University of Iowa | VVC-U of Iowa-5 |
| Lenti-U6-sgMettl3/Cre | This paper | N/A |
| Lenti-U6-sgNeoR1/Cre | Laboratory of Dr. Monte Winslow | N/A |
| E.coli BL21 expressing GST-0 | Laboratory of Dr. Hua Lu and Shelya Zeng | N/A |
| E.coli BL21 expressing GST-p53 | Laboratory of Dr. Hua Lu and Shelya Zeng | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 2000 | Thermo Fisher Scientific | 11668019 |
| Doxorubicin hydrochloride | Sigma-Aldrich | D1515 |
| Puromycin dihydrochloride | Sigma-Aldrich | P7255 |
| Cycloheximide | Sigma-Aldrich | C7698 |
| Glutathione Sepharose 4B | Cytiva | 17-0756-01 |
| GST-0 | This paper | N/A |
| GST-p53 | This paper | N/A |
| 6X-His-HDM2 | Laboratory of Dr. Hua Lu and Shelya Zeng | N/A |
| Critical commercial assays | ||
| Clarity Western ECL Substrate | Bio-Rad | 1705060 |
| ECL Prime Western Blotting System | Millipore-Sigma | GERPN2232 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | 23227 |
| TRIzol reagent | Thermo Fisher | 15596026 |
| PowerUP SYBR green master mix | Thermo Fisher | 4367660 |
| M-MLV Reverse Transcriptase | Thermo Fisher | 28025013 |
| iScript Supermix cDNA synthesis kit | Bio-Rad | 1708841 |
| Gateway BP Clonase II Enzyme Mix | Thermo Fisher | 11789-020 |
| Gateway LR Clonase II Enzyme Mix | Thermo Fisher | 11791-020 |
| Q5 Site-directed mutagenesis kit | NEB | E0554S |
| TNT SP6 High-Yield Wheat Germ Protein Expression System | Promega | L3260 |
| Deposited data | ||
| M6A-eCLIP-seq and RNA-seq | This paper | GEO:GSE169343 |
| shLuc and shMettl3 MEF RNA-seq | This paper | GEO:GSE193341 |
| Mendeley Data | This paper | Mendeley Data: https://dx.doi.org/10.17632/gdpmnddt7v.1 |
| Mouse p53 ChIP-seq | Kenzelmann-Broz et al. (2013) | GEO:GSE46240 |
| E1A;HRasG12V p53WT and p53 null MEF RNA-seq | Valente et al. (2020) | GEO:GSE136355 |
| Achilles DepMap (CCLE_Depmap_21Q4 release) | Meyers et al. (2017) | DepMap: http://depmap.org |
| Scripts used in the study | This paper | GitHub: https://github.com/CancerCompBioLab/Mettl3_paper |
| Experimental models: Cell lines | ||
| Mouse: E1A;HRasG12V p53WT MEFs | Laboratory of Dr. Laura Attardi | N/A |
| Mouse: E1A;HRasG12V p53−/− MEFs | Laboratory of Dr. Laura Attardi | N/A |
| Mouse: E1A;HRasG12V p53WT MEFs shLuc | This paper | N/A |
| Mouse: E1A;HRasG12V p53WT MEFs shMettl3-1 | This paper | N/A |
| Mouse: E1A;HRasG12V p53WT MEFs shMettl3-2 | This paper | N/A |
| Mouse: E1A;HRasG12V p53−/− MEFs shLuc | This paper | N/A |
| Mouse: E1A;HRasG12V p53−/− MEFs shMettl3-1 | This paper | N/A |
| Mouse: E1A;HRasG12V p53−/− MEFs shMettl3-2 | This paper | N/A |
| Mouse: Flp-In™-3T3 | Thermo Fisher | R76107 |
| Mouse: Flp-In-3T3 p53-LAP | This paper | N/A |
| Mouse: Flp-In-3T3 p53−/− | This paper | N/A |
| Mouse: Embryonic Stem cells Wild-type | Laboratory of Dr. Howard Y. Chang | N/A |
| Mouse: Embryonic Stem cells Mettl3−/− | Laboratory of Dr. Howard Y. Chang | N/A |
| Human: A549 lung cancer cell line: TP53WT | Stanford University School of Medicine laboratories | N/A |
| Human: A549 lung cancer cell line: TP53−/− | This manuscript | N/A |
| Human: H1299 lung cancer cell line | Stanford University School of Medicine laboratories | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: ICR-Scid: IcrTac:ICR-Prkdcscid | Taconic Biosciences | ICRSC-M |
| Mouse: KrasLSL-G12D mice: B6.129S4-Krastm4Tyj/J | Laboratory of Dr. Tyler Jacks | N/A |
| Mouse: Rosa26LSL-tdTomato | Madisen et al. (2010) | N/A |
| Mouse: H11LSL-Cas9: B6;129-Igs2tm1(CAG-cas9+)Mmw/J | Chiou et al. (2015); Laboratory of Dr. Monte Winslow | 026816 |
| Mouse: Mettl14fl/fl | Wang et al. (2018); Stanford University School of Medicine laboratories | N/A |
| Oligonucleotides | ||
| See Table S4 for primers | This paper | N/A |
| Recombinant DNA | ||
| pENTR p53 | This paper | N/A |
| pG-LAP7/puro destination vector | Laboratory of Dr. Peter Jackson | N/A |
| pG-LAP7/puro p53 (p53-LAP) | This paper | N/A |
| pENTR Mettl3 | This paper | N/A |
| pG-LAP2 Mettl3WT (FLAG-METTL3 WT) | This paper | N/A |
| pG-LAP2 Mettl3APPA (FLAG-METTL3 APPA) | This paper | N/A |
| pCS2-HA destination vector | Laboratory of Dr. Peter Jackson | N/A |
| pCS2-HA-Mettl3 (HA-METTL3) | This paper | N/A |
| pcDNA3.1–3XHA-p53 | Laboratory of Dr. Laura Attardi | N/A |
| pcDNA3.1–3XHA-p53 Δ1–42 | This paper | N/A |
| pcDNA3.1–3XHA-p53 Δ1–61 | This paper | N/A |
| pX330 p53 | Addgene | 59910 |
| shRNA vectors with shRNAs targeting Luciferase, Mettl3, METTL3 | Laboratory of Dr. Pedro Batista | N/A |
| Lenti-U6-sgMettl3 | This paper | N/A |
| Lenti-U6-sgNeoR1 | Laboratory of Dr. Monte Winslow | N/A |
| pX458 TP53 series | This paper | N/A |
| Software and algorithms | ||
| Enrichr | Chen et al. (2013), Kuleshov et al. (2016) | https://maayanlab.cloud/Enrichr/ |
| Metascape | Zhou et al. (2019) | http://metascape.org/gp/index.html#/main/step1 |
| Byonic | Protein Metrics | https://proteinmetrics.com/resources/byonic-advanced-peptide-and-protein-identification-software/ |
| Image Lab | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
| NDPview 2.0 | Hamamatsu | https://www.hamamatsu.com/us/en/product/type/U12388-01/index.html |
| QPath | Univ of Edinburgh | https://qupath.github.io/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Umi_tools (v0.5.1) | Smith et al. (2017) | https://github.com/CGATOxford/UMI-tools |
| Cutadapt (v2.7) | Martin (2011) | https://github.com/marcelm/cutadapt |
| STAR (v2.6.0c) | Dobin et al. (2013) | https://github.com/alexdobin/STAR |
| CLIPper | Lovci et al. (2013) | https://github.com/YeoLab/clipper |
| GENCODE (release M21) | Frankish et al. (2019) | https://www.gencodegenes.org/human/ |
| HOMER (v3.1) | Heinz et al. (2010) | http://homer.ucsd.edu/homer/motif/motifDatabase.html |
| limma (v3.46) | Ritchie et al. (2015) | https://bioconductor.org/packages/release/bioc/html/limma.html |
| genefu (v2.22.1) | Gendoo et al. (2016) | http://bioconductor.org/packages/release/bioc/html/genefu.html |
| DISCOVER package | Canisius et al. (2016) | https://ccb.nki.nl/software/discover/ |
| DEseq2 | Love et al. (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Fastp | Chen et al. (2018) | https://github.com/OpenGene/fastp |
| Featurecounts (1.5.3) | Liao et al. (2014) | https://rnnh.github.io/bioinfo-notebook/docs/featureCounts.html |